

Abstract
Polyphosphoinositides (PPIs) and in particular phosphatidylinositol-(4,5)-bisphosphate (PI4,5P2), control many cellular events and bind with variable levels of specificity to hundreds of intracellular proteins in vitro. The much more restricted targeting of proteins to PPIs in cell membranes is thought to result in part from the formation of spatially distinct PIP2 pools, but the mechanisms that cause formation and maintenance of PIP2 clusters are still under debate. The hypothesis that PIP2 forms sub-micron-size clusters in the membrane by electrostatic interactions with intracellular divalent cations is tested here using lipid monolayer and bilayer model membranes. Competitive binding between Ca2+ and Mg2+ to PIP2 is quantified by surface pressure measurements and analyzed by a Langmuir competitive adsorption model. The physical chemical differences among three PIP2 isomers are also investigated. Addition of Ca2+, but not Mg2+, Zn2+ or polyamines to PIP2-containing monolayers induces surface pressure drops coincident with the formation of PIP2 clusters visualized by fluorescence, atomic force and electronic microscopy. Studies of bilayer membranes using steady-state probe-partitioning fluorescence resonance energy transfer (SP-FRET) and fluorescence correlation spectroscopy (FCS) also reveal Me2+-induced cluster formation or diffusion retardation which follows the trend: Ca2+ ≫ Mg2+ > Zn2+, and polyamines have minimal effects. These results suggest that divalent metal ions have substantial effects on PIP2 lateral organization at physiological concentrations, and local fluxes in their cytoplasmic levels can contribute to regulating protein-PIP2 interactions.
INTRODUCTION
Polyphosphoinositides (PPIs) affect numerous physiological functions including cytoskeleton remodeling,1 ion channel and transporter activation,2 peripheral membrane protein docking,3 and vesicle traffic.4 The most abundant PPI, phosphatidylinositol-4,5-bisphosphate (PI4,5P2) which accounts for less than 1 % of total phospholipid, when immobilized on liposomes or beads binds nearly 280 intracellular proteins5 but in vivo is highly selective in binding specific proteins in particular locations and times within the cell. Much more is known about the biochemical interactions of PIP2 with isolated purified proteins than about how signaling events are regulated locally by PIP2. Even the existence of local membrane domains enriched in PIP2 is a matter of dispute6 in part because the strong electrostatic repulsions between highly anionic head groups of PIP2 might be expected to prevent its specific lateral organization within the lipid bilayer. Consequently, most studies of PIP2-protein binding treat the lipid essentially as a monomer that is randomly distributed and freely diffusible within the bilayer and is constrained only when bound to a protein.7
In addition to protein-lipid binding, other mechanisms for generating local enrichment of PIP2 have also been hypothesized to explain the formation of spatially distinct PIP2 pools in the plasma membrane. As reviewed elsewhere,6,8,9 these mechanisms include hydrogen bond networking through polar lipid head groups10,11; partitioning into cholesterol-rich, raft-like domains12,13; partitioning away from cholesterol-rich domains14,15; local production from PI4P by PI4P-5K1,16; electrostatic sequestering7,17; and protein fence models.18,19 The electrostatic sequestering model describes how a significant fraction of PIP2 is sequestered by peptides or proteins with polybasic domains, such as MARCKS.20 The sequestered PIP2 is not able to interact with other target proteins, but can be liberated when Ca2+/calmodulin reverses the binding of MARCKS. This hypothesis clearly elaborates how proteins with polybasic domains lead to the sequestering of PIP2 and motivates the hypothesis that protein-PIP2 interactions in turn might be affected by electrostatic interactions between PIP2 and other intracellular multivalent cations such as polyamines and divalent metal ions.
As a parallel and complementary model for the electrostatic sequestering hypothesis, we test here the hypothesis that formation of PIP2-rich clusters or microdomains is triggered and stabilized by the electrostatic interactions between PIP2 and physiologically common cations, mostly divalent cations and polyamines. In this study the term cluster refers to membrane regions enriched in PIP2 due to counterion-mediated attractive interactions between these lipids, as opposed to the term domain, which often refers to lipid demixing within membranes caused, for example, by cholesterol–dependent phase transitions in which PIP2 might enrich in a specific phase but is not specifically required for its formation. As suggested in a previous study14, divalent cations such as Ca2+ induce like-charge attraction between highly charged PIP2 in lipid model membranes. This report quantifies differences in the clustering capacity of divalent metal ions, measures the size distribution of PIP2 nano- and micro-domains, and identifies differences among the three naturally occurring PIP2 isomers. These data document the significant effect of intracellular ions on PIP2 structure in the membrane that could impact protein-PIP2 interactions, and suggest a potential feedback loop between local PIP2 and Ca2+ signaling.
EXPERIMENTAL SECTION
Lipids and Reagents
Natural PIP2 (porcine brain L-α-phosphatidylinositol-4,5-bisphosphate), synthetic PIP2 analogs (dioleoyl phosphatidylinositol-(x,y)- bisphosphate) and neutral phospholipids such as SOPC (1-stearoyl-2-oleoyl-sn-glycero-3-phosphocholine), DOPC (1,2-dioleoyl-sn-glycero-3-phosphocholine) and Rho-DOPE (1,2-dioleoyl-sn-glycero-3-phosphoethanolamine-N-(lissamine rhodamine B sulfonyl)) were from Avanti (Alabaster, AL). Fluorescently-labeled PIP2 analogs GloPIPs BODIPY-TMR PI(4,5)P2 (C16) and GloPIPs BODIPY-FL PI(4,5)P2 (C16) are purchased from Echelon Biosciences (Salt Lake City, UT). Lipids were dissolved in chloroform/methanol 2:1 mixed solvent, and the concentrations of unlabeled lipid stock solutions were routinely monitored by a phosphorus assay as described elsewhere.21 The concentrations of fluorescently labeled lipids were calibrated by their fluorescence intensity. Subphase reagents HEPES, EDTA, CaCl2, MgCl2, NaCl, sucrose and glucose were purchased from Fisher Scientific (Hampton, NH); other subphase reagents such as ethylenediamine (EDA), diethyl-enetriamine (DETA), triethylenetetramine (TETA), ZnCl2 and cholestanol were purchased from Sigma-Aldrich (St. Louis, MO) and dithiothreitol (DTT) was purchased from Research Product Int. Corp. (Mt. Prospect, IL).
Binding Affinity Measurements
Similar to the cation binding affinity studies performed by Ohki et. al.22-24, a simplified Ca2+-binding affinity assay was carried out as described previously.14 Limited by the complexity in the binding stoichiometry of highly charged PIP2 molecules, which have net charges that can vary from -3 to -5 under most experimental conditions25,26, only a global binding constant is reported using a Langmuir adsorption model. The measurement is performed on a MicroTroughX Langmuir trough (Kibron Inc. Helsinki, Finland) controlled by the FilmWare 3.57 software package (Kibron). Monolayer subphases were prepared with 10 mM HEPES, 1 μM EDTA, and 5 mM DTT at pH 7.4 dissolved in 18.2 MΩ ddH2O. For each measurement, 7 nmol of pre-mixed lipid was deposited on 30 mL buffered solution, and the monolayer surface pressure was monitored with a surface probe using the Wilhelmy method.27 When the surface pressure reached equilibrium at 20 mN/m, concentrated cation stock solution (less than 0.3% of subphase volume fraction) was injected into the subphase and gently mixed without perturbing the monolayer. The surface pressure change was then recorded until the surface pressure again reached equilibrium. For other cations, such as Mg2+ and polyamines, a competitive Ca2+-binding assay was carried out by titrating Ca2+ in the presence of the other cations at various concentrations. The surface pressure measurement was analyzed using the Langmuir competitive adsorption model.28,29
Imaging Supported Lipid Monolayers
Supported lipid monolayers were prepared by transferring monolayers from the Langmuir trough onto glass coverslips using the Langmuir-Schaeffer method. For fluorescence microscopy, lipid monolayers were doped with 0.1 mol% BODIPY-FL PIP2 and Rho-DOPE and examined under an inverted microscope (Leica, DM IRBE) with a 100X oil objective. AFM images of air-dried supported lipid monolayers were taken using tapping mode AFM (Digital Instruments, Santa Barbara, CA) and processed by Nanoscope® IIIa software (v. 5.12; Digital Instruments). For fluid phase AFM, the transferred lipid sample was again immersed in its subphase solution and imaged by the Bioscope AFM (Digital Instruments, Santa Barbara, CA). Air-dried supported lipid monolayer samples were further processed for EM imaging. Samples were unilaterally coated with a thin layer of platinum (1 nm) from a 20° angle and carbon (5 nm) from an ~80° angle with an Auto306 vacuum evaporator (Edwards, UK). The coated sample was floated on a diluted hydrofluoric acid solution to separate from the coverslip, and transferred onto formvar-coated EM grids. Samples were analyzed using a JEM-1011 transmission electron microscope (JEOL USA, Peabody, MA) at an accelerating voltage of 100 kV. Images were captured by an ORIUS 835.10W CCD camera (Gatan, Warrendale, PA)
Infrared Spectroscopy of Supported Lipid Monolayers
Attenuated total reflection Fourier transform infrared (ATR-FTIR) spectra were collected using a Magna-IR 860 spectrometer with a Harrick’s Horizon attachment and equipped with Opus software. Pure PIP2 monolayers were transferred onto a germanium internal reflection element (IRE) and immersed in their subphase buffer solution. The infrared spectra of supported PIP2 monolayers with different divalent cation concentrations were monitored at room temperature in the range of 650-4000 cm-1, using a resolution of 1 cm-1.
PIP2 Phase-Partitioning on LUVs
Steady-state probe-partitioning Förster resonance energy transfer (SP-FRET)30 was used to probe PIP2 phase demixing within a bilayer membrane. For this purpose, two different fluorescent PIP2 analogs were used: BODIPY-FL and BODIPY-TMR PI(4,5)P2. 5.2 mol% PIP2-containing LUVs (including 0.3 mol% each fluorescent PIP2 analog) were prepared using a mini-extruder (Avanti, Alabaster, AL). While the probe-probe distance is expected to be 120 Å and the Förster distance for the selected FRET pair is about 57 Å31, an increase in FRET efficiency is expected as titrated cations induce the formation of PIP2-rich clusters. Holding the PIP2 mole fraction and overall PIP2 concentration in the LUV as constants, the fluorescence spectra for D-A (donor with acceptor), D (donor only) and A (acceptor only) were collected independently using a LS-50B Luminescence Spectrometer (Perkin-Elmer, Beaconsfield, UK). The spectra were combined linearly to calculate the concentration-dependent fluorescence intensity change and FRET efficiency change, and to rule out potential artifacts of fluorescence decay due to environmental changes. For polyamine titrations, the pH of each polyamine stock solution was adjusted to 7.4, and 5 mM DTT was added to stock solutions to keep polyamines from oxidizing.
Nanocluster Formation and Diffusion Retardation on GUVs
5 mol% PIP2-containing GUVs were prepared by electroswelling.32 0.1 nmol of lipid mixture solution was spread and dried on indium-doped tin oxide (ITO) glasses (Delta Technologies, Loveland, CO) on a hotplate at 60 °C. Following vacuum drying for 2 hrs, the ITO was assembled with another clean ITO with a Fastwell silicon spacer (Grace Bio-Labs, Bend, OR) and filled with a 300 mM sucrose solution. The samples were then left in a homemade heating block, heated at 60 °C and applied with a 1V AC field at 5 Hz for two hours using a function generator (B & K Precision, Yorba Linda, CA) and monitored by an oscilloscope (B & K Precision).
GUVs were asymmetrically labeled with approximately 0.5 and 0.01 mol% BODIPY-TMR PIP2 for imaging and FCS, respectively. GUVs, diluted in isotonic glucose solutions containing 10 mM HEPES and various multivalent cations, were added to vacuum grease-sealed chambers covered with clean coverslips. The samples were kept in the dark for at least 30 mins to allow the GUVs to settle down to the glass surface. The diffusion of PIP2 in model membranes was studied by fluorescence correlation spectroscopy (FCS). The experimental setup, sample preparation, data acquisition, and analysis protocols are described elsewhere.33 GUVs sealed in the chamber were allowed to sit for 30 min prior to fluorescence intensity fluctuation measurements. A 514 nm laser was focused near the top center of the GUVs to avoid lipid-solid support interaction. Each FCS curve was obtained by correlating the fluorescence signal for a duration of about 30 s and fit by a multi-component two dimensional diffusion equation to yield its characteristic diffusion times (τD). For each condition, 11 to 25 auto-correlation curves were collected from multiple vesicles.
RESULTS
Competitive Binding of Me2+ to PIP2
Previous work, confirmed here (data not shown) reported that Ca2+ induces PIP2 clustering on a lipid monolayer accompanied by a significant surface pressure drop, which could be recovered by adding EDTA into the subphase solution.14 The binding affinity of Ca2+ can be determined directly through titration of surface pressure measurements. In contrast to Ca2+, Mg2+ has minimal effect on surface pressure upon binding to PIP2 under the same experimental condition. Therefore, binding affinities of Mg2+ to PIP2 were investigated by surface pressure measurements through competitive titration with Ca2+ (Fig 1A). The competitive Langmuir adsorption model is described in Eq. 1-3. From a conventional adsorption equilibrium equation and mass balance, [S]total = [S-Ca2+] + [S-Mg2+] + [S], where [S] is the # of free binding sites over the system volume, [S-Ca2+] and [S-Mg2+] indicates the concentrations of binding sites occupied by Ca2+ and Mg2+, respectively. The competitive binding of Ca2+ and Mg2+ to free binding sites on the membrane can be expressed as:
| (1) |
Figure 1.
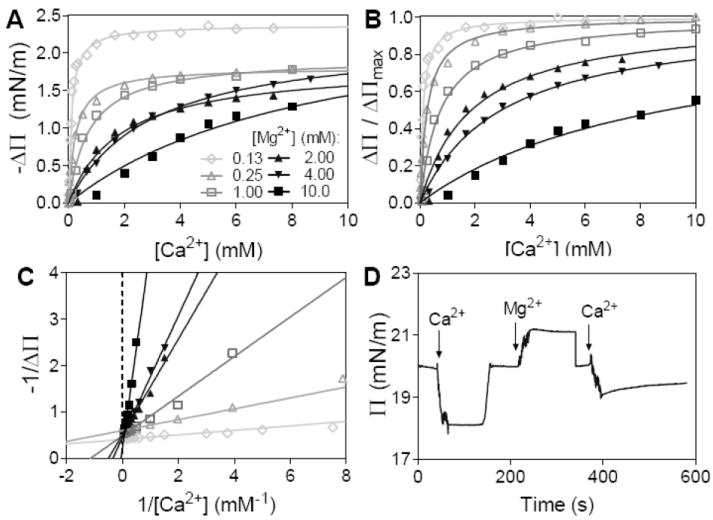
Competitive binding of Mg2+ and Ca2+ to PIP2-containing monolayers. (A) Langmuir adsorption isotherms of calcium binding to L-α-PI(4,5)P2 under different Mg2+ concentration. (B) Same isotherms normalized to their ΔП Max. (C) Langmuir plot of calcium adsorption isotherms. (D) Time course of surface pressure change when 1 mM Ca2+, 10 mM Mg2+, and 9 mM Ca2+ were added sequentially to a monolayer. (Surface pressures were reset to 20 mN/m using the motorized barriers each time before adding divalent cations.) For all surface pressure experiments, 25 mol% PIP2 in SOPC monolayers were used on 10 mM HEPES buffered subphase with 1 μM EDTA and 5 mM DTT, at pH 7.4, room temperature.
The term at the bottom right can be described as a conditional Ca2+ dissociation constant (KcD,Ca) which depends on Mg2+ concentration. By assuming that the degree of normalized surface pressure drop is proportional only to the coverage fraction θ of Ca2+ over free binding sites, i.e. θ=[S-Ca2+]/[S]-total=ΔΠ/ΔΠMax, the surface pressure change can now be linked to the Mg2+-dependent conditional PIP2-binding affinity of Ca2+ (KcD,Ca) as described by Eq. 2:
| (2) |
KD,Ca and KcD,Ca can be measured independently under conditions without and with Mg2+, respectively, and the apparent binding affinity of Mg2+ (KD,Mg) can also be determined. Langmuir adsorption isotherms of Ca2+ binding to PIP2 under different magnesium concentrations are shown in Figure 1A and normalized in Figure 1B. The data are fit by Eq. 2 and without any constraints using GraphPad Prism v.5.03 (Graph-Pad software, La Jolla, CA). For 25 mol% PIP2 at pH 7.4 (10 mM HEPES, 1 μM EDTA and 5 mM DTT), the averaged ΔΠmax is determined to be 2.2 ± 0.4 mN/m. The double-reciprocal Langmuir plot indicates Mg2+ as a competitive inhibitor in preventing Ca2+ from condensing PIP2-containing model membranes (Fig. 1C). Such competitive binding is also shown in a time-course experiment: the surface pressure drop induced by 1 mM Ca2+ can be partially recovered by sequentially adding 10 mM Mg2+ into the subphase. The recovery of surface pressure is not due to electrostatic screening of the PIP2 headgroup since adding more Ca2+ until the total calcium concentration reaches 10 mM leads again a drop in surface pressure (Fig. 1D). Such competitive binding and its antagonistic effect on tuning surface pressure can be well described by Eq. 3:
| (3) |
Using natural extracted PI(4,5)P2, the determined apparent dissociation constants for Ca2+ and Mg2+ are 4.6 ± 1.3 and 7.7 ± 1.8 μM, respectively. The fact that both divalent cations show similar binding affinity to PIP2 monolayers is consistent with the argument that the binding is mainly driven through electrostatic interactions.34
Intrinsic Binding Constants of Ca2+ and Mg2+ to PIP2-containing Membranes
The affinity of counterions to monolayers containing charged lipids is the result of both intrinsic affinity to the lipid monomer and electrostatic attraction due to the surface potential caused by the charged lipids at the interface. An intrinsic binding constant should be independent of surface potential which varies with the charge density, the pH, and the ionic strength.35 As the charge density (φPIP2), the pH, and the monovalent salt concentration are fixed for all monolayer studies, the surface potential can be simplified as a function of divalent cation concentration. Therefore, the effective association constant (Ka) at any given [M2+] can be described as:
| (4) |
Here KI,M is the intrinsic binding constant of the divalent cation M2+ to PIP2. Eq. 5 holds true at any given [Ca2+], and considering that [Ca2+]free ≈[Ca2+]T at calcium concentration > 1 μM, Ka([Ca2+]) can be approximated by dθ/d[Ca2+]T as described in eq.6:
| (5) |
| (6) |
The measured KD,Ca can therefore be interpreted as the effective dissociation constant as [Ca2+] approaches 0. Combining eq.(4) and (6), the apparent KD,Ca can be shown to depend only on the initial surface potential, ψ0([Mg2+]), and the intrinsic binding constant (KI,Ca) can be calculated if the surface potential of a PIP2-containing monolayer with no magnesium is known. On the other hand, when there is magnesium in the solution, the binding constant can be described as:
| (7) |
Therefore, competitive binding between calcium and magnesium can be described in a surface potential-dependent manner. Since only the boundary potential, a combination of dipole potential and surface potential, is directly measureable from a lipid monolayer,36 the surface potential can be estimated by zeta potential studies of LUVs with the same lipid composition. The zeta potential of PIP2-containing LUVs have been measured at only slightly different ionic conditions,37,38 and a reasonable estimate of the surface potential of 25 mol% PIP2 LUVs in 10 mM HEPES at pH7.4 is around -80 mV. Combining this value of surface potential with the measurement of effective binding constants determined from Figure 1, leads to estimated intrinsic binding constants for Ca2+ and Mg2+ at around 360 and 220 M-1, respectively. These values are consistent with those reported previously.38
Ca2+/Mg2+ Selectivity of Different PIP2 Isomers
To determine if there is structural specificity between PIP2 isomers in binding to divalent cations, we apply the same competitive binding assay to three different synthetic PIP2 analogs: DOPI(3,4)P2, DOP(3,5)P2 and DOPI(4,5)P2. All 25 mol% PIP2s-containing lipid monolayers show similar affinities for calcium (Fig. 2A). However, the conditional binding affinities of PIP2s in binding to Ca2+ becomes different at millimolar magnesium (Fig. 2B). The Ca2+-induced surface pressure drop is inhibited by Mg2+ to different degrees, depending on the PIP2 isomer (Fig. 2C). While the three PIP2 isomers show the same KD,Ca but their different conditional Ca2+ binding affinity at millimolar magnesium implies they have different KD,Mg. The binding affinities for Ca2+ and Mg2+ and their selectivity ratio (KD,Mg/KD,Ca) for natural PI(4,5)P2 and synthetic PIP2 isomers are summarized in Fig. 2D & E. In short, L-α-PI(4,5)P2 has slightly lower cation binding affinities compared to DOPI(4,5)P2 at the same condition. This difference is likely due to the fact that L-α-PI(4,5)P2 is a lipid mixture in which the majority contains the highly unsaturated arachidonyl chain that leads to a slightly higher area per molecule and therefore lower charge density. The fact that natural and synthetic PI(4,5)P2 have similar selectivity ratios suggests that the packing of lipids with cations is mainly determined by the head group conformation. Moreover, PI(4,5)P2 among three PIP2 isomers shows the highest Ca2+- binding preference and PI(3,5)P2 has a preference for Mg2+ over Ca2+. These subtle physical chemical differences among the three PIP2 isomers might be relevant to the mechanisms of their different physiological roles.
Figure 2.
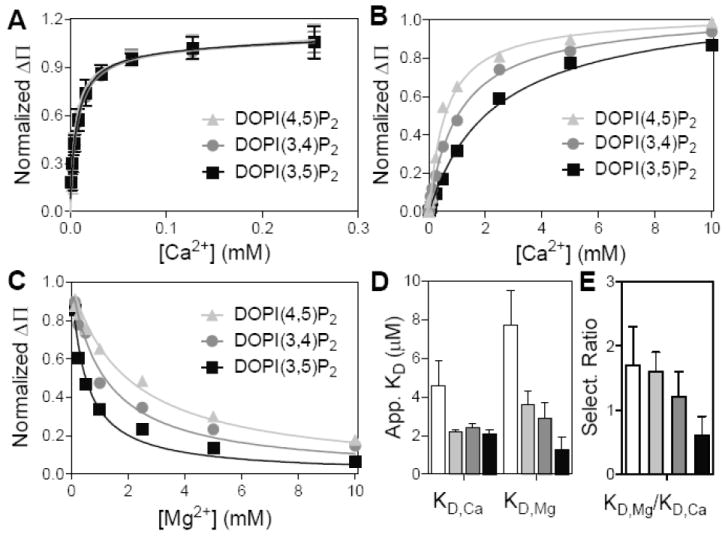
Me2+-binding selectivities of PIP2 isomers. (A) The binding of Ca2+ to three PIP2 isomers without Mg2+. Mean ± SE, n = 3. (B) Similar Ca2+ binding curve at 1 mM Mg2+. (C) Surface pressure drop induced by 1 mM Ca2+ under different Mg2+ concentrations. Data were fit by the Langmuir adsorption model (Eq. 2) to find (D) the apparent KD for Ca2+ and Mg2+, and (E) the selective ratio in binding to PIP2s. The color coding is consistent through (A) to (E), while the white bar in last two panels represents L-α-PI(4,5)P2.
Formation of PIP2-rich Clusters
Limitations due to drifting motion, long working distance, and optical diffraction prevent visualization of PIP2-rich nano-size clusters by fluorescence microscopy of free-standing monolayers at the air-water interface. As a first step to look for cluster forming conditions and cluster size distributions, we use the Langmuir-Schaeffer method to create supported lipid monolayers. A 50 mol% PIP2/SOPC monolayer is doped with a trace amount of BODIPY-FL PIP2 and Rho-DOPE and transferred onto a glass coverslip before and after 1 mM Ca2+ is added into the subphase solution. Since the BODIPY fluorophores are labeled on the hydrocarbon tails of PIP2, the head group interaction with calcium is expected to be unperturbed. Examined by fluorescence microscopy, the fluorescently-labeled PIP2 is strongly phase separated from the Rho-DOPE doped background lipids after addition of millimolar Ca2+ (Fig. 3, A-C), and the cluster formation is reversed by adding 10 mM EDTA (Fig. 3D).
Figure 3.

Ca2+ induces phase separation of L-α-PIP2 in background SOPC on supported lipid monolayers. Fluorescence images of 50 mol% PIP2 dual labeled with (A) 0.1 mol% Rhodamine-DOPE and (B) 0.1 mol% C16 BODIPY-FL PI(4,5)P2, were merged in (C), showing Ca2+-induced phase demixing. (D) The phase demixing is reversed by adding excess EDTA. Without fluorescent lipids, Ca2+-induced phase demixing is shown by tapping mode AFM with (E) 2 mol%, (F) 25 mol%, and (G) 50 mol% PIP2. (H) Under the same PIP2 fraction and cation concentration, Mg2+-induced PIP2 clusters are much smaller. The transferred lipid monolayers immersed in buffer (I) with and (J) without Ca2+ are examined also by fluid-phase contact mode AFM. Divalent cation concentration is 1 mM through (A) to (I), while 10 mM EDTA is also added in (D) to test reversibility. Supported lipid monolayers are transferred at constant surface pressure (20 mN/m) unless otherwise indicated.
Potential artifacts from using fluorescent PIP2 analogs are examined. Surface pressure drop measurements using 25 mol% labeled PIP2 instead of natural PIP2 show that labeled PIP2 has somewhat higher affinity for Ca2+ compared to natural PIP2 under the same condition, while the surface pressure drop is smaller. (Supplementary Fig. S1A) AFM imaging of the same transferred monolayer shows that the clusters formed by labeled PIP2 are smaller than the clusters formed by natural PIP2. (Supplementary Fig. S1B&C) Therefore, the formation of PIP2-rich clusters detected by fluorescence microscopy and the surface pressure drop is not due to non-specific interactions between labeled PIP2 molecules.
Further investigations using tapping mode AFM on supported lipid monolayers with different PIP2 fraction show the cluster size increases as the PIP2 mole fraction increases, suggesting that these submicron-size clusters are PIP2-rich (Fig. 3E-G). Reversibility of PIP2 cluster formation after adding excess EDTA is confirmed using AFM (data not shown). The correlation between PIP2 cluster formation and surface pressure change was pointed out previously by Levental et. al.14, and this coincidence is further established by the calcium dependence of cluster formation imaged by AFM (Fig. S2). On the other hand, Mg2+-induced PIP2 clusters appear to be much smaller under the same conditions, indicating a weaker surface condensing ability (Fig. 3H). In order to rule out the possibility that these clusters are transferring or drying defects, an imaging study was also performed by fluid phase contact mode AFM. The transferred samples are immersed in the subphase solutions under conditions with or without calcium. Submicron-size clusters are found only when there is Ca2+ in the subphase solution (Fig. 3I & J).
PIP2 Cluster Formation under Near-physiological Conditions
We further investigated if PIP2-rich clusters form at physiologically reasonable surface pressures, calcium concentrations, monovalent salt concentrations, and PIP2 levels. Cluster formation is first tested at high monovalent salt concentration (Fig. 4A & B) with micromolar Ca2+. Clusters are visible at micromolar Ca2+ when there is merely 1 mol% PIP2 on the supported lipid monolayers under both high surface pressure and high monovalent salt concentration conditions (Fig. 4D). No clusters are found when the subphase solution is free of calcium (Fig 4C).
Figure 4.

Formation of submicron-size L-α-PIP2 clusters in background SOPC at near-physiological conditions. Cluster formation is tested under (A, B) high ionic strength with 25 mol% PIP2 and (C, D) high surface pressure and high ionic strength with 1 mol% PIP2. 10 mol% PIP2 monolayers under different ionic conditions are imaged by both (E, G) AFM and (F, H) TEM. The radius distribution of PIP2-rich clusters in (F) is shown as an inset. Scale bars are 1 μm unless otherwise indicated. (High П: lipid monolayer transferred at 35 mN/m; high I: 150 mM KCl in the subphase.)
Since intracellular [Mg2+] is about 3 orders of magnitude higher than the intracellular [Ca2+] and they both bind to PIP2 with similar affinity, we tested whether millimolar Mg2+ prevents PIP2 from forming nano-size clusters in micromolar Ca2+. Both AFM and TEM studies show that micromolar Ca2+ induces formation of clusters with a radius distribution of 40 ± 11 nm (Fig. 4F, inset) in lipid monolayers. The clusters induced by Ca2+ become flatter and fewer when the monolayer subphase also contains millimolar Mg2+, but they do not disappear (Fig. 4G & H).
Dehydration occurs during titration by Ca2+ but not Mg2+
ATR-FTIR spectroscopy revealed further differences between Ca2+ and Mg2+ binding to PIP2. Pure PIP2 monolayers, transferred onto germanium IREs, were immersed in buffer solution and titrated with divalent cations (Fig. 5A & B). A significant change occurs in the water-related peak intensity. The O-H stretching peak at 3350 cm-1 decreases during titration by Ca2+, but it slightly increases during titration by Mg2+. This result suggests that partial dehydration takes place when Ca2+, but not Mg2+, binds to the PIP2 head groups and is consistent with a differential scanning calorimetry study of Ca2+-PIP2 micelle interactions.39 These results suggest that water between PIP2 head groups might be excluded as a result of PIP2-water hydrogen bonding network disruption11 and divalent cation bridge-binding.40 However, the antisymmetric PO2-stretching at the region 1220-1250 cm-1, which indicates the hydration status of the lipid head group,41 shows no detectable peak shift. Moreover, an increase in surface pressure is expected if the PIP2-water hydrogen bonding network is disrupted, but the opposite is observed as Ca2+ was added. These results suggest a second possible explanation: partial dehydration of the hydration shell of calcium, but not magnesium, upon binding to the lipids. Considering that Ca2+ and Mg2+ have similar hydrated ionic radii42 but very different responses, the difference in hydration is likely to result from the 20% lower dehydration energy of Ca2+ compared to that of Mg2+.43 These two events, loss of water from the lipid and dehydration of the ion are not be mutually exclusive, as suggested in the summary diagram (Fig. 5C)
Figure 5.
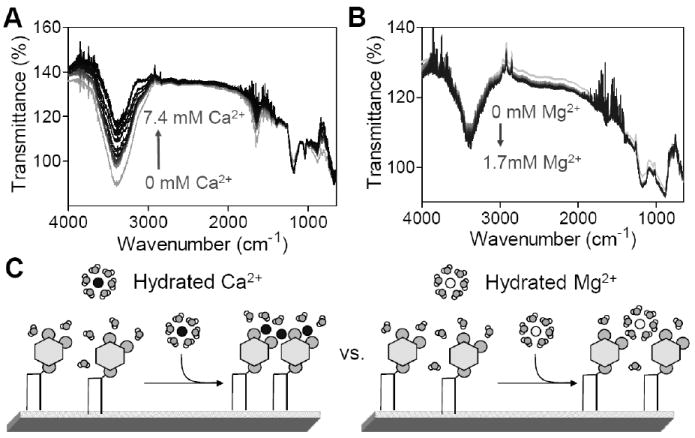
Dehydration upon titration by Ca2+, but not Mg2+. ATR-FTIR spectra are collected during optical titration with (A) Ca2+ and (B) Mg2+ using pure PIP2 supported monolayers. (C) Cartoon pictures show the putative differences between Ca2+ and Mg2+ when interacting with PIP2 head groups.
FRET as a Tool for Monitoring PIP2 Cluster Formation in LUVs
Cation-induced PIP2 cluster formation in LUVs was studied by SP-FRET. The tested ions are compared in two different categories: divalent cations and polyamines. For divalent cations, the ions tested include three physiologically important cations: Ca2+, Mg2+, and Zn2+. Cation concentration-dependent FRET efficiency measurements effectively quantify the degree of PIP2 cluster formation. The trend in inducing PIP2 cluster formation in a bilayer membrane follows the order: Ca2+≫Mg2+>Zn2+ (Fig. 6A). The concentration-dependent fluorescence changes for individual titrations, including EDTA4- back titration with pre-existing 100 μM Ca2+, are also reported (Fig. 6B-D). The apparent binding of Ca2+ determined by FRET appears to be lower, ca. 220 μM, than that measured by surface pressure changes in monolayers. This difference is rationalized by the lower PIP2 fraction (5 mol%) compared to 25 mol% in the monolayer (Supplementary Fig. S3). The surface potential is estimated to be -47 mV lower in a 25 mol% PIP2-containing monolayer, consistent with the PIP2 fraction-dependent zeta potential study carried out by Toner et. al.38
Figure 6.
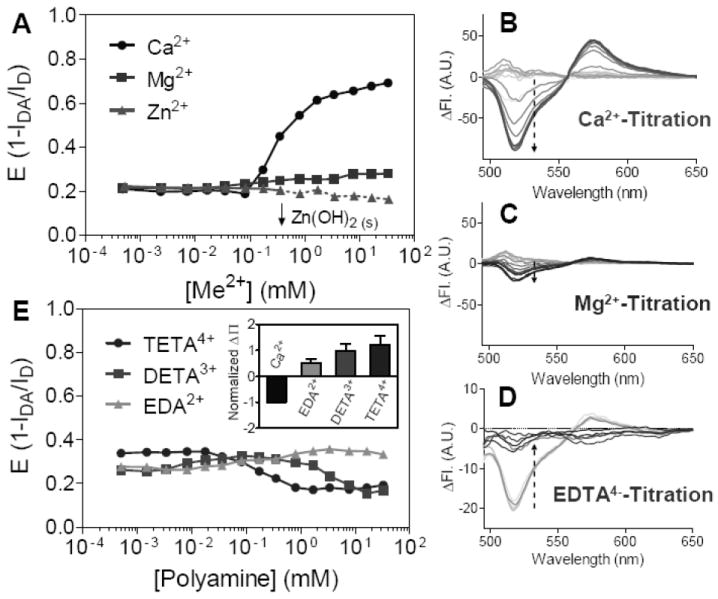
Effects of different cations in SP-FRET on LUVs. (A) Concentration-dependent SP-FRET of BODIPY-FL and BODIPY-TMR PIP2 are measured with physiological divalent cations. Arrow indicates the concentration at which insoluble Zn(OH)2 forms. Fluorescence intensity changes are shown for titration with (B) Ca2+ and (C) Mg2+. (D) Reversibility is tested by titrating with EDTA4- with pre-existing 100 μM Ca2+. (E) The same experiment was repeated using polyamines with charges ranging from +2 to +4; (inset) normalized surface pressure responses of 25 mol% PIP2 monolayers as 1 mM multivalent cations are added. LUVs were excited at 470 nm with overall lipid concentration and PIP2 mole fraction held constant at 5 mol%, including 0.3 mol% each fluorescent PIP2 analog. PIP2/DChol/SOPC = 5/15/80 for all PIP2-containing LUVs. Buffer: 10mM HEPES, 5mM DTT, 1 μM EDTA, pH 7.4.
In contrast to the expectation if binding of counterions was purely electrostatic, polyamines with charges more than +2 are not stronger than divalent metal cations in bridging PIP2 head groups11 (Fig. 6E). EDA (+2 polyamine) can be treated as a weak divalent cation (similar to Mg2+), but polyamines with higher charges and longer backbones are not any stronger in condensing PIP2-containing membranes. This result is consistent with surface pressure measurements of lipid monolayers, in which the addition of polyamines usually leads to an increase of surface pressure, rather than a decrease. As shown in Fig. 6E inset, more charges in the polyamine lead to greater surface pressure increase, suggesting that the sequestering of PIP2 through polybasic domains of a peptide is very different from the PIP2 condensing effect induced by divalent metal cations.
Diffusion Retardation of PIP2 on GUVs
Because SP-FRET studies do not provide information about cluster size distribution on bilayer membranes, we performed fluorescence correlation spectroscopy to study the diffusion of PIP2 in GUVs. In asymmetrically labeled GUVs, rounded and fluid-like PIP2-rich clusters are clearly seen when there is 2 μM Ca2+ in the GUV solution, but not in the control or with 1 mM Mg2+ (Fig. 7A-C). Since nm scale clusters form after addition of Mg2+ to PIP2-containing monolayers, it is possible that Mg2+-induced PIP2 clusters in GUVs are too small to be detected by optical microscopy. Therefore, FCS was used to determine if there are nano-size PIP2 clusters by studying cation-induced diffusion retardation of PIP2. The auto-correlation curves for PIP2 diffusion with different multivalent cations under near-physiological concentrations are shown in Fig. 7D. The data are fit by a two-dimensional diffusion equation as shown in eq 9:
| (9) |
Figure 7.
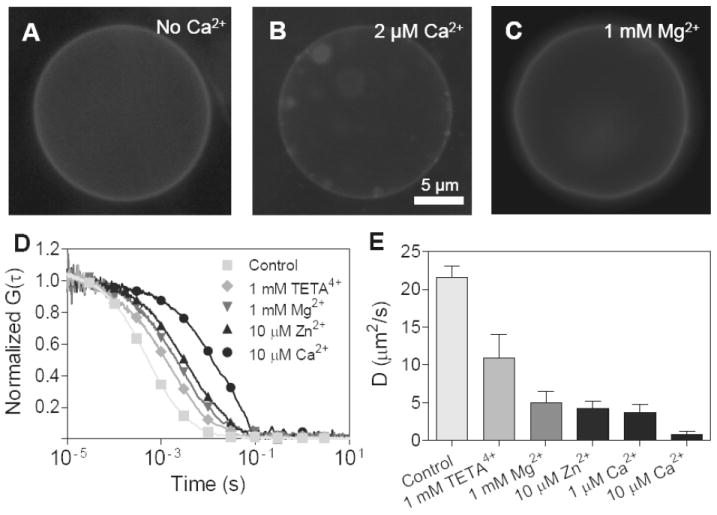
Nano-sized cluster formation and diffusion retardation on PIP2-containing GUVs. Images show the lateral inhomogeneity of asymmetrically labeled BODIPY-TMR PIP2 on GUVs under the following ionic conditions: (A) no divalent cations; (B) 2 μM Ca2+; (C) 1 mM Mg2+. (D) Representative autocorrelation curves of fluorescently labeled PIP2 studied by FCS. (E) PIP2 diffusion coefficient determined by model fitting. Mean ± SE, n > 11. GUVs with PIP2/SOPC= 5/95 were used for imaging and PIP2/DChol/SOPC = 5/15/80 were used for FCS.
The extracted diffusion coefficients under different ionic conditions are shown in Fig. 7E after averaging multiple measurements (n>10). The results show that millimolar magnesium effectively slows down the diffusion of fluorescently labeled PIP2 by 4-fold, showing that the binding of Mg2+ exerts a substantial effect on the membrane consistent with the existence of nano-size Mg2+-induced PIP2 clusters. The diffusion retardation is much more sensitive to Ca2+ than to other divalent cations. The diffusion of PIP2 is slowed down to 0.8 ± 0.4 μm2/s by 10 μM Ca2+, which is very close to the diffusion co-efficient of PIP2 on the inner leaflet of a plasma membrane.44 The fact that TETA4+ has a less significant effect in retarding PIP2 diffusion is consistent with the hypothesis that polyamines with fewer charges (less than +7) are not able to sequester PIP2 since they can not form complexes with more than one PIP2 molecule.9 On the other hand, this result supports the idea that the major difference between divalent cations and polyamines may come from their different packing configuration with anionic lipid head groups.34
DISCUSSION
Electrostatic interactions between PPIs and divalent cations first attracted attention in late 50s.45,46 Binding affinities of common divalent cations for PIP2 were first determined by pH titration in aqueous PIP2 micelle solutions,47 and followed by several studies measuring the binding affinity by quantifying the partitioning of radioactive 45Ca on a PIP2-containing water-methanol-chloroform solution,48 lipid monolayer,49 or erythrocyte ghost membranes.50 More recently, the adsorption of divalent cations to PIP2 was determined through electrophoretic mobility and surface potential measurements using PPI-containing vesicles and well described by Poisson-Boltzmann modified Gouy-Chapman theory.38 Although the reported affinities are slightly different depending on the experimental setup, and the intrinsic binding constants are not available for each case, conclusions are consistent throughout these studies: Mg2+ binds to PIP2 with a similar or slightly lower affinity compared to that of Ca2+, and an imaging study of Ca2+- and Mg2+-induced lateral aggregation of PIP2 on GUVs,51 also suggests that Mg2+ can be considered as a “weaker” Ca2+.
The competitive binding of Ca2+ and Mg2+ has not been not closely examined and their effects on the lateral organization of PIP2 on a membrane are uncharacterized. The finding that Ca2+ and Mg2+ have different surface pressure effects after binding to PIP2 is unexpected and leads to the development of a competitive PIP2-binding assay. This assay is used here to quantify the relative affinities of Mg2+ and Ca2+ for PPIs and to compare their selective binding to different PIP2 isomers.
The fact that PIP2 isomers have different divalent cation binding selectivity has several implications. The difference between isomers implies that binding to divalent ions is not entirely determined by electrostatics. The opposite preference for Ca2+ or Mg2+ between PI(4,5)P2 and PI(3,5)P2 and the different effects on surface pressures of monolayers containing these lipids might relate to the physiological functions of the lipids. Although the physical chemical differences between PIP2 isomers are subtle both in binding affinity measurements and in ionization state measured by 31P-NMR,26 these results might help explain how PPI selectivity is achieved, especially for PI(3,5)P2 for which no obvious proteins ligands have yet been identified to explain its biological function.
The Ca2+-induced surface pressure drop is PIP2 fraction-dependent (data not shown) and large enough to cause an area mismatch between inner and outer leaflets of the plasma membrane sufficient to induce membrane curvature. In this context it is interesting that Ca2+ and polyamines have opposite effects on surface pressure and therefore curvature. The same PIP2 fraction dependency is also seen in PIP2-rich cluster formation, suggesting that the formation of PIP2-rich clusters might directly account for the Ca2+-induced surface pressure drop, or equivalently to the decrease in molecular area at constant pressure. The finite size and stability of PIP2-Ca2+ clusters is unexpected since a simple electrostatic model predicts that such clusters should coarsen and eventually phase-separate.34 Evidently additional repulsive interactions between clusters arise during their formation in mixed lipids membranes.
The different surface pressure response of Ca2+ and Mg2+ upon binding to PIP2 and the fact that Mg2+ has comparably small effects on PIP2 cluster formation, as examined by AFM, SP-FRET and FCS, can be rationalized by the ATR-FTIR study. The ATR-FTIR spectroscopy shows dehydration as Ca2+ binds to PIP2 head groups, but not when Mg2+ binds. While dehydration has been reported to be due to the loss of water in between charged lipid head groups, partial dehydration of Ca2+ has also been widely reported upon binding to anionic lipid head groups both experimentally52,53 and computationally.54,55 As suggested in the summary diagram (Fig. 5C), the interaction between Ca2+ and PIP2 can be considered as a two step processes: the initial binding is driven by electrostatic interactions, and the Ca2+-induced condensing of PIP2 head groups is entropy-driven by hydrated water release.
The probe-partitioning FRET on lipid bilayers also supports the proposed two-step mechanism and shows further differences between multivalent polyamines and divalent metal ions. Competitive binding between Ca2+ and spermine4+ to PIP2 has been reported based on their antagonistic effects on lipid scrambling.56 An interesting observation suggests that the electrostatic interaction between PIP2 and short chain polyamines, which in some sense is similar to the interaction of PIP2-binding proteins with polybasic domains, fails to induce PIP2 clustering as divalent cations do. Instead of reducing the electrostatic repulsion between PIP2 head groups, the association of polyamines effectively pushes PIP2 molecules away from each other (Fig. 6E and inset). This result leads to two new points of view in protein-lipid interaction: 1. the membrane docking of polyamines/polybasic peptides may not lead to strong lateral segregation of PIP2, or not as strong as occurs in the interaction with divalent cations. 2. Divalent cation-induced PIP2 clustering may dominant the electrostatic interaction between PIP2 and polybasic peptides.
An alternative way of looking into the difference between divalent cation-induced PIP2-clustering and polyamine-induced PIP2-sequestering comes from the FCS measurements (Fig. 7D & E). TETA4+ at millimolar concentration slows down the diffusion of PIP2 by roughly 50%, but it is still more than 10 times faster than the slow diffusion induced by a few micromolar Ca2+. The size of a cation-induced PIP2 cluster can be estimated by the Saffman-Delbrück model57 or its extended form, the Hughes-Pailthorpe-White (HPW) model,58 with appropriate parameters.59 The estimated PIP2 cluster sizes from both the HPW model and the SD model are not significantly different as the object diffuses faster than 2 μm2/s. The calculated cluster size with 10 μM Ca2+ is around 80 and 130 nm from the SD and HPW models, respectively, which is very close to the size of Ca2+-induced PIP2 clusters observed in supported lipid monolayers (Figure 4). On the other hand, the calculated cluster size with 1 mM TETA4+ is less than 0.5 Å, suggesting there are no PIP2-rich clusters under this ionic condition.
A comparison between studies using lipid monolayer and bilayer model membranes is not simple and direct. An obvious example comes from cholesterol-dependent phase demixing: a binary lipid mixture, cholesterol and DOPC, phase demixed on a lipid monolayer but not on a lipid bilayer. Important differences between monolayers and bilayers include electrostatic potential and bending stiffness. The restricted binding and reduced packing flexibility of lipids in monolayers might explain why the PIP2-rich clusters seem to be rounder and fluid-like in the GUVs but appear more solid-like in a supported lipid monolayer.
If the results using purified systems can be extended to the cell membrane, then the ability of PIP2-interacting proteins to bind their membrane targets would be affected as PIP2 local concentration and surface potential are changed by divalent cations, as suggested by numerous studies. For example, Mg2+ induces the inhibition of KCNQ K+ channels,60 Kir2.161 and TRPM7 channel proteins62 all of which are PIP2-activated and the inhibition can be reversed by adding exogenous PIP2. Another example is Ca2+-induced PKCα C2 domain membrane docking, which has been suggested to work through a target-activated messenger affinity (TAMA) mechanism due to the increased local concentration of anionic lipids together with Ca2+.63 The mechanisms of these phenomena are not completely understood since not enough is known about the interaction between PIP2 and divalent cations. Potentially, local fluxes of pH, Ca2+, and factors that change PIP2 electrostatics as well as localized activation of enzymes that produce or degrade PIP2 can alter the lateral distribution of PIP2 and affect PIP2-protein interactions.
CONCLUSION
Multivalent cations including metal ions and polyamines have significant and specific effects on the distribution of polyphosphoinositides in lipid monolayers and bilayers. Most notably, Ca2+ has a strong condensing effect on PIP2-containing membranes that coincides with appearance of nm-scaled clusters. Mg2+ has much weaker effects on monolayer surface pressure and cluster formation, and polyamines expand, rather than condense, PIP2-containing monolayers. The preferential binding of Ca2+ and Mg2+ for the three naturally occurring PIP2 isomers might relate to their highly distinct biological functions.
Ca2+ and Mg2+ have very different effects on surface pressures and PIP2-rich cluster formation, even though the binding of both ions to PIP2 is electrostatic driven and both ions have similar binding affinity. Their difference in inducing PIP2-rich clusters is potentially explained by their difference in dehydration enthalpy, as supported by the ATR-FTIR study.
Supplementary Material
Acknowledgments
This work is supported by NIH grants GM083272 (PAJ), HL067286 (Y-H W) and GM087253 (TS).
Footnotes
Supporting Information
Ca2+-binding affinity measurements as well as AFM imaging for cluster formation using 25 mol% labeled PIP2-containing monolayer is included. Ca2+-dependence of cluster formation and the explanation for decreased apparent KD in FRET Measurement as a result of reduced surface potential is included. This information is available free of charge via the Internet at http://pubs.acs.org/.
References
- 1.Janmey PA, Lindberg U. Nature Reviews Molecular Cell Biology. 2004;5:658–666. doi: 10.1038/nrm1434. [DOI] [PubMed] [Google Scholar]
- 2.Suh BC, Hille B. Annual Review Of Biophysics. 2008;37:175–195. doi: 10.1146/annurev.biophys.37.032807.125859. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.Varnai P, Rother KI, Balla T. Journal Of Biological Chemistry. 1999;274:10983–10989. doi: 10.1074/jbc.274.16.10983. [DOI] [PubMed] [Google Scholar]
- 4.Lariccia V, Fine M, Magi S, Lin MJ, Yaradanakul A, Llaguno MC, Hilgemann DW. Journal Of General Physiology. 2011;137:111–132. doi: 10.1085/jgp.201010468. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Cadmel B, Schleber C, Condron M, Patsiouras H, Connolly L, Catimel J, Nice EC, Burgess AW, Holmes AB. Journal Of Proteome Research. 2008;7:5295–5313. doi: 10.1021/pr800540h. [DOI] [PubMed] [Google Scholar]
- 6.van Rheenen J, Achame EM, Janssen H, Calafat J, Jalink K. Embo Journal. 2005;24:1664–1673. doi: 10.1038/sj.emboj.7600655. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Gambhir A, Hangyas-Mihalyne G, Zaitseva I, Cafiso DS, Wang JY, Murray D, Pentyala SN, Smith SO, McLaughlin S. Biophysical Journal. 2004;86:2188–2207. doi: 10.1016/S0006-3495(04)74278-2. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Hilgemann DW. Pflugers Archiv-European Journal Of Physiology. 2007;455:55–67. doi: 10.1007/s00424-007-0280-9. [DOI] [PubMed] [Google Scholar]
- 9.McLaughlin S, Wang JY, Gambhir A, Murray D. Annual Review Of Biophysics And Biomolecular Structure. 2002;31:151–175. doi: 10.1146/annurev.biophys.31.082901.134259. [DOI] [PubMed] [Google Scholar]
- 10.Redfern DA, Gericke A. Biophysical Journal. 2004;86:2980–2992. doi: 10.1016/S0006-3495(04)74348-9. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Levental I, Cebers A, Janmey PA. Journal Of The American Chemical Society. 2008;130:9025–9030. doi: 10.1021/ja800948c. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Pike LJ, Casey L. Journal Of Biological Chemistry. 1996;271:26453–26456. doi: 10.1074/jbc.271.43.26453. [DOI] [PubMed] [Google Scholar]
- 13.Pike LJ, Miller JM. Journal Of Biological Chemistry. 1998;273:22298–22304. doi: 10.1074/jbc.273.35.22298. [DOI] [PubMed] [Google Scholar]
- 14.Levental I, Christian DA, Wang YH, Madara JJ, Discher DE, Janmey PA. Biochemistry. 2009;48:8241–8248. doi: 10.1021/bi9007879. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Levental I, Byfield FJ, Chowdhury P, Gai F, Baumgart T, Janmey PA. Biochemical Journal. 2009;424:163–167. doi: 10.1042/BJ20091283. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Doughman RL, Firestone AJ, Anderson RA. Journal Of Membrane Biology. 2003;194:77–89. doi: 10.1007/s00232-003-2027-7. [DOI] [PubMed] [Google Scholar]
- 17.Golebiewska U, Gambhir A, Hangyas-Mihalyne G, Zaitseva I, Radler J, McLaughlin S. Biophysical Journal. 2006;91:588–599. doi: 10.1529/biophysj.106.081562. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Vanmeer G, Simons K. Embo Journal. 1986;5:1455–1464. doi: 10.1002/j.1460-2075.1986.tb04382.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Golebiewska U, Kay JG, Masters T, Grinstein S, Im W, Pastor RW, Scarlata S, McLaughlin S. Molecular Biology Of The Cell. 2011;22:3498–3507. doi: 10.1091/mbc.E11-02-0114. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.McLaughlin S, Murray D. Nature. 2005;438:605–611. doi: 10.1038/nature04398. [DOI] [PubMed] [Google Scholar]
- 21.Rouser G, Fleische S, Yamamoto A. Lipids. 1970;5:494–496. doi: 10.1007/BF02531316. [DOI] [PubMed] [Google Scholar]
- 22.Ohki S. Biochimica Et Biophysica Acta. 1982;689:1–11. doi: 10.1016/0005-2736(82)90182-1. [DOI] [PubMed] [Google Scholar]
- 23.Ohki S, Ohshima H. Biochimica Et Biophysica Acta. 1985;812:147–154. doi: 10.1016/0005-2736(85)90532-2. [DOI] [PubMed] [Google Scholar]
- 24.Ohshima H, Ohki S. Journal Of Colloid And Interface Science. 1985;103:85–94. [Google Scholar]
- 25.Levental I, Janmey PA, Cebers A. Biophysical Journal. 2008;95:1199–1205. doi: 10.1529/biophysj.107.126615. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Kooijman EE, King KE, Gangoda M, Gericke A. Biochemistry. 2009;48:9360–9371. doi: 10.1021/bi9008616. [DOI] [PubMed] [Google Scholar]
- 27.Kates M. Techniques of Lipidology. 2. Elsevier Science Publishers B.V; Amsterdam: 1986. [Google Scholar]
- 28.Huang CP, Huang CP, Morehart AL. Water Research. 1991;25:1365–1375. [Google Scholar]
- 29.Sheng PX, Ting YP, Chen JP. Industrial & Engineering Chemistry Research. 2007;46:2438–2444. [Google Scholar]
- 30.Buboltz JT. Physical Review E. 2007;76 doi: 10.1103/PhysRevE.76.021903. [DOI] [PubMed] [Google Scholar]
- 31.Karolin J, Johansson LBA, Strandberg L, Ny T. Journal Of The American Chemical Society. 1994;116:7801–7806. [Google Scholar]
- 32.Mathivet L, Cribier S, Devaux PF. Biophysical Journal. 1996;70:1112–1121. doi: 10.1016/S0006-3495(96)79693-5. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.Smith-Dupont KB, Guo L, Gai F. Biochemistry. 2010;49:4672–4678. doi: 10.1021/bi100426p. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.Ellenbroek WG, Wang YH, Christian DA, Discher DE, Janmey PA, Liu AJ. Biophysical Journal. 2011;101:2178–2184. doi: 10.1016/j.bpj.2011.09.039. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35.Hauser H, Darke A, Phillips MC. European Journal Of Biochemistry. 1976;62:335–344. doi: 10.1111/j.1432-1033.1976.tb10165.x. [DOI] [PubMed] [Google Scholar]
- 36.Brockman H. Current Opinion In Structural Biology. 1999;9:438–443. doi: 10.1016/S0959-440X(99)80061-X. [DOI] [PubMed] [Google Scholar]
- 37.Ohki S, Muller M, Arnold K, Ohshima H. Colloids And Surfaces B-Biointerfaces. 2010;79:210–218. doi: 10.1016/j.colsurfb.2010.03.052. [DOI] [PubMed] [Google Scholar]
- 38.Toner M, Vaio G, McLaughlin A, McLaughlin S. Biochemistry. 1988;27:7435–7443. doi: 10.1021/bi00419a039. [DOI] [PubMed] [Google Scholar]
- 39.Takizawa T, Hayashi K, Yabuki S, Nakata Y. Thermochimica Acta. 1988;123:247–253. [Google Scholar]
- 40.Hirai M, Takizawa T, Yabuki S, Nakata Y, Hirai T, Hayashi K. Journal Of The Chemical Society-Faraday Transactions. 1996;92:1493–1498. [Google Scholar]
- 41.Pohle W, Gauger DR, Fritzsche H, Rattay B, Selle C, Binder H, Bohlig H. Journal Of Molecular Structure. 2001;563:463–467. [Google Scholar]
- 42.Volkov AG, Paula S, Deamer DW. Bioelectrochemistry And Bioenergetics. 1997;42:153–160. [Google Scholar]
- 43.Wulfsberg G. Principles of Descriptive Chemistry. Brooks/Cole Publishing; Monterey CA: 1987. [Google Scholar]
- 44.Golebiewska U, Nyako M, Woturski W, Zaitseva I, McLaughlin S. Molecular Biology Of The Cell. 2008;19:1663–1669. doi: 10.1091/mbc.E07-12-1208. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 45.Edsall JT, Wyman J. Biophysical Chemistry. Vol. 1 Academic; New York: 1958. [Google Scholar]
- 46.Tanford C. Physical Chemistry of Macromolecules. Wiley; New York: 1961. [Google Scholar]
- 47.Hendrickon HS, Fullington JG. Biochemistry. 1965;4:1599–1605. doi: 10.1021/bi00884a021. [DOI] [PubMed] [Google Scholar]
- 48.Dawson RMC. Biochemical Journal. 1965;97:134–138. doi: 10.1042/bj0970134. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 49.Hauser H, Dawson RMC. European Journal Of Biochemistry. 1967;1:61–69. doi: 10.1007/978-3-662-25813-2_11. [DOI] [PubMed] [Google Scholar]
- 50.Buckley JT, Hawthorn Jn. Journal Of Biological Chemistry. 1972;247:7218–7223. [PubMed] [Google Scholar]
- 51.Carvalho K, Ramos L, Roy C, Picart C. Biophysical Journal. 2008;95:4348–4360. doi: 10.1529/biophysj.107.126912. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 52.Sinn CG, Antonietti M, Dimova R. Colloids And Surfaces A-Physicochemical And Engineering Aspects. 2006;282:410–419. doi: 10.1016/j.colsurfa.2006.02.061. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 53.Sulpice JC, Zachowski A, Devaux PF, Giraud F. Journal Of Biological Chemistry. 1994;269:6347–6354. [PubMed] [Google Scholar]
- 54.Vernier PT, Ziegler MJ, Dimova R. Langmuir. 2009;25:1020–1027. doi: 10.1021/la8025057. [DOI] [PubMed] [Google Scholar]
- 55.Potoff JJ, Issa Z, Manke CW, Jena BP. Cell Biology International. 2008;32:361–366. doi: 10.1016/j.cellbi.2008.03.002. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 56.Sulpice JC, Moreau C, Devaux PF, Zachowski A, Giraud F. Biochemistry. 1996;35:13345–13352. doi: 10.1021/bi960624a. [DOI] [PubMed] [Google Scholar]
- 57.Saffman PG, Delbruck M. Proceedings Of The National Academy Of Sciences Of The United States Of America. 1975;72:3111–3113. doi: 10.1073/pnas.72.8.3111. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 58.Hughes BD, Pailthorpe BA, White LR. Journal Of Fluid Mechanics. 1981;110:349–372. [Google Scholar]
- 59.Gambin Y, Lopez-Esparza R, Reffay M, Sierecki E, Gov NS, Genest M, Hodges RS, Urbach W. Proceedings Of The National Academy Of Sciences Of The United States Of America. 2006;103:2098–2102. doi: 10.1073/pnas.0511026103. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 60.Suh BC, Hille B. Journal Of General Physiology. 2007;130:241–256. doi: 10.1085/jgp.200709821. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 61.Ballester LY, Vanoye CG, George AL. Channels. 2007;1:209–217. doi: 10.4161/chan.4770. [DOI] [PubMed] [Google Scholar]
- 62.Gwanyanya A, Sipido KR, Vereecke J, Mubagwa K. American Journal Of Physiology-Cell Physiology. 2006;291:C627–C635. doi: 10.1152/ajpcell.00074.2006. [DOI] [PubMed] [Google Scholar]
- 63.Corbin JA, Evans JH, Landgraf KE, Falke JJ. Biochemistry. 2007;46:4322–4336. doi: 10.1021/bi062140c. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.