

Abstract
Small GTPases mediate transmembrane signaling and regulate the actin cytoskeleton in eukaryotic cells. Here, we characterize the auditory pathology of adult male CBA/J mice exposed to traumatic noise (2–20 kHz; 106 dB; 2 h). Loss of outer hair cells was evident 1 h after noise exposure in the basal region of the cochlea and spread apically with time, leading to permanent threshold shifts of 35, 60, and 65 dB at 8, 16, and 32 kHz. Several biochemical and molecular changes correlated temporally with the loss of cells. Immediately after exposure, the concentration of ATP decreased in cochlear tissue and reached a minimum after 1 h while the immunofluorescent signal for p-AMPKα significantly increased in sensory hair cells at that time. Levels of active Rac1 increased, whereas those of active RhoA decreased significantly 1 h after noise attaining a plateau at 1–3 h; the formation of a RhoA-p140mDia complex was consistent with an activation of Rho GTPase pathways. Also at 1–3 h after exposure, the caspase-independent cell death marker, Endo G, translocated to the nuclei of outer hair cells. Finally, experiments with the inner ear HEI-OC1 cell line demonstrated that the energy-depleting agent oligomycin enhanced both Rac1 activity and cell death. The sum of the results suggests that traumatic noise induces transient cellular ATP depletion and activates Rho GTPase pathways, leading to death of outer hair cells in the cochlea.
Introduction
Noise-induced hearing loss (NIHL) is one of the most frequent disabilities in industrialized societies (Oishi and Schacht, 2011). Loss of hair cells in the auditory end organ after noise exposure is well documented (Henderson et al., 2006); however, the comprehensive mechanisms of noise-induced loss of hair cells and hearing are unknown. In the inner ear, calcium is elevated in hair cells following noise exposure (Fridberger et al., 1998; Oliver et al., 2001), probably through influx via calcium channels since channel blockers protect against NIHL (Heinrich et al., 1999). Traumatic noise also activates the calcium-dependent phosphatase calcineurin (Minami et al., 2004) and mediates mitochondria-related cell death (Vicente-Torres and Schacht, 2006).
Energy depletion is associated with calcium influx which, in turn, may affect mitochondrial metabolism (Arnold, 2005) and directly modulate apoptotic pathways, such as c-Jun N-terminal kinase signaling (Størling et al., 2005). Calcium homeostasis and energy metabolism are intertwined in complex feedback loops and it is not known whether energy depletion causes calcium influx or calcium influx perturbs energy metabolism. Extracellular ATP concentrations were elevated in guinea pig cochlear fluids after a short 15 min noise exposure (Muñoz et al., 2001). Conversely, ATP depletion following traumatic noise exposure that leads to permanent hearing loss is reported in rat cochlear tissue (Vlajkovic et al., 2004). Likewise, the ATP content of the organ of Corti in situ decreased under ischemic conditions (Thalmann et al., 1972). We hypothesize here that noise trauma is accompanied by metabolic overstimulation, causing transient cellular energy depletion and consequent activation of Rho GTPase pathways, leading to death of hair cells.
This hypothesis is based on the well characterized activation by metabolic stress of AMP-activated protein kinase (AMPK), a cellular energy sensor that detects and reacts to the AMP/ATP balance of the cell (Winder and Thomson, 2007). In response to energy depletion, the γ-subunit of AMPK allosterically binds AMP, exposing the threonine 172 (T172) residue of the catalytic α-subunit. Subsequent phosphorylation of T172 by upstream kinases, such as liver kinase B1, directs the cell to switch off energy-consuming activities and switch on energy-generating processes to help restore the energy balance of the cell (Hardie, 2003; Winder and Thomson, 2007). Downstream targets of AMPK also include small GTPases such as Rac1 (Levine et al., 2007; Lee et al., 2008) with possible effects in cellular structure or redox balance.
Small GTPases are the major modulators of the actin cytoskeleton (Burridge and Wennerberg, 2004; Cullen, 2006; Rao, 2008). As molecular switches, Rho GTPases exist in two states: an inactive state bound to GDP and an active state in which GDP is replaced by GTP. Among the Rho family of GTPases, Rac1, RhoA, and Cdc42 play crucial roles in cellular processes for assembly and disassembly of the actin cytoskeleton. RhoA regulates F-actin formation via its downstream targets, also called Rho-effectors, such as Rho-associated coiled-coil containing kinase, myosin light chain phosphatase, and LIM kinase (Spiering and Hodgson, 2011). In addition, RhoA also regulates the formin family of proteins. There are 15 identified mammalian formins and the two most extensively studied are the mammalian diaphanous formins (mDia) 1 and 2. mDia1 can be activated by GTP-RhoA (Watanabe et al., 1999; Goode and Eck, 2007). Along with its function in actin polymerization, mDia1 has been associated with microtubule stabilization. NADPH oxidase, a reactive oxygen species (ROS)-producing enzyme complex, is also considered a Rac1 effector protein (Nimnual et al., 2003; Sarfstein et al., 2004; Ueyama et al., 2006; Flinder et al., 2011), and the formation of a Rac1-p67phox complex indicates activated NADPH oxidase (Hordijk, 2006).
In this study, hair cell death after a broadband noise exposure that causes a permanent hearing loss in adult CBA/J mice was first characterized. Then the concentration of ATP in the cochlear tissue, phosphorylation of 5′ AMPK in outer hair cells following noise exposure, and Rho GTPase-linked signaling pathways were investigated. Finally, activation of Rac1 by the ATP-depleting agent oligomycin, an ATP synthase inhibitor, was assessed in HEI-OC1 cells.
Materials and Methods
Materials.
ECL and Supersignal Dura-Enhanced Chemiluminescence Plus (ECL-Plus) for Western blotting detection and PageRuler Plus protein ladder were purchased from Fisher Scientific. BenchMark Protein ladders were purchased from Invitrogen Life Technologies. The antibodies used in this study were as follows: polyclonal rabbit anti-RhoA (Santa Cruz Biotechnology #SC179), polyclonal rabbit anti-Nox3 (Santa Cruz Biotechnology #SC67005), polyclonal rabbit anti-endonuclease G (Millipore #AB3639), monoclonal mouse anti-Rac1 (Millipore #05-389), polyclonal rabbit anti-GAPDH (Millipore #ABS16), monoclonal rabbit anti-phospho-AMPKα (Thr172) (Cell Signaling Technology #2535), monoclonal mouse anti-p67phox (BD Biosciences #610912), monoclonal mouse anti-mDia1 (BD Bioscience #610848), and polyclonal rabbit anti-Myosin VII (Proteus Bioscience #25-6790). Secondary antibodies for Western blotting were obtained from Jackson ImmunoResearch Laboratories, and secondary Alexa 488 or Alexa 594 fluorescent antibodies, Alexa 488 phalloidin, propidium iodide, and Hoechst 33342 from Invitrogen. Complete mini EDTA-free protease inhibitor mixture tablets were purchased from Roche Diagnostic GmbH and ATP depletion was measured by an ATP Bioluminescence Assay Kit HS II (Roche). Lab-Tek chamber permanox slides were purchased from Fisher Scientific. Culture media were purchased from Invitrogen. All other reagents were purchased from Sigma-Aldrich.
In vivo experiments in mice
Animals.
Male CBA/J mice at 12 weeks of age (Harlan Sprague Dawley) had free access to water and a regular mouse diet (Purina 5025), and were kept at 22 ± 1°C under a standard 12 h light/dark cycle to acclimate for 1 week before the experiments. All research protocols were approved by the Institutional Animal Care and Use Committee at the Medical University of South Carolina (MUSC). Animal care was under the supervision of Division of Laboratory Animal Resources at MUSC.
Noise exposure.
Mice in separate stainless steel wire cages (9 × 9 × 9 cm) were exposed to a broadband noise (BBN) with a frequency spectrum from 2 to 20 kHz at 106 dB sound pressure level (SPL) for 2 h to induce permanent threshold shifts or 92 dB SPL to induce temporary threshold shifts (TTS). The sound exposure chamber is fitted with a loudspeaker (model 2450H; JBL) driven by a power amplifier (model XLS 202D; Crown Audio) fed from a CD player (model CD-200; Tascam TEAC American). Audio CD sound files are created and equalized with audio editing software (Audition 3; Adobe System). Sound levels are calibrated with a sound level meter (model 1200; Quest Technologies) at multiple locations within the sound chamber to ensure uniformity of the sound field, and are measured before and after exposure to ensure stability. Control mice were kept in silence within the same chamber for 2 h.
Auditory brainstem response.
Mice were anesthetized with an intraperitoneal injection of xylazine (20 mg/kg) and ketamine (100 mg/kg), and then placed in a sound-isolated and electrically shielded booth (Acoustic Systems). Body temperature was monitored and maintained near 37°C with a heating pad. Acoustic stimuli were delivered monaurally to a Beyer earphone attached to a customized plastic speculum inserted into the ear canal. Subdermal electrodes were inserted at the vertex of the skull, under the left ear, and under the right ear (ground). Auditory brainstem responses (ABRs) were measured at 8, 16, and 32 kHz. Tucker Davis Technology System III hardware and SigGen/Biosig software were used to present the stimuli (15 ms duration tone bursts with 1 ms rise–fall time) and record the response. Up to 1024 responses were averaged for each stimulus level. Thresholds were determined for each frequency by reducing the intensity in 10 dB increments and then in 5 dB steps near threshold until no organized responses were detected. Thresholds were estimated between the lowest stimulus level where a response was observed and the level without response. All ABR measurements were conducted by the same experimenter blinded to the treatment conditions.
Surface preparation and diaminobenzidine staining of hair cells.
The temporal bones were removed immediately following euthanasia and perfused through the scala with a solution of 4% paraformaldehyde (PFA) in PBS, pH 7.4, and kept in this fixative overnight at 4°C. The cochleae were then rinsed in PBS. Before decalcification of each cochlea in a 4% solution of sodium EDTA (adjusted with HCl to pH 7.4), the apical otic capsule was removed. The EDTA solution was changed daily for 3 d at 4°C. Following decalcification, the cochleae were placed into 3% hydrogen peroxide for 2.5 h to quench endogenous peroxidases. After incubation in a solution for blocking nonspecific antibody binding overnight at 4°C, the tissues were incubated with a primary antibody (rabbit polyclonal anti-myosin VII at a 1:100 dilution) for 4 d at 4°C on a Nutator mixer, washed in PBS, and then incubated overnight at 4°C with biotinylated goat anti-rabbit antibody at a 1:100 dilution. The specimens were rinsed again and then incubated in ABC solution (PK-4001; Vector Laboratories) overnight. Following another washing, the cochleae were incubated in diaminobenzidine (DAB) for 3 h, as necessary for sufficient staining intensity, followed by washing to stop the DAB reaction. Finally, the cochleae were microdissected under a microscope into apical, middle, and basal segments and mounted on slides with Fluoromount-G mounting media. Images were taken with a Zeiss AxioCam MRc5 camera with Axioplan 2 imaging software under Zeiss microscope. A sample size of five mice was used for each experimental group.
Hair cell counts on cochlear epithelia from the adult mouse.
Images from the apex through the base of the surface preparation with DAB-stained preparations were captured using a 40× lens on the Zeiss microscope. The lengths of the cochlear epithelia were measured and recorded in millimeters. Both outer and inner hair cells were counted from the apex to the base along the entire length of the mouse cochlear epithelium. Percentages of hair cell loss in each 0.5 mm length of epithelium were plotted as a function of the cochlear length as a cytocochleogram.
Immunocytochemistry for cochlear surface preparations.
Following decalcification with 4% EDTA, cochleae were used for immunocytochemistry on surface preparations. The cochleae were dissected under a microscope by removing the softened otic capsule, stria vascularis, Reissner's membrane, and tectorial membrane. The remaining tissue, including the modiolus and cochlear sensory epithelium, was permeabilized in 3% Triton X-100 solution for 30 min at room temperature. The specimens were washed three times with PBS, and blocked with 10% goat serum for 30 min at room temperature, followed by incubation with anti-Endo G (1:500), p-AMPKα (1:50), or anti-Nox3 (1:50) in darkness at 4°C for 72 h. After washing three times (10 min each), the tissues were incubated with the Alexa 488-conjugated secondary antibody at a concentration of 1:200 at 4°C overnight in darkness. After washing three times, tissues were incubated with Hoechst 33342 (2 μg/ml in PBS) or propidium iodide (1 μg/ml in PBS) at room temperature for 30 min. After the final wash with PBS, the tissue was dissected by removing the modiolus. The epithelia were divided into three segments (apex, middle, and base). Specimens were mounted on slides with antifade mounting media. Control incubations were routinely processed without primary antibody treatments.
For detection of apoptotic and necrotic outer hair cell nuclei, surface preparations of the cochlear sensory epithelium were stained with propidium iodide (1 μg/ml in PBS) for 1 h after incubation in 3% Triton X-100 for 30 min at room temperature. Immunolabeled images were taken using a Zeiss laser confocal microscope (Zeiss LSM 510; Carl Zeiss Microimaging). For each group, a sample size at least of three was used in all immunocytochemistry analyses.
Quantification of the immunofluorescence signals from surface preparations.
Immunofluorescence (red) of p-AMPKα or Nox3 on surface preparations was quantified from original confocal images, each taken with a 63× magnification lens under identical conditions and equal parameter settings for laser gains and photomultiplier tube gains, using ImageJ software (National Institutes of Health (NIH), Bethesda, MD). The cochleae from the noise-exposed and unexposed groups were fixed and stained simultaneously with identical solutions and processed in parallel. All the surface preparations were counterstained with phalloidin (green) for labeling hair cell structure to identify the comparable parts of the hair cells for capture of confocal images. The borders of each individual outer hair cell were outlined based on the phalloidin staining. The immunofluorescence of p-AMPKα or Nox3 of hair cells was measured in the upper basal cochlear surface preparations in 0.12 mm segments, each containing ∼60 outer hair cells. The intensity of the background fluorescence was subtracted and average fluorescence per cell was then calculated. The relative fluorescence was quantified by normalizing the ratio of average fluorescence of noise-exposed hair cells to the average fluorescence of the unexposed hair cells. Each condition was replicated in four different animals.
Extraction of total protein.
Cochleae were rapidly removed and dissected in ice-cold 10 mm PBS at pH 7.4. To extract total protein, tissues from the cochleae of one mouse were homogenized in ice-cold radioimmunoprecipitation assay (RIPA) lysis buffer containing RIPA lysis buffer base (50 mm Tris-HCl, 1% IGEPAL, 0.25% Na-deoxycholate, 150 mm NaCl, 1 mm EDTA, 1 mm PMSF, 1 mm NaF) plus Phosphatase Inhibitor Cocktails II and III, and Roche Protease Inhibitor by using a glass/glass micro Tissue Grind pestle and vessel for 30 s. After 30 min on ice, tissue debris was removed by centrifugation at 10,000 × g at 4°C for 10 min and the supernatants were retained as the total protein fractions. Protein concentrations were determined using the Bio-Rad Protein Assay dye reagent (Bio-Rad) with bovine serum albumin (BSA) as a protein standard. Two cochleae from the same mouse were pooled for each sample.
Intracellular ATP measurement.
Intracellular ATP content in the extracts from two entire cochleae of one mouse was measured with an ATP Bioluminescence Assay Kit HS II according to the manufacturer's instructions (Roche). This assay kit is developed specifically for the detection of ATP with the highest sensitivity. It uses the ATP dependency of the light-emitting luciferase-catalyzed oxidation of luciferin. The values were calibrated against an ATP standard curve. Two cochleae from the same mouse were pooled for each sample. A total of seven samples were analyzed at each time point.
Pull-down assay to detect active Rac1 and RhoA.
Mice were decapitated by cervical dislocation and cochleae were rapidly removed from the temporal bone and immediately frozen in liquid nitrogen. Six cochleae from three mice were pooled for each sample and homogenized in RIPA buffer with a Tissue-Tearor for 30 s on ice. After centrifugation at 15,000 × g at 4°C for 10 min, the supernatant was retained. After measurement of the protein concentration, 300 μg of total protein for each sample was used for a pull-down assay with Rac1 and RhoA activation kits (Stressgen). Subsequently, Western blotting was performed with an anti-Rac1 or anti-RhoA antibody. A sample size of three was used for each time point.
Immunoprecipitation to assess for Rac1/p67 or RhoA/p104-mDia complexes.
Ig proteins were precleared from 300 μg total cochlear protein by incubation with 0.25 μg of the control mouse IgG and 20 μl of agarose-conjugated protein A or G at 4°C for 30 min. After centrifugation at 1000 × g for 30 s at 4°C to remove the bead-bound IgG, the cleared lysate was incubated with the primary antibody Rac1 or RhoA (2 μg) for 6 h at 4°C, and then 20 μl of agarose-conjugated protein A was added. After incubation at 4°C on a rotating device overnight, the pellets were collected by centrifugation at 1000 × g for 1 min at 4°C. The pellets were washed three times with lysis buffer; after the final wash the pellets were resuspended in 20 μl of electrophoresis buffer (including 0.5 m Tris-HCl, 4% SDS, 20% glycerol, 0.02% bromophenol blue, and 10% β-mercaptoethanol). Subsequently, Western blotting was performed with an anti-p67phox or anti-mDia1 antibody. Six cochleae from three mice were pooled for each sample. A total of at least three samples were used for each group.
Western blot analysis.
Protein samples (50 μg) were separated by SDS-PAGE. After electrophoresis, the proteins were transferred onto a nitrocellulose membrane (Pierce) and blocked with 5% solution of nonfat dry milk in PBS-0.1% Tween 20 (PBS-T). The membranes were incubated with anti-Rac1 (1:2000), or anti-Rho A (1:500) for 2 h, and then washed three times (10 min each) with PBS-T. Membranes were incubated with an appropriate secondary antibody at a concentration of 1:10,000 for 1 h. Following extensive washing of the membrane, the immunoreactive bands were visualized by ECL or ECL-Plus. The membranes were then stripped and restained for GAPDH at a concentration of 1:20,000 as a control for sample loading.
X-ray films of Western blots were scanned and analyzed using ImageJ software (NIH). First, the background staining density for each band was subtracted from the band density. Next, the probing protein/GAPDH ratio was calculated from the band densities run on the same gel to normalize for differences in protein loading. Finally, the difference in the ratio of the control and experimental bands was tested for statistical significance. At least three samples were used for each group in all Western blotting experiments.
In vitro experiment with HEI-OC1 cells
HEI-OC1 cell culture.
HEI-OC1 cells (obtained from Dr. F. Kalinec, House Ear Institute, Los Angeles, CA) (Kalinec et al., 2003) were plated on 100 mm Falcon Primeria plates in 8–10 ml of DMEM with 10% fetal bovine serum (FBS), 10 μg of γ-interferon/L, and 50 U of penicillin. The cells were allowed to proliferate in an incubator with 10% CO2 at 33°C (permissive conditions). The cultures were allowed to grow to 70–80% confluency before being used for the experiments.
ATP depletion with oligomycin treatment in HEI-OC1 cells.
HEI-OC1 cells were chemically depleted of ATP by treatment with 1 μm oligomycin in nonglucose DMEM without FBS in an incubator with 10% CO2 at 33°C. Time points were determined based on the variant assays.
Extraction of total protein from HEI-OC1 cells for pull-down assay to detect active Rac1.
Cell cultures from four culture plates were pooled per sample. Cell cultures treated with or without 1 μm oligomycin were rapidly rinsed twice with ice-cold 10 mm PBS and then ice-cold RIPA lysis buffer containing RIPA lysis buffer base (50 mm Tris-HCl, 1% IGEPAL, 0.25% Na-deoxycholate, 150 mm NaCl, 1 mm EDTA, 1 mm PMSF, 1 mm NaF) plus Phosphatase Inhibitor Cocktails II and III, and Roche Protease Inhibitor was added to the plates. Cells were scraped from the bottom of the dishes and moved to conical tubes. After 30 min on ice, tissue debris was removed by centrifugation at 10,000 × g at 4°C for 10 min and the supernatants were retained as the total protein fractions. Protein concentrations were determined using the Bio-Rad Protein Assay dye reagent (Bio-Rad) with BSA as a protein standard. Total protein (800 μg) for each sample was used for a pull-down assay with Rac1 activation kits (Stressgen). Three samples were used for each treatment group.
ATP measurement after oligomycin treatment in HEI-OC1 cells.
Cell cultures from one culture plate per sample were treated with or without 1 μm oligomycin. Intracellular ATP content in the extracts from HEI-OC1 cells was measured with an ATP Bioluminescence Assay Kit HS II as described for in vivo samples. Three samples were used at each time point (control, 30 min, 1 h, and 3 h).
Quantification of ATP depletion-induced HEI-OC1 cell death by flow cytometry assay.
BD ApoAlert Annexin V-FITC Apoptosis Kit (BD Biosciences Clontech) was used per the manufacturer's instructions. HEI-OC1 cells were treated with 1 μm oligomycin or without oligomycin for 24 h in nonglucose DMEM without FBS in an incubator with 10% CO2 at 33°C. First, ∼5 × 105 cells were collected and rinsed with binding buffer, then resuspended in 200 μl of binding buffer. Next, 5 μl of Annexin V and 10 μl of propidium iodide were added to the cells and incubated at room temperature for 10 min in the dark. Another 300 μl of binding buffer was added to bring the reaction volume up to 500 μl before the cells were counted using a single laser emitting excitation light at 488 nm. Cell death via either apoptosis or necrosis was determined by positive propidium iodide or Annexin V staining. Viable cells were stained double negative. A sample size of three was used for each treatment group.
Immunocytochemistry for cultured HEI-OC1 cells.
HEI-OC1 cells were cultured in Lab-Tek chamber permanox slides. After the cells reached ∼60% confluency, they were divided into control group (cells treated with glucose-free medium) and ATP-depletion group (cells treated with 1 μm oligomycin in glucose-free medium). Cultures were rinsed with PBS three times, then fixed immediately with 2% PFA for 20 min and incubated in 0.5% Triton X-100 for 15 min at room temperature. After washing three times with PBS, a blocking solution of 3% goat serum was added to the sections for 30 min at room temperature, followed by the primary antibody at a concentration of 1:500 for anti-Endo G for 3 h. The cultures were then washed three times with PBS and incubated with secondary antibody (Alexa 488- or Alexa 546-conjugated) for 1 h at room temperature in darkness. Next, the cultures were labeled with propidium iodide (1 μg/ml in PBS) for 30 min in darkness. After washing with PBS, the fixed cultures were mounted and images were taken with a Zeiss laser confocal microscope. Three samples were used for each group.
Statistical analysis.
Data were statistically evaluated by one-way ANOVA with Student–Newman–Keuls multiple-comparison tests using Primer of Biostatistics software (McGraw-Hill Software) or two-tailed one sample t test using GraphPad Software.
Results
Permanent NIHL and hair cell loss in the adult CBA/J mouse
Exposure of 12-week-old male CBA/J mice to BBN at 106 dB SPL for 2 h resulted in permanent threshold shifts of ∼35, 60, and 65 dB at 8, 16, and 32 kHz, respectively, by 2 weeks after the exposure (Fig. 1A). Loss of hair cells largely in the basal and middle regions of the epithelium corresponded with the ABR functional measurements. No hair cells were missing in the apical region. Outer hair cell loss began ∼2.5 mm from the apex and increased to complete loss at 4 mm through the end of the epithelium. Interestingly, in the 2.5–4 mm region, outer hair cell loss ranged from partial to complete, while inner hair cells were still present. Inner hair cell loss began at 4.5 mm and reached an average of 80% missing cells at the end of the basal region (5.5 mm) (Fig. 1B,C). No inner hair cells remained in the hook region (data not shown).
Figure 1.
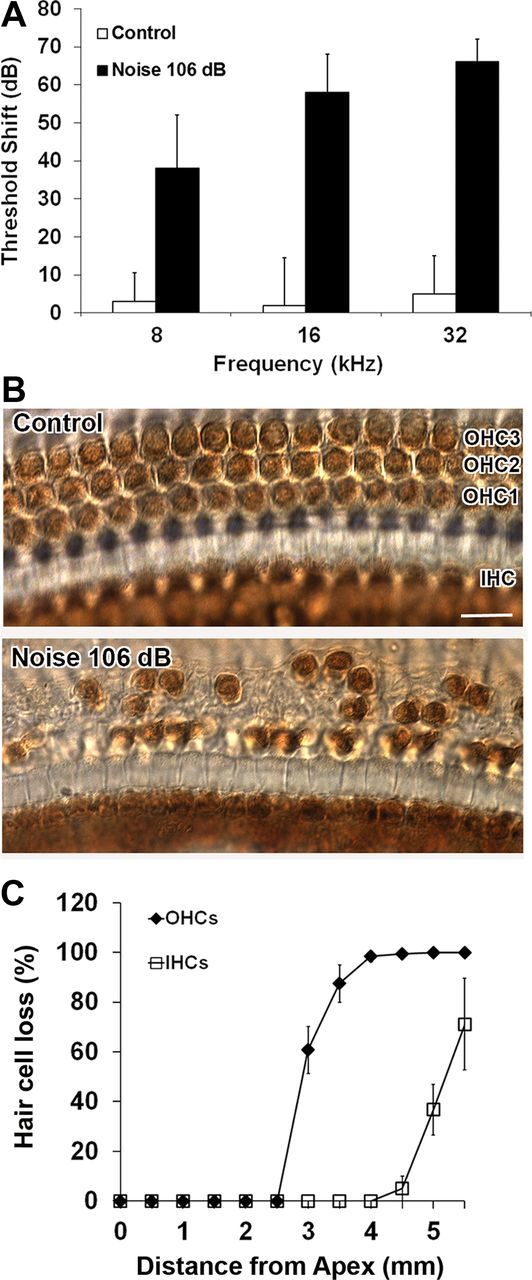
A, ABR threshold shifts. The bar graph represents the permanent NIHL in 3-month-old male CBA/J mice resulting 2 weeks after exposure to 2–20 kHz BBN at 106 dB SPL for 2 h. Permanent threshold shifts were greatest at high frequencies and decreased toward lower frequencies. Data are presented as means + SD; n = 5 for each condition. B, The representative images illustrate hair cell losses in the middle region (3 mm from apex) corresponding with the ABR functional measurements shown in the bar graph in A. OHC1, 2, and 3 indicate outer hair cell rows 1, 2, and 3; IHC indicates the one row of inner hair cells. These images are representative of five mice analyzed for each condition. Scale bar, 10 μm. C, Quantification of hair cell loss. Both outer and inner hair cells were counted from apex to base along the entire length of the mouse cochlear epithelium (see Materials and Methods). Both outer and inner hair cell loss increased in an apex-to-base gradient. Data are presented as means ± SD; n = 5 for each condition. OHC, outer hair cells; IHC, inner hair cells.
Traumatic noise triggers both apoptotic and necrotic outer hair cell death
We applied morphological criteria in conjunction with propidium iodide uptake to determine the extent of apoptosis and necrosis (Cole and Perez-Polo, 2002). No cells with apoptotic or necrotic nuclei were seen in normal controls or immediately after noise exposure. However, 1 h after noise exposure, outer hair cells in the basal segment showed both types of cell death with the majority exhibiting signs of necrosis characterized by swollen nuclei (Fig. 2A, arrowheads). Some cells exhibited morphological features of apoptosis with chromatin condensation (Fig. 2A, arrows). Over time, the prevalence of apoptosis increased and that of necrosis decreased.
Figure 2.

A, Apoptotic and necrotic outer hair cell death in the basal segment of the cochlear epithelium. Control cochleae and those harvested immediately after noise exposure showed no outer hair cells displaying features of apoptotic or necrotic death. At 1 and 3 h postnoise exposure cells with swollen necrotic (arrowheads) or fragmented apoptotic nuclei (arrows) were present, but more cells with apoptotic nuclei were observed at 3 h than 1 h postnoise exposure. Red, propidium iodide staining for nuclei. B, Translocation of Endo G to the nuclei of some outer hair cells in the basal segment of the cochlear epithelium. Immunofluorescence of Endo G on surface preparations from control mice showed Endo G in outer hair cells, but not in their nuclei. Immediately, 1 h, and 3 h postexposure, Endo G translocated into some nuclei of outer hair cells (arrows). Green, Endo G; blue, Hoechst 33342 staining for nuclei. All images were taken from the upper basal turn and each figure is representative of three individual mice for each condition. “Immediately” indicates samples obtained immediately after 2 h BBN noise exposure at 106 dB; 1 and 3 h indicate samples obtained 1 and 3 h postnoise exposure. Scale bars: 10 μm.
Translocation of the mitochondrial protein Endo G into the nucleus indicates activation of caspase-independent cell death pathways (Li et al., 2001; Lockshin and Zakeri, 2002). Endo G has been shown to translocate to the nuclei of outer hair cells after noise exposure in a guinea pig model (Yamashita et al., 2004b). In our control mice, Endo G staining was present in the cytoplasm of outer hair cells but absent from the nuclei, as seen on surface preparations of the cochlear epithelium (Fig. 2B, control). Endo G was still present in the cytoplasm of the majority of outer hair cells in the basal region immediately after noise exposure; however, at 1 and 3 h postexposure, Endo G translocated into the nuclei of some of the cells with condensed nuclei (Fig. 2B, arrows).
Traumatic noise induces transient ATP depletion
To test our hypothesis that transient energy depletion is linked to hair cell death, we first measured the intracellular ATP concentrations in cochlear extracts. The concentration of ATP in control cochleae was 11.4 μmol/g protein. Immediately after noise exposure, the concentration of ATP decreased significantly to nearly half of control levels and continued to decline to 3.4 μmol/g protein at 1 h postnoise exposure. The level of ATP slowly recovered but remained significantly lower than in control mice at 6.0 μmol/g protein at 5 h postnoise exposure (p < 0.05; n = 7) (Fig. 3).
Figure 3.

ATP levels in the cochlear tissue of the inner ear. The level of ATP decreased significantly immediately after noise exposure and was lowest at 1 h postnoise exposure. The ATP level recovered slowly, but remained significantly lower than those of control mice at 5 h postnoise exposure. Data are presented as means + SD; *p < 0.05, n = 7.
AMPK is an energy sensor; when cellular ATP levels decrease, the levels of AMP increase and AMPK is activated. We assessed the activation of AMPK in hair cells by immunofluorescence with a monoclonal p-AMPKα antibody. Only a single band was detected in homogenates of total cochlear tissue by Western blot when probing with the p-AMPKα antibody. Quantitative analysis of the band densities showed no significant changes between control and immediately, 1 h, and 3 h post-traumatic noise exposure groups (n = 4) (Fig. 4B). However, when probing individual cell populations by immunofluorescence, p-AMPKα increased in sensory hair cells 1 h after traumatic noise exposure (Fig. 4A). Quantitative analysis of the p-AMPKα-associated fluorescence in outer hair cells revealed a significant increase. The ratio of relative fluorescence of control to 1 h posttraumatic noise samples was 1:3 (p < 0.01; n = 4). For comparison, some mice were exposed to BBN at 92 dB for 2 h resulting in only TTS (Fig. 5A). Under the TTS conditions, no increase in immunofluorescence of p-AMPKα in sensory hair cells appeared (Fig. 5B). Quantitative analysis of the immunofluorescence of p-AMPKα in outer hair cells showed no difference between control and 1 h post-TTS noise (p > 0.05; n = 3). These results indicate that energy depletion in sensory hair cells was associated specifically with traumatic noise that induced permanent hearing loss.
Figure 4.

p-AMPKα increased transiently in sensory hair cells after traumatic noise exposure. A, Representative images revealed an increase in fluorescence of p-AMPKα antibody in outer hair cells 1 h post-traumatic noise exposure. These images were taken from the upper basal turn and each figure is representative of four individual mice analyzed for each condition. Scale bar, 10 μm. Quantification of the p-AMPKα-associated fluorescence (see Materials and Methods) showed a significant increase in outer hair cells 1 h post-traumatic noise exposure (p < 0.01, n = 4). B, Western blots showed a single protein band of p-AMPKα in cochlear homogenates with no difference between each group. Data are presented as means + SD, n = 4. I, 1 h, and 3 h indicate immediately, 1 h, and 3 h after BBN exposure at 106 dB for 2 h.
Figure 5.
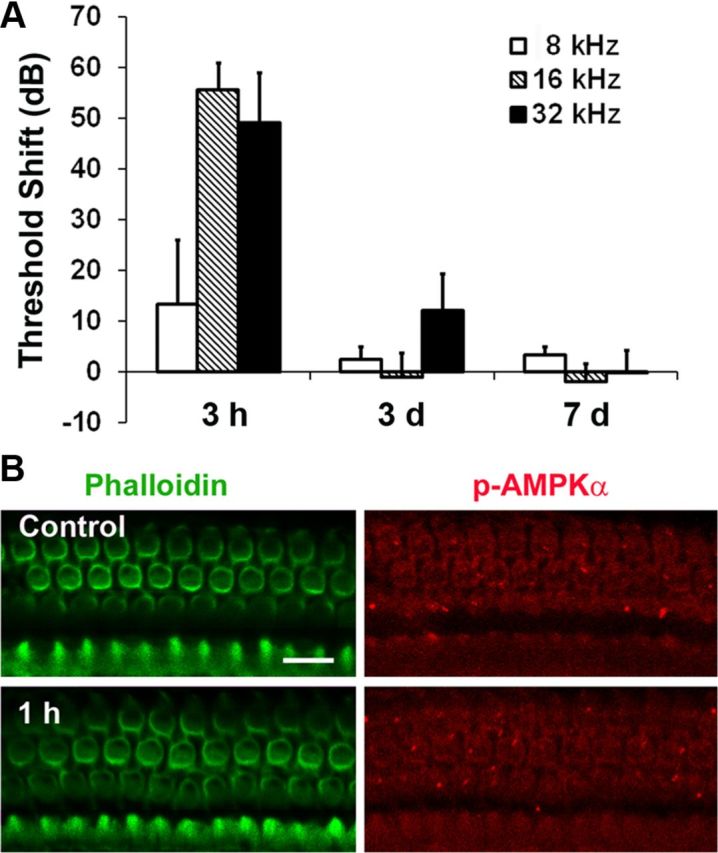
A, Noise-induced TTS. The bar graph represents TTS in 12-week-old male CBA/J mice after exposure to 2–20 kHz BBN at 92 dB SPL for 2 h. Hearing loss recovered completely 1 week after noise exposure. Data are presented as means + SD; n = 5 for each condition; 3 h, 3 d, and 7 d indicate the time after TTS-induced noise exposure. B, Representative images showed no difference in p-AMPKα-associated fluorescence in outer hair cells 1 h post-TTS noise exposure (p > 0.05, n = 3). These images were taken from the upper basal turn of the cochlear epithelium. Scale bar, 10 μm.
Traumatic noise increases Rac1 and decreases RhoA activities
We analyzed active Rac1 (GTP-Rac1) and active RhoA (GTP-RhoA) by pull-down assay of whole cochlear homogenates to investigate whether Rho GTPase pathways are involved in hair cell damage after noise exposure. GTP-Rac1 increased 1 h after noise exposure and subsequently reached a plateau above control levels that was maintained for 3 h. Quantification of the ratio of the protein band densities of the GTP-Rac1 to total Rac1 showed that the increase from 1 to 3 h after traumatic noise exposure was significant (p < 0.05; n = 3; Fig. 6A). Corresponding with the increase in active Rac1, GTP-RhoA decreased 1 h after noise exposure and showed lower band densities until 3 h. Quantification of the band densities of GTP-RhoA to total RhoA confirmed the significance of this decrease (p < 0.01; n = 4; Fig. 6B). In contrast to the changes in active Rac1 and RhoA, the levels of total Rac1 and RhoA were not affected by noise exposure (p > 0.05; n = 3; Fig. 6).
Figure 6.
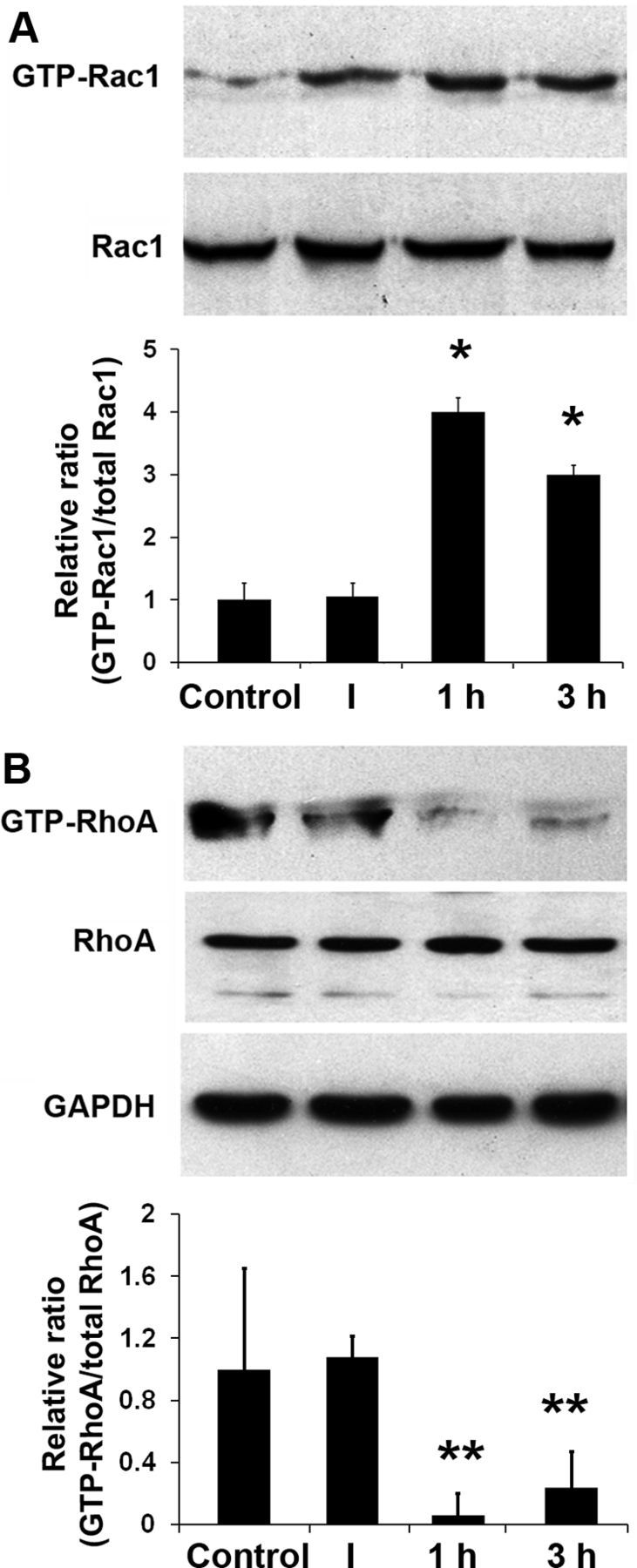
Noise trauma increases activated Rac1 and decreases activated RhoA. A, GTP-Rac1 increased at 1 and 3 h after noise exposure. Quantification of the chemiluminescent bands showed that the ratio of GTP-Rac1 to total Rac1 increased significantly at 1 and 3 h post-traumatic noise exposure. Data are presented as means + SD; *p < 0.05, n = 3 at each time point. B, In contrast, GTP-RhoA levels were reduced at 1 and 3 h post-traumatic noise exposure. Data are presented as means + SD; **p < 0.01; n = 4 at each time point. Total Rac1 and RhoA did not change (p > 0.05; n = 3). GAPDH was used as a control to compare protein loading. I, 1 h, and 3 h indicate immediately, 1 h, and 3 h after noise exposure.
Noise trauma increases NADPH oxidase via activation of Rac1
Traumatic noise is known to induce ROS formation in outer hair cells (Yamashita et al., 2005); we investigated whether ROS formation by traumatic noise might be caused via the induction of NADPH oxidase activity by Rac1 activation. Since we observed that active Rac1, but not total Rac1, increases in the inner ear after noise exposure, we analyzed Rac1-p67phox complexes by immunoprecipitating Rac1 from cochlear extracts and immunoblotting for p67phox. Noise trauma significantly increased the formation of Rac1-p67phox complexes (Fig. 7A) but did not change the level of total p67phox (p > 0.05; n = 3; Fig. 7B). Quantitative analysis of the band densities of the Rac1/p67phox complexes to total Rac1 showed a statistically significant increase 3 h post-traumatic noise exposure. The ratio of the band densities of control to 3 h post-traumatic noise exposure was 1:2 (p < 0.05; n = 3).
Figure 7.

A, Noise trauma increases formation of Rac1/p67phox complexes. Complexes of Rac1 with p67phox (a subunit of NADPH oxidase) were detected by immunoprecipitation (IP) with an antibody against Rac1, followed by immunoblotting (IB) with an antibody for p67phox. The membrane was then immunoblotted with a Rac1 antibody as a control for the amount of Rac1 in the IP assay. The band densities of complexes of Rac1/p67phox quantified (see Materials and Methods) were stronger after noise treatment compared with the control group (p < 0.05; n = 3). B, Levels of p67phox in the cochlear total protein extracts were detected by Western blot and showed no change (p > 0.05; n = 3). GAPDH serves as a loading control.
Additionally, the amount of Nox3 in cochlear homogenates was investigated because Nox3 has been reported as an NADPH subunit localized to the sensory epithelia of the inner ear (Bánfi et al., 2004). Probing Western blots with a Nox3 antibody showed a single protein band for Nox3 in cochlear tissue and no statistical differences in the expression of Nox3 between control and 1 or 3 h after noise exposure (n = 7; Fig. 8B). Nox3 immunofluorescence on cryosections illustrated fluorescence localization to spiral ganglion cells, but without quantitative differences between treatment groups (data not shown), which is consistent with the results from Western blots (Fig. 8B). Finally, immunofluorescence of Nox3 on surface preparations was stronger in sensory hair cells 1 h postnoise exposure than in control preparations (Fig. 8A). Quantitative analysis of the immunofluorescence of Nox3 in outer hair cells showed a statistically significant increase in Nox3 expression 1 h after traumatic noise exposure. The ratio of the relative immunofluorescence of control to 1 h post-traumatic noise exposure samples was 1:2 (p < 0.05; n = 4). These results suggest that noise trauma can activate NADPH oxidase via GTP-Rac1.
Figure 8.

A, Expression of Nox3 increased in sensory hair cells after traumatic noise exposure. Representative images from surface preparations showed an increase in fluorescence (red) of Nox3 antibody in hair cells 1 h postnoise exposure. These images were taken from the upper basal turn and each figure is representative of four individual mice for each condition. Scale bar, 10 μm. Quantification of Nox3-associated fluorescence in outer hair cells showed a significant increase (p < 0.05; n = 4). B, Western blotting of Nox3 in total cochlear extracts showed a single band for Nox3 with no significant difference between control, 1 h, and 3 h post-BBN exposure at 106 dB for 2 h. Data are presented as means + SD, n = 7.
Traumatic noise diminishes both RhoA/p140mDia complex and total p140mDia
The mammalian homolog of diaphanous (mDia) is a Rho effector that regulates F-actin polymerization. Complexes of RhoA-p140mDia were detected by immunoprecipitation with an antibody to RhoA, followed by immunoblotting with an antibody to p140mDia. Total p140mDia was detected by direct immunoblot. Consistent with the decrease in active RhoA, the amount of RhoA-p140mDia complexes decreased after traumatic noise (Fig. 9A). Quantitative analysis of the band densities of RhoA-p140mDia complexes to total RhoA revealed a statistically significant decrease in both at 3 h after traumatic noise exposure. The ratio of the band densities of control to 3 h postnoise exposure was 1:0.7 (p < 0.05; n = 3). Furthermore, the total level of p140mDia was also decreased 3 h after traumatic noise (Fig. 9B). Quantitative analysis of the band densities of p140mDia to GAPDH (used as a loading control for normalization) revealed a significant decrease in the total amount of p140mDia protein 3 h post-traumatic noise. The relative ratio of the band densities of control to 3 h post-traumatic noise was 1:0.4 (p < 0.01; n = 3).
Figure 9.

A, Noise trauma decreases the formation of RhoA/p140mDia complexes. Complexes of RhoA with p140mDia were detected by immunoprecipitation (IP) with an antibody against RhoA, followed by immunoblotting (IB) with an antibody to p140mDia. The membrane was then immunoblotted with the RhoA antibody as a control for the amount of RhoA in the IP assay. The band densities of complexes of RhoA/p140mDia decreased after noise exposure compared with the control group (p < 0.05; n = 3). B, Total p140mDia decreased 3 h postnoise exposure. The total amount of p140mDia in the cochlear homogenates was detected by IB with p140mDia. GAPDH serves as a loading control. The band densities of p140mDia decreased postnoise exposure compared with controls (p < 0.01; n = 3).
ATP-depletion agent oligomycin induces cell death and enhances Rac1 activity in inner ear HEI-OC1 cells
Oligomycin is an inhibitor of ATP synthase. We treated HEI-OC1 cells with oligomycin to block ATP generation and to establish further evidence that transient energy depletion activates small GTPase pathways. HEI-OC1 is a conditionally immortalized mouse cell line derived from the postnatal organ of Corti that displays a variety of markers for sensory hair cells, including math1, myosin7a, and prestin, in addition to markers for nonsensory cells. Although they are not adult mouse hair cells, they have been extensively used to elucidate pathways of hair cell pathology (Kim et al., 2010). Therefore, these cells offer valuable information as to whether ATP depletion alone results in Rac1 activation. The concentration of ATP decreased by 50% after 1 h of 1 μm oligomycin treatment in HEI-OC1 cells (Fig. 10A). Continued treatment for 24 h induced significantly higher HEI-OC1 cell death than in the control group determined by a flow cytometry assay (Fig. 10C). The cell death proportion increased from 2% in the control group to 25% in the 1 μm oligomycin group (p < 0.05; n = 5). In addition, Endo G remained in the cytoplasm with no nuclear staining in control cells, while energy depletion with 1 μm oligomycin treatment for 1 h resulted in diffuse Endo G staining in the cytosol and nuclear translocation. (Fig. 10B). A pull-down assay from HEI-OC1 cell homogenates for active Rac1 after 1 μm oligomycin treatment for 1 h had higher band densities of GTP-Rac1. The ratio of the band densities of GTP-Rac1 to total Rac1 showed a statistically significant increase after 1 μm oligomycin treatment for 1 h. The relative ratio of the band density of control to oligomycin treatment was 1:1.5 (p < 0.05; n = 3). The total level of Rac1 remained unchanged during 1 μm oligomycin treatment (Fig. 11).
Figure 10.

A, ATP depletion induced by oligomycin treatment in HEI-OC1 cells. The concentration of ATP was measured over time in HEI-OC1 cells treated with 1 μm oligomycin using the ATP Bioluminescence Assay Kit HS II (see Materials and Methods). Data are presented as means ± SD; n = 3; *p < 0.05 after 1 and 3 h of treatment. B, Endo G staining in the nuclei of some cells 1 h after 1 μm oligomycin treatment (arrows). Endo G is stained green, and nuclei red with propidium iodide (PI). C, Oligomycin induces cell death. Following incubation with 1 μm oligomycin for 24 h, cell death of HEI-OC1 cells was detected by a flow cytometry assay. Oligomycin treatment significantly increased the amount of cell death compared with the control treatment (p < 0.05; n = 5).
Figure 11.

ATP depletion increases active Rac1 in HEI-OC1 cells. ATP depletion via treatment with 1 μm oligomycin for 1 h led to increased levels of active Rac1 as compared with control-treated cells. Quantitative comparison of the band densities of GTP-Rac1 to total Rac1 showed a significant increase of GTP-Rac1 in the oligomycin-treated group (p < 0.05; n = 3). Transient ATP depletion did not influence the level of total Rac1. GAPDH is used as a loading control.
Discussion
Our results suggest that traumatic noise-induced outer hair cell death is mediated by Rho GTPase activation. Furthermore, our data are consistent with the notion that activation of the Rho GTPases in the inner ear by traumatic noise is linked to the transient cellular ATP depletion.
The validity and limitations of various estimates of the energy state of inner ear tissues have been extensively discussed by Thalmann et al. (1982). Our quantitative ATP measurements make a clear case for noise-induced reduction of ATP associated with noise conditions that lead to permanent, but not temporary, hearing loss. It is reasonable to assume that mitochondrial damage is responsible for this decrease in ATP levels, as the majority of ATP is produced in the mitochondria of essentially all cells, including outer hair cells. In addition to the possibility that calcium dysregulation impairs mitochondrial function, traumatic noise can also trigger local vasoconstriction in the inner ear, leading to hypoperfusion (Miller et al., 2003). Transient ischemia can decrease ATP levels in the organ of Corti, as already observed by Thalmann et al. (1972) in situ. ATP depletion has also been confirmed in vivo in the cochlear spiral ligament (Nagashima et al., 2011) and in the inner ear tissue of rats after traumatic noise exposure (Vlajkovic et al., 2004). Mitochondrial involvement in the events following traumatic noise exposure is also implicated by mitochondria-mediated hair cell death (Vicente-Torres and Schacht, 2006), as well as by the observation that supplementation with creatine, an energy buffer, can reduce both temporary and permanent NIHL (Minami et al., 2007). In addition, the results from the current study are consistent with the notion that ATP depletion invokes AMPKα-mediated pathways.
RhoA and Rac1 are molecular switches with opposite actions that participate in an antagonistic feedback loop. The increased GTP-Rac1 levels and decreased GTP-RhoA caused by noise are likely directly linked, since RhoA acts as an antagonist of active Rac1. The activation of Rac1 may also lead to an increase in the activity of NADPH oxidase (Takeya and Sumimoto, 2003; Takeya et al., 2003; Flinder et al., 2011). Although ROS formation in outer hair cells as a result of noise trauma has been well documented (Yamashita et al., 2004a; Henderson et al., 2006), the results of this study go further to suggest a mechanism responsible for ROS generation. Increases in the amount of Rac1/p67phox complexes and of Nox3 in outer hair cells indicate that noise trauma can activate NADPH oxidase via Rac1-GTP. Thus, activation of NADPH oxidase may be a culprit behind ROS formation in the inner ear in addition to mitochondrial deficiencies. We should note in this context that both enhanced extracellular ATP and intracellular ATP depletion have been reported to activate Rac1 (Hallett et al., 2003; Zhang et al., 2010). Such discrepancies are possible due to the responses in different cell types and under varied experimental conditions. The activation of Rac1 by either the presence or absence of ATP may suggest a situation-dependent duality of Rac1 function.
The presence of traumatic noise-induced apoptotic and necrotic hair cell death in this mouse model is in agreement with earlier studies in a variety of other species (Hu et al., 2002; Yamashita et al., 2005). Endo G is a mitochondrial protein whose translocation to the nucleus indicates activation of caspase-independent cell death pathways (Li et al., 2001; Lockshin and Zakeri, 2002). Endo G translocation into the nuclei of some of the outer hair cells with condensed nuclei after traumatic noise exposure in the CBA/J mice is consistent with earlier observations in a guinea pig model (Yamashita et al., 2004b).
The novel finding that levels of active GTP-Rac1 were increased and levels of GTP-RhoA were reduced by traumatic noise in the cochlea is an interesting parallel to kanamycin-induced inner ear stress (Jiang et al., 2006) suggesting that traumatic noise and ototoxic drugs induce similar effects on Rho GTPase pathways in the cochlea.
Furthermore, actin rearrangements appear to change the gross morphology of hair cell structure after these stressors. p140mDia is a likely mechanistic link between the small GTPases and actin rearrangements. p140mDia is a Rho effector (Watanabe et al., 1999) that controls stress fiber formation, focal adhesions, and actin polymerization. The decrease in expression of p140mDia after traumatic noise suggests decreased gene expression, implying that traumatic noise affects the actin cytoskeleton via Rho gene transcription. Interestingly, mutation in the human homolog of the p140mDia gene leads to production of truncated diaphanous protein and nonsyndromic hearing loss DFNA1 (Lynch et al., 1997).
The in vivo data provided here suggest a link between ATP depletion and the activation of Rho-family GTPases. This result is underscored by the cell culture model data, which indicate that ATP depletion contributes to the activation of Rac1. Intriguingly, in both in vivo and in vitro models, cell death is preceded by translocation of Endo G into nuclei, further attesting to the validity of the cell culture as a novel model to investigate consequences of noise trauma.
In conclusion, this study lends strong support to the hypothesis that traumatic noise leads to dysfunction of energy metabolism causing transient cellular energy depletion in cochlear sensory hair cells and activation of Rho-family GTPase pathways in the cochlea. The Rho GTPase pathways, in turn, lead to ROS formation via NADPH oxidase, resulting in hair cell death (Fig. 12).
Figure 12.

Summary of the molecular pathway. Traumatic noise causes transient cellular energy depletion in the inner ear, as shown by an increase in expression of p-AMPKα in sensory hair cells, and, consequently, activates Rho GTPase pathways, as determined by GTP-Rac1 and GTP-RhoA analysis. The increase in activated Rac1 leads to ROS formation, as shown by an increase in Nox3 expression through activation of NADPH oxidase (as determined by assessment of Rac1/p67 phox complexes), thus promoting hair cell death. Additionally, increased active Rac1 also leads to a decrease in activated RhoA levels, which decreases the formation of complexes of RhoA/p140mDia and the levels of diaphanous protein and leads to hair cell death.
Footnotes
This work was conducted in Medical University of South Carolina (MUSC) Walton Research Building in renovated space supported by Grant C06 RR014516. Animals were housed in MUSC Children's Research Institute animal facilities supported by Grant C06 RR015455 from the Extramural Research Facilities Program of the National Center for Research Resources. The research project described was supported by Grant R01 DC009222 from the National Institute on Deafness and Other Communication Disorders–National Institutes of Health. We thank Drs. Jochen Schacht and Bradley Schulte for their valuable comments on the manuscript.
References
- Arnold S. Estrogen suppresses the impact of glucose deprivation on astrocytic calcium levels and signaling independently of the nuclear estrogen receptor. Neurobiol Dis. 2005;20:82–92. doi: 10.1016/j.nbd.2005.02.002. [DOI] [PubMed] [Google Scholar]
- Bánfi B, Malgrange B, Knisz J, Steger K, Dubois-Dauphin M, Krause KH. NOX3, a superoxide-generating NADPH oxidase of the inner ear. J Biol Chem. 2004;279:46065–46072. doi: 10.1074/jbc.M403046200. [DOI] [PubMed] [Google Scholar]
- Burridge K, Wennerberg K. Rho and Rac take center stage. Cell. 2004;116:167–179. doi: 10.1016/s0092-8674(04)00003-0. [DOI] [PubMed] [Google Scholar]
- Cole KK, Perez-Polo JR. Poly(ADP-ribose) polymerase inhibition prevents both apoptotic-like delayed neuronal death and necrosis after H(2)O(2) injury. J Neurochem. 2002;82:19–29. doi: 10.1046/j.1471-4159.2002.00935.x. [DOI] [PubMed] [Google Scholar]
- Cullen PJ. Decoding complex Ca2+ signals through the modulation of Ras signaling. Curr Opin Cell Biol. 2006;18:157–161. doi: 10.1016/j.ceb.2006.02.012. [DOI] [PubMed] [Google Scholar]
- Flinder LI, Timofeeva OA, Rosseland CM, Wierød L, Huitfeldt HS, Skarpen E. EGF-induced ERK-activation downstream of FAK requires rac1-NADPH oxidase. J Cell Physiol. 2011;226:2267–2278. doi: 10.1002/jcp.22563. [DOI] [PubMed] [Google Scholar]
- Fridberger A, Flock A, Ulfendahl M, Flock B. Acoustic overstimulation increases outer hair cell Ca2+ concentrations and causes dynamic contractions of the hearing organ. Proc Natl Acad Sci U S A. 1998;95:7127–7132. doi: 10.1073/pnas.95.12.7127. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Goode BL, Eck MJ. Mechanism and function of formins in the control of actin assembly. Annu Rev Biochem. 2007;76:593–627. doi: 10.1146/annurev.biochem.75.103004.142647. [DOI] [PubMed] [Google Scholar]
- Hallett MA, Dagher PC, Atkinson SJ. Rho GTPases show differential sensitivity to nucleotide triphosphate depletion in a model of ischemic cell injury. Am J Physiol Cell Physiol. 2003;285:C129–138. doi: 10.1152/ajpcell.00007.2003. [DOI] [PubMed] [Google Scholar]
- Hardie DG. the AMP-activated protein kinase cascade: the key sensor of cellular energy status. Endocrinology. 2003;144:5179–5183. doi: 10.1210/en.2003-0982. [DOI] [PubMed] [Google Scholar]
- Heinrich UR, Maurer J, Mann W. Ultrastructural evidence for protection of the outer hair cells of the inner ear during intense noise exposure by application of the organic calcium channel blocker diltiazem. ORL J Otorhinolaryngol Relat Spec. 1999;61:321–327. doi: 10.1159/000027693. [DOI] [PubMed] [Google Scholar]
- Henderson D, Bielefeld EC, Harris KC, Hu BH. The role of oxidative stress in noise-induced hearing loss. Ear Hear. 2006;27:1–19. doi: 10.1097/01.aud.0000191942.36672.f3. [DOI] [PubMed] [Google Scholar]
- Hordijk PL. Regulation of NADPH oxidases: the role of Rac proteins. Circ Res. 2006;98:453–462. doi: 10.1161/01.RES.0000204727.46710.5e. [DOI] [PubMed] [Google Scholar]
- Hu BH, Henderson D, Nicotera TM. F-actin cleavage in apoptotic outer hair cells in chinchilla cochleas exposed to intense noise. Hear Res. 2002;172:1–9. doi: 10.1016/s0378-5955(01)00361-6. [DOI] [PubMed] [Google Scholar]
- Jiang H, Sha SH, Schacht J. Rac/Rho pathway regulates actin depolymerization induced by aminoglycoside antibiotics. J Neurosci Res. 2006;83:1544–1551. doi: 10.1002/jnr.20833. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kalinec GM, Webster P, Lim DJ, Kalinec F. A cochlear cell line as an in vitro system for drug ototoxicity screening. Audiol Neurootol. 2003;8:177–189. doi: 10.1159/000071059. [DOI] [PubMed] [Google Scholar]
- Kim HJ, Lee JH, Kim SJ, Oh GS, Moon HD, Kwon KB, Park C, Park BH, Lee HK, Chung SY, Park R, So HS. Roles of NADPH oxidases in cisplatin-induced reactive oxygen species generation and ototoxicity. J Neurosci. 2010;30:3933–3946. doi: 10.1523/JNEUROSCI.6054-09.2010. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lee YM, Lee JO, Jung JH, Kim JH, Park SH, Park JM, Kim EK, Suh PG, Kim HS. Retinoic acid leads to cytoskeletal rearrangement through AMPK-Rac1 and stimulates glucose uptake through AMPK-p38 MAPK in skeletal muscle cells. J Biol Chem. 2008;283:33969–33974. doi: 10.1074/jbc.M804469200. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Levine YC, Li GK, Michel T. Agonist-modulated regulation of AMP-activated protein kinase (AMPK) in endothelial cells. Evidence for an AMPK → Rac1 → Akt → endothelial nitric-oxide synthase pathway. J Biol Chem. 2007;282:20351–20364. doi: 10.1074/jbc.M702182200. [DOI] [PubMed] [Google Scholar]
- Li LY, Luo X, Wang X. Endonuclease G is an apoptotic DNase when released from mitochondria. Nature. 2001;412:95–99. doi: 10.1038/35083620. [DOI] [PubMed] [Google Scholar]
- Lockshin RA, Zakeri Z. Caspase-independent cell deaths. Curr Opin Cell Biol. 2002;14:727–733. doi: 10.1016/s0955-0674(02)00383-6. [DOI] [PubMed] [Google Scholar]
- Lynch ED, Lee MK, Morrow JE, Welcsh PL, León PE, King MC. Nonsyndromic deafness DFNA1 associated with mutation of a human homolog of the Drosophila gene diaphanous. Science. 1997;278:1315–1318. [PubMed] [Google Scholar]
- Miller JM, Brown JN, Schacht J. 8-iso-prostaglandin F(2alpha), a product of noise exposure, reduces inner ear blood flow. Audiol Neurootol. 2003;8:207–221. doi: 10.1159/000071061. [DOI] [PubMed] [Google Scholar]
- Minami SB, Yamashita D, Schacht J, Miller JM. Calcineurin activation contributes to noise-induced hearing loss. J Neurosci Res. 2004;78:383–392. doi: 10.1002/jnr.20267. [DOI] [PubMed] [Google Scholar]
- Minami SB, Yamashita D, Ogawa K, Schacht J, Miller JM. Creatine and tempol attenuate noise-induced hearing loss. Brain Res. 2007;1148:83–89. doi: 10.1016/j.brainres.2007.02.021. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Muñoz DJ, Kendrick IS, Rassam M, Thorne PR. Vesicular storage of adenosine triphosphate in the guinea-pig cochlear lateral wall and concentrations of ATP in the endolymph during sound exposure and hypoxia. Acta Otolaryngol. 2001;121:10–15. doi: 10.1080/000164801300006209. [DOI] [PubMed] [Google Scholar]
- Nagashima R, Yamaguchi T, Kuramoto N, Ogita K. Acoustic overstimulation activates 5′-AMP-activated protein kinase through a temporary decrease in ATP level in the cochlear spiral ligament prior to permanent hearing loss in mice. Neurochem Int. 2011;59:812–820. doi: 10.1016/j.neuint.2011.08.015. [DOI] [PubMed] [Google Scholar]
- Nimnual AS, Taylor LJ, Bar-Sagi D. Redox-dependent downregulation of Rho by Rac. Nat Cell Biol. 2003;5:236–241. doi: 10.1038/ncb938. [DOI] [PubMed] [Google Scholar]
- Oishi N, Schacht J. Emerging treatments for noise-induced hearing loss. Expert Opin Emerg Drugs. 2011;16:235–245. doi: 10.1517/14728214.2011.552427. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Oliver D, Ludwig J, Reisinger E, Zoellner W, Ruppersberg JP, Fakler B. Memantine inhibits efferent cholinergic transmission in the cochlea by blocking nicotinic acetylcholine receptors of outer hair cells. Mol Pharmacol. 2001;60:183–189. doi: 10.1124/mol.60.1.183. [DOI] [PubMed] [Google Scholar]
- Rao PV. The pulling, pushing and fusing of lens fibers: a role for Rho GTPases. Cell Adh Migr. 2008;2:170–173. doi: 10.4161/cam.2.3.6495. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sarfstein R, Gorzalczany Y, Mizrahi A, Berdichevsky Y, Molshanski-Mor S, Weinbaum C, Hirshberg M, Dagher MC, Pick E. Dual role of Rac in the assembly of NADPH oxidase, tethering to the membrane and activation of p67phox: a study based on mutagenesis of p67phox-Rac1 chimeras. J Biol Chem. 2004;279:16007–16016. doi: 10.1074/jbc.M312394200. [DOI] [PubMed] [Google Scholar]
- Spiering D, Hodgson L. Dynamics of the Rho-family small GTPases in actin regulation and motility. Cell Adh Migr. 2011;5:170–180. doi: 10.4161/cam.5.2.14403. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Størling J, Zaitsev SV, Kapelioukh IL, Karlsen AE, Billestrup N, Berggren PO, Mandrup-Poulsen T. Calcium has a permissive role in interleukin-1beta-induced c-jun N-terminal kinase activation in insulin-secreting cells. Endocrinology. 2005;146:3026–3036. doi: 10.1210/en.2005-0036. [DOI] [PubMed] [Google Scholar]
- Takeya R, Sumimoto H. Molecular mechanism for activation of superoxide-producing NADPH oxidases. Mol Cells. 2003;16:271–277. [PubMed] [Google Scholar]
- Takeya R, Ueno N, Kami K, Taura M, Kohjima M, Izaki T, Nunoi H, Sumimoto H. Novel human homologues of p47phox and p67phox participate in activation of superoxide-producing NADPH oxidases. J Biol Chem. 2003;278:25234–25246. doi: 10.1074/jbc.M212856200. [DOI] [PubMed] [Google Scholar]
- Thalmann I, Marcus NY, Thalmann R. Adenine nucleotides of the organ of Corti under metabolic stress. Laryngoscope. 1982;92:199–203. doi: 10.1002/lary.1982.92.2.199. [DOI] [PubMed] [Google Scholar]
- Thalmann R, Miyoshi T, Thalmann I. The influence of ischemia upon the energy reserves of inner ear tissues. Laryngoscope. 1972;82:2249–2272. doi: 10.1288/00005537-197212000-00013. [DOI] [PubMed] [Google Scholar]
- Ueyama T, Geiszt M, Leto TL. Involvement of Rac1 in activation of multicomponent Nox1- and Nox3-based NADPH oxidases. Mol Cell Biol. 2006;26:2160–2174. doi: 10.1128/MCB.26.6.2160-2174.2006. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Vicente-Torres MA, Schacht J. A BAD link to mitochondrial cell death in the cochlea of mice with noise-induced hearing loss. J Neurosci Res. 2006;83:1564–1572. doi: 10.1002/jnr.20832. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Vlajkovic SM, Housley GD, Muñoz DJ, Robson SC, Sévigny J, Wang CJ, Thorne PR. Noise exposure induces up-regulation of ecto-nucleoside triphosphate diphosphohydrolases 1 and 2 in rat cochlea. Neuroscience. 2004;126:763–773. doi: 10.1016/j.neuroscience.2004.04.023. [DOI] [PubMed] [Google Scholar]
- Watanabe N, Kato T, Fujita A, Ishizaki T, Narumiya S. Cooperation between mDia1 and ROCK in Rho-induced actin reorganization. Nat Cell Biol. 1999;1:136–143. doi: 10.1038/11056. [DOI] [PubMed] [Google Scholar]
- Winder WW, Thomson DM. Cellular energy sensing and signaling by AMP-activated protein kinase. Cell Biochem Biophys. 2007;47:332–347. doi: 10.1007/s12013-007-0008-7. [DOI] [PubMed] [Google Scholar]
- Yamashita D, Jiang HY, Schacht J, Miller JM. Delayed production of free radicals following noise exposure. Brain Res. 2004a;1019:201–209. doi: 10.1016/j.brainres.2004.05.104. [DOI] [PubMed] [Google Scholar]
- Yamashita D, Miller JM, Jiang HY, Minami SB, Schacht J. AIF and EndoG in noise-induced hearing loss. Neuroreport. 2004b;15:2719–2722. [PubMed] [Google Scholar]
- Yamashita D, Jiang HY, Le Prell CG, Schacht J, Miller JM. Post-exposure treatment attenuates noise-induced hearing loss. Neuroscience. 2005;134:633–642. doi: 10.1016/j.neuroscience.2005.04.015. [DOI] [PubMed] [Google Scholar]
- Zhang Y, Gong LH, Zhang HQ, Du Q, You JF, Tian XX, Fang WG. Extracellular ATP enhances in vitro invasion of prostate cancer cells by activating Rho GTPase and upregulating MMPs expression. Cancer Lett. 2010;293:189–197. doi: 10.1016/j.canlet.2010.01.010. [DOI] [PubMed] [Google Scholar]