

Abstract
Diabetes is associated with β-cell failure. But it remains unclear whether the latter results from reduced β-cell number or function. FoxO1 integrates β-cell proliferation with adaptive β-cell function. We interrogated the contribution of these two processes to β-cell dysfunction, using mice lacking FoxO1 in β-cells. FoxO1 ablation caused hyperglycemia with reduced β-cell mass following physiologic stress, such as multiparity and aging. Surprisingly, lineage-tracing experiments demonstrated that loss of β-cell mass was due to β-cell dedifferentiation, not death. Dedifferentiated β-cells reverted to progenitor-like cells expressing Neurogenin3, Oct4, Nanog, and L-Myc. A subset of FoxO1-deficient β-cells adopted the α-cell fate, resulting in hyperglucagonemia. Strikingly, we identify the same sequence of events as a feature of different models of murine diabetes. We propose that dedifferentiation trumps endocrine cell death in the natural history of β-cell failure, and suggest that treatment of β-cell dysfunction should restore differentiation, rather than promoting β-cell replication.
Introduction
The pathogenesis of β-cell dysfunction in type 2 diabetes remains controversial (Talchai et al., 2009). β-cells of diabetics respond poorly to a glucose challenge and fail to mount an appropriately timed response (Ferrannini, 2010). In addition, physiologic adaptation of β-cell function to conditions like pregnancy or aging–mainly achieved by modulating β-cell replication–is most taxing for pre-diabetic individuals, indicating that not only is the endocrine islet’s homeostatic function poor, but so is its ability to cope with metabolic or environmental stressors (Accili et al., 2010).
During the course of diabetes, and in animal models of β-cell dysfunction, deficits in adaptive β-cell mass are largely viewed as arising from an unbalanced rate of self-renewal vs. apoptosis (Butler et al., 2007; Rieck et al., 2009). However, the increased rate of apoptosis in diabetic islets is modest relative to the impairment of β-cell function (Butler et al., 2007), while individual variations in β-cell mass are extensive (Rahier et al., 2008). In addition to cell-autonomous defects that can be demonstrated long before disease onset (Weyer et al., 1999), there are cell-nonautonomous contributors to β-cell dysfunction: hyperglycemia, dyslipidemia, hormones, cytokines, changes in endothelial cell function and vascular flow (Ferrannini, 2010).
The failure of sustainable β-cell function under metabolic stress (e.g., during pregnancy or aging) (Rankin and Kushner, 2010; Rieck et al., 2009) has been attributed to different biological mechanisms. Autophagy, a physiologic process of organelle maintenance, can morph into a disease process under conditions of nutrient oversupply–as seen in diabetes (Hur et al., 2010). Activation of the unfolded protein response affects insulin secretion, with subsequent changes in β-cell mass( Matsuda et al., 2010). The limited glycolytic capacity of the β-cell can result in the generation of reactive oxygen species, and the ensuing oxidative stress can uncouple glucose sensing from insulin secretion( Robertson, 2004).
In addition to processes impinging on β-cell survival and hence on islet mass, β-cell dedifferentiation can be observed in vitro (Weinberg et al., 2007) and during ex vivo culture of human islets (Gershengorn et al., 2004). Evidence that it might occur in common forms of β-cell failure has been inferred from partial pancreatectomy studies (Jonas et al., 1999), but has not been shown to occur in type 2 diabetes, nor has its mechanism been explored.
Recent studies have shone light on fate conversion of pancreatic cells under genetically extreme conditions, such as: (i) α-cell to β-cell in response to forced expression of Pax4 (Collombat et al., 2007) or extreme ablation of β-cells (Thorel et al., 2010); (ii) β-cell to duct epithelium in response to Akt gain-of-function (Elghazi et al., 2009); (iii) β-cell to α-cell in response to ablation of the DNA methyltransferase Dnmt1 in β-cells (Dhawan et al., 2011; Papizan et al., 2011); and (iv) exocrine-to-endocrine cells in response to over-expression of transcription factors, Pdx1, MafA, and Neurog3 (Zhou et al., 2008).
Transcription factor FoxO1 integrates signals regulating β-cell mass (Kitamura et al., 2002; Okamoto et al., 2006) and stress response (Kawamori et al., 2006; Kitamura et al., 2005; Martinez et al., 2006). FoxO1 has been found to determine cell fate in differentiating adipocytes (Nakae et al., 2003), myoblasts (Kitamura et al., 2007), and enteroendocrine cells (Talchai et al., 2012). But considerably less is known about FoxO1 requirements in maintaining cell differentiation. In light of its role, straddling diverse aspects of β-cell physiology, we used mice lacking FoxO1 in β-cells to probe mechanisms of β-cell failure in type-2 diabetes. Surprisingly, our studies reveal that β-cell dedifferentiation and conversion into other endocrine cell types are under-recognized mechanisms of β-cell failure in type 2 diabetes and that FoxO1 is required to enforce the β-cell fate under metabolic stress.
Results
FoxO1 Ablation Increases Susceptibility To Metabolic Stress
To determine FoxO1 activity during diabetes progression, we surveyed FoxO1 immunoreactivity in β-cells of mice with insulin-resistant diabetes: GIRKO (Lin et al., 2011), as well as db/db mice. In addition to euglycemic mice (glucose ≤150 mg/dl), we studied mice with mild fasting hyperglycemia (150–250 mg/dl), and severe hyperglycemia (≥500 mg/dl). In euglycemia, FoxO1 showed cytoplasmic localization in β-cells (Figure 1A). In contrast, with mild hyperglycemia, FoxO1 could be found in a distinctive punctate nuclear pattern in β-cells, consistent with its nuclear translocation in response to oxidative stress (Kitamura et al., 2005). In this condition, we also saw partial loss of insulin and FoxO1 expressing cells (Figure 1A). As hyperglycemia increased, loss of FoxO1 immunoreactivity paralleled loss of insulin content (Figure 1A), consistent with previous observations (Kitamura et al., 2005; Lin et al., 2011; Xuan et al., 2010). Nonetheless, we don’t know whether loss of FoxO1 is a cause or an effect of β-cell failure, nor do we know what happened to the ‘missing’ β-cells.
Figure 1. FoxO1 Localization During Diabetes Progression And Foxo1 Knockout.

(A) Immunofluorescence with insulin (green), and FoxO1 (red) in euglycemic (glucose=100 mg/dl), mildly hyperglycemic (glucose=150–250 mg/dl) db/db mice, and severely hyperglycemic GIRKO mice (glucose > 500 mg/dl) (n = 10). Insets show representative β-cells.
(B) Lineage tracing analysis of recombination at the Foxo1 locus in IKO:RosaGfp and control mice (Ins2-Cre:Foxo1fl/+;RosaGfp or Ins2-Cre:Foxo1+/+;RosaGfp) using Gfp (green, indicates recombination) and FoxO1 antibodies (red) in pancreata from 3-month-old mice (n = 3).
(C) Islet morphology and immunohistochemistry with insulin (red) and glucagon (Gcg, green) in multiparous or aging (16- to 20-month-old) wild-type and IKO mice (n = 4).
(D) Fasting blood glucose.
(E) β-cell mass.
(F) α-cell mass.
(G) Fed plasma insulin.
(H) Fed plasma glucagon.
(I) Pancreatic insulin content.
(J) Pancreatic glucagon content. (n = 8–16 for blood metabolite analyses, n = 4 for morphometry and n=8 for pancreatic hormone content). Data represent means ± SEM. * = p <0.05, and ** = p <0.01 by Student’s t-test.
To address these questions, we utilized mice with somatic deletion of Foxo1 in β-cells (IKO) (Kitamura et al., 2009). To document Foxo1 deletion in Cre-expressing cells, we generated IKO:Rosa26-Gfp and control Ins2-cre:Foxo1fl/+:Rosa26-Gfp mice (Talchai et al., 2012). As expected, Gfp+ cells lacked FoxO1 immunoreactivity (Figure 1B), while measurements of Foxo1 mRNA in collagenase-purified islets demonstrated a ~70% decrease compared with wild-type controls, with residual mRNA probably arising from islet vasculature and connective tissue (Figure S1A). In basal conditions, IKO mice showed normal body weight (Figure S1B), islet architecture, β-cell morphology by electron microscopy (EM), and levels of mRNA encoding β-cell markers Pdx1, MafA, Nkx6.1, NeuroD1, Insulin1, Insulin2, Prohormone convertase 1 (Pcsk1), Pcsk2, Glucokinase (Gck), and Glucose transporter 2 (Glut2). Interestingly, they showed increased mRNA encoding Foxo3a and Foxo4 (Figures S1C–S1H). IKO mice displayed normal glucose tolerance, insulin and glucagon secretion (Figures S1I–S1Q). In addition, fasting blood glucose was normal, as were fed insulin and glucagon levels and pancreatic content of insulin and glucagon (Figures 1D–1J).
To assess the consequences of FoxO1 ablation in the β-cell response to physiologic stress, we studied multiparous females (Rieck et al., 2009) and aging males (Rankin and Kushner, 2010) as pathophysiologic models of β-cell stress. In both models, we saw a ~30% decrease of β-cell mass and a ~50% increase in α-cell mass, associated with fasting hyperglycemia, decreased fed insulin levels and pancreatic insulin content, and increased fed glucagon levels and pancreatic glucagon content (Figures 1C–1J). We further examined β-cell and α-cell function in vivo, and found that both multiparous and aging IKO mice had impaired glucose tolerance, decreased insulin secretion and elevated glucagon secretion in response to glucose and arginine compared to wild-type controls (Figures S1I–S1P). To distinguish between an intrinsic defect of insulin and glucagon secretion in multiparous IKO mice and secondary changes brought about by glucose toxicity (Robertson, 2004), we isolated islets and compared insulin secretion. We observed reduced insulin release in response to different glucose concentrations in islets from IKO multiparae (Figures S1Q and S1R), consistent with a cell-autonomous effect of the Foxo1 mutation.
We confirmed these findings in two additional models of β-cell dysfunction: (i) hyperglycemia–induced by chemical ablation of β-cells with low-dose streptozotocin (STZ) (Movassat et al., 1997), and (ii) insulin resistance–caused by insulin receptor haploinsufficiency (InsR+/−) (Kido et al., 2000). In both instances, we saw decreased β-cell mass, altered pancreatic glucagon and insulin content, abnormal fed plasma insulin and glucagon, with fasting hyperglycemia (Figures S1S–S1Y). The constellation of hyperglycemia, hyperglucagonemia and relative hypoinsulinemia with decreased insulin–to–glucagon ratios is reminiscent of human type-2 diabetes (Dunning and Gerich, 2007).
Cell-Autonomous Dedifferentiation Of FoxO1-Deficient β-Cells
To examine whether β-cell dysfunction was due to loss of β-cell number or β-cell function, we employed lineage tracing to mark cells in which Ins2-cre had been active. In these experiments, we used a sensitive approach based on automated localization of confocal immunofluorescence, and compared virgin female and multiparous IKO mice with matched wild-type controls using immunohistochemistry with Gfp and insulin (Figure 2A green and red, respectively). The activation frequency of Ins2-cre in β-cells–as defined by the ratio of co-localization area (yellow) to insulin-immunoreactive area (red)–was 75–88% in virgin IKO and wild-type mice (Figures 2A, 2B, S2A). We then assessed the proportion of green (Gfp+), red (Ins+), and yellow cells (Gfp+/Ins+) in IKO vs. wild-type multiparae. The predictions were that (i) if reduced β-cell mass in IKO multiparae was due to decreased cell-autonomous survival, the number of yellow cells should decrease; (ii) if reduced β-cell mass was due to impaired repopulation of the islets by newly formed β-cells that escaped Cre-mediated recombination, the number of red cells should decrease, and (iii) if both mechanisms contributed to the reduction, red and yellow cells should decrease (Figure 2A).
Figure 2. Lineage-Tracing Of β-Cells In Multiparae.

(A) Experimental design and expected outcomes of lineage-tracing experiments.
(B) Immunofluorescence with Gfp (green) and insulin (red) in IKO:RosaGfp and control RosaGfp virgins.
(C) Immunofluorescence with Gfp (green) and insulin, Pdx1, or MafA antibodies (red) in IKO:RosaGfp and control RosaGfp multiparae. Middle and right panels show representative islets with moderate (middle) and extreme degrees (right) of loss of insulin immunoreactivity.
(D) Immunofluorescence with Gfp (green) and insulin, Pdx1, or MafA (red), showing degranulated (orange) and dedifferentiated (green) β-cells inIKO:RosaGfp multiparae (n = 4 for each group).
(E) Electron microscopy showing a representative wild-type β-cell (left) and a degranulated (orange arrow, dg) or a dedifferentiated β-cell (green arrow, dd) in IKO multiparae.
(F–I) Immunohistochemistry with Gfp (green) and Pcsk2, Glut2, Gck, and Pcsk1 (red) in recombined β-cells of multiparous IKO:RosaGfp and control RosaGfp mice. In some sections, DNA is counterstained with DRAQ5 (blue or white). Insets represent individual color channels.
(J) qPCR analysis of Pdx1, MafA, Nkx6.1, NeuroD1, Pcsk2, Gck, and Glut2 in islets isolated from control and IKO mice (n=4 for histology, and n=4 for qPCR). Data show means ± SEM. * = P <0.05, and ** = p <0.01 by Student’s t-test.
Instead, we observed similar numbers of red cells in the two genotypes (Figure S2A), while–to our surprise–multiparous IKO mice showed a large increase in the number of green cells, i.e., recombined β-cells that no longer produced insulin. There were large inter-islet variations, even within the same pancreas. In some islets we observed residual punctate insulin immunoreactivity, resulting in faint merged fluorescence (i.e., intensity of red color ≤40% of average wild-type β-cells) (Figure 2C, middle panels), while in others insulin immunoreactivity could no longer be detected, resulting in completely green cells (Figure 2C, right hand panels). Double immunohistochemistry with Gfp and Pdx1, or Gfp and MafA revealed that wholly green cells expressed neither β-cell transcription factor (Figure 2C), while cells with residual faint yellow immunoreactivity showed persistent expression of both proteins (Figure 2D). Thus, based on the proposed criteria for the progression of β-cell failure (Weir and Bonner-Weir, 2004), it appears that faintly yellow cells are partly degranulated β-cells, while green cells are dedifferentiated β-cells (Weinberg et al., 2007). The two cell types combined represented ~25% of total recombined cells in IKO islets, thus accounting nearly entirely for the loss of β-cell mass in Foxo1-ablated multiparae (Figure S2A). To extend our findings, we performed lineage-tracing studies in aging IKO (> 16-month-old), and IKO/InsR+/− males, age–matched with multiparous females (8 month-old). We confirmed the occurrence of degranulation and dedifferentiation in both models (Figure S2B). These data suggest that dedifferentiation is a shared cell-autonomous mechanism of β-cell loss, rather than a specific response to multiple pregnancies.
The presence of similar numbers of β-cells that escaped recombination in IKO and controls (Figure S2A, red bars) demonstrates that the two groups did not differ by their regenerative abilities, i.e., that the difference in β-cell mass is due to cell-autonomous effects of Foxo1 ablation.
The data are consistent with the explanation that FoxO1-deficient β-cells have normal survival, but become dedifferentiated. Accordingly, islet EM showed cells with scattered residual EM-dense granules, typical of degranulated β-cells, as well as cells with empty granules in multiparous IKO mice (Figure 2E). In addition, double immunostaining with Gfp and various β-cell markers demonstrated a near-complete loss of C-peptide and Pcsk1 (encoding prohormone convertase 1), and substantial decreases of Glut2, Gck, and Pcsk2 immunoreactivities in multiparous and in aging IKO mice (Figures 2F-2I, S2C, and S2D). Further, mRNAs encoding Pdx1, Nkx6.1, NeuroD1, MafA, Pcsk2, Gck, and Glut2 were decreased in islets isolated from IKO multiparae (Figure 2J), providing a potential explanation for the secretory defects observed in vivo (Figures 1 and S1).
FoxO1 Ablation Does Not Alter β-Cell Death Or Self-Renewal In Vivo
Apoptosis contributes to β-cell loss (Butler et al., 2007). In light of the possible proapoptotic role of FoxO1, we surveyed apoptotic cells by quantification of cleaved caspase-3 and by TUNEL assays, but found similar numbers in multiparous IKO and wild-type mice (Figures 3A and 3B). Interestingly, no pancreatic hormone colocalized with apoptotic nuclei, suggesting that apoptosis occurred in dedifferentiated cells.
Figure 3. Analysis Of β-Cell Survival In FoxO1-Deficient Mice.
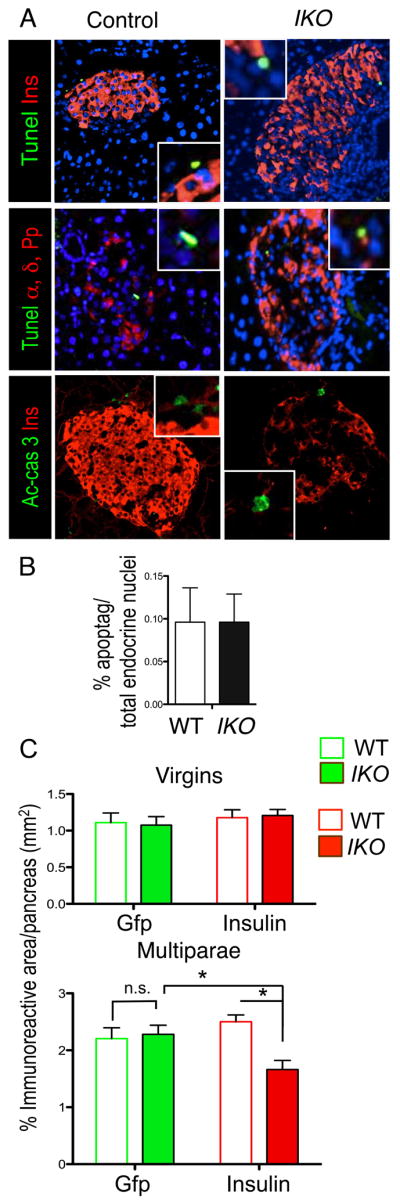
(A) TUNEL assay or active-caspase 3 (green) in insulin- and glucagon-immunoreactive cells (red) of multiparous wild-type and IKO mice.
(B) Quantification of apoptotic nuclei as detected by TUNEL assays in wild-type and IKO mice (n = 8).
(C) Quantification of Gfp (green) or insulin immunoreactive area (red). The pixel area was converted to mm2 and plotted as percentage of total pancreas area (also expressed in mm2) from virgins and multiparae (n= 4 for each group).
Data show means ± SEM. * = P<0.05 by Student’s t -test.
To quantitate the contribution of dedifferentiation vs. death to β-cell loss, we compared the ratios of Gfp and insulin immunoreactive areas to total pancreatic area in virgin and multiparous mice from both genotypes. In basal conditions, we observed no difference between virgin animals of the two genotypes, indicating that they possess the same complement of β-cells and similar recombination rates. If the reduced β-cell mass in multiparous IKO was due to cell death, we should have observed a reduction of both Gfp- and insulin-immunoreactive areas as a fraction of total pancreatic area in IKO vs. wild-type mice. But we observed that only the insulin-immunoreactive area was reduced in multiparous IKO mice, while the Gfp-immunoreactive area was similar between the two genotypes (Figure 3C). These data show that loss of β-cell mass in IKO multiparae is due to loss of insulin production in β-cells, not to cell death.
We also investigated the possibility that FoxO1-ablated β-cells had abnormal turnover. But immunohistochemistry with the cell cycle antigen Ki67 under basal conditions, or during and after pregnancy showed no difference in labeling between the two genotypes. We obtained similar data in glucagon-producing cells (Figures S3A–S3C), ruling out a contribution of altered cell replication to decreased β-cell mass and increased α-cell mass.
FoxO1-Deficient β-Cells Revert To An Uncommitted Endocrine Progenitor-Like Stage
We probed the differentiation stage of FoxO1-deficient, insulin-depleted β-cells, using markers of pre-endocrine progenitors (Sox9), endocrine progenitors (Neurog3), and committed endocrine cells (ChromograninA, ChgA) (Seymour et al., 2007). FoxO1-deficient Gfp+ cells were Sox9−/ChgA+, indicating that they are endocrine pre-β-cells, and not pre-endocrine progenitors (Figure 4A). Neurog3 detection in adult islets is problematic at best (Wang et al., 2009). Accordingly, Neurog3 immunoreactivity was hardly detectable in islets from virgin 3-month-old mice of either genotype (Figure S4A), but clearly visible in fetal pancreas (Figure S4B). We detected a stark increase of Neurog3 immunoreactivity in FoxO1-ablated cells of multiparous, aging, and InsR+/−/IKO mice (Figures 4A and S4C). Double immunohistochemistry demonstrated that cells expressing high levels of Neurog3 (Neurog3high) contained no insulin, Pdx1, and MafA while mature β-cells showed the opposite pattern (Figure 4B and S4D) (Wang et al., 2009). These findings were corroborated by increases in Neurog3 mRNA and protein levels in islets from IKO mice (Figures 4C and S4E).
Figure 4. Staging Of Differentiation Of FoxO1-Deficient β-Cells.

(A) Immunofluorescence with Gfp (green, indicating recombined β-cells) and insulin (red, left panels), or ChgA (red, central panels), Neurog3 (red, central right panels), Sox9 (red, right panels) in multiparous IKO:RosaGfp and control RosaGfp mice. DNA is counterstained with DAPI.
(B) Immunofluorescence with insulin (white), and Neurog3 (red, left panels), Pdx1 or MafA (green), and Neurog3 (red, central and right panels) in multiparous IKO and wild-type mice.
(C) Neurog3 immunoblotting in the indicated tissues. Data represent means ± SEM (n=4). * = P <0.05 by Student’s t-test.
Multipotency Of Dedifferentiated β-Cells
To investigate the cause of β cell dedifferentiation, we studied plasticity and pluripotentcy of FoxO1-deleted cells. In human embryonic stem cells, FOXOs regulate pluripotency via OCT4, NANOG, and SOX2 (Zhang et al., 2011), and in human fetal pancreas, OCT4 is co-expressed with NEUROG3 and NESTIN (Wang et al., 2009). In addition, pluripotent bone marrow stromal cells co-expressing OCT4, NANOG, and NEUROG3 can be differentiated into insulin+ cells (Zhao et al., 2008). Interestingly, during embryonic development and post-natal maintenance of β-cells, L-Myc is temporally co-regulated with Neurog3+ progenitors and their descendants (White et al., 2008). L-Myc is required to induce cellular reprogramming (Nakagawa et al., 2010). Accordingly, we found that Gfp-immunoreactive β-cells from wild-type mice express low levels of Oct4 and L-Myc, and little if any Nanog, while lineage-traced IKO β-cells expressed high levels of all three proteins (Figure 5A), as did human embryonic stem cells and mouse embryonic pancreas (Figures S5A and S5B). Sox2 was not detectable in islets from either genotype, consistent with the finding that Foxo1 deletion did not affect β-cell proliferation (Figure 5A) (Basu-Roy et al., 2012). Furthermore, in IKO islets from multiparae, we detected cells co-expressing Neurog3high and L-Myc. L-Myc and Oct4 were hardly detectable in β-cells from wild-type mice, but highly enriched in insulin-depleted IKO islets (Figures S5B and S5C). These results are supported by the findings that mRNA encoding Oct4, Nanog, and L-Myc are expressed at comparable levels in islets from IKO multiparae and flow-sorted Neurog3-Gfp+ cells isolated at E14.5, although they are both considerably lower than embryonic stem cells (Figure S5D). The findings indicate that ‘empty’ β-cells in IKO mice are not degranulated β-cells, but represent a distinctive pre-β-cell differentiation stage.
Figure 5. Multipotency And Plasticity Of Dedifferentiated β-Cells.

(A) Immunofluorescence with Gfp (green, indicating recombined β-cells) and Oct4 (red, left panels), or L-Myc (red, central panels), Nanog (red, central right panels), Sox2 (red, right panels) in multiparous IKO:RosaGfp and control RosaGfp mice.
(B) Immunofluorescence with Gfp (green) (recombined cells) and a cocktail of glucagon (labeling α-cells), pancreatic polypeptide (Pp cells), and somatostatin (δ-cells) antibodies (red) in multiparous and aging IKO:RosaGfp and control RosaGfp mice. Orange arrows indicate merged fluorescence (M); red arrows indicate pre-existing α, δ, and Pp-cells. Insets illustrate representative cells. In the central right panel, the yellow area shows automatic localization assigned by software analysis of confocal images. Boxed regions (non-β-cells) are shown at higher magnification on top. The far right panel demonstrates a representative z-stack of confocal images showing co-localization of merged fluorescence to the same cell.
(C) Immunohistochemistry with Gfp (green, indicating recombined β-cells), and Pdx1, or MafA (magenta) and glucagon (Gcg, red) in IKO and wild-type multiparae.
(D) Immunohistochemistry with glucagon (red) and nestin or vimentin (blue) in IKO and wild-type multiparae, including representative z-stacks of confocal images (n = 4–6 in all panels).
Conversion of dedifferentiated β-cells into other hormone-producing cells
We further hypothesized that the multipotency of Ins−/Neurog3high/Oct4+/L-Myc+ cells in IKO islets increased their plasticity. Given the rise in α-cell mass and pancreatic glucagon content in multiparous and in aging IKO mice, we asked whether β-cell dedifferentiation was associated with conversion into other endocrine pancreatic cell types. To this end, we used lineage tracing to follow the fates of dedifferentiated β-cells by confocal immunofluorescence colocalization of Gfp with islet hormones other than insulin (Figure 5B, green and red, respectively). If insulin-depleted β-cells converted into other islet cell types, we expected to detect FoxO1-ablated β-cells (green) that became immunoreactive with glucagon, somatostatin, or pancreatic polypeptide (Pp) and thus gave rise to yellow immunoreactivity (Figures 5B and S5E). For simplicity, we refer to yellow cells as α, δ, and Pp cells. Indeed, we observed a population of Gfp-labeled cells decorated by antibodies to pancreatic hormone other than insulin; this population accounted for a ~35% increase in α, δ, and Pp cell mass in IKO mice (Figures 5B, S5E, and S5F). Following the fate of lineage-traced β-cells, we detected Gfp+/glucagon+ cells that were immunoreactive with Pdx1 or MafA in IKO multiparae (Figure 5C). This result demonstrates that some dedifferentiated β-cells gave rise to glucagon-containing cells. These data indicate that there is conversion of former β-cells into α, δ, and Pp cells.
Importantly, none of the converted cells co-expressed insulin, supporting the view that regression to a pre-β-cell (possibly Neurog3+) state is a pre-requisite for conversion into non-β-cells. Indeed, lineage tracing showed that recombined FoxO1-deficient cells from multiparous and from aging mice have increased expression of MafB (Figure S5F), a factor involved in the determination of pre-hormone producing (uncommitted) as well as glucagon-producing cells (Hang and Stein, 2011). This result provides an explanation for the hyperglucagonemia and increased pancreatic glucagon content seen in IKO multiparae (Figures 1F, 1H, and 1J).
Plasticity Of Converted α And δ-Cells In IKO Mice
We further analyzed the similarities between glucagon-immunoreactive cells in IKO mice and bona fide α-cells. We detected increased transcript levels encoding glucagon and MafB, but not α-cell markers Arx, Pax6, and Brn4 in IKO islets compared to controls (Figure S5G). Consistent with the lineage tracing data, we found ectopic expression of β-cell transcription factors Pdx1, MafA, and Nkx6.1 in converted α and δ cells (Figures S5I–S5K). These data provide further support for β-cell conversion into non-β, hormone-producing cells. Similarly, marker analysis of insulin-expressing cells in IKO multiparae identified not only reduced numbers of Pdx1+, MafA+, and Nkx6.1+ cells, but also Pdx1+, MafA+, or Nkx6.1+ cells that failed to express insulin (Figure S5I–S5K). The latter cell type might represent α-cells converting into β-cells (Thorel et al., 2010). But it’s unlikely, given that we observed reduced β-cells, expanded α, δ, and Pp cells, and increased pancreatic glucagon content, and we did not detect insulin/glucagon double-positive cells.
In addition, we did not find co-localization of FoxO1 with α, δ, and Pp cells, thus ruling out a cell non-autonomous role of ectopically expressed FoxO1 (Figures S5L and S5M). The mRNA analysis of α cell markers, and the ectopic expression of β-cell transcription factors in α–, δ–cells indicate that conversion into glucagon- or somatostatin-producing cells was due to increased plasticity rather than transdifferentiation. These data indicate that lack of FoxO1 in β-cells curtails insulin expression, and renders key β-cell transcription factors, Nkx6.1, Pdx1, MafA, and Neurog3 (Zhou et al., 2008), insufficient to maintain β-cell identity.
β-cells, islets, and pancreas can de-differentiate and activate expression of mesenchymal markers, such as vimentin and nestin, in vitro (Russ et al., 2011; Zulewski et al., 2001), but this phenomenon has not been described in vivo. Using immunohistochemistry with nestin or vimentin, we investigated whether conversion from β- to α-cells required co-expression of mesenchymal markers in vivo. We observed occasional colocalization of the two proteins with glucagon in IKO multiparae, but not in wild-type controls (Figure 5D). The data suggest that expression of mesenchymal markers influences conversion to α-cells.
Relevance Of The Findings To Type 2 Diabetes
Is dedifferentiation of β-cells a unique finding in Foxo1 knockouts, or is it a common path to β-cell failure? Using insulin-resistant diabetic GIRKO (Lin et al., 2011) and db/db mice, we measured β-cell differentiation using ChgA, FoxO1, and insulin immunoreactivity. In moderately diabetic mice, insulin+/ChgA+ cells decreased by 15–45%, whereas in severely diabetic mice 90–95% of islet cells were ChgA+ but only 10–15% were insulin+ or hormone+ (Figures 6A and 1A). Abundant ChgA+/hormone− cells persisted in severely hyperglycemic mice (Figures 6A, 6B, and S6A), providing a practical illustration of ‘empty β-cells’ during diabetes progression. These cells were also decorated by endocrine marker Synaptophysin, and showed progressive loss of FoxO1 during diabetes development (Figures S6B–S6D), leading to a large increase of Synaptophysin+/FoxO1− cells in severely diabetic mice. These data provide evidence that a large proportion of endocrine mass is retained despite severe hyperglycemia (500 mg/dL), and that FoxO1 is required to enforce β-cell identity in insulin-resistant diabetes.
Figure 6. β-Cell Dedifferentiation In Diabetes (GIRKO Mice).

(A) Immunohistochemistry with insulin (green), combined glucagon (gcg), pancreatic polypeptide (Pp), somatostatin (Sms) (blue) and ChgA (red).
(B) Quantification of area of ChgA-immunoreactive area (red), 4-hormone-immunoreactive area (insulin, glucagon, Pp, somotatostatin) (yellow), and their ratios (grey). The pixel area was converted to mm2 and plotted as percentage of total pancreatic area (also expressed in mm2) (n= 5 for each group). Data show means ± SEM. * = P<0.05 by Student’s t -test.
(C) Immunohistochemistry with insulin (white) and Neurogenin3, L-Myc, or Oct4 (red).
(D) Immunohistochemistry with glucagon (white) and MafA (red) (top panels); or glucagon (green) and vimentin (red) (middle panels); or glucagon (green) and nestin (red) (bottom panels) (n=4 for each group).
Similar to mice with FoxO1 ablation, diabetes in db/db mice was associated with a marked increase in Neurog3, Oct4, and L-Myc immunoreactivities that mirrored loss of insulin and β-cell markers, MafA and Pdx1(Figure s6 C, and S6E–S6G). In addition, we confirmed the increased plasticity of islets in db/db mice, as we detected glucagon -containing cells that reacted with MafA, vimentin, or nestin (Figure 6D).
These data indicate that genetic ablation of FoxO1 phenocopies the molecular abnormalities associated with diabetes progression, and support the conclusions that β-cell dedifferentiation is pathogenic in the β-cell dysfunction of type 2 diabetes and that the gradual loss of FoxO1 expression during diabetes progression causes β-cell dysfunction (Figure 7).
Figure 7. Proposed Mechanism Of β-Cell Failure.

Healthy β-cells produce insulin and have cytoplasmic FoxO1 (yellow). In the early phases of metabolic stress, insulin production (green) is maintained, but FoxO1 undergoes nuclear translocation to enforce the β-cell fate (red). If the stress persists, FoxO1 expression declines (blue nucleus) as Neurog3, Oct4, Nanog, and L-Myc are reactivated, and β-cell transcription factors are unable to forestall a drop in insulin production (grey). The outcome is twofold: most former β-cells revert to an uncommitted endocrine progenitor stage (grey). A subset of cells undergoes conversion into other hormone-producing cells (orange).
Discussion
The main conclusions of this work are: (i) β-cell dedifferentiation occurs commonly in type 2 diabetes and is associated with an acquired loss of FoxO1 function; (ii) there exists an inverse relationship between FoxO1 and β-cell differentiation as diabetes unfolds, with a striking increase of Neurog3, Oct4, Nanog, and L-Myc expression ; (iii) FoxO1 is required to maintain β-cell identity and prevent β-cell conversion into non-β pancreatic endocrine cells in response to chronic pathophysiologic stress.
Dedifferentiation As A Mechanism Of β-Cell Loss
Our study provides direct in vivo evidence that β-cell dedifferentiation with loss of FoxO1 is a shared mechanism of β-cell failure in diverse models of metabolic stress. This discovery represents a departure from the view that β-cell failure is caused by a reduction of β-cell mass secondary to apoptosis (Butler et al., 2007). While the metabolic abnormalities in mice with β-cell-specific FoxO1 knockout are not as marked as those seen in the other diabetic models, this might be due to compensatory increases in other FoxOs (Figure S1G). Nevertheless, the impairment β-cell mass and function caused by lack of FoxO1 is consistently observed in other models of diabetes (Kobayashi et al., 2012).
We delineate stages in the progression of β-cell dysfunction during type 2 diabetes (Weir and Bonner-Weir, 2004) in greater detail than hitherto achieved, and provide surprising evidence for the reemergence of endocrine progenitor-like cells during adulthood as a result of β-cell dysfunction. We find degranulated β-cells, with decreased insulin content and preserved expression of β-cell markers, Pdx1 and MafA. Although this state had been postulated based on mRNA measurements (Jonas et al., 1999), our data provide anatomical, ultramorphological, and functional evidence for these cells.
The ‘Selfish’ β-Cell
The identification of a set of dedifferentiated β-cells (ChgA+/Neurog3high/L-Myc+/Oct4+) that no longer contribute to insulin production in diabetic islets supports the view that a progressive decline of β-cell function, or ‘β-cell exhaustion’ antedates the physical demise of β-cells (Ferrannini, 2010). Importantly, we show that β-cell dedifferentation is a regression to an endocrine progenitor-like stage rather than a degenerative stage. We speculate that there is an advantage to β-cells in adopting the dedifferentiated fate, and suggest that this ‘selfish’ behavior facilitates their survival.
We propose that during metabolic stress, FoxO1 is required to restrict the β-cell fate by promoting genes required for β-cell identity and by preventing reactivation of embryonic endocrine progenitor genes. Our work expands the implications of the increased plasticity of pancreatic endocrine cells observed as a survival mechanism in models of extreme β-cell ablation (Thorel et al., 2010), by suggesting that plasticity occurs in common forms of β-cell dysfunction, and is caused by the downregulation of FoxO1 that follows hyperglycemia-induced oxidative stress (Kitamura et al., 2005). The loosening of β-cell fate observed following loss of FoxO1 is consistent with the phenotype of Akt gain-of-function (Elghazi et al., 2009). But because Akt has multiple substrates in addition to FoxO1, Akt gain-of-function results in an even greater diversity of cellular fates, while the effect of FoxO1 ablation is limited to endocrine lineages.
Similarly, reactivation of Neurog3 has been seen in the context of extreme depletion of exocrine tissue (Xu et al., 2008), or β-cells (Thorel et al., 2010). In normal pancreatic endocrine differentiation, Neurog3 inhibits its own expression (Wang et al., 2008). In adult islets, increased Neurog3 levels can decrease β-cell survival (Dror et al., 2007). The rise of Neurog3 in FoxO1-deficient β-cells is consistent with a repressor role of FoxO1 in the regulation of Neurog3 expression in gut and pancreatic endocrine differentiation (Al-Masri et al., 2010; Talchai et al., 2012). These data uncover a role of FoxO1 to inhibit Neurog3 reactivation during diabetes development, and illustrate that Neurog3 overexpression is associated with β-cells dysfunction.
The model arising from our observations can help explain why decreases in β-cell mass occur slowly as a function of diabetes duration (Rahier et al., 2008). It is consistent with and provides an explanation for the slow progression and temporary reversibility of β-cell dysfunction, vindicating the concept of ‘β-cell rest’ as a diabetes treatment (Weng et al., 2008).
Conversion Of Different Cell Types In Diabetic Islets
Genetic loss of FoxO1 by conditional knockout phenocopies key aspects of the natural history of islet dysfunction in diabetic mice. First, canonical β-cell transcription factors, Pdx1, MafA, and Nkx6.1 are insufficient to maintain β-cell fate in an altered metabolic environment. Secondly, our lineage-tracing studies show that a subset of dedifferentiated β-cells undergoes conversion into other endocrine cell types, providing a potential explanation for the reciprocal relationship between β-cells and α-cells in diabetes. While quantitatively small when compared to the total number of dedifferentiated β-cells, converted β-cells account for the relative increase in α-cells, and thus provide a potential explanation for the baffling alterations in insulin-to-glucagon ratios and hyperglucagonemia observed in human type 2 diabetes (Dunning and Gerich, 2007). Third, we provide in vivo evidence for the longstanding in vitro and ex vivo observations of activation of mesenchymal markers in endocrine cells during metabolic stress (Russ et al., 2011; Zulewski et al., 2001).
Conclusions
Our findings indicate that there is an ample time window between functional depletion of insulin in β-cells, and their demise. These observations offer a glimmer of hope that salvaging dedifferentiated β-cells might provide an approach to treating β-cell dysfunction in diabetes. This idea is supported by the observations that a fraction of dedifferentiated β-cells can produce other types of pancreatic hormones, indicating that they can become hormone-producing cells, and providing impetus to develop approaches to re-differentiate them into functional β-cells. Our work highlights a FoxO1-dependent mechanism that prevents dedifferentiation and provides insight into maintenance of β-cell function in type 2 diabetes.
Methods
Antibodies, Western Blotting, And Immunohistochemistry
We fixed and processed tissues for immunohistochemistry as described (Kitamura et al., 2009). We applied perfused fixation (Padilla et al., 2010), and antigen retrieval for nuclear transcription factor detection (Nacalai USA). A list of antibodies used in these studies is provided in Supplemental Online Methods. We used FITC-, Cy3-, and Alexa-conjugated donkey secondary antibodies (Jackson Immunoresearch Laboratories, and Molecular Probes), or peroxidase staining as described (Kitamura et al., 2009). Nuclear extracts for western blotting were prepared from 50–100 islets from 4–6 mice from each condition (Wang et al., 2009). 10 μg of proteins were loaded into each well. We used Apoptag In Situ apoptosis detection kit for TUNEL assays (Millipore) and stained nuclei with DAPI or DRAQ5 (Cell Signaling). Image acquisition, analysis, and β-cell mass morphometry have been described (Xuan et al., 2010). For electron microscopy, α-cell mass morphometry and immunofluorescent colocalization analysis were performed as described (Gao et al., 2007). Coexpression of Gfp and insulin or glucagon, somatostatin, and Pp cells, was verified and quantified as ratio of overlapping area/total immunoreactive area (single-stained regions–overlapping area) as automatically assigned by confocal microscopy and Laser Scanning Microscope Software (Zeiss LSM 510 and 710). 4 pancreatic sections from 4 mice from each genotype were sampled 150 μm apart, and used for a two-tailed paired Student’s t test analysis. Total pancreas area was captured at 100X, and immunostaining area at 200X. The ratio of immunoreactive area/pancreas area was converted from pixel to mm2 and plotted as mm2.
Mice
Ins2-cre (Herrera, 2000), Rosa26-eGfp (Gu et al., 2002), InsR+/− (Accili et al., 1996) and Foxo1flox/lox mice have been described (Paik et al., 2007). We performed genotyping as described (Kitamura et al., 2009).
Metabolic Studies
We injected STZ i.p. at a dose of 50mg/kg for five consecutive days in 3–4 month-old male mice and began to analyze them 21 days later (Movassat et al., 1997). We analyzed multiparous females (defined as having carried at least three pregnancies to term) at 8 months of age, and age-matched virgin control females. We analyzed IKO:InsR+/− mice and controls at 8 months of age. For aging studies, we analyzed 16–20 month-old male mice. We performed glucose tolerance tests in overnight-fasted mice by intraperitoneal injection of glucose (2 g/kg) (Xuan et al., 2010). We performed GSIS and arginine-stimulated insulin secretion tests as described (Kido et al., 2000). We measured glucagon by radioimmunoassay and insulin by ELISA (Millipore).
RNA Procedures
We used standard techniques for mRNA isolation and quantitative PCR. References to PCR primer sequences are provided in the Supplemental Online Methods.
Statistical Analyses
We used two-tailed Student’s t-test for data analysis. One asterisk: P< 0.05; two asterisks: P< 0.01 by Student’s t-test. We present data as means ± SEM.
Supplementary Material
Highlights.
Dedifferentiation, not apoptosis, is the main cause of β-cell failure
FoxO1 enforces the β-cell fate during β-cell adaptation to metabolic stress
Dedifferentiated β-cells are prone to converting into α, δ, and Pp endocrine cells
Acknowledgments
Supported by the Druckenmiller Fellowship of the New York Stem Cell Foundation to C.T., by NIH grants DK64819, DK58282, DK63608 (Columbia University Diabetes Research Center), the Brehm Coalition, and the Russell Berrie Foundation. We thank Drs. J.Y. Kim-Muller, T. Mastracci, H. Hua, (Columbia University Medical Center) and S. Cinti (Università Politecnica delle Marche) for advice and reagents, and Q. Xu for excellent technical support. We thank members of the Accili laboratory for insightful discussion and critical reading of the manuscript.
Footnotes
Publisher's Disclaimer: This is a PDF file of an unedited manuscript that has been accepted for publication. As a service to our customers we are providing this early version of the manuscript. The manuscript will undergo copyediting, typesetting, and review of the resulting proof before it is published in its final citable form. Please note that during the production process errors may be discovered which could affect the content, and all legal disclaimers that apply to the journal pertain.
References
- Accili D, Ahren B, Boitard C, Cerasi E, Henquin JC, Seino S. What ails the beta-cell? Diabetes Obes Metab. 2010;12(Suppl 2):1–3. doi: 10.1111/j.1463-1326.2010.01296.x. [DOI] [PubMed] [Google Scholar]
- Accili D, Drago J, Lee EJ, Johnson MD, Cool MH, Salvatore P, Asico LD, Jose PA, Taylor SI, Westphal H. Early neonatal death in mice homozygous for a null allele of the insulin receptor gene. Nat Genet. 1996;12:106–109. doi: 10.1038/ng0196-106. [DOI] [PubMed] [Google Scholar]
- Al-Masri M, Krishnamurthy M, Li J, Fellows GF, Dong HH, Goodyer CG, Wang R. Effect of forkhead box O1 (FOXO1) on beta cell development in the human fetal pancreas. Diabetologia. 2010;53:699–711. doi: 10.1007/s00125-009-1632-0. [DOI] [PubMed] [Google Scholar]
- Basu-Roy U, Seo E, Ramanathapuram L, Rapp TB, Perry JA, Orkin SH, Mansukhani A, Basilico C. Sox2 maintains self renewal of tumor-initiating cells in osteosarcomas. Oncogene. 2012;31:2270–2282. doi: 10.1038/onc.2011.405. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Butler PC, Meier JJ, Butler AE, Bhushan A. The replication of beta cells in normal physiology, in disease and for therapy. Nat Clin Pract Endocrinol Metab. 2007;3:758–768. doi: 10.1038/ncpendmet0647. [DOI] [PubMed] [Google Scholar]
- Collombat P, Hecksher-Sorensen J, Krull J, Berger J, Riedel D, Herrera PL, Serup P, Mansouri A. Embryonic endocrine pancreas and mature beta cells acquire alpha and PP cell phenotypes upon Arx misexpression. J Clin Invest. 2007;117:961–970. doi: 10.1172/JCI29115. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Dhawan S, Georgia S, Tschen SI, Fan G, Bhushan A. Pancreatic beta cell identity is maintained by DNA methylation-mediated repression of Arx. Developmental cell. 2011;20:419–429. doi: 10.1016/j.devcel.2011.03.012. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Dror V, Nguyen V, Walia P, Kalynyak TB, Hill JA, Johnson JD. Notch signalling suppresses apoptosis in adult human and mouse pancreatic islet cells. Diabetologia. 2007;50:2504–2515. doi: 10.1007/s00125-007-0835-5. [DOI] [PubMed] [Google Scholar]
- Dunning BE, Gerich JE. The role of alpha-cell dysregulation in fasting and postprandial hyperglycemia in type 2 diabetes and therapeutic implications. Endocr Rev. 2007;28:253–283. doi: 10.1210/er.2006-0026. [DOI] [PubMed] [Google Scholar]
- Elghazi L, Weiss AJ, Barker DJ, Callaghan J, Staloch L, Sandgren EP, Gannon M, Adsay VN, Bernal-Mizrachi E. Regulation of pancreas plasticity and malignant transformation by Akt signaling. Gastroenterology. 2009;136:1091–1103. doi: 10.1053/j.gastro.2008.11.043. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ferrannini E. The stunned beta cell: a brief history. Cell metabolism. 2010;11:349–352. doi: 10.1016/j.cmet.2010.04.009. [DOI] [PubMed] [Google Scholar]
- Gao N, White P, Doliba N, Golson ML, Matschinsky FM, Kaestner KH. Foxa2 controls vesicle docking and insulin secretion in mature Beta cells. Cell Metab. 2007;6:267–279. doi: 10.1016/j.cmet.2007.08.015. [DOI] [PubMed] [Google Scholar]
- Gershengorn MC, Hardikar AA, Wei C, Geras-Raaka E, Marcus-Samuels B, Raaka BM. Epithelial-to-mesenchymal transition generates proliferative human islet precursor cells. Science. 2004;306:2261–2264. doi: 10.1126/science.1101968. [DOI] [PubMed] [Google Scholar]
- Gu G, Dubauskaite J, Melton DA. Direct evidence for the pancreatic lineage: NGN3+ cells are islet progenitors and are distinct from duct progenitors. Development. 2002;129:2447–2457. doi: 10.1242/dev.129.10.2447. [DOI] [PubMed] [Google Scholar]
- Hang Y, Stein R. MafA and MafB activity in pancreatic beta cells. Trends in endocrinology and metabolism. 2011;22:364–373. doi: 10.1016/j.tem.2011.05.003. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Herrera PL. Adult insulin- and glucagon-producing cells differentiate from two independent cell lineages. Development. 2000;127:2317–2322. doi: 10.1242/dev.127.11.2317. [DOI] [PubMed] [Google Scholar]
- Hur KY, Jung HS, Lee MS. Role of autophagy in beta-cell function and mass. Diabetes Obes Metab. 2010;12(Suppl 2):20–26. doi: 10.1111/j.1463-1326.2010.01278.x. [DOI] [PubMed] [Google Scholar]
- Jonas JC, Sharma A, Hasenkamp W, Ilkova H, Patane G, Laybutt R, Bonner-Weir S, Weir GC. Chronic hyperglycemia triggers loss of pancreatic beta cell differentiation in an animal model of diabetes. The Journal of biological chemistry. 1999;274:14112–14121. doi: 10.1074/jbc.274.20.14112. [DOI] [PubMed] [Google Scholar]
- Kawamori D, Kaneto H, Nakatani Y, Matsuoka TA, Matsuhisa M, Hori M, Yamasaki Y. The forkhead transcription factor Foxo1 bridges the JNK pathway and the transcription factor PDX-1 through its intracellular translocation. J Biol Chem. 2006;281:1091–1098. doi: 10.1074/jbc.M508510200. [DOI] [PubMed] [Google Scholar]
- Kido Y, Burks DJ, Withers D, Bruning JC, Kahn CR, White MF, Accili D. Tissue-specific insulin resistance in mice with mutations in the insulin receptor, IRS-1, and IRS-2. The Journal of clinical investigation. 2000;105:199–205. doi: 10.1172/JCI7917. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kitamura T, Kitamura YI, Funahashi Y, Shawber CJ, Castrillon DH, Kollipara R, DePinho RA, Kitajewski J, Accili D. A Foxo/Notch pathway controls myogenic differentiation and fiber type specification. The Journal of clinical investigation. 2007;117:2477–2485. doi: 10.1172/JCI32054. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kitamura T, Kitamura YI, Kobayashi M, Kikuchi O, Sasaki T, Depinho RA, Accili D. Regulation of pancreatic juxtaductal endocrine cell formation by FoxO1. Molecular and cellular biology. 2009;29:4417–4430. doi: 10.1128/MCB.01622-08. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kitamura T, Nakae J, Kitamura Y, Kido Y, Biggs WH, 3rd, Wright CV, White MF, Arden KC, Accili D. The forkhead transcription factor Foxo1 links insulin signaling to Pdx1 regulation of pancreatic beta cell growth. The Journal of clinical investigation. 2002;110:1839–1847. doi: 10.1172/JCI200216857. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kitamura YI, Kitamura T, Kruse JP, Raum JC, Stein R, Gu W, Accili D. FoxO1 protects against pancreatic beta cell failure through NeuroD and MafA induction. Cell metabolism. 2005;2:153–163. doi: 10.1016/j.cmet.2005.08.004. [DOI] [PubMed] [Google Scholar]
- Kobayashi M, Kikuchi O, Sasaki T, Kim HJ, Yokota-Hashimoto H, Lee YS, Amano K, Kitazumi T, Susanti VY, Kitamura YI, et al. FoxO1 as a double-edged sword in the pancreas: analysis of pancreas- and beta-cell-specific FoxO1 knockout mice. American journal of physiology Endocrinology and metabolism. 2012;302:E603–613. doi: 10.1152/ajpendo.00469.2011. [DOI] [PubMed] [Google Scholar]
- Lin HV, Ren H, Samuel VT, Lee HY, Lu TY, Shulman GI, Accili D. Diabetes in mice with selective impairment of insulin action in Glut4-expressing tissues. Diabetes. 2011;60:700–709. doi: 10.2337/db10-1056. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Martinez SC, Cras-Meneur C, Bernal-Mizrachi E, Permutt MA. Glucose Regulates Foxo1 Through Insulin Receptor Signaling in the Pancreatic Islet {beta}-cell. Diabetes. 2006;55:1581–1591. doi: 10.2337/db05-0678. [DOI] [PubMed] [Google Scholar]
- Matsuda T, Kido Y, Asahara S, Kaisho T, Tanaka T, Hashimoto N, Shigeyama Y, Takeda A, Inoue T, Shibutani Y, et al. Ablation of C/EBPbeta alleviates ER stress and pancreatic beta cell failure through the GRP78 chaperone in mice. J Clin Invest. 2010;120:115–126. doi: 10.1172/JCI39721. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Movassat J, Saulnier C, Portha B. Insulin administration enhances growth of the beta-cell mass in streptozotocin-treated newborn rats. Diabetes. 1997;46:1445–1452. doi: 10.2337/diab.46.9.1445. [DOI] [PubMed] [Google Scholar]
- Nakae J, Kitamura T, Kitamura Y, Biggs WH, 3rd, Arden KC, Accili D. The forkhead transcription factor Foxo1 regulates adipocyte differentiation. Developmental cell. 2003;4:119–129. doi: 10.1016/s1534-5807(02)00401-x. [DOI] [PubMed] [Google Scholar]
- Nakagawa M, Takizawa N, Narita M, Ichisaka T, Yamanaka S. Promotion of direct reprogramming by transformation-deficient Myc. Proceedings of the National Academy of Sciences of the United States of America. 2010;107:14152–14157. doi: 10.1073/pnas.1009374107. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Okamoto H, Hribal ML, Lin HV, Bennett WR, Ward A, Accili D. Role of the forkhead protein FoxO1 in beta cell compensation to insulin resistance. The Journal of clinical investigation. 2006;116:775–782. doi: 10.1172/JCI24967. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Padilla SL, Carmody JS, Zeltser LM. Pomc-expressing progenitors give rise to antagonistic neuronal populations in hypothalamic feeding circuits. Nature medicine. 2010;16:403–405. doi: 10.1038/nm.2126. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Paik JH, Kollipara R, Chu G, Ji H, Xiao Y, Ding Z, Miao L, Tothova Z, Horner JW, Carrasco DR, et al. FoxOs Are Lineage-Restricted Redundant Tumor Suppressors and Regulate Endothelial Cell Homeostasis. Cell. 2007;128:309–323. doi: 10.1016/j.cell.2006.12.029. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Papizan JB, Singer RA, Tschen SI, Dhawan S, Friel JM, Hipkens SB, Magnuson MA, Bhushan A, Sussel L. Nkx2.2 repressor complex regulates islet beta-cell specification and prevents beta-to-alpha-cell reprogramming. Genes Dev. 2011;25:2291–2305. doi: 10.1101/gad.173039.111. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Rahier J, Guiot Y, Goebbels RM, Sempoux C, Henquin JC. Pancreatic beta-cell mass in European subjects with type 2 diabetes. Diabetes Obes Metab. 2008;10(Suppl 4):32–42. doi: 10.1111/j.1463-1326.2008.00969.x. [DOI] [PubMed] [Google Scholar]
- Rankin MM, Kushner JA. Aging induces a distinct gene expression program in mouse islets. Islets. 2010;2:345–352. doi: 10.4161/isl.2.6.13376. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Rieck S, White P, Schug J, Fox AJ, Smirnova O, Gao N, Gupta RK, Wang ZV, Scherer PE, Keller MP, et al. The transcriptional response of the islet to pregnancy in mice. Molecular endocrinology. 2009;23:1702–1712. doi: 10.1210/me.2009-0144. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Robertson RP. Chronic oxidative stress as a central mechanism for glucose toxicity in pancreatic islet beta cells in diabetes. J Biol Chem. 2004;279:42351–42354. doi: 10.1074/jbc.R400019200. [DOI] [PubMed] [Google Scholar]
- Russ HA, Sintov E, Anker-Kitai L, Friedman O, Lenz A, Toren G, Farhy C, Pasmanik-Chor M, Oron-Karni V, Ravassard P, et al. Insulin-producing cells generated from dedifferentiated human pancreatic beta cells expanded in vitro. PLoS One. 2011;6:e25566. doi: 10.1371/journal.pone.0025566. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Seymour PA, Freude KK, Tran MN, Mayes EE, Jensen J, Kist R, Scherer G, Sander M. SOX9 is required for maintenance of the pancreatic progenitor cell pool. Proceedings of the National Academy of Sciences of the United States of America. 2007;104:1865–1870. doi: 10.1073/pnas.0609217104. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Talchai C, Lin HV, Kitamura T, Accili D. Genetic and biochemical pathways of beta-cell failure in type 2 diabetes. Diabetes Obes Metab. 2009;11(Suppl 4):38–45. doi: 10.1111/j.1463-1326.2009.01115.x. [DOI] [PubMed] [Google Scholar]
- Talchai C, Xuan S, Kitamura T, Depinho RA, Accili D. Generation of functional insulin-producing cells in the gut by Foxo1 ablation. Nat Genet. 2012;44:406–412. doi: 10.1038/ng.2215. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Thorel F, Nepote V, Avril I, Kohno K, Desgraz R, Chera S, Herrera PL. Conversion of adult pancreatic alpha-cells to beta-cells after extreme beta-cell loss. Nature. 2010;464:1149–1154. doi: 10.1038/nature08894. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wang S, Hecksher-Sorensen J, Xu Y, Zhao A, Dor Y, Rosenberg L, Serup P, Gu G. Myt1 and Ngn3 form a feed-forward expression loop to promote endocrine islet cell differentiation. Dev Biol. 2008;317:531–540. doi: 10.1016/j.ydbio.2008.02.052. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wang S, Jensen JN, Seymour PA, Hsu W, Dor Y, Sander M, Magnuson MA, Serup P, Gu G. Sustained Neurog3 expression in hormone-expressing islet cells is required for endocrine maturation and function. Proceedings of the National Academy of Sciences of the United States of America. 2009;106:9715–9720. doi: 10.1073/pnas.0904247106. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Weinberg N, Ouziel-Yahalom L, Knoller S, Efrat S, Dor Y. Lineage tracing evidence for in vitro dedifferentiation but rare proliferation of mouse pancreatic beta-cells. Diabetes. 2007;56:1299–1304. doi: 10.2337/db06-1654. [DOI] [PubMed] [Google Scholar]
- Weir GC, Bonner-Weir S. Five stages of evolving beta-cell dysfunction during progression to diabetes. Diabetes. 2004;53(Suppl 3):S16–21. doi: 10.2337/diabetes.53.suppl_3.s16. [DOI] [PubMed] [Google Scholar]
- Weng J, Li Y, Xu W, Shi L, Zhang Q, Zhu D, Hu Y, Zhou Z, Yan X, Tian H, et al. Effect of intensive insulin therapy on beta-cell function and glycaemic control in patients with newly diagnosed type 2 diabetes: a multicentre randomised parallel-group trial. Lancet. 2008;371:1753–1760. doi: 10.1016/S0140-6736(08)60762-X. [DOI] [PubMed] [Google Scholar]
- Weyer C, Bogardus C, Mott DM, Pratley RE. The natural history of insulin secretory dysfunction and insulin resistance in the pathogenesis of type 2 diabetes mellitus. J Clin Invest. 1999;104:787–794. doi: 10.1172/JCI7231. [DOI] [PMC free article] [PubMed] [Google Scholar]
- White P, May CL, Lamounier RN, Brestelli JE, Kaestner KH. Defining pancreatic endocrine precursors and their descendants. Diabetes. 2008;57:654–668. doi: 10.2337/db07-1362. [DOI] [PubMed] [Google Scholar]
- Xu X, D’Hoker J, Stange G, Bonne S, De Leu N, Xiao X, Van de Casteele M, Mellitzer G, Ling Z, Pipeleers D, et al. Beta cells can be generated from endogenous progenitors in injured adult mouse pancreas. Cell. 2008;132:197–207. doi: 10.1016/j.cell.2007.12.015. [DOI] [PubMed] [Google Scholar]
- Xuan S, Szabolcs M, Cinti F, Perincheri S, Accili D, Efstratiadis A. Genetic analysis of type-1 insulin-like growth factor receptor signaling through insulin receptor substrate-1 and -2 in pancreatic beta cells. The Journal of biological chemistry. 2010;285:41044–41050. doi: 10.1074/jbc.M110.144790. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zhang X, Yalcin S, Lee DF, Yeh TY, Lee SM, Su J, Mungamuri SK, Rimmele P, Kennedy M, Sellers R, et al. FOXO1 is an essential regulator of pluripotency in human embryonic stem cells. Nature cell biology. 2011;13:1092–1099. doi: 10.1038/ncb2293. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zhao M, Amiel SA, Ajami S, Jiang J, Rela M, Heaton N, Huang GC. Amelioration of streptozotocin-induced diabetes in mice with cells derived from human marrow stromal cells. PLoS One. 2008;3:e2666. doi: 10.1371/journal.pone.0002666. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zhou Q, Brown J, Kanarek A, Rajagopal J, Melton DA. In vivo reprogramming of adult pancreatic exocrine cells to beta-cells. Nature. 2008;455:627–632. doi: 10.1038/nature07314. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zulewski H, Abraham EJ, Gerlach MJ, Daniel PB, Moritz W, Muller B, Vallejo M, Thomas MK, Habener JF. Multipotential nestin-positive stem cells isolated from adult pancreatic islets differentiate ex vivo into pancreatic endocrine, exocrine, and hepatic phenotypes. Diabetes. 2001;50:521–533. doi: 10.2337/diabetes.50.3.521. [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.