

Abstract
Common variable immunodeficiency disorders (CVID), the most frequent cause of symptomatic primary immunodeficiency, are defined by impaired antibody production. Notwithstanding, T cell activation and granulomatous manifestations represent the main causes of CVID morbidity even in patients receiving immunoglobulin (Ig) G replacement therapy. Additionally, gut pathology is a frequent feature of CVID. In this study, we investigated monocyte imbalances and their possible relationship with increased microbial translocation in CVID patients. Monocyte subsets were defined according to CD14 and CD16 expression levels and evaluated in terms of human leucocyte antigen D-related (HLA-DR), CD86 and programmed death-1 molecule ligand 1 (PD-L1) expression by flow cytometry, in parallel with the quantification of plasma lipopolysaccharide (LPS) and serum levels of soluble CD14 (sCD14), LPS-binding protein (LBP) and anti-LPS antibodies. CVID patients (n = 31) featured significantly increased levels of serum sCD14 and an expansion of CD14brightCD16+ monocytes in direct correlation with T cell and B cell activation, the latter illustrated by the frequency of the CD21lowCD38low subset. Such alterations were not observed in patients lacking B cells due to congenital agammaglobulinaemia (n = 4). Moreover, we found no significant increase in circulating LPS or LBP levels in CVID patients, together with a relative preservation of serum anti-LPS antibodies, in agreement with their presence in commercial IgG preparations. In conclusion, CVID was associated with monocyte imbalances that correlated directly with T cell activation markers and with B cell imbalances, without an association with plasma LPS levels. The heightened monocyte activated state observed in CVID may represent an important target for complementary therapeutic strategies.
Keywords: common variable immunodeficiency, CVID, monocyte activation, plasma LPS, T cell activation
Introduction
Common variable immunodeficiency disorders (CVID) represent the most frequent cause of symptomatic primary immunodeficiency [1]. CVID are defined by quantitative and qualitative reduction in antibody production due to heterogeneous defects in mature B cells [1,2]. Besides imbalances in B cell subsets, CVID are associated frequently with persistent T cell activation and loss of naive T cells, which are reported even in patients receiving replacement therapy with immunoglobulin (Ig) G [1–4]. Indeed, non-infectious complications, such as lymphoproliferation and granulomatous disease, are currently the main causes of morbidity and mortality in CVID [1,2]. The mechanisms underlying the chronic immune activation associated with CVID remain largely unclear. Another main cause of CVID morbidity is gastrointestinal pathology, with or without malabsorption [1,2]. The impairment in IgA production and other CVID-associated mucosal alterations have been shown to be often associated with increased intestinal permeability [5]. Thus, it is plausible that increased levels of microbial translocation, particularly of bacterial products such as lipopolysaccharide (LPS), with consequent monocyte stimulation, may contribute to the chronic immune activation observed in CVID patients, as reported for human immunodeficiency virus (HIV-1)-infected individuals [6].
Monocytes are important orchestrators of the immune system, linking innate and adaptive immune responses. Blood monocytes have the ability to migrate into tissues, where they can differentiate and give rise to distinct functional cell types, such as macrophages and dendritic cells (DCs) [7]. They have long been recognized as the main promoters of inflammatory responses to pathogens and to be involved in many inflammatory diseases [7]. Recent studies have called attention to their suppressive and regulatory functions, suggesting that monocytes play a central role in the modulation of immune responses, particularly in the context of chronic immune activation [8]. These data point to the heterogeneity of monocyte subsets, with distinct functions and phenotypes [9–11]. Recently, an effort was made in putting forward a consensus nomenclature that proposed three main monocyte subsets defined according to the expression levels of CD14 and CD16, as follows: classical CD14brightCD16- monocytes, the most abundant monocyte population found in peripheral blood; intermediate CD14brightCD16+; and non-classical CD14dimCD16+ subsets [12]. The intermediate CD14brightCD16+ monocyte subset has been shown consistently to expand in many proinflammatory clinical settings [12,13], and particularly in association with HIV infection [14,15].
CVID has been associated with defective in vitro maturation of DCs from monocytes, at least in a subset of patients [16,17], and with disturbances in the monocyte responses upon LPS stimulation in vitro[18]. Additionally, CVID patients were shown to have reduced numbers of circulating DCs [19,20], with perturbed differentiation and function, namely reduced expression levels of the co-stimulatory molecules CD86 and CD80, an impaired ability to produce interleukin (IL)-12 upon stimulation [21–23], as well as lower antigen-presenting capacity in mixed lymphocyte reactions [22,23].
Monocyte-related alterations, namely an increased frequency of CD16-positive monocytes [24] and decreased numbers of myeloid DCs [25], have also been reported in patients lacking B cells due to blockade in early B cell development in the bone marrow associated with genetic defects in Bruton's tyrosine kinase (Btk) leading to congenital agammaglobulinaemia [26]. Nevertheless, we and others have shown that, in contrast to CVID, congenital agammaglobulinaemia is not associated with a significant increase in T cell activation markers [3,18,27].
In this study, we investigated the monocyte compartment in patients with CVID and provide evidence that CVID was associated with increased markers of monocyte activation in direct correlation with the expansion of activated T cell subsets, irrespective of plasma LPS levels. Monocytes may thus be important contributors to the inflammatory milieu that leads to T cell activation, lymphoproliferative manifestations and granuloma formation associated with CVID [1,28].
Material and methods
Cohort characterization
The study involved 31 patients with CVID, diagnosed according to the European Society for Immunodeficiencies criteria (http://www.esid.org), namely IgG and IgA and/or IgM levels at least two standard deviations below the mean for age, impaired antibody response to vaccines, absent/low isohaemagglutinins and exclusion of defined causes of hypogammaglobulinaemia. Patients with congenital agammaglobulinaemia, presenting less than 1% of B cells within total peripheral lymphocytes (n = 4), were also included. These cohorts have been described previously [3]. The clinical and epidemiological characterization of these cohorts is summarized in Table 1. Twenty-nine CVID and all congenital agammaglobulinaemia patients were receiving IgG replacement therapy, adjusted to maintain preinfusion Ig levels above 650 mg/dl. The two CVID patients not receiving IgG featured levels of total serum IgG of 227 and 473 mg/dl. All patients were free from symptomatic infections at the time of collection of the blood samples, which was always performed immediately before the immunoglobulin infusions in the patients receiving intravenous administration. Four CVID patients were receiving steroid therapy at the time of the study. Fifteen healthy individuals were studied in parallel. All subjects gave written informed consent for blood sampling and processing. The study was approved by the Ethical Boards of the Faculdade de Medicina da Universidade de Lisboa and of the Hospital de Santa Maria, and performed in accordance with the 1964 Declaration of Helsinki and its later amendments.
Table 1.
Clinical and epidemiological data of the studied cohorts
| Healthy | CVID | Congenital agammaglobulinaemia† | |
|---|---|---|---|
| Number (male/female) | 15 (5/10) | 31 (11/20) | 4 (4/0) |
| Age (years) | 39 ± 11 | 40 ± 13 | 26 ± 5 |
| Clinical manifestationsठ| |||
| Autoimmune disease | n.a. | 17/31 (55%) | 0 |
| Adenopathies | n.a. | 10/31 (32%) | 0 |
| Lymphoid proliferation | n.a. | 18/20¶ (86%) | 0 |
| Granulomas | n.a. | 3/20¶ (15%) | 0 |
| Chronic diarrhoea | n.a. | 15/31 (48%) | 0 |
| Splenomegaly | n.a. | 16/31 (52%) | 0 |
| EUROclass classification | |||
| smB+21norm | n.a. | 5/29 (17%) | n.a. |
| smB+21lo | n.a. | 9/29 (31%) | n.a. |
| smB-21norm | n.a. | 6/29 (21%) | n.a. |
| smB-21lo | n.a. | 9/29 (31%) | n.a. |
| smB-Trnorm | n.a. | 9/29 (31%) | n.a. |
| smB-Trhi | n.a. | 6/29 (21%) | n.a. |
| IgG replacement therapy§ | |||
| Intravenous | n.a. | 24/31 (77%) | 4 (100%) |
| Subcutaneous | n.a. | 5/31 (16%) | 0 |
| Length of IgG therapy (years) | n.a. | 7 ± 6 | 16 ± 11 |
Two of the congenital agammaglobulinaemia patients had a known genetic defect in the Bruton's tyrosine kinase (Btk) gene; namely, one had the IVS17-1G→C mutation and the other presented with the R288Q mutation; in the other two patients, mutations in the Btk gene have been excluded and evaluation of autosomal recessive forms is ongoing.
Diagnostic criteria: autoimmune disease – clinical data, given the impairment in antibody production; bronchiectasis – computed tomography; splenomegaly – longitudinal spleen diameter superior to 15 cm (computed tomography or ultrasonography); adenopathies – lymph node larger than 1 cm diameter in two or more lymphatic chains in clinical and/or imaging exams; lymphoid proliferation and granulomas – diffuse lymphocytic infiltrates or granulomas on gastrointestinal, lymph node or pulmonary biopsies.
Percentage within total cohort evaluated in brackets.
Total number of individuals with biopsies. CVID: common variable immunodeficiency; n.a., not applicable; smB: switched-memory B cells; Tr: transitional B cells.
Cell staining and flow cytometric analysis
Phenotypic analysis was performed using whole blood samples collected immediately before IgG administration. After staining with monoclonal antibodies and red blood cells lysis using BD fluorescence activated cell sorter (FACS) lysing solution (BD Biosciences, San Jose, CA, USA), samples were acquired on a FACSCalibur flow cytometer (BD Biosciences). The following anti-human monoclonal antibodies were used, with the clone and the respective directly conjugated fluorochrome specified in brackets: CD16 [3G8; fluorescein isothiocyanate (FITC)], CD3 [SK7; peridinin chlorophyll (PerCP)], CD4 (SK3; PerCP), CD8 (SK1; PerCP), CD8 [RPA-T8; allophycocyanin (APC)], CD38 [HB7; phycoerythrin (PE)], CD86 (FUN-1; PE), IgD (IA6-2; PE), IgM (G20-127; APC), human leucocyte antigen D-related (HLA-DR) (L243; FITC and PerCP), interferon (IFN)-γ (4S.B3; FITC), from BD Biosciences; CD4 (RPA-T4, FITC and PerCP-Cy5·5), CD8 (RPA-T8; FITC and PE), CD14 (61D3; PE-Cy7 and APC), CD19 (HIB19; PerCP-Cy5·5 and PE-Cy7), CD27 (O323; FITC, PE and APC), CD45RA (HI100; FITC and APC), PD-L1 (MIH1; APC) and tumour necrosis factor (TNF)-α (MAb11; PE), from eBiosciences; and CD21 (BL13; FITC) from IO Test (Beckman Coulter, Brea, CA, USA). Data were analysed using CellQuest (BD Biosciences) and FlowJo (Tree Star Inc., Ashland, OR, USA) softwares. Lymphocyte and monocyte subsets were defined within manually set lymphogate/monogate. Results were expressed as percentage of cells that stained positive for a given marker, or as its mean fluorescence intensity (MFI) within the defined population. Total cell numbers were calculated by multiplying the percentage of each population within total lymphocytes/monocytes by the peripheral blood lymphocyte/monocyte count obtained at the clinical laboratory on the day of sampling.
Analysis of cytokine production
Cytokine production was assessed at the single-cell level, as described previously [3]. Briefly, freshly isolated peripheral blood mononuclear cells cultured in the presence of Brefeldin A (10 µg/ml; Sigma-Aldrich, St Louis, MO, USA) for 4 h at 37°C with 5% CO2, were stimulated with LPS (100 ng/ml; Sigma-Aldrich) for assessing TNF-α production by monocytes or with phorbol myristate acetate (50 ng/ml; Sigma-Aldrich) and ionomycin (500 ng/ml; Calbiochem, Merck Biosciences, Nottingham, UK) for assessing the lymphocyte production of IFN-γ.
Quantification of soluble CD14 (sCD14), LPS-binding protein (LBP), LPS and IgG endotoxin core antibodies (EndoCAb)
The human EndoCAb® assay kit and the enzyme-linked immunosorbent assay (ELISA) kit for Endoblock LBP (HyCult Biotechnology, Uden, the Netherlands) were used to quantify the serum concentrations of IgG EndoCAb and LBP, respectively. Serum sCD14 levels were quantified by ELISA using the human sCD14 Quantikine (R&D Systems, Minneapolis, MN, USA). Plasma LPS levels were quantified using the limulus amoebocyte assay (Cambrex, East Rutherford, NJ, USA); plasma samples were diluted and pretreated as described previously [6]. All samples were assayed in duplicate, according to the manufacturer's instructions.
Statistical analysis
Statistical analyses were performed using GraphPad Prism 5·0 (GraphPad Software, La Jolla, CA, USA). Two group comparisons were performed using the Mann–Whitney U-test. Spearman's coefficient was used to determine the significance of the correlation between two variables. Results are expressed as mean ± standard error of the mean (s.e.m.), and P-values <0·05 were considered significant.
Results
CVID was associated with monocyte activation
The monocyte compartment was investigated in a previously described cohort of 31 patients with CVID (Table 1) [3].
CVID patients showed significantly higher levels of sCD14 than healthy subjects (Fig. 1a), suggesting significant levels of monocyte activation [6,29].
Fig. 1.
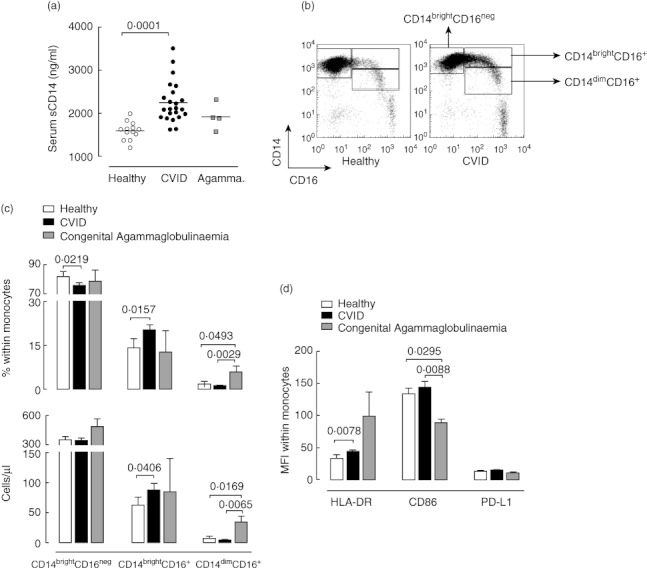
Monocyte activation markers in common variable immunodeficiency (CVID) and congenital agammaglobulinaemia. (a) Serum levels of soluble CD14 (sCD14). Each dot represents one individual, with bars indicating mean values. Open circles refer to healthy individuals, solid circles to CVID patients, and grey squares to congenital agammaglobulinaemia (Agamma) patients. (b) Illustrative flow cytometric analysis of monocyte subsets according to CD14 and CD16 expression levels in representative CVID (right panel) and healthy (left panel) individuals. (c) Frequency and absolute counts of monocyte subsets as defined in (b). (d) Mean fluorescence intensity (MFI) of human leucocyte antigen D-related (HLA-DR), CD86 and programmed death-1 molecule ligand 1 (PD-L1) within total monocytes. Healthy individuals are represented in open bars, CVID patients in solid bars and congenital agammaglobulinaemia patients in grey bars. Bars indicate mean ± standard error of the mean. P-values of statistically significant differences are shown.
Monocyte subsets were defined by flow cytometric whole blood analysis through the differential expression of CD14 and CD16, as illustrated in Fig. 1b. Although the number of total circulating monocytes in CVID patients was similar to age-matched healthy individuals (493·3 ± 35·81 cells/µl in controls versus 504·7 ± 40·25 cells/µl in CVID patients; P = 0·9810), we found a significant increase in both the frequency and absolute numbers of CD14brightCD16+ monocytes in CVID patients in comparison with controls (Fig. 1c), a subset associated with inflammatory settings [11–15,30].
In agreement with a state of monocyte activation in CVID patients, the levels of expression of HLA-DR within total monocytes were significantly higher than in controls (Fig. 1d), due mainly to increased levels of HLA-DR within the expanded CD14brightCD16+ monocyte subset (MFI in healthy individuals 133·0 ± 11·34 versus 187·2 ± 13·28 in CVID patients; P = 0·0052). Conversely, no significant alterations in the expression levels of the co-stimulatory molecule CD86 were found in CVID patients (Fig. 1d).
We found no increase in the expression levels of PD-L1 on monocytes in CVID patients compared to healthy subjects (Fig. 1d), suggesting that this inhibitory pathway was probably not induced significantly in CVID, despite the evidence for immune activation.
We also investigated the monocyte compartment in a group of patients we have described previously [3] with congenital agammaglobulinaemia (Table [1]) and who lack circulating B cells due to defects in early B cell development, and found no significant increase in serum sCD14 or in the frequency of CD14brightCD16+ monocytes in these patients (Fig. 1a,c).
Btk deficiency, a main cause of congenital agammaglobulinaemia, has been shown to be associated with an increased frequency of CD16-expressing monocytes [24], but no distinction was made at that time between CD14brightCD16+ and CD14dimCD16+ subsets, which are believed to possess distinct functional properties [30]. We found that the increase in CD16 was restricted to CD14dimCD16+ monocytes (Fig. 1c). This subset was expanded significantly in patients with congenital agammaglobulinaemia, both in terms of frequency and absolute counts, in relation to both healthy individuals and CVID patients (Fig. 1c).
Although the results should be interpreted cautiously, given the small number of patients with congenital agammaglobulinaemia evaluated, our data showed that, in contrast to CVID, there was no expansion of the CD14brightCD16+ subset and no increase in sCD14 levels.
Overall, we provided evidence for significant levels of monocyte activation being a feature of CVID.
Monocyte activation was unrelated to plasma LPS levels in CVID
Our findings of no up-regulation of markers of monocyte activation in patients lacking B cells raise the possibility that factors other than defective mucosal IgA production, and the possible related increase in microbial translocation, are contributing to the CVID-associated monocyte activation.
CVID patients showed low to undetectable levels of plasma LPS, a bacterial product that increases in the peripheral blood as a result of microbial translocation in the gut (Fig. 2a). Moreover, CVID patients with gastrointestinal manifestations, such as chronic diarrhoea or evidence of malabsorption, did not feature higher levels of plasma LPS than healthy controls (Fig. 2a).
Fig. 2.
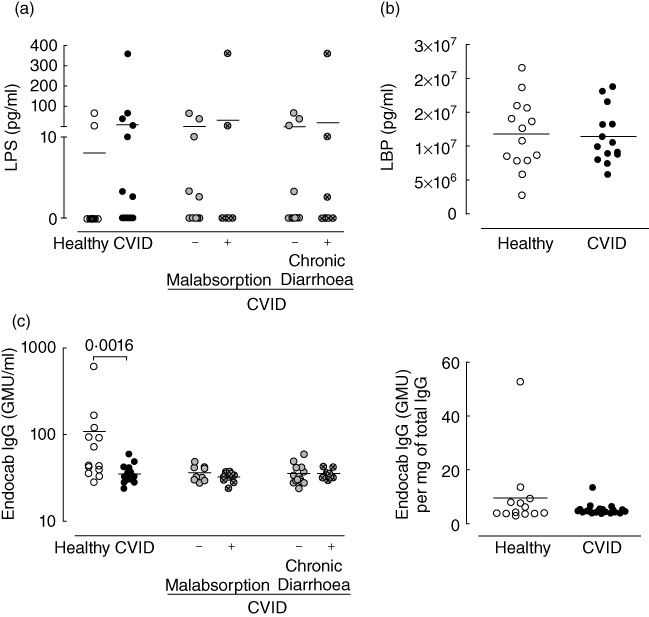
Plasma lipopolysaccharide (LPS) levels and related molecules in common variable immunodeficiency (CVID). (a) Plasma LPS levels. (b) Serum LPS-binding protein (LBP) levels. (c) Anti-LPS antibodies (left) and its ratio relative to total immunoglobulin (Ig) G (right). Each dot represents one individual: healthy individuals are represented in open circles, CVID patients in solid circles. Bars indicate mean. P-values of statistically significant differences are shown.
We also confirmed that monocytes from CVID patients were not impaired in their ability to produce TNF-α upon in vitro LPS stimulation (percentage of monocytes expressing TNF-α: 55·75 ± 3·56 in 26 CVID patients and 65·96 ± 4·24 in eight healthy controls; P = 0·2820), as reported previously [18]. These data, showing that monocytes from CVID patients were not refractory to additional LPS stimulation in vitro, further suggest a lack of previous exposure to increased plasma LPS.
We measured the levels of LPS-binding protein (LBP), a molecule that binds circulating LPS [29], and found them to be similar in healthy individuals and CVID patients (Fig. 2b), further suggesting that the degree of microbial translocation was not substantial.
Another important pathway contributing to LPS clearance from circulation involves antibodies against the LPS core oligosaccharide (EndoCAb) [6]. It is possible that IgG replacement therapy provides adequate amounts of EndoCAb levels, restoring the humoral response to LPS that patients are probably unable to mount.
We measured EndoCAb IgG levels in five IgG lots administered to the patients (two lots of intravenous Octagam® 5%, two lots of subcutaneous Vivaglobin® 16% and one lot of subcutaneous Gammanorm® 16·5%) and found these antibodies to be present in significant amounts [values between 120 GMU (IgG median units)/ml and 250 GMU/ml]. Additionally, the amount of EndoCAb IgG within total IgG was similar in the serum of CVID patients and healthy individuals, even though total serum IgG was significantly lower in CVID patients than in controls (Fig. 2c). All patients assessed were receiving IgG replacement therapy, with the exception of one, who still presented detectable amounts of EndoCAb IgG (29·78 GMU/ml; total IgG 473 mg/dl and EndoCAb IgG within total IgG 6·13 GMU/mg).
Thus, given the reduced plasma LPS and the unaltered serum LBP levels that we documented in CVID patients, IgG replacement therapy might be sufficient to limit the levels of LPS in peripheral blood. These results are against a major role of the gut-associated microbial translocation to the documented monocyte activation.
In summary, the alterations observed in the monocyte populations from CVID patients seemed to be unrelated to plasma LPS levels.
Monocyte activation was associated directly with T cell disturbances in CVID patients
We reported above that, in contrast to CVID, patients with congenital agammaglobulinaemia showed no increase in markers associated with monocyte activation. Our results are particularly interesting, as we had reported previously the lack of T cell activation and significant T cell imbalances in these patients with congenital agammaglobulinaemia [3].
Accordingly, in CVID patients the expansion of CD14brightCD16+ monocytes was associated directly with T cell activation, and correlated inversely with the frequency of naive CD4 T cells (Fig. 3). The expansion of this population in CVID patients was also associated with the frequency of IFN-γ-producing CD4 T cells (Fig. 3), another measure of T cell activation that we have shown previously to be increased significantly in this CVID cohort in comparison to healthy individuals [3].
Fig. 3.
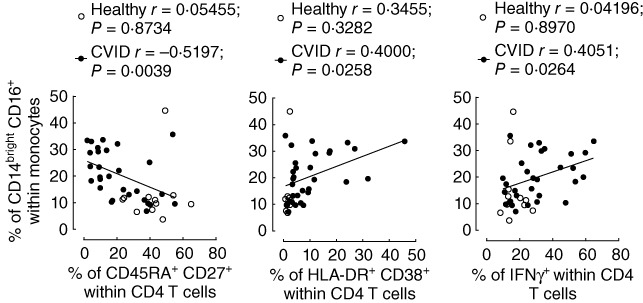
Relationship between markers of monocyte and T cell activation in common variable immunodeficiency (CVID). Correlation between the frequency of CD14brightCD16+ monocytes and the frequency of naive (CD45RA+CD27+, left panel), activated human leucocyte antigen D-related (HLA-DR)+CD38+ (middle panel) or interferon (IFN)-γ-producing cells (right panel) within CD4 T cells in CVID patients (solid circles) and in healthy individuals (open circles). Each dot represents one individual. Spearman's correlation coefficients are shown.
Importantly, as described above, we found no significant up-regulation of the inhibitory molecule PD-L1 in CVID patients compared to healthy subjects, both within whole monocytes (Fig. 1d) and in the three monocyte subsets (data not shown). We further investigated a possible association between PD-L1 levels in monocytes and T cell imbalances and found that PD-L1 levels within the expanded CD14brightCD16+ monocyte subset increased in direct correlation with CD4 T cell activation (r = 0·6056, P = 0·0003) and the loss of naive CD4 T cells (r = −0·5202, P = 0·0038) in CVID patients. Nevertheless, PD-L1 up-regulation in the CD14brightCD16+ monocyte subset of CVID patients was apparently not as marked as that observed in other persistent inflammatory settings, such as HIV-1 infection [31–33], suggesting an impairment of the PD-L1 pathway in CVID, possibly translating into a reduced ability to control immune activation. This is particularly relevant given the fact that the PD-1/PD-L1 pathway is involved in peripheral tolerance and autoimmunity [34] and that the expansion of CD14brightCD16+ monocytes was significantly higher in CVID patients with autoimmune disease (P = 0·0328). Of note, this expansion was not more marked in CVID patients with granuloma or lymphoproliferation (P > 0·05). Additionally, significant correlations were observed between the expression levels of the co-stimulatory molecule CD86 within CD14brightCD16+ monocytes and T cell imbalances (r = 0·3706, P = 0·0402 with CD4 T cell activation; r = −0·4300, P = 0·0199 with the frequency of naive CD4 T cells) in CVID patients, reinforcing the relevance of addressing the regulation of co-stimulatory and inhibitory pathways in future CVID studies.
Altogether, our data showed that monocyte activation in CVID was associated directly with markers of T cell activation.
CVID patients grouped according to the EuroClass classification featured distinct monocyte imbalances
B cell imbalances have been used as an attempt to classify CVID patients into more homogeneous subgroups. The EuroClass classification has thus been proposed based in three main B cell abnormalities observed in CVID: the reduction in switched-memory B cells and the expansions of either transitional or CD21lowCD38low B cells [35]. When our cohort was stratified according to the EuroClass, we found no differences regarding CD86 expression within CD14brightCD16+ monocytes among the different subgroups (P > 0·1900). However, PD-L1 expression within this subset was significantly higher in patients with reduced switched-memory B cells and expansion of transitional B cells (smB-Trhi) compared to those who did not present that expansion (smB-Trnorm) (Fig. 4a). In addition, we found that the expansion of CD14brightCD16+ monocytes was particularly relevant in patients who did not have reduced switched-memory B cells but had expanded CD21lowCD38low B cells (smB+21lo) compared to patients who did not (smB+21norm) (Fig. 4b). In agreement, the frequency of CD14brightCD16+ monocytes was associated directly with the frequency of CD21lowCD38low B cells in CVID patients (Fig. 4c).
Fig. 4.
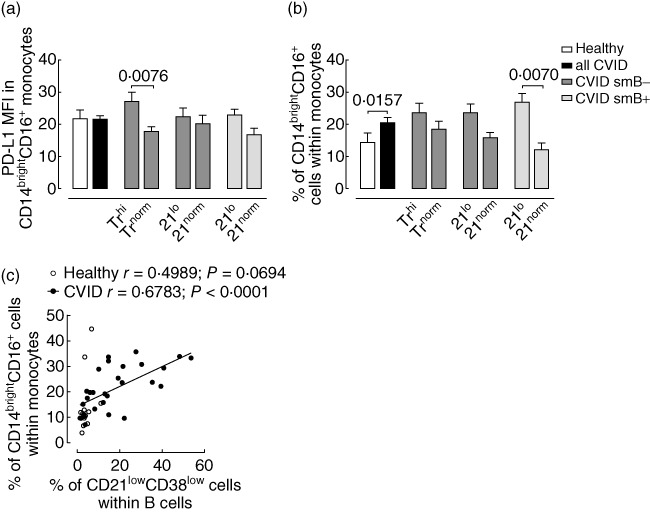
Markers of monocyte activation in common variable immunodeficiency (CVID) grouped according to the EuroClass classification. (a) Mean fluorescence intensity (MFI) of programmed death-1 molecule ligand 1 (PD-L1) within CD14brightCD16+ monocytes. Healthy individuals are represented in open bars and CVID patients in solid bars. (b) Frequency of CD14brightCD16+ monocytes. Bars indicate mean ± standard error of the mean. P-values of statistically significant differences are shown. (c) Correlation between the frequency of CD14brightCD16+ monocytes and the frequency of CD21lowCD38low B cells in CVID patients (solid circles) and in healthy individuals (open circles). Each dot represents one individual. Spearman's correlation coefficients are shown.
In conclusion, we showed that monocyte imbalances were particularly marked in CVID patients lacking switched-memory B cells and/or having an abnormal expansion of the CD21lowCD38low B cells.
Discussion
In this study we have shown that CVID is associated with increased serum levels of sCD14 and an expansion of CD14brightCD16+ monocytes, in direct correlation with T cell and B cell imbalances. Moreover, monocyte hyperactivation occurred irrespective of plasma LPS levels, suggesting that monocytes may represent important targets for therapies complementary to IgG replacement aiming to control the granulomatous and lymphoproliferative manifestations in CVID.
Such monocyte activation markers were not observed in patients lacking peripheral B cells due to congenital agammaglobulinaemia who, notably, also did not present major T cell imbalances. Although the number of patients with congenital agammaglobulinaemia was small, our results are in agreement with a previous study that also reported a lack of major alterations in monocyte function as assessed in vitro in patients with X-linked agammaglobulinaemia [18].
CD14 shedding from the membrane of monocytes is generally accepted as a marker of monocyte activation, with levels of sCD14 being increased in inflammatory and autoimmune diseases in the context of acute inflammation and sepsis, as well as in HIV-infected patients [6,29]. CD14brightCD16+ monocytes have been shown to have a high inflammatory potential, based on transcriptome analysis [11,30], and to be expanded in inflammatory conditions [12,13], being associated with disease progression in chronic HIV-1 infection [14,15].
We showed that the expression of HLA-DR within total monocytes was significantly higher in CVID than in controls, compatible with a state of monocyte activation. This is in agreement with the study by Cambronero et al. which showed an up-regulation of HLA-DR within the whole monocyte population in CVID patients [18]. It is worth noting that this increase was due mainly to the expression of HLA-DR within the expanded CD14brightCD16+ monocyte subset, supporting its enhanced inflammatory capacity. Conversely, expression levels of the co-stimulatory molecule CD86 were similar in CVID patients and in healthy individuals. CD86, a marker of monocyte differentiation and acquisition of antigen-presentation properties, has been shown to be expressed at reduced levels in monocyte-derived DCs generated in vitro, supporting an impaired function as antigen-presenting cells in CVID [16,36].
Monocytes may also up-regulate inhibitory molecules, which are thought to play an important role in the regulation of immune responses and in limiting immunopathology. PD-L1 (B7-H1) binds programmed death-1 molecule (PD-1), an inhibitory receptor expressed on many cells, particularly activated T and B lymphocytes, establishing a pathway of central relevance in inflammatory states [31,32]. Both PD-L1 and PD-1 can be up-regulated upon activation, with PD-1 expression on T cells having been associated with functional exhaustion in the context of chronic viral infection, due to persistent antigen exposure [37–39]. Of note, PD-L1 expression has been shown to be up-regulated on monocytes from HIV-infected patients [31,32]. We found that monocyte activation in CVID was not accompanied by significant up-regulation of the inhibitory molecule PD-L1, which may favour the deleterious impact of activated monocytes.
In addition, we showed that monocyte imbalances were unrelated to plasma LPS levels in CVID patients, who appear to have a relatively good preservation of the pathways involved in LPS clearance, including anti-LPS antibodies provided by the IgG replacement therapy, and normal levels of LBP. Serum LBP quickly binds LPS molecules in circulation [29]. LBP levels have been shown to increase in sepsis associated with LPS-containing microorganisms (Gram-negative bacteria) and in HIV infection [6,29], in which case a direct correlation with the degree of microbial translocation, estimated by plasma LPS levels, has been reported [15]. Furthermore, both LPS and LBP levels are increased in inflammatory bowel disease, in correlation with disease activity, and recover to normal levels following treatment [40]. The fact that LBP levels were similar in healthy individuals and CVID patients, and that there was no increase in plasma LPS, further suggests that the degree of microbial translocation was not significant. Thus, our data raise the possibility that IgG replacement therapy may provide sufficient antibodies against LPS to control the putative increase in microbial translocation. Longitudinal studies assessing patients prior to and after commencing replacement therapy with IgG will be of great relevance to understand its potential role in regulating microbial translocation, which may have implications in other clinical settings, such as HIV/acquired immune deficiency syndrome (AIDS).
Additionally, we show that the expansion of CD14brightCD16+ monocytes was particularly relevant in CVID patients with autoimmunity and that the frequency of this monocyte population was correlated directly with the frequency of CD21lowCD38low B cells. The expansion of this B cell subset has also been associated with autoimmunity in CVID [35], supporting a role for CD14brightCD16+ monocytes in the course of the disease. We should point out that our CVID cohort presents an unusually high frequency of autoimmune manifestations, probably related to a reference bias associated with an immunology department in a central hospital.
Although several studies have acknowledged the relevance of immune activation in CVID pathophysiology, little is known about what drives and sustains it, and whether it is related to the increased frequency of infections or to altered response to pathogens. Our data are in agreement with a possible multi-factorial event impacting on several compartments of the immune system. Irrespective of the monocyte alterations that we describe contributing to the inflammatory process itself or being a marker of underlying inflammation, our finding of major monocyte imbalances in particular subgroups of CVID patients may help in defining new lines of investigation.
In conclusion, our data show that CVID was associated with monocyte alterations that correlated directly with T cell activation markers and with B cell imbalances, without a relationship to plasma LPS levels, supporting a potential clinical relevance of therapeutic strategies targeting monocytes to control the inflammatory manifestations in CVID patients.
Acknowledgments
This work was supported by grants from ‘Fundação para a Ciência e a Tecnologia’ (FCT) and by ‘Programa Operacional Ciência e Inovação 2010’ (POCI2010) to R.M.M.V. and A.E.S. R.R.B. received a scholarship from FCT and SPS from AstraZeneca. The authors acknowledge Octapharma for kindly providing the EndoCAb® Assay Kits.
The authors acknowledge Russell Foxall and Íris Caramalho for helpful discussion and critical reading of the manuscript.
Disclosure
The authors declare that they have no conflict of interest.
References
- 1.Resnick ES, Moshier EL, Godbold JH, Cunningham-Rundles C. Morbidity and mortality in common variable immune deficiency over 4 decades. Blood. 2012;119:1650–7. doi: 10.1182/blood-2011-09-377945. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2.Park MA, Li JT, Hagan JB, Maddox DE, Abraham RS. Common variable immunodeficiency: a new look at an old disease. Lancet. 2008;372:489–502. doi: 10.1016/S0140-6736(08)61199-X. [DOI] [PubMed] [Google Scholar]
- 3.Barbosa RR, Silva SP, Silva SL, et al. Primary B-cell deficiencies reveal a link between human IL-17-producing CD4 T-cell homeostasis and B-cell differentiation. PLoS ONE. 2011;6:e22848. doi: 10.1371/journal.pone.0022848. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Giovannetti A, Pierdominici M, Mazzetta F, et al. Unravelling the complexity of T cell abnormalities in common variable immunodeficiency. J Immunol. 2007;178:3932–43. doi: 10.4049/jimmunol.178.6.3932. [DOI] [PubMed] [Google Scholar]
- 5.Malamut G, Verkarre V, Suarez F, et al. The enteropathy associated with common variable immunodeficiency: the delineated frontiers with celiac disease. Am J Gastroenterol. 2010;105:2262–75. doi: 10.1038/ajg.2010.214. [DOI] [PubMed] [Google Scholar]
- 6.Brenchley JM, Price DA, Schacker TW, et al. Microbial translocation is a cause of systemic immune activation in chronic HIV infection. Nat Med. 2006;12:1365–71. doi: 10.1038/nm1511. [DOI] [PubMed] [Google Scholar]
- 7.Auffray C, Sieweke MH, Geissmann F. Blood monocytes: development, heterogeneity, and relationship with dendritic cells. Annu Rev Immunol. 2009;27:669–92. doi: 10.1146/annurev.immunol.021908.132557. [DOI] [PubMed] [Google Scholar]
- 8.Said EA, Dupuy FP, Trautmann L, et al. Programmed death-1-induced interleukin-10 production by monocytes impairs CD4+ T cell activation during HIV infection. Nat Med. 2010;16:452–9. doi: 10.1038/nm.2106. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Passlick B, Flieger D, Ziegler-Heitbrock HW. Identification and characterization of a novel monocyte subpopulation in human peripheral blood. Blood. 1989;74:2527–34. [PubMed] [Google Scholar]
- 10.Ancuta P, Rao R, Moses A, et al. Fractalkine preferentially mediates arrest and migration of CD16+ monocytes. J Exp Med. 2003;197:1701–7. doi: 10.1084/jem.20022156. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Zawada AM, Rogacev KS, Rotter B, et al. SuperSAGE evidence for CD14++CD16+ monocytes as a third monocyte subset. Blood. 2011;118:e50–61. doi: 10.1182/blood-2011-01-326827. [DOI] [PubMed] [Google Scholar]
- 12.Ziegler-Heitbrock L, Ancuta P, Crowe S, et al. Nomenclature of monocytes and dendritic cells in blood. Blood. 2010;116:e74–80. doi: 10.1182/blood-2010-02-258558. [DOI] [PubMed] [Google Scholar]
- 13.Moniuszko M, Bodzenta-Lukaszyk A, Kowal K, Lenczewska D, Dabrowska M. Enhanced frequencies of CD14++CD16+, but not CD14+CD16+, peripheral blood monocytes in severe asthmatic patients. Clin Immunol. 2009;130:338–46. doi: 10.1016/j.clim.2008.09.011. [DOI] [PubMed] [Google Scholar]
- 14.Han J, Wang B, Han N, et al. CD14(high)CD16(+) rather than CD14(low)CD16(+) monocytes correlate with disease progression in chronic HIV-infected patients. J Acquir Immune Defic Syndr. 2009;52:553–9. doi: 10.1097/qai.0b013e3181c1d4fe. [DOI] [PubMed] [Google Scholar]
- 15.Ancuta P, Kamat A, Kunstman KJ, et al. Microbial translocation is associated with increased monocyte activation and dementia in AIDS patients. PLoS ONE. 2008;3:e2516. doi: 10.1371/journal.pone.0002516. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Scott-Taylor TH, Green MR, Eren E, Webster AD. Monocyte derived dendritic cell responses in common variable immunodeficiency. Clin Exp Immunol. 2004;138:484–90. doi: 10.1111/j.1365-2249.2004.02640.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Scott-Taylor TH, Green MR, Raeiszadeh M, Workman S, Webster AD. Defective maturation of dendritic cells in common variable immunodeficiency. Clin Exp Immunol. 2006;145:420–7. doi: 10.1111/j.1365-2249.2006.03152.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Cambronero R, Sewell WA, North ME, Webster AD, Farrant J. Up-regulation of IL-12 in monocytes: a fundamental defect in common variable immunodeficiency. J Immunol. 2000;164:488–94. doi: 10.4049/jimmunol.164.1.488. [DOI] [PubMed] [Google Scholar]
- 19.Martinez-Pomar N, Raga S, Ferrer J, et al. Elevated serum interleukin (IL)-12p40 levels in common variable immunodeficiency disease and decreased peripheral blood dendritic cells: analysis of IL-12p40 and interferon-gamma gene. Clin Exp Immunol. 2006;144:233–8. doi: 10.1111/j.1365-2249.2006.03063.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Viallard JF, Camou F, Andre M, et al. Altered dendritic cell distribution in patients with common variable immunodeficiency. Arthritis Res Ther. 2005;7:R1052–5. doi: 10.1186/ar1774. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Bayry J, Lacroix-Desmazes S, Kazatchkine MD, et al. Common variable immunodeficiency is associated with defective functions of dendritic cells. Blood. 2004;104:2441–3. doi: 10.1182/blood-2004-04-1325. [DOI] [PubMed] [Google Scholar]
- 22.Nourizadeh M, Aghamohammadi A, Moazzeni SM, et al. Altered dendritic cell function in response to sera of common variable immunodeficiency patients. Inflamm Res. 2007;56:527–32. doi: 10.1007/s00011-007-7081-7. [DOI] [PubMed] [Google Scholar]
- 23.Cunningham-Rundles C, Radigan L. Deficient IL-12 and dendritic cell function in common variable immune deficiency. Clin Immunol. 2005;115:147–53. doi: 10.1016/j.clim.2004.12.007. [DOI] [PubMed] [Google Scholar]
- 24.Amoras AL, da Silva MT, Zollner RL, Kanegane H, Miyawaki T, Vilela MM. Expression of Fc gamma and complement receptors in monocytes of X-linked agammaglobulinaemia and common variable immunodeficiency patients. Clin Exp Immunol. 2007;150:422–8. doi: 10.1111/j.1365-2249.2007.03512.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Yong PF, Workman S, Wahid F, Exley A, Webster AD, Ibrahim MA. Selective deficits in blood dendritic cell subsets in common variable immunodeficiency and X-linked agammaglobulinaemia but not specific polysaccharide antibody deficiency. Clin Immunol. 2008;127:34–42. doi: 10.1016/j.clim.2007.12.007. [DOI] [PubMed] [Google Scholar]
- 26.Conley ME. Genetics of hypogammaglobulinemia: what do we really know? Curr Opin Immunol. 2009;21:466–71. doi: 10.1016/j.coi.2009.07.003. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Martini H, Enright V, Perro M, et al. Importance of B cell co-stimulation in CD4(+) T cell differentiation: X-linked agammaglobulinaemia, a human model. Clin Exp Immunol. 2011;164:381–7. doi: 10.1111/j.1365-2249.2011.04377.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Mechanic LJ, Dikman S, Cunningham-Rundles C. Granulomatous disease in common variable immunodeficiency. Ann Intern Med. 1997;127:613–7. doi: 10.7326/0003-4819-127-8_part_1-199710150-00005. [DOI] [PubMed] [Google Scholar]
- 29.Kitchens RL, Thompson PA. Modulatory effects of sCD14 and LBP on LPS–host cell interactions. J Endotoxin Res. 2005;11:225–9. doi: 10.1179/096805105X46565. [DOI] [PubMed] [Google Scholar]
- 30.Cros J, Cagnard N, Woollard K, et al. Human CD14dim monocytes patrol and sense nucleic acids and viruses via TLR7 and TLR8 receptors. Immunity. 2010;33:375–86. doi: 10.1016/j.immuni.2010.08.012. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Trabattoni D, Saresella M, Biasin M, et al. B7-H1 is up-regulated in HIV infection and is a novel surrogate marker of disease progression. Blood. 2003;101:2514–20. doi: 10.1182/blood-2002-10-3065. [DOI] [PubMed] [Google Scholar]
- 32.Wang X, Zhang Z, Zhang S, et al. B7-H1 up-regulation impairs myeloid DC and correlates with disease progression in chronic HIV-1 infection. Eur J Immunol. 2008;38:3226–36. doi: 10.1002/eji.200838285. [DOI] [PubMed] [Google Scholar]
- 33.Tendeiro R, Foxall RB, Baptista AP, et al. PD-1 and its ligand PD-L1 are progressively up-regulated on CD4 and CD8 T-cells in HIV-2 infection irrespective of the presence of viremia. AIDS. 2012;26:1065–71. doi: 10.1097/QAD.0b013e32835374db. [DOI] [PubMed] [Google Scholar]
- 34.Keir ME, Butte MJ, Freeman GJ, Sharpe AH. PD-1 and its ligands in tolerance and immunity. Annu Rev Immunol. 2008;26:677–704. doi: 10.1146/annurev.immunol.26.021607.090331. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35.Wehr C, Kivioja T, Schmitt C, et al. The EUROclass trial: defining subgroups in common variable immunodeficiency. Blood. 2008;111:77–85. doi: 10.1182/blood-2007-06-091744. [DOI] [PubMed] [Google Scholar]
- 36.Trujillo CM, Muskus C, Arango J, Patino PJ, Montoya CJ. Quantitative and functional evaluation of innate immune responses in patients with common variable immunodeficiency. J Investig Allergol Clin Immunol. 2011;21:207–15. [PubMed] [Google Scholar]
- 37.Day CL, Kaufmann DE, Kiepiela P, et al. PD-1 expression on HIV-specific T cells is associated with T-cell exhaustion and disease progression. Nature. 2006;443:350–4. doi: 10.1038/nature05115. [DOI] [PubMed] [Google Scholar]
- 38.Petrovas C, Casazza JP, Brenchley JM, et al. PD-1 is a regulator of virus-specific CD8+ T cell survival in HIV infection. J Exp Med. 2006;203:2281–92. doi: 10.1084/jem.20061496. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39.Trautmann L, Janbazian L, Chomont N, et al. Upregulation of PD-1 expression on HIV-specific CD8+ T cells leads to reversible immune dysfunction. Nat Med. 2006;12:1198–202. doi: 10.1038/nm1482. [DOI] [PubMed] [Google Scholar]
- 40.Pastor Rojo O, Lopez San Roman A, Albeniz Arbizu E, de la Hera Martinez A, Ripoll Sevillano E, Albillos Martinez A. Serum lipopolysaccharide-binding protein in endotoxemic patients with inflammatory bowel disease. Inflamm Bowel Dis. 2007;13:269–77. doi: 10.1002/ibd.20019. [DOI] [PubMed] [Google Scholar]