

Abstract
A variety of genetic and molecular alterations underlie the development and progression of colorectal neoplasia (CRN). Most of these cancers arise sporadically due to multiple somatic mutations and genetic instability. Genetic instability includes chromosomal instability (CIN) and microsatellite instability (MSI), which is observed in most hereditary non-polyposis colon cancers (HNPCCs) and accounts for a small proportion of sporadic CRN. Although many biomarkers have been used in the diagnosis and prediction of the clinical outcomes of CRNs, no single marker has established value. New markers and genes associated with the development and progression of CRNs are being discovered at an accelerated rate. CRN is a heterogeneous disease, especially with respect to the anatomic location of the tumor, race/ethnicity differences, and genetic and dietary interactions that influence its development and progression and act as confounders. Hence, efforts related to biomarker discovery should focus on identification of individual differences based on tumor stage, tumor anatomic location, and race/ethnicity; on the discovery of molecules (genes, mRNA transcripts, and proteins) relevant to these differences; and on development of therapeutic approaches to target these molecules in developing personalized medicine. Such strategies have the potential of reducing the personal and socio-economic burden of CRNs. Here, we systematically review molecular and other pathologic features as they relate to the development, early detection, diagnosis, prognosis, progression, and prevention of CRNs, especially colorectal cancers (CRCs).
Keywords: Early detection, molecular diagnosis, prognosis, molecular staging, colorectal cancer
1. Introduction
Colorectal neoplasias (CRNs) are a major medical concern worldwide, and they have a substantial impact on the costs of healthcare. In particular, colorectal adenocarcinomas (CRCs) are responsible for considerable morbidity and mortality. In the United States (US) in 2009, there were an estimated 146,970 new cases and 49,980 deaths from CRCs [1]. They are the second leading cause of cancer deaths in the US. The disease is heterogeneous, especially with respect to the anatomic location of the tumor, and there are racial differences and genetic and dietary interactions that influence its development. The colon accounts for 70% of large intestinal cancer. Approximately 95% of CRCs are believed to have evolved from adenomatous polyps. CRCs arising de novo from the epithelium are only 5% of the total. A variety of genetic and molecular changes occur as these polyps transform from benignity to malignancy [2].
Colonoscopy has allowed a change in CRC intervention, from one of early detection to one of prevention. It leads to removal of polyps before molecular changes occur and before lesions reach the threshold of malignancy. While the cost of monitoring patients by colonoscopy may seem extensive, it is a fraction of the cost of treating invasive cancer. After diagnosis and treatment, the tumor markers, CEA and Ki-67, can be used to follow these patients in addition to colonoscopy. New markers and genes associated with the development of CRCs are being discovered at an accelerated rate. This article reviews molecular and other pathologic features as they relate to the development, early detection, diagnosis, prognosis, progression, and prevention of CRNs, especially CRCs.
2. Risk factors
Risk factors in colorectal carcinogenesis include a family history of CRNs, development of polyps, inflammatory bowel disease (e.g., ulcerative colitis), obesity, tobacco and alcohol abuse, high stress, and factors associated with the Western diet. Patients who exercise strenuously and take non-steroidal anti-inflammatory drugs daily have a reduced likelihood of developing CRCs [3,4].
2.1. Dietary factors
Diets high in red and processed meats and refined sugar and low in fruits, vegetables, and fiber are thought to increase the risk for CRCs. Of special concern are the products of pyrolysis (i.e., chemicals resulting from overcooking, especially if they stay with the meat or if the gravy and drippings are consumed). Dietary consumption of starch; calcium; vitamins B, C, D, and E; β-carotene (a precursor of vitamin A); and folate appear to be protective. Nevertheless, there is a general lack of understanding of factors in the diet that cause increased risk or are protective for CRNs; the risk may involve the interaction of dietary components and bile acids with intestinal bacteria, and intestinal mucus with dietary carcinogens [5–7]. There is the contention that dietary fiber decreases transit time in the intestine, thus minimizing long-term contact between the dietary carcinogens and the intestinal mucosa [8]. Multiple dietary components acting concomitantly may be required for the development of CRNs.
2.2. Chronic inflammation
Chronic inflammation of the colorectum, similar to that in ulcerative colitis, is a risk factor for CRC. Grizzle et al. [382] have proposed that such tumors are the results of longstanding, continuous damage, inflammation, and repair (LOCDIR). The concept is that LOCDIR changes cellular features of the epithelium, causing loss of cellular differentiation (loss of cellular mucus) and development of cellular atypia and mutations at multiple sites. DNA damage, with microsatellite instability (MSI) and genomic instability, may arise within a year. Kruppel-like Factor 6 (KLF-6) is frequently inactivated in CRC [9]. As cellular atypia increase, there may be progress from low- to high-grade dysplasia. After 10 or more years, carcinomas may develop without an exophytic feature. After 10 years of ulcerative colitis, the risk of colon cancer is 20 to 30 times that for a matched population. As an effective preventive measure, most patients with ulcerative colitis undergo total colectomy with ileostomy. A more controversial but also effective procedure is proctocolectomy with distal rectal mucosectomy. Although Crohn’s disease had long been thought to have no association with the development of CRCs, it is now known that there is an 8% risk of developing CRC over a 20-year period [10]. The problem of chronic inflammation with healing and epithelial changes at the cellular and molecular levels may be involved, since most of these cancers occur in the strictured areas of the intestines.
2.3. Environmental exposures
Environmental exposures have various effects on the development of CRNs, depending upon the individual’s metabolic enzymes that are involved in detoxification of carcinogens or activation of procarcinogens. For example, individuals with the E487K polymorphism of aldehyde dehydrogenase 2 (ALDH2) have reduced ALDH2 activity. Alcohol consumption is of greater risk for individuals heterozygous for ALDH2 [11]. Similarly, 5,10-methylene tetrahydrofolate reductase polymorphisms coupled with low folate levels increase the risk of CRC development associated with alcohol consumption [12–14]. Also, polymorphisms in L-arylamine-N-acetyltransferase, cytosolic glutathione S-transferase, and cystathionine β-synthase increase the risk of cancer, and, in some cases, the age at onset of CRCs [15, 16].
3. CRC Models, hereditary and sporadic
3.1. Hereditary CRNs
Most CRCs are sporadic; fewer than 5% are familial. There are two recognized familial forms of CRNs. These involve the inheritance of a single copy of a mutated gene, APC, which causes the development of familial adenomatous polyposis [17] or one mutated copy of a mismatch repair gene, such as MSH2 or MLH-1, which causes hereditary nonpolyposis CRC (HNPCC) with MSI [18–20]. Cowden disease (an autosomal dominant hamartomatous syndrome often associated with breast cancers, thyroid disease, trichlemmomas, and gastrointestinal polyps and cancers) does not appear to be a strong contributor to familial forms of CRNs. The general development of these forms of hereditary CRCs is illustrated in Fig. 1. Various mutations in APC also cause familial CRCs in the Gardner syndrome (epidermal inclusion cysts, bone osteomas, and colonic polyps), attenuated adenomatous polypoposis coli, and the Turcot syndrome [21–23]. Other familial forms of CRC occur in MU-TYH (MYH)-associated polyposis (mutations in MU-TYH [24]); juvenile polyposis syndrome (mutations in SMAD4, BMPR1a or PTEN) [25,26]; the CpG island methylator phenotype (CIMP) [27]; and CRNs associated with glioblastoma multiforme (mutations in multiple pathways, including p53 and PMS2) [28]. Thus, development of CRNs is frequently associated with mutations/methylations or dysregulation of the following genes: APC, MSH2, MLH1, PMS1, PMS2, MSH6, PTEN, SMAD4, MUTYH (MYH), BMPR1α, LKB1/STK11, and B-RAF. The progression of CRNs, as will be discussed, is associated with mutations or dysregulation of DCC, K-ras, Bcl2, and p53.
Fig. 1.
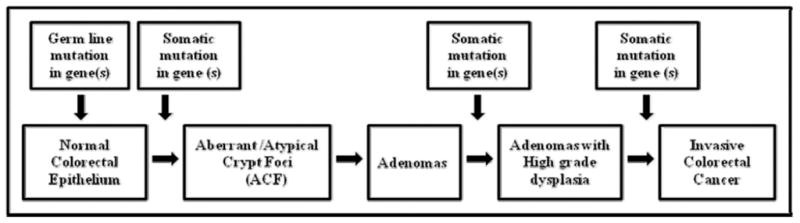
The general development of hereditary CRCs.
3.2. Sporadic CRNs
The model presented by Fearon and Vogelstein has been the paradigm of the genetic alterations involved in development of CRCs [2]. The pattern of CRC development, as exhibited in FAP, is the best model for the development of most common cases of sporadic CRCs; however, the order and pattern of development of mutations are more variable in sporadic cases. Most sporadic CRCs develop due to multiple somatic mutations and genetic instability, which are broadly grouped into the category of chromosomal instability (CIN); a smaller proportion follows the MSI observed in most HNPCCs.
Epigenetic changes secondary to hypermethylation of CpG islands of the promoters of mismatch repair genes, especially hypermethylation leading to the inactivation of both copies of hMLH1, are thought to cause sporadic tumors with MSI. This is consistent with the high rate of MSI (up to 15%) observed in sporadic CRCs. Other genes confer a risk for the development of CRC. The transforming growth factor-β (TGFβ) signal transduction pathway is thought to be important in the development of multiple types of cancer. In HNPCC, MSI frequently knocks out functioning TGFβR2 [29]. Similarly, a polymorphic allele of TGFβR1 (TGFβR1*6A) has an increased frequency in patients with CRC, and germ-line allele-specific expression of TGFβR1 is associated with an increased risk of CRC [30–33].
4. Lesions of CRNs
4.1. Preinvasive neoplasia
CRCs arise primarily from polyps, which are mucosal pedunculated and sessile lesions found on the luminal aspect of the colorectum. Frequently, a polyp component is present at the edge of a CRC. All adenomas have a malignant potential; frequency of transformation increases as histopathological features change in tubular (5%), tubulovillous (22%), and villous (40%) adenomas [34]. There is a significantly higher risk of these polyps containing malignant cells when the size increases to greater than 2 cm.
4.1.1. Aberrant crypt foci (ACF)
Although polyps are thought to arise from ACF, transitional lesions between ACF and polyps have not been described. ACF, first identified by Bird [35–37] in carcinogen-induced CRNs of rodents, represent an early histopathologic change in the development of CRNs. Subsequently, ACF were identified in human colonic mucosa, where the lesions were associated with early cellular mutations in K-ras [37–39]. ACF develop following increased cellular proliferation and cellular accumulation. Some of the histologic and molecular changes of ACF are listed in Table 1.
Table 1.
Features of aberrant crypt foci
| Histo-morphology | Tumors identified in 25% of animals with associated ACF changes at 20 weeks (A) |
| Increased staining with methylene blue (gross) (A&H) | |
| Increased size of the crypt (A&H) | Enzymatic alterations |
| Tendency to form associated groups of dysplastic glands (A&H) | Hexosamidase decreased (A) or increased (H) |
| Slit-like irregular lumens (A&H) | Increased stromal gamma-glutamyltransferase (A) |
| Development of carcinoma in situ (A&H) | Increased stromal alkaline phosphatase (A) |
| Mucin depletion | Decreased stromal acid phosphatase (A) |
| Decreased mucin (A) | Variable (usually increased) stromal 5′-nucleotidase (A) |
| Increased sialomucins and decreased sulfomucins (A) | |
| Increased PAS reactive material (A) | Mutations/genomic changes |
| K-ras codons 12 & 13 (A&H) | |
| Increased proliferation indices | p53 (uncommon) (A&H) |
| BrdU incorporation (A&H) | APC (H) |
| Mitoses | Increased genomic instability (H) |
| PCNA (A) | |
| ACF in cases with CRC | Molecular changes/increased oncofetal markers |
| Dysplasia identified: 6% severe, 8% moderate 40% mild (H): carcinoma in situ identified (H) | Altered pattern of expression of p21W af–1 |
| Increased expression of CEA (H) |
A = Animal models; H = Humans.
4.1.2. Adenomatous polyps/adenomas
Most polyps develop in the colorectum. About 40% of patients over 50 years of age have at least one colorectal polyp, and the number increases with age [40]. Typically, 65–85% of polyps are tubular, 10–25% tubulovillous, and 5–10% villous. Flat adenomas, which may develop into CRNs, are rare and occur characteristically in families with inheritable CRNs. About 90% of the preinvasive neoplastic lesions of the colorectum are thought to be polyps or polyp precursors, such as ACF. Most tubular polyps are considered to be neoplastic, and are designated as adenomas or adenomatous polyps. Periodic removal of all polyps from the colorectum reduces the development of CRCs [40–42].
In our study of 90 cases of CRCs, along with their contiguous adenomatous components (CAdCs), patients whose CAdCs and contiguous CRCs demonstrated p53 nuclear accumulation (p53nac) had shorter median survival (35.9 months) than those whose CAdCs and CRCs did not (80.56 months). Patients whose CAdCs had p53nac and lacked Bcl-2 expression had a lower median survival (15.74 months) compared to patients whose CAdCs did not demonstrate p53nac but had increased expression of Bcl-2 (71.8 months). These findings suggest that, for adenomas demonstrating p53nac but lacking Bcl-2 expression, their contiguous CRCs are more likely to become aggressive [43].
4.1.3. Other pre-invasive neoplastic lesions of the colorectum
In contrast to adenomatous polyps, hyperplastic polyps, unless they are associated with familial syndromes, have little to no potential to develop into CRNs. Polyps of patients with the hyperplastic polyposis syndrome may harbor foci of epithelial dysplasia, and hence these patients are considered at risk for the development of CRNs. A mixture of epithelial serration and dysplasia occurs in mixed polyps or serrated adenomas, which may be precursors to adenomatous polyps. Serrated adenomas may exhibit mutations in K-ras or BRAF as well as MSI secondary to methylation of hMLH1, while flat adenomas, which may develop into CRNs, are rare and occur characteristically in families with inheritable CRNs.
4.2. Transition from pre-invasive neoplasia to invasive neoplasia
Pre-invasive neoplasias, including carcinoma in situ (high grade dysplasia) do not metastasize. After a tumor reaches 2 mm in size, it requires vascularization to survive. Through angiogenesis, capillaries develop in the tumor, and when malignancy develops, cells can enter the circulation for microinvasion. As microinvasion begins, typically in adenomas, metastasis remains unlikely, either because of the limited number of neoplastic cells exposed to lymphatic channels and the reduced number of lymphatic channels present in the colorectal mucosa or lamina propria or because the volume of malignant cells is small. In the colorectal mucosa, lymphatics are found only at the base of the mucosa, immediately above the muscularis mucosa. Some capillaries, however, are in the lamina propria. Until there is invasion through the muscularis mucosa, the tumor is a Tis component in the TNM staging system (Stage 0); when the tumor invades through the muscularis mucosa, it becomes T1 (also Stage I). Invasion into the muscularis propria without metastases leads to T2 lesions, and invasion without metastasis across the muscularis propria to the serosa results in T3 lesions (Stage IIA). Invasion, without metastasis, that perforates the serosa (visceral peritoneum) results in T4 or Stage IIB lesions. Of note, Stage I and II lesions may be cured by surgery.
Why do metastases not occur until tumors are larger? Perhaps they appear more frequently as CRCs increase in size due to interaction of cancer cells with greater numbers of lymphatics and blood vessels. This would lead to a greater likelihood of metastatic spread to local lymph nodes (Stage III) and then to distant metastases (Stage IV) (Fig. 2). However, per ‘size category,’ HNPCCs are less aggressive than FAP lesions or sporadic CRCs. Alternatively, metastatic spread could be facilitated by metastasis-inducing genes, such as osteopontin, MEK1, and CXCR4 [44]. The main genes that affect metastatic spread, however, typically inhibit the process, as reviewed by Stafford et al. [45]. These genes can be grouped based on their location: membrane-bound, cytoplasmic, or nuclear. Further, some microRNAs (e.g., miR-146) may suppress the metastatic spread of breast cancer [46].
Fig. 2.
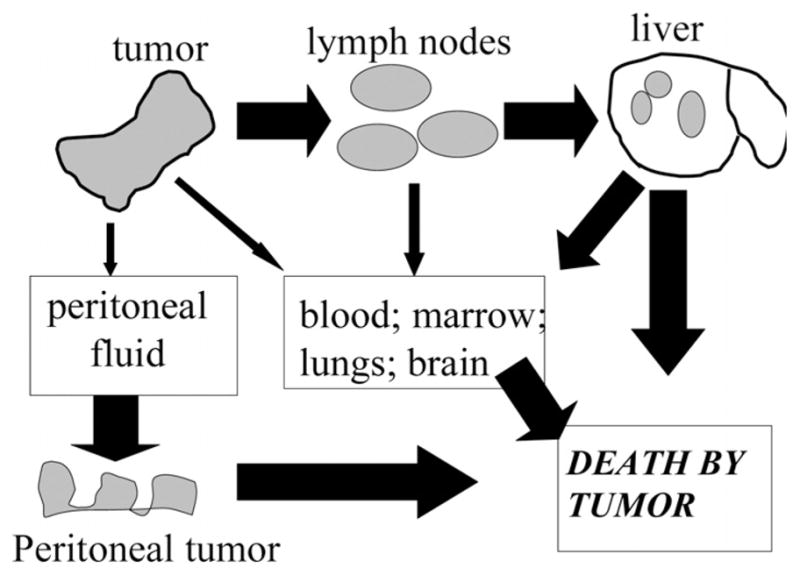
Pathways by which CRCs cause death. *The size of the arrow is proportional to the likelihood of the pathway.
An alternative to accepted patterns of metastases and growth in general is the hypothesis of self-seeding, developed by Norton and colleagues [47]. According to this concept, cells from non-metastatic tumors circulate in blood/lymph fluid and return to the peripheral area of the primary tumor, seeding its periphery. When the surface area of the primary tumor and its volume reach a critical level, the tumor cells no longer completely return to the primary tumor, and metastases occur. Further, when the primary tumor is surgically removed, seeding of the tumor by the circulating cells can no longer occur, leading to metastases; however, neoadjuvant therapy may reduce metastatic spread [48, 49].
4.3. Invasive neoplasia
4.3.1. Distribution of neoplasia in the colorectum
CRC is a heterogeneous disease, especially with respect to the anatomic location of the tumor. Based on the embryologic development of these areas from the midgut (the proximal colon) and hindgut (the distal colorectum), the colorectum can be divided into the proximal colon (cecum, ascending colon, and proximal 2/3 of the transverse colon), distal colon (1/3 of transverse colon at the splenic flexure, descending colon, and sigmoid colon), and the rectum [50]. There are anatomic, functional, and molecular differences between the proximal and distal colorectum, including differences in the phenotypic expression of various biomarkers [51]. Proximal and distal CRCs often differ in their response to screening tests, the stage at which they are diagnosed, their etiology, and their subsequent effect on mortality [52–63]. Relative to proximal CRCs, distal CRCs have a higher incidence of abnormalities in the p53 gene [56,59–61], more allelic deletions of the 17p gene [54,56,60], and lower rates of MSI [63,64]. Furthermore, the prognostic significance of p53nac is greater for adenocarcinomas of the proximal than the distal colon [65–67]. In contrast, p53nac is significantly associated with a higher incidence of local recurrence in distal but not proximal CRCs [68].
Rectal tumors are considered apart from CRCs. This is because the proximal and distal colons are different embryologically and have dissimilar characteristics (Table 2). The therapeutic approach to these tumors is different from that for colon tumors. Over the last several decades, there has been an apparent shift in the most frequent location of occurrence of colon cancers from the distal to proximal areas of the colon. This may be due to a change in methods of diagnosis, especially with the increased use of colonoscopy, which can detect cancers earlier than barium enemas, used in the past, or sigmoidoscopy, which views only the distal colon and rectum. The first column of Table 3 shows the distribution of colon and rectal adenocarcinomas in the period 1941–1956 [69]. We have noted a shift to more proximal lesions, especially in African-Americans.
Table 2.
Features of proximal versus distal colon versus rectum
| Characteristics | Proximal colon | Distal colon | Rectum |
|---|---|---|---|
| Embryological development | Midgut | Hindgut | Hindgut |
| Vascular supply | Superior mesenteric | Inferior mesenteric | Inferior mesenteric |
| Primary function | Fluid transport from lumen; fecal material is liquid | Some fluid transport from lumen plus storage; semisolid fecal material | Fecal storage; feces is solid |
| Primary pattern of mucin | Sulformucins in top 2/3 of crypt; acidic sialomucins in bottom 1/3 of crypts. | N- and O-acetylated sialomucins in superficial epithelial cells and top portion of crypt; sulfomucins at base of crypts. | N- and O-acetylated sialo- mucins in superficial epithelial cells and top portion of crypt; sulfomucins at base of crypts. |
| Blood group expression | Appropriate expression of A, B, H and Leb blood groups | No expression of appropriate A, B, H, and Leb blood groups | No expression of appropriate A, B, H, and Leb blood groups |
| Location of CRC with micro- satellite instability, i.e. hereditary non-polyposis colon cancer | Usual location of MSI tumors | MSI tumors much less frequent | Infrequent |
| Aberrant crypt foci associated with CRC | 50% of cases | 100% of cases | 100% of cases |
| Pattern of mucin of tumors | O-acetylated sialomucins not increased | O-acetylated sialomucins increased | O-acetylated sialomucins increased |
| K-ras mutations in tumors | Less common | More common | More common |
| p53 mutations – nuclear accumulation | Less common than distal | Majority of distal tumors | More common |
| Risk of cancer development secondary to polymorphic enzymes of cytosolic glutathione S-transferase | Increased | Not increased | Not increased |
Table 3.
Distribution of CRCs based on patient ethnicity and tumor location at UAB
| Location | Copeland et al. [69]. % | UAB Caucasian- American % | UAB African- American % |
|---|---|---|---|
| Cecum | 5.2 | 22.7 | 21.8 |
| Ascending | 5.3 | 16.9 | 12.8 |
| Transverse | 8.4 | 8.5 | 8.2 |
| Descending | 6.9 | 15.7 | 7.1 |
| Sigmoid | 30.8 | 22.5 | 23.9 |
| Rectum | 39.8 | 13.6 | 26.1 |
| Proximal colon – all components | 18.9 | 48.2 | 42.8 |
| Distal colon – all components | 41.3 | 38.2 | 31.0 |
4.3.2. Racial and sexual differences in CRCs
Based on SEER results, both colorectal incidence and mortality are higher in African-Americans than Caucasian-Americans and other racial ethnic groups (Tables 3 and 4) [1,70]. In all racial/ethnic groups, males have a higher incidence and mortality. Over the last decade, incidence and mortality rates for CRCs in males have declined in every racial/ethnic group of interest. For females, the incidence over the last decade has decreased in African-Americans, Caucasian-Americans, and Asian Americans; mortality has decreased for all racial and ethnic groups [1, 70].
Table 4.
CRC incidence and mortality rates, 2001–2005 (1, 12, 13)
| RACE/ETHNICITY | INCIDENCE*
|
MORTALITY*
|
||
|---|---|---|---|---|
| Male | Female | Male | Female | |
| African-American | 71.2 | 54.5 | 31.8 | 22.4 |
| Caucasian-American | 58.9 | 43.2 | 22.1 | 15.3 |
| Hispanic/Latino | 47.3 | 32.8 | 16.5 | 10.8 |
| Asian American/Pacific | 48 | 35.4 | 14.4 | 10.2 |
| American Indian/Alaska | 46 | 41.2 | 20.5 | 14.2 |
| Native | ||||
| Overall** | 59.2 | 43.8 | 22.7 | 15.9 |
Based on cases per 100,000 population.
A variety of reasons have been proposed for the racial disparity in survival differences between African-American and Caucasian-American patients, reviewed in Alexander et al. [52]. These include differences in age, advanced stage at diagnosis, treatment differences, socioeconomic factors, access to health care, biologic differences, and approaches to analysis [71,72]. Recent studies shed light on disparities in access to care and on the molecular characteristics of CRCs in different patient populations, especially African-Americans as compared to non-Hispanic, Caucasian-American patients. This new information will help guide future clinical research and could aid in reducing the difference in cancer outcomes.
There are racial differences in the anatomic locations of primary CRCs [55,73]. African-Americans have an almost equal distribution of adenocarcinomas in the proximal and distal colorectum, but, for Caucasians, distal tumors predominate [74]. In our study of racial differences in post-surgical survival [52], there were poorer 5-year (p = 0.01) and 10-year (p = 0.02) survivals for African-Americans than for Caucasian-Americans with CRCs; however, there were no appreciable differences between African-Americans and Caucasian-Americans with regard to age (p = 0.47), hospital treatment modality (p = 0.37), tumor stage (p = 0.77), or tumor grade (p = 0.17). The only statistical difference between Caucasian-Americans and African-Americans was the anatomic site (p = 0.05), with African-Americans having primarily proximal colon tumors and Caucasian-Americans having primarily distal tumors. Similar outcome differences were observed at 5 years and 10 years, accounting for overall survival with hazard ratios of 1.67 (95% CI 1.21–2.33) and 1.52 (95% CI, 1.12–2.07), respectively. With respect to 5- and 10-year survivals, African-Americans with Stage II disease had hazard ratios of 2.53 (95% CI, 1.31–4.86) and 1.82 (95% CI, 1.04–3.18), respectively.
Our study of post-surgical survival suggests that poorer survival is based upon the biology of the lesion. In our separate study on patients drawn from different institutions, however, medical treatment provided and stage at diagnosis were associated with differences in outcome between Caucasian-Americans and African-Americans; the differences were eliminated for patients treated by the Department of Veterans Affairs [75]. Of note, Caucasian-Americans present with a greater proportion of tumors at local and regional stages [76].
Our studies indicate that there may be biologic differences between CRCs arising in African-Americans and those in Caucasians. Our studies have established that p53nac detected by immunohistochemistry (IHC) is a stronger prognostic biomarker for proximal CRCs in Caucasians than in African-Americans [66,77]. In contrast, high-grade CRCs lead to a shorter time of survival for African-Americans than for Caucasian-Americans [78]. As we recently reported, the incidence of single nucleotide polymorphisms (SNPs) at codon 72 of the p53 gene was higher in African-Americans than in Caucasian-Americans, and the proline/proline phenotypes of p53 contributed to aggressive progression of CRCs in African-Americans [79]. This polymorphism (proline/proline) has a greater effect on stability of p53 in African-Americans than in Caucasian-Americans, leads to higher grade tumors in African-Americans, and contributes to aggressive progression of CRC [79].
A practical problem is that variations of biomarkers with race and anatomic location of the tumor affect the numbers of specimens that must be studied for validation of molecular markers. For example, in the US, racial and ethnic differences require, at a minimum, population studies of Caucasian-American, African-American, Native American, Asian-Indian subcontinent, Asian-Chinese/Japanese/other, and Hispanic sub-populations. Similarly, because of differing dietary requirements, religious subgroups may require separate studies, as do groups with relatively uniform genetic patterns (e.g., Ashkenazi Jews in the study of breast or ovarian cancers).
5. Prognostic molecular markers
Although the molecular phenotypes of early lesions of hereditary CRNs have been described as derived from either the suppressor gene pathway of FAP or a mutator phenotype causing MSI, few advances have been made in the phenotypic description of sporadic CRCs, especially the molecular changes involved in the invasive stages of these lesions. Our goal has been to identify reliable molecular information that can be used to predict the clinical outcomes of CRCs. Specifically, we aim to characterize the components of molecular phenotypes of CRCs that identify aggressive subtypes. Our laboratory has described phenotypic molecular patterns of invasive CRNs that correlate with clinical outcomes.
Early discovery of CRCs has relied on the relatively insensitive and non-specific detection of occult blood or blood products in the stool or on the expensive and invasive method of endoscopy. However, the detection in stools of atypical molecules, such as mutated p53 or K-ras, of oncofetal proteins, viz., carcinoembryologic antigen (CEA), or of long DNA, may be more specific. The sensitivity and specificity of such methods of molecular detection may be improved by measuring multiple molecular markers of colorectal epithelial cells, which can be obtained from feces.
Traditional clinical counseling of patients and their families and therapeutic decisions have been based upon the pathologic and/or clinical stages of CRCs. In the future, however, the molecular phenotypes of CRCs may be used to determine the ‘molecular stage’ of CRCs and thus aid in counseling and therapeutic decisions, especially if adjuvant therapy is to be used for subgroups of Stage I, II, or III lesions [80].
Various molecular phenotypes are associated with aggressive subtypes of CRCs, including molecular markers that are independent of pathologic stage. These include nuclear accumulation of p53 (p53nac), as identified by IHC; and the phenotypic expression of Bcl-2, MUC-1 (mucin core protein), or p27kip–1, a cell cycle inhibitor.
5.1. Nuclear accumulation of p53 (p53nac)
As determined by our methods of IHC, CRCs with cells demonstrating p53nac typically have single-point mutations in p53, as determined by sequencing the complete p53 gene or analysis by single-strand conformation polymorphism [81,82]. In our initial studies, we reported that, in our overall patient population, p53nac was associated with a poor prognosis; however, on subsequent evaluations, we determined that p53nac was important prognostically, primarily for proximal tumors of Caucasian patients [66] (Fig. 3). The literature, as reviewed by Manne et al. [83], Grizzle et al. [65, 81], and McLeod and Murray [84], is not clear with respect to the prognostic importance of p53 in CRCs. This confusion may have resulted in part because of variations of p53nac in mixed populations, including differences in race and anatomic location of tumors, as well as by the methods used for analysis and evaluation. The method of analysis of p53 is especially important, since different points of cut-off in evaluations may vary, depending upon whether or not antigen recovery is accomplished prior to staining [65,66,81,82, 84,85].
Fig. 3.

Survival analysis of Caucasian (N = 346) and African-American patients (N = 241) with CRCs based on p53nac and tumor location. A = Caucasians (N = 142) with proximal CRCs; B = Caucasians (N = 204) with distal CRCs; C = African-Americans (N = 120) with proximal CRCs; D = African-Americans (N = 121) with distal CRCs.
5.1.1. p53 mutations
For patients in geographic and/or ethnic/racial populations, there are differences in the incidence of p53 abnormalities in CRCs. For example, the p53 mutational spectrum of CRCs is different in Japanese patients compared to patients in the US, i.e., the mutation pattern at specific hot spots in the p53 gene varies with populations. C:G to T:A transitions at CpG sites are predominant in US patients, but are less common in Japanese patients [86]. Similarly, there are racial/ethnic variations in the hot spots of point mutations. For example, the codon [175] hot spot does not occur in the Finish population, but there is a codon [213] hot spot. In our studies, we found that the incidence of an SNP at codon 72 of the p53 gene is higher in African-American than in Caucasian patients and is associated with higher mortality [79]. Our further analysis indicated that most (79%) proximal CRCs from African-Americans exhibited wild-type p53, whereas proximal CRCs from Caucasians exhibited a higher incidence of point mutations (52%), at ‘hot spot’ codons (175, 245, 248, 249 and 273). These mutations were located in the L3 domain of p53 (codons 245, 248, and 249). Univariate survival analysis of proximal CRCs of Caucasians demonstrated that mutations in the L3 domain of p53 are more lethal than the wild-type p53 [83]. In contrast, in an European population, p53nac in distal tumors is associated with a higher incidence of local recurrence than p53nac in proximal tumors [68].
5.1.2. p53 polymorphisms
In addition to gene mutations, p53 polymorphisms are risk factors for human malignancies, including CRCs [87,88]. To date, only five polymorphisms have been found in the coding region: four in exon 4 at codons 34, 36 [88], 47 [89] and 72 [90], and one in exon 6 at codon 213 [91]. p53 polymorphisms are also found in the intronic regions: two in intron 1 [92,93], one in intron 2 [94], one in intron 3 [95], two in intron 6 [96,97], five in intron 7 [98,99], and one in intron 9 [100]. Of these, only those at codon 72 have been well characterized. The alleles of the polymorphism in codon 72 encode an arginine (CGC, Arg72) and a proline residue (CCC, Pro72). Earlier studies have noted an association between codon 72 polymorphism and risk of CRCs, although the findings were not conclusive [87,101,102]. The polymorphism at codon 72 (CGC to CCC, substitution of an arginine residue with a proline), however, is a risk factor, and tumors exhibiting Pro72 polymorphisms have increased incidences of missense point mutations in the p53 gene [103]. The Arg/Pro polymorphic site is located in a proline-rich region (residues 64–92) of the p53 protein; this region is required for the growth suppression and apoptosis mediated by p53 but not for cell cycle arrest (reviewed in [104]). Indeed, Dumont et al. reported that, compared to Pro72, the Arg72 form of p53 has a 15-fold enhanced capacity to induce apoptosis and that this increase is due to more efficient localization of the Arg72 form to mitochondria than the Pro72 form [105]. In contrast, for the Pro/Pro phenotype, there is an increased capacity for cancer cell proliferation [105] and cancer progression [103].
5.2. Bcl-2
Several oncogenes and tumor suppressor genes, such as Bcl-2 and p53, are involved in regulating programmed cell death and cellular proliferation. Dys-regulation and/or alteration of the Bcl-2 and p53 genes occur during the development and progression of CRCs [106–111]. In addition, lack of Bcl-2 expression correlates with local invasion, metastasis, and recurrence [112–114]. Our laboratory initially found, in our complete population, that the phenotypic expression of Bcl-2 was important prognostically and that the combination of p53nac plus Bcl-2 was even more prognostically useful [85]. In a more detailed study with a larger population, we subsequently found that Bcl-2 was important in all CRCs (including Caucasian and African-American populations). The strongest correlation of the expression of Bcl-2 with clinical outcome was primarily for Stage II CRCs (Fig. 4). Our recent studies have demonstrated that Bcl-2 expression status aids in predicting the recurrence [115] and survival of CRC patients, especially in Stage II disease.
Fig. 4.

Survival analysis of CRC patients based on Bcl-2 expression and tumor stage. A = All stages included (N = 490); B = Stage I (N = 103); C = Stage II (N = 182); D = Stage III (N = 148).
Patients with CRCs that exhibit high levels of Bcl-2 expression have been reported to have a good clinical outcome [106,116,117]; however, some studies have not observed such an association [107,112,118,119]. Conversely, for a small group of CRC patients (n = 48), Bhatavdekar et al. [120] correlated high Bcl-2 expression with a poor prognosis. Although the reasons for this controversy (e.g., ethnic/religious and dietary differences) are not known, we hypothesize that the prognostic importance of Bcl-2 expression in CRCs is limited to specific subgroups of patients, similar to the clinical usefulness of p53nac in CRCs, which varies with racial/ethnic groups and with anatomic location of the tumor.
To establish the prognostic usefulness of Bcl-2 in CRCs, we performed a meta-analysis of reported studies on the relationship of Bcl-2 expression and overall survival. We requested original data on all comparable studies from authors who had evaluated the prognostic usefulness of Bcl-2 in predicting overall survival. Some authors, for various reasons, could not supply us with their data. Ultimately, nine studies incorporating a total of 1,989 patients provided either the hazard ratio and 95% confidence interval or information from which the hazard ratio and its variance could be estimated. The meta-analysis indicated that Bcl-2 is a useful prognostic marker for CRCs. The results are outlined in Table 5.
Table 5.
Results of the meta-analysis of lack of Bcl-2 expression and its effect on CRC mortality
| Estimates of effect | Number of studies | Summary HR (95% CI) |
|---|---|---|
| Studies with adjusted & unadjusted estimates | 9 | 2.62 (1.93, 3.54) |
| Studies with adjusted estimates? | 6 | 1.69 (1.06, 2.32) |
A separate meta-analysis was conducted for studies that reported adjusted hazard ratios (HR) and 95% confidence intervals (95%).
5.3. MUC-1
A characteristic feature of glandular epithelial tissues is synthesis and secretion of mucins, which are large glycoproteins that protect epithelial surfaces. Alterations in mucins with regard to the rate of their production and the extent of their glycosylation have been reported for human malignancies [121], including CRNs [122,123]. Of the various mucin antigens, MUC-1 is best characterized. Higher expression of MUC-1 correlates with increased incidences of regional lymph node and liver metastases [124–126]. Although expression of MUC-1 is associated with aggressiveness of CRCs, its prognostic usefulness in CRNs has not been evaluated adequately. Studies from our laboratory [126] as well as others [124,125] have suggested that increased expression of the core peptide of MUC-1 is associated with a poor prognosis for CRCs.
In an evaluation of the prognostic importance of MUC-1 and MUC-2 for CRCs, we found that the phenotypic expression of MUC-2 was not useful prognostically, but the expressions of MUC-1 and MUC-2 were useful markers in defining the mucinous subtype. In contrast, the phenotypic expression of MUC-1 was important prognostically for CRCs of Caucasians but not of African-Americans [126]. We found no variation in the prognostic importance of MUC-1 based on anatomic location of the tumor [126].
5.4. Proliferation and p27kip–1 expression
p27kip–1 inhibits the activity of cyclin-dependent kinases (cdks), and, like p21waf–1, prevents progression of cells into the S phase of the cell cycle. Decreased p27kip–1 protein expression is associated with large CRCs, positive lymph nodes, and poor patient survival [127–129]. Furthermore, a small study (n = 41) demonstrated that p27kip–1 is a predictive indicator for tumor metastasis and clinical outcome in right-sided colon tumors [130]. Further studies from our group have suggested that decreased expression of p27kip–1 is an indicator of poor prognosis of patients with Stage III CRCs [131] (Fig. 5).
Fig. 5.
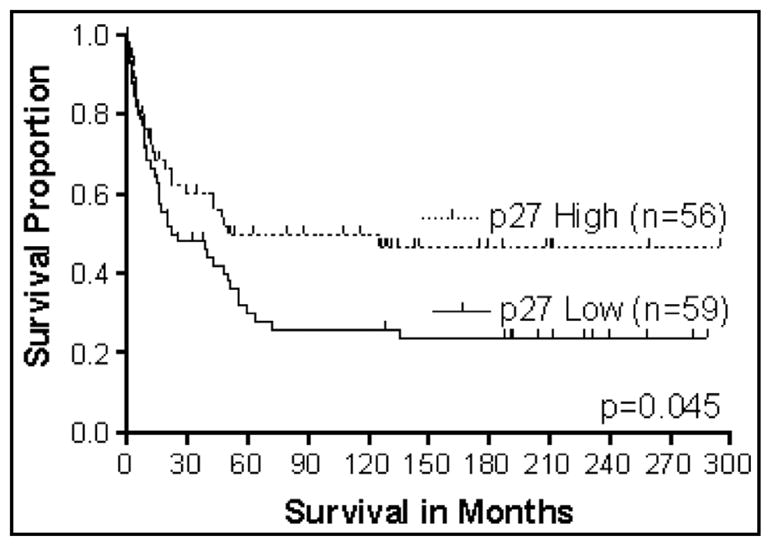
Survival analysis of stage III CRC patients based on p27 expression (N = 115).
We performed a preliminary analysis of the importance of the proliferative index biomarkers, Ki67/MIB-1 and p27kip–1, for CRCs of 48 African-American and 54 Caucasian patients, which had been analyzed previously for p53. In this study, similar proportions of CRCs, collected from African-Americans and Caucasians, exhibited increased expression of p27kip–1 (50% and 54%, respectively) and Ki67 (54% and 50%, respectively). Univariate Kaplan-Meier survival analyses demonstrated that African-Americans with CRCs exhibiting higher expression of p27kip–1 without p53nac had a marginally better overall survival (log rank test, P = 0.055) than any other combination of p27kip–1 and p53nac. In Caucasians, lower expression of p27kip–1 with p53nac demonstrated the lowest probability of overall survival (log rank test, P = 0.011). No prognostic value was found for p27kip–1 or Ki-67 alone or the combinations of Ki-67 with p27kip–1 or p53nac (our unpublished data). Further studies are needed to understand the prognostic importance of p27kip–1 in CRCs based on the anatomic locations of the tumors and patient ethnicity.
5.5. Microsatellite instability (MSI) in CRCs
Mismatch repair deficiency and associated MSI, which are characteristics of HNPCC [18,132] and a subset (~12%) of sporadic CRCs [133,134], can be caused by genes that are either mutated [135] or silenced [136]. Phenotypically, MSI cancers are predominantly right-sided and more likely to be poorly differentiated, mucinous, and larger, and to have a low incidence of metastasis at presentation and a better outcome [137–139].
5.5.1. MSI in CRC and race/ethnicity
The incidence of MSI-High (MSI-H) tumors is 3-fold higher in African-American CRC patients relative to a nonracially selected but serially collected population [140]. Most of these tumors are proximal and well differentiated and have high levels of mucin production [140]. Furthermore, for African-American patients, this higher incidence of MSI correlates with higher incidences of MLH1, MSH2, and BRAF abnormalities [141]. These findings may reflect dietary differences or genetic polymorphisms that may be common in this population. There is also a similar incidence of MSI in CRCs of non-Hispanic Caucasians and in Omani CRC patients, but most of the MSI CRCs are located in the left-side of the colon [142].
5.6. MCC Gene (Mutated in CRCs)
In cases of CRC from FAP, the MCC gene is on chromosome 5q21 in close proximity to the APC gene [143]. It is in the region that frequently exhibits loss of heterozygosity (LOH) in CRCs [144]. The protein encoded by the MCC gene has 829 amino acids and shares a short region of similarity with the muscarinic acetylcholine receptor known to regulate specificity of G protein coupling [143]. Somatic point mutations of MCC were identified in 6 of 90 (15%) sporadic CRCs, but similar mutations were not found in the germ lines of FAP patients ([145]. Also, LOH at the MCC locus or in its vicinity was demonstrated in 22 of 40 (55%) sporadic CRCs [146]. However, for two unrelated patients with FAP, MCC was located outside the deleted regions [147]. Mutations of MCC also have been detected in other neoplasms. The roles of MCC in FAP and/or sporadic CRN are yet to be identified. MCC promoter methylation is a frequent early event in a subset of CRCs and precursor lesions associated with the serrated CRC pathway. MCC is a nuclear, β-catenin-interacting protein that can act as a tumor suppressor in the serrated CRC pathway by inhibiting Wnt/β-catenin signal transduction [148].
5.7. DCC Gene (Deleted in CRCs)
Although Fearon et al. [149] identified the DCC tumor suppressor gene on chromosome 18q, earlier studies implicated LOH in chromosome 18q in the development of sporadic [150] and familial CRCs [151,152]. The protein product of the DCC gene (about 180kD) has structural homologies with neural adhesion protein (NCAM). Most human tissues express the DCC protein at low levels; however, there is higher expression in the brain and normal colorectal epithelium [149,153]. The frequency of allelic loss of 18q is higher in larger adenomas (50%) and primary CRCs (70%) [150,154] than in small and mildly to moderately dysplastic adenomas ([155]. The DCC protein may be involved in cell adhesion, in invasion of CRC cells [149], and/or in terminal differentiation. Allelic loss of the DCC locus is higher in primary CRCs of patients with liver metastasis (95%) than in patients without liver metastasis (40%) [156], suggesting that, in CRCs, allelic loss of DCC is associated with the acquisition of metastatic capacity.
Similarly, expression of DCC mRNA is less in CRCs than in adenomas, indicating that inactivation of the DCC gene is associated with the progression of colorectal tumors [149,157,151–159]. The transmembrane location of the DCC protein is consistent with its involvement in cell-cell and cell-substratum interactions and in the regulation of cell growth and metastasis [149,156, 160]. Based on results with murine models, however, others have questioned involvement of the DCC gene in cell growth, differentiation, morphogenesis, and/or tumorigenesis [161] as well as its involvement in the pathogenesis of CRCs in humans [162].
In CRCs, there are point mutations in the DCC gene [163]. Direct evidence to support its function as a tumor suppressor, however, has come from a study with nude mouse, which demonstrated suppression of the malignant phenotype of xenografts of transformed human keratinocytes by restoration of DCC expression [164]. The tumorigenicity of human CRC cells is suppressed by introduction of a normal copy of chromosome 18q [165], but not by the transfer of a normal copy of the DCC gene. Furthermore, in neuronal cells, the DCC gene is involved in the regulation of cell migration and differentiation [166–168].
Descriptions of the intensity of immunostaining for the DCC protein in human neoplastic tissues have not been consistent [169–171]. Use of an antigen retrieval technique, however, enhances detection of the DCC protein in archival tissues of CRCs and shows that expression of the DCC protein is usually an “all-or-none” phenomenon, with a granular cytoplasmic pattern that is homogeneous throughout the tumor [172]. The cells of the crypts and of the luminal surface of normal colorectal epithelium also express the protein, but the non-epithelial cells do not [172].
There are lower five-year survival rates for patients with CRCs that exhibit allelic loss of chromosome 18q as compared to patients whose tumors do not (for stage II, 54% versus 93%, and for stage III, 32% versus 52%) [173]. The expression of DCC, as determined by IHC, in stage II and III CRCs was a strong positive predictor for patient survival; the five-year survival rate for patients with CRCs that expressed the DCC protein was 94%, compared to 62% for CRCs that did not [172]. In CRCs, UNC5C and DCC methylation is associated with loss of gene expression [174,175].
5.8. Transforming growth factor beta (TGFβ)
The TGFβ family of cytokines consists of more than two dozen secreted peptides that are essential for normal development and which have various functions, including the regulation of cellular proliferation, differentiation, cell-cell recognition, and apoptosis. The TGFβ cytokines signal by simultaneously contacting two transmembrane serine/threonine kinases, the type I and type II receptors. Another group of proteins, the MAD-related family, function in the signaling pathways of TGFβ receptors [174–177].
The TGFβ cytokines typically exert an antiproliferative effect, and there is frequently loss of the antiproliferative response to TGFβ in the progression of neoplasia. In CRCs that are replication-error positive, this lack of response is a result of mutations in genes of the type II TGFβ receptor [178]. Other CRCs have receptors and cells resistant to TGFβ-induced growth inhibition, contributing to malignant progression [179]. In sporadic CRCs, there is a mutation in one of the MAD-related genes, MADR2. This mutation renders the gene non-functional, indicating a role for MADR2 as a tumor suppressor gene [180].
In contrast, TGFβ-1, as detected by IHC, is expressed in most CRCs, and intense staining for TGFβ-1 correlates with advanced Dukes’ stage and metastases of primary tumors [181,182]. Messenger RNA for TGFβ-1 expression in CRC cells and elevated plasma levels of messenger RNA for TGFβ-1 are associated positively with increased Dukes’ stage [183]. These findings suggest a correlation between over-expression of the TGFβ-1 gene and progression of CRN.
Other functions for TGFβ in colon tumorigenesis include stimulation of angiogenesis by inducing thymidine phosphorylase [184]; regulation of extracellular matrix adhesion molecules, including carcinoembryonic antigen [185]; and enhancement of the secretion of gelatinase B, a matrix-degrading enzyme [186]. A meta-analysis revealed that the TGFB1 509 C allele is a risk factor for development of CRCs in Asians but not Europeans [187]. A prospective study demonstrated the influence of polymorphisms in the TGFB1 pathway on CRC progression [188].
5.9. β-catenin
The β-catenin signaling is involved in colorectal carcinogenesis, but the contribution of β-catenin nuclear translocation to tumor progression is not known [189]. Nuclear translocation of β-catenin, however, may be involved in the development of intramucosal and invasive colonic cancers but not adenomas [190]. Of the 38 primary CRCs, 11 (29%) showed nuclear accumulation of β-catenin with cytoplasmic staining. Nuclear accumulation positivity was more frequently associated with lymph node metastasis than negative nuclear accumulation [100% (11/11) vs. 67% (18/27), p = 0.04]. There was a significant difference in median survival time between the nuclear β-catenin-positive group (1130 days) and the negative group (2102 days: p = 0.037). All of the patients (9/9) in the former group died when the recurrence was in the liver, but 42% (8/19) in the latter group survived, even if the recurrence was in the liver (p = 0.03) [191]. Another study involved IHC evaluation of β-catenin and p53 expression in 141 cases of primary sporadic CRCs and 45 matched metastases [192]. The combination of low β-catenin and high p53 expression in primary CRCs may be a prognostic factor in predicting the progression of the disease, the occurrence of metastasis, and a more severe outcome [192].
5.10. SMAD-4
The Smad-4 (mothers against decapentaplegic homolog 4) gene is located at the chromosome 18q21.1 locus, and its product belongs to the Smad family of proteins. Smad-4 is an intracellular transducer that mediates the TGF-β-Smad-dependent signaling pathway. It also functions in translocating TGF-β complex signals into the nucleus, leading to inhibition of growth of colon epithelial cells by regulating cell proliferation, differentiation, and apoptosis [193–195]. For Smad-4 [149,194], LOH on chromosome 18q21, found at several stages of CRCs, is associated with poor prognosis in lymph node-negative (stage II) CRC patients [149, 173,196,197]. However, the prognostic value of LOH of Smad4, especially in lymph node-positive (stage III) patients remains controversial [173,196,198–200]. There is a higher incidence of Smad4 mutations in CRCs [201,202], especially in those with distant metastasis (35%) relative to local tumors (~10%) [201,202]. Other studies, however, have found Smad4 mutations only in a small proportion of CRCs [203,204]. In addition, animal studies show that Smad4 inactivation is involved in the malignant transformation of gastrointestinal adenomas [205,206], and there is a reduction in Smad4 mRNA levels during tumor progression [207]. Low immunophenotypic expression of Smad4 correlates with poor patient survival in CRCs [203,208–212]. In mice, TGF-β/Smad signaling suppresses progression and metastasis of CRC cells and tumors, and loss of Smad4 may be the underlying mechanism for the functional shift of TGF-β from a tumor suppressor to a tumor promoter. This knowledge may aid in the development of CRC therapeutics involving inhibitors of TGF-β signaling [213].
5.11. Molecular staging involving a combination of molecular markers
It is unlikely that only one or two molecular markers will be adequate for developing an effective approach to molecular staging of CRCs; a combination of multiple molecular markers is likely to be necessary. Due to limited data on p27kip–1, a candidate prognostic marker, this combination for Caucasians would, at present, be limited to p53, MUC-1, and Bcl-2. Because of variation with race and location, the combination of p53nac and Bcl-2 was of use for prognosis, but only for Caucasians overall with CRCs in the proximal and distal subgroups [81].
Like Bcl-2, MUC-1 interacts prognostically with p53nac so that, for Caucasians, the combination of p53nac plus MUC-1 is valuable for prognosis [126]. Because both MUC-1 and p53 are useful prognostically for Caucasian patients with CRCs, we evaluated the association of the combination of different markers with prognosis. The survival analysis for the combination of three markers (p53nac, Bcl-2 and MUC-1) demonstrated that p53nac-negative, MUC-1 negative, Bcl-2 positive was the best phenotype combination; the worst combination was p53nac-positive, MUC-1-positive, Bcl-2- negative [81]. Similarly, our unpublished findings on evaluation of a combination of 4 markers (p53nac, Bcl-2, p27kip–1, and MUC-1) demonstrated that p53nac-negative, MUC-1 negative, Bcl-2 positive, and p27kip–1 positive was the best phenotypic combination, as compared to p53nac positive, MUC-1 positive, Bcl-2 negative, p27kip–1 negative phenotypes (Fig. 6). Based on these findings, the aggressive subtype of CRCs can be identified by the molecular phenotypes. These phenotypes are given in Table 6.
Fig. 6.
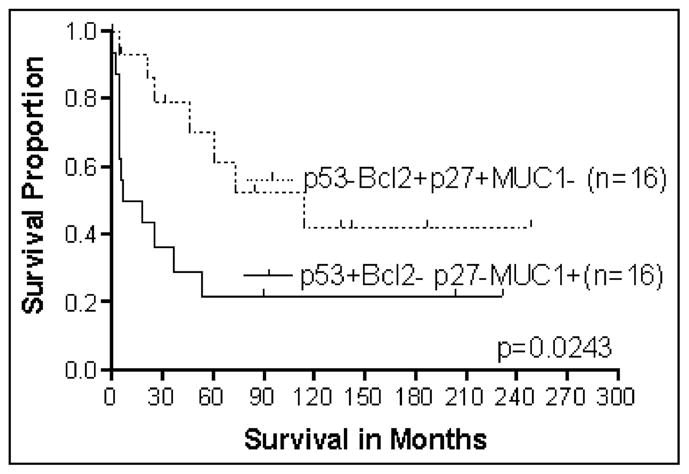
Survival analysis of Caucasian patients with CRCs based on multiple markers (p53nac, Bcl-2, p27 and MUC1) expression (N = 32).
Table 6.
Molecular phenotypes of aggressive subtypes of CRCs
| CRCs in caucasians
|
CRCs in African-Americans
|
||
|---|---|---|---|
| Proximal | Distal | Proximal | Distal |
| p53nac, + MUC-1 | + MUC-1, –Bcl-2 | – Bcl-2 (Weak) | – Bcl-2 |
| – Bcl-2–, – p27kip–1 | – p27kip–1 | ||
5.12. Markers independent of stage
The most valuable prognostic molecular markers are those that provide information relating to the clinical aggressiveness of tumors beyond that indicated by the tumor stage or components of the stage. Such molecular markers are probably related more to the biology of the tumor than to how advanced the lesion is at surgery. An example of a molecular pathway affecting the aggressiveness of tumors is that tumors derived from the mutator phenotype pathway and which exhibit MSI are less aggressive than other CRCs, even though they tend to be of greater size when they are surgically removed.
Prognostic molecular markers, independent of stage, can not only provide substantial information prior to surgical removal and staging of the tumor, they may add additional information: a molecular component that can be added to pathologic/clinical stage in the calculation of hazard ratios. In addition, such molecular markers can aid in the decision concerning whether or not to use adjuvant and/or novel therapies for early lesions, especially Stage II lesions.
To date, the molecular markers p53, p27kip–1, Bcl-2 and MUC-1 show promise as being useful prognostically for CRCs, but each awaits more complete validation. In our view, no one molecular marker will be adequate for evaluating all CRCs. Rather, groups of molecular markers will be useful for specific subsets of CRCs, as subdivided by race and/or anatomic location of the primary tumor.
Variation of the usefulness of prognostic biomarkers with stage is most likely related to how factors controlling mitoses are regulated. Since multiple genes control metastases, unless a prognostic biomarker also is related to a gene regulating metastasis, the biomarker is unlikely to be independent of stage. Instead, the prognostic gene will first influence the rate at which the lesion increases in size up to and until metastasis. Because this influence on prognosis will continue, to some degree, after metastasis, the biomarker may be less indicative of tumor progression, which will then be controlled primarily by a gene modulating metastasis.
It is likely that genes controlling metastases to local lymph nodes are different from those controlling metastases from lymph nodes to the liver. This may explain differences in prognostic genes related to nodal metastasis, but not distant metastasis.
6. Novel prognostic markers
6.1. MUC4
MUC4 is aberrantly expressed in CRCs, but its prognostic value is unknown. We evaluated 132 CRCs for the IHC expression of MUC4 and found that patients with early-stage tumors (I and II) with high MUC4 expression had a shorter disease-specific survival than those with low expression (log-rank, p = 0.007). Tumors from patients with advanced-stage CRCs (III and IV) did not demonstrate such a difference (log-rank, p = 0.108) (Fig. 7) [214]. The multivariate regression models generated separately for early- and advanced-stage patients confirmed that increased expression of MUC4 is an independent indicator of a poor prognosis only for patients with early-stage CRCs (hazard ratio, 3.77; confidence interval, 1.46–9.73). Hence, increased MUC4 expression is a predictor of poor survival of patients with early stage CRCs [214].
Fig. 7.

Survival analysis of CRC patients (N = 132) based on MUC4 expression. A = stage I and II (N = 75); B = Stage III and IV (N = 57).
6.2. Rabphilin-3A-like (RPH3AL)
The RPH3AL protein has structural similarities with rabphillin-3A, RIM, and synaptotagmin-like proteins (Slp1 to 5), which contain modules (C2 domains) for protein and phospholipid interaction. Since RPH3AL lacks these domains, it was originally named Noc2 (no C2 domains). The RPH3AL gene was originally discovered as a pancreatic islet gene encoding the RAS-associated protein (Rab)-binding domain of rabphilin [215]. Although the precise functions of RPH3AL are not known, the gene is involved in the regulation of endocrine endocytosis through its interactions with the cytoskeleton [215,216]. Our recent study showed that Noc2, Rab3A, Rab27A, and RIM2, together with the islet cell hormones, insulin and glucagon, are present in normal and endocrine tumor tissues of human pancreas [217]. For CRCs, ours [218,219] and one other study [220] of this tumor type have involved mutational analysis of the RPH3AL gene; they have demonstrated six missense mutations resulting in amino acid substitutions in the coding region of this gene. This suggested a tumor suppressor role for RPH3AL, but only in a few cases (our study, 5 of 95, 5%; and other study, 6 out of 50, 12%) [218,220]. We, however, have detected LOH at this gene locus in a considerable proportion of CRCs (58%). The incidence of LOH was associated with depth of invasion of the tumor, nodal metastasis, distant metastasis, high-grade tumor differentiation, and short survival of patients [218,219]. Furthermore, our studies with CRCs have identified five missense mutations in RPH3AL and several SNPs and have demonstrated the clinical relevance of an SNP at 5′UTR-25 [221].
6.3. miRNAs
MicroRNAs (miRNAs) are a family of small (20 to 24-nucleotide), non-coding RNAs that regulate gene expression at the post-transcriptional level [222]. Since approximately half of the known human miRNAs are located at cancer-associated regions of the genome, miRNAs may function in the pathogenesis of various human cancers [223]. Many proteins involved in key signaling pathways of CRCs, such as members of the Wnt/β-catenin and phosphatidylinositol-3-kinase (PI-3-K) pathways, KRAS, p53, extracellular matrix regulators, and epithelial-mesenchymal transition (EMT) transcription factors, are altered and are apparently regulated in CRCs by miRNAs [224]. In cultured cells, miR-135a and miR-135b decrease translation of the APC transcript. Of note, these molecules are up-regulated in colorectal adenomas and carcinomas, and they correlate with low APC levels [225]. The KRAS oncogene is a direct target of the let-7 miRNA family [226] and miR-143 [227]. Inhibition of KRAS expression by miR-143 blocked constitutive phosphorylation of MAPK [227]. Studies based on microRNA arrays found a consistent loss of miR-126 expression in colon cancer lines as compared to normal human colon epithelia; reconstitution of miR-126 resulted in a growth reduction [228]. miR-21 is the miRNA most frequently up-regulated in CRCs. It is hypothesized that suppression of PTEN controlled by miR-21 is associated with augmentation of PI-3-K signaling and progression of CRCs [229–231].
Several groups have described aspects of the connection between p53 and the miRNA network [232]. Members of the conserved miR-34a-c family are direct transcriptional targets of p53. Genes responsive to miR-34a are those that regulate cell-cycle progression, cellular proliferation, apoptosis, DNA repair, and angiogenesis [233]. The high frequency of their methylation in 1p36, the genomic interval harboring miR-34a, in CRCs and its contribution to the p53 network imply that miR-34a, b, and c function as tumor suppressors that can be lost during development of CRCs [222]. Up-regulation of miR-21 in CRC cells increases their migratory and invasive capacities in a manner similar to that of glioblastomas [234]. Furthermore, miR-21 acts on PDCD4, a tumor suppressor gene that is an independent prognostic factor in resected CRCs and negatively regulates intravasation through the invasion-related urokinase receptor gene (uPAR) [235].
As determined with a mouse model of colon cancer, the angiogenic activity of c-Myc is due, at least in part, to downstream activation of the miR-17-92 cluster d [236]. Over-expressed cyclooxygenase-2 (COX-2) contributes to the growth and invasiveness of tumor cells in patients with CRCs. For CRC cell lines, there is an inverse correlation between COX-2 and miR-101 expression, and the direct translational inhibition of COX-2 mRNA is mediated by miR-101 [237].
Various approaches have been used to predict and identify functional SNPs within miRNA binding sites and to evaluate their biological relevance [238]. Regarding CRCs, of eight candidate genes predicted by computer simulation, the genes for CD86, a co-stimulatory ligand on lymphocytes, and for the insulin receptor carry SNPs that are associated with the risk of sporadic CRCs (odds ratios 2.74 and 1.94, respectively) [239]. The biological relevance of these SNPs, however, has not been confirmed by functional studies in cultured cells.
CRC patients have a serum miRNA profile different from that of healthy subjects [240]. In all cases, 69 miRNAs were detected in the sera from CRC patients but not in sera from healthy subjects. CRC patients share several serum miRNAs (miR-134, miR-146a, miR-221, miR-222, and miR-23a) with lung cancer patients.
CRC patients with tumors expressing high levels of miR-200c have a poorer prognosis, regardless of tumor stage; and p53 mutations, commonly found in CRCs, are associated with greater than two-fold miR-200c overexpression [241]. miR-21 overexpression correlates with the established prognostic factors (nodal stage, metastatic disease, and UICC stage) [231]. In tumors of CRC patients, 37 miRNAs are differentially expressed [230]. From this group, miR-20a, miR-21, miR-106a, miR-181b, and miR-203 were found, by Cox regression analysis, to be associated with poor survival and were selected for validation. Validation was performed by measuring expression of miRNAs by use of real-time PCR in tumor and paired non-tumor tissues [230]. In the validation set, only high expression of miR-21 was associated with a poor prognosis; this association was independent of age, sex, and tumor location. Multivariate analysis further revealed that high miR-21 expression in tumors was associated with poor survival, independent of the tumor stage. Similar findings were also found in our preliminary studies [242]. In patients who received adjuvant therapy, high miR-21 expression indicated a poor response to therapy [230]. Patients having Stage II CRC tumors with high expression of miR-320 or miR-498 had shorter progression-free survival than those whose tumors showed low expression [243].
7. Predictive markers
Adjuvant therapy with 5-fluorouracil (5FU)/leucovorin (LV) or 5FU/LV/irinotecan moderately reduces recurrence rates and the risk of dying from resectable CRCs (Stage III) [244–246]. The combination of 5FU/LV/oxaliplatin (FOLFOX) prolongs the survival in second-line treatment of patients with disseminated disease [247]. Data from MOSAIC [248,249] and QUASAR [250] trials on use of FOLFOX demonstrate a relapse/progression-free survival benefit for patients with Stage II or III CRCs. Current treatment recommendations in the US, however, include using FOL-FOX not only for Stage III disease but also for Stage II disease, if high-risk clinical features are presented. Improved surgical techniques [251–254], pre- or post-surgical radiotherapy [253,255], and post-surgical radiotherapy and chemotherapy have decreased failure rates for rectal cancers [256–259]. Innovative treatment modalities, such as anti-angiogenic and antimetastatic agents [260–262], farnesyl transferase inhibitors [263], novel vaccines [264,265], and gene therapy approaches are in clinical trials [266,267]. Findings of several retrospective studies and prospective clinical trials have been inconclusive, because patients received different 5FU regimens, and each regimen involved a different mechanism of action. Indeed, there is a reported survival benefit of 5FU-based treatment in patients with Stage III [268] or Stage IV [269] disease of the distal colon; however, such a benefit was not observed in another study [270]. The role of adjuvant treatment in patients with Stage II CRCs remains controversial [271].
The anti-tumor activity of 5FU has been related to its induction of apoptosis by damaging DNA and to its alteration of the expression profiles of pro- and anti-apoptotic molecules (Bax, Bcl-2, and p53). Chemo-resistance may depend on the function and relations between pro- and anti-apoptotic proteins [272–274]. However, our experience in assessing the prognostic value of p53nac [275,276], and expression of Bcl-2 [115,277,278], Bax [279,280], p27kip–1 [81,131], MUC1 [126], suppressin [281], MSI [282,283], and the Rabphillin-3A-like gene [284–286] in CRCs has suggested that these molecular markers have different biologic consequences based on tumor location, pathologic stage, and race/ethnicity of patients who experience only surgery.
7.1. p53
Mutant forms of the p53 protein can be detected by routine IHC; but native p53 protein can not. The association of p53 abnormalities in CRCs with survival has been studied extensively, but studies in relation to abnormal expression of p53 and the therapy responses are few. Two retrospective studies, however, demonstrated that patients whose Stage III tumors did not exhibit p53 abnormalities benefited from 5FU-based treatments [287,288].
7.2. Bcl-2
In CRCs, the lack of Bcl-2 expression correlates with tumor invasion, metastasis, and recurrence [112, 113]. Studies by Sinicrope et al. [289,290] and by us [277,278] indicate that patients with CRCs expressing Bcl-2 have a better clinical outcome than patients with tumors that do not (also reviewed in [115,277,278, 291]). There was, however, no significant association between Bcl-2 expression and the clinical outcome for patients with metastatic CRCs who received 5FU/LV treatment [292].
7.3. Bax
Bax/Bcl-2 heterodimers protect cells from apoptosis [293]. In contrast, an abundant amount of Bax causes formation of Bax homodimers that make the cells susceptible to apoptosis [106,294]. Furthermore, patients with hepatic metastases from CRCs having increased expression of Bax had higher survival rates [274]. Experiments involving cells in culture demonstrate that the lack of Bax expression abolishes the apoptotic response to chemopreventive and anti-inflammatory drugs [295].
7.4. p21waf–1
p21waf–1, a gene activated by p53, is a target of p53 that binds to and inhibits the function of the cyclin-cdks complexes and thus halts cell cycle progression [296, 297]. The p21 gene is induced by DNA-damaging agents that trigger G1 arrest of cells, both in a p53-dependent and -independent manner [298,299]. Studies involving use of IHC or hybridization in situ have demonstrated that increased expression of p21waf–1 in CRCs is associated with better survival [300–302]; however, its predictive value is unknown.
7.5. p27kip–1
This protein also appears to have functions in the cell cycle and in cell differentiation [303]. Overexpression of p27kip–1 augments apoptosis induced in cultured cells by 5FU [304]. Decreased expression of the p27kip–1 protein is associated with large-sized CRCs and with lymph node metastases. Low expression is an indicator of a poor clinical outcome [127,128,305–307], but only for Stage III tumors [131].
7.6. Ki67
IHC detection of cell cycle-associated Ki67 has been utilized to assess proliferation in human malignancies. Studies designed to examine the potential association of proliferation with tumor progression, biologic behavior, or patient prognosis generally have been inconclusive, and in some instances, contradictory [300,308]. There are, however, few studies that have focused on the predictive value of Ki-67 in CRCs.
7.7. Thymidylate synthase (TS)
TS is a target for 5FU inhibition of cell proliferation. Its levels predict response to therapies such as 5FU/LV, methotrexate-5FU, and raltitrexed [309–311]. The expression of TS, measured by IHC, is an independent prognosticator of disease-free and overall survival of patients, particularly those with rectal tumors [312]. TS expression is higher in left-sided CRCs and is associated with poor patient survival [313]. Nuclear expression of TS in normal colonic mucosa appears to be a predictive factor that can be used to identify a sub-set of patients at risk of developing severe gastrointestinal toxicities related to 5FU-based therapies [314].
7.8. Thymidine phosphorylase (TP)
TP is an enzyme involved in the metabolism of 5FU, catalyzing the reversible phosphorolysis of thymidine to thymine and 2-deoxyribose-1-phosphate. In CRCs, decreased expression of TP, like TS, is associated with a better response to 5FU therapy [310,315].
7.9. Dihydropyrimidine dehydrogenase (DPD)
DPD, the rate-limiting enzyme in 5FU metabolism, converts 5FU to dihydrofluorouracil [316]. Cancer patients deficient in DPD may experience profound systemic toxicity in response to 5FU [317]. Furthermore, for CRCs, high levels of DPD expression, measured as either mRNA or protein, correlate with resistance to 5FU [315,318,319].
7.10. Epidermal growth factor receptor (EGFR)
This protein, a member of the tyrosine kinase family, is often over-expressed in epithelial tumors, including CRCs. Therapeutic agents targeted to EGFR include the monoclonal antibody (MAb) C225 and Iressa (ZD1839). These agents have only modest antitumor activity when administered as single agents but display considerable synergism with cytotoxic drugs [320–323].
7.11. VEGF
Vascular endothelial growth factor (VEGF) is involved in tumor angiogenesis. The expression of VEGF in Stage II colonic cancers predicts for recurrence after surgery [324]. Results of a randomized phase II trial of metastatic CRCs suggest that administration of an MAb against VEGF in combination with chemotherapy results in improved response rates and a longer time to progression compared to chemotherapy alone [325].
7.12. MUC1
In CRCs, higher expression of MUC1 correlates with an increased incidence of regional lymph node metastasis and liver metastasis [125,126,326]. Its abundance and interaction with class I MHC molecules make it a potential target for immunotherapy. A phase I study of metastatic or locally advanced colonic carcinoma reveals that immunization by a MUC1-mannan fusion protein elicits an increased humoral response in patients with advanced-stage CRCs and suggests that MUC1 vaccine therapy may be useful for patients with advanced disease [327].
7.13. KRAS
A KRAS mutation is present in 30–49% of CRCs. Activating mutations of the KRAS gene are biomarkers for resistance to cetuximab or panitumumab. Anti-EGFR therapies have been approved for the treatment of metastatic CRCs, but only on condition that the mutation state of the KRAS gene is first determined. This is because the combination of chemotherapy with anti-EGFR is expected to increase the response rate only for patients with the wild-type KRAS gene [328]. In addition, among CRCs carrying wild-type KRAS, mutation of BRAF or PIK3CA or loss of PTEN expression may be associated with resistance to EGFR-targeted MAb treatment. These additional biomarkers require further validation before being incorporated into clinical practice. The use of KRAS mutations as selection biomarkers for treatment with the anti-EGFR MAb (e.g., panitumumab or cetuximab) is a major step toward individualized treatment for patients with metastatic CRCs [224].
7.14. BRAF mutations
Mutations in the BRAF gene may be useful for predicting the efficacy/response to EGFR inhibitors, such as cetuximab and panitumumab, as is expression of KRAS gene mutations [329]. In general, mutations in either KRAS or BRAF exclude mutations in other genes. Further, patients bearing metastatic CRCs with mutations in the BRAF gene and who had wild-type KRAS did not respond to treatment with these EGFR inhibitors and were more likely to experience disease progression [330].
7.15. Microsatellite instability (MSI)
Adjuvant therapy with 5FU benefited patients with CRCs exhibiting chromosomal instability but not those with tumors exhibiting MSI [270]. In this clinical trial, 5FU-based adjuvant therapy actually decreased the survival of patients with CRCs exhibiting MSI. These findings are counterintuitive to the findings that MSI-H in sporadic CRCs is associated with a less aggressive phenotype and with improved survival when compared with stage-matched microsatellite-stable and MSI-Low CRCs [331,332]. Nevertheless, the predictive value of MSI-H remains controversial [333,334]. Genome-wide surveys show associations of the locus at 8q24.21 for CRC patients under 50 years age [335] and the locus at 11q23.1 for female Lynch syndrome patients [336]. Larger studies are required to establish the value of MSI as a predictive marker.
7.16. miRNAs
Let-7g and miR-181b are indicators for response to chemotherapy based on the fluoropyrimidine S-1 [337].
8. Candidate molecular markers of CRC progression
Some molecular markers have potential usefulness for assessing the clinical outcomes in many cancers, including CRCs; however, the following markers need further validation in larger cohorts of CRC samples to establish their value in assessing clinical outcomes.
8.1. CDKN1A
CDKN1A (p21, Cip1), a protein encoded by the CD-KN1A gene located on chromosome 6 (6p21.2) [338], binds to and inhibits the activity of cyclin-CDK2 or -CDK4 complexes, and thus functions as a regulator of cell cycle progression at the G1 phase [338]. Expression of this gene is tightly controlled by p53, through which this protein mediates the p53-dependent G1 phase arrest in response to stress stimuli [339]. In CRCs, decreased expression of the CDKN1A protein is associated with a poor clinical outcome [340]. No studies, however, have evaluated the expression of CD-KN1A in CRCs with an emphasis on its predictive capacity.
8.2. CDK4
This catalytic subunit of the protein kinase complex is associated with progression of the G1 phase of the cell cycle. Its activity is restricted to the G1-S phase, which is controlled by the regulatory subunits, D-type cyclins and the CDK inhibitor p16 (INK4a). CDK4 is responsible for phosphorylation of the retinoblastoma gene product (Rb). For a variety of cancers, mutations in this gene and in genes for its related proteins, D-type cyclins, p16 (INK4a), and Rb, are associated with tumorigenesis [341]. In familial melanoma, there are germline mutations in the p16INK4a binding domain of CDK4 [342].
8.3. GADD45
In humans and rodents, the GADD (growth arrest and DNA damage) genes are induced in response to DNA damage [343]. GADD45, the only member of the family that is induced by ionizing radiation or by genotoxic stress in the presence of wild-type 53 [344, 345], encodes a nuclear acidic protein of 165 amino acids [346], which is widely expressed in normal tissues and particularly in quiescent cells [347]. The GADD45 protein interacts directly with the essential replication factor, proliferating cell nuclear antigen, to block DNA replication and to repair damaged DNA [348]. As determined by studies with cultured cells, GADD45 regulates cell growth by arresting cells in the G2-phase of the cell cycle [349]. No mutations in the GADD45 gene are noted in various human malignancies or cancer cell lines [350,351]; however, a small proportion of pancreatic cancers exhibit mutations in this gene [352].
8.4. TP53I3
The gene PIG3 (p53-induced gene 3) is induced by p53 but not by p53 mutants unable to induce apoptosis, suggesting that the PIG3 protein is involved in p53-mediated cell death [353,354]. It contains a p53 consensus binding site in its promoter region and a downstream pentanucleotide microsatellite sequence. By interacting with the downstream pentanucleotide microsatellite sequence, p53 transcriptionally activates this gene. The microsatellite is polymorphic, with a varying number of pentanucleotide repeats directly correlating with the extent of transcriptional activation by p53. This polymorphism may be associated with differential susceptibility to cancer [338].
8.5. BMP4
BMP4 belongs to the TGF-β superfamily of proteins. It is involved in bone and cartilage development, especially tooth and limb development and fracture repair, and in muscle development, bone mineralization, and uteric bud development [355]. BMP-4 enhances growth factor-induced proliferation of mammary epithelial cells [356]. In ovarian cancer cells, autocrine BMP4 signaling regulates expression of the ID3 proto-oncogene [357].
8.6. FLRT3
Fibronectin Leucine-rich Repeat Transmembrane 3 (FLRT3) is a type I transmembrane protein that contains extracellular leucine-rich repeats and the small GTPase, Rnd1 [358]. During embryogenesis, FLRT3 and Rnd1 together modulate cell adhesion by controlling cell surface levels of cadherin. Over-expression of FLRT3 or Rnd1 reduces cadherin-mediated cell adhesion, whereas their loss increases such adhesion [359]. In human melanoma cells, FLRT3, RUNX2, MIG-6, and SMURF2 are downstream targets of the p14ARF tumor suppressor [360].
8.7. BCL2-like 14
BCL2-like 14 (an apoptosis facilitator), also known as BCL2L14, is a human gene encoding a protein that belongs to the BCL2 family. BCL2 family members form hetero- or homodimers and act as anti- or pro-apoptotic regulators; they are involved in a variety of cellular activities [361,362]. For this gene, there are four alternatively spliced transcript variants, which encode three distinct isoforms [361,362]. Its over-expression induces apoptosis in cells.
8.8. DAPK2
Death-associated protein kinase 2 (DAPK2), is also known as death-associated protein kinase-related protein 1 (DRP-1). When over-expressed in cancer cells, DAPK2, a Ca2/calmodulin (CaM)-regulated serine/threonine kinase, functions as a mediator of programmed cell death [363,364]. Over-expression of this gene induces cellular apoptosis through its multiple polyadenylation sites [338].
8.9. MT1A
Metallothioneins (MTs), which are low-molecular-weight and sulfhydryl-rich proteins, are factors in regulatory processes, including cell growth and multiplication [365]. MTs are involved in carcinogenesis, as indicated by results of studies on malignant tumors of the large bowel, liver, and stomach. The positive correlation between MT expression and expression of Ki-67 and PCNA antigens points to a function of the proteins in cell proliferation [366]. IHC studies have revealed that 26–100% of invasive ductal breast cancers express the MT protein. The MT-1F and MT-2A iso-forms are associated with higher histological grades of breast cancers. There is higher expression of MT-1E mRNA in estrogen receptor-negative tumors compared to their estrogen receptor-positive counterparts. MT expression in breast cancer is associated with a poorer prognosis; MT expression may protect breast cancer cells from chemotherapeutic agents [367].
8.10. ANXA5
Annexin A5 (ANXA5) is a Ca2+-binding protein involved in membrane organization and dynamics. As determined by a proteomic investigation, there is increased expression of ANX5 in the invasive front of head and neck squamous cell carcinomas [364]. This protein also has possible functions in cell proliferation and invasiveness [368].
8.11. MAD2L2
The mitotic arrest deficient-2-like 2 (MAD2L2) gene encodes the spindle checkpoint protein MAD2L2 (or MAD2B), a component of a surveillance system that delays anaphase until all chromosomes are correctly oriented. Defects in this mitotic checkpoint contribute to genetic instability, i.e., to numerical and structural aberrations of chromosomes [369]. In CRCs, over-expression of MAD2L2 correlates with a poor prognosis [297]. Expression changes in MAD2L2 are involved in renal carcinogenesis, which is characterized by multiple chromosomal abnormalities [370].
8.12. IL1R2
Interleukin 1 receptor, type II (IL1R2), also known as CD121b (Cluster of Differentiation 121b), belongs to the interleukin-1 receptor family. This protein binds interleukin-α (IL1A), interleukin-β (IL1B), and interleukin 1 receptor, type I (IL1R1/IL1RA), and acts as a decoy receptor that inhibits the activity of its ligands. Interleukin 4 antagonizes the activity of interleukin 1 by inducing the expression and release of this cytokine [371].
8.13. CACYBP
Calcyclin binding protein (CACYBP) may be involved in calcium-dependent ubiquitination and subsequent proteosomal degradation of target proteins. It probably serves as a molecular bridge in ubiquitin E3 complexes and participates in the ubiquitin-mediated degradation of β-catenin [372]. Over-expressed Ca-cyBP/SIP leads to suppression of growth of renal cell carcinomas [373]. Although CacyBP/SIP is implicated in p53-induced β-catenin degradation, its precise function is unknown. CacyBP/SIP, which is detected in all types of tumor tissues, is highly expressed in nasopharyngeal carcinoma, osteogenic sarcoma, and pancreatic cancer [374].
8.14. ICAM2
Intercellular adhesion molecule 2 (ICAM2), also known as CD102 (Cluster of Differentiation 102), mediates adhesive interactions related to antigen-specific immune responses, NK-cell mediated clearance, lymphocyte recirculation, and other cellular interactions associated with immune response and surveillance [375, 376].
8.15. CD47
The CD47 gene encodes an unusual member of the immunoglobulin superfamily of membrane proteins, which is involved in the increase in intracellular calcium concentration that occurs upon cell adhesion to an extracellular matrix. The encoded protein, a receptor for the C-terminal cell-binding domain of thrombospondin, may be involved in membrane transport and signal transduction [377]. CD47 may inhibit NK cell-mediated cytotoxicity to cancer cells, providing a mechanism of immune escape for cancers [378]. In chronic lymphocytic leukemia, CD47 ligation induces caspase-independent cell death [379].
9. Future directions
The analysis of a single biomarker may be useful in predicting therapeutic efficacy or patient survival, but it is insufficient to identify the complex relationships among the various parameters that affect the clinical outcomes of patients. By ignoring such relationships and sequential patterns, an important source of potentially useful information is discarded. Nevertheless, to select such information from complex data will require powerful statistical tools. Current statistical methods are inadequate to handle the vast amount of data necessary for comprehensive evaluation and integration of molecular and clinicopathologic factors in estimating clinical outcomes [380,381]. Therefore, newer data-mining tools, including neural networks and decision trees [80], are needed to identify relationships and associations which, in turn, are useful predicting clinical outcomes [72].
As demonstrated by computer models designed to predict the aggressiveness of CRCs, molecular and demographic features of patients, especially age (≤ 65 years), have a substantial impact on their clinical outcomes [72]. Future efforts should be directed toward development of models of CRCs in which age and other demographic features, together with pathologic, molecular, and clinical stages, are used to predict clinical outcomes and to aid in selection of appropriate therapies for subgroups of patients. In addition, as the number of molecular markers increase, the number of patient cases and controls required for validation also increase. This may be the limiting factor.
10. Summary of chapter
As for other cancers, CRCs are heterogeneous. This is true with respect to tumor stage, tumor grade, tumor location within the colorectum, genetics, and racial differences in the hosts. Some molecular markers associated with CRCs have different biologic consequences based on tumor location, pathologic stage, and race/ethnicity of the patients. This leads to the conclusions that: 1) potential confounding variables such as tumor stage, location, and patient race/ethnicity should be taken into consideration; 2) analysis of selected combinations of markers would be more informative than analysis of any one marker; 3) the proximal colon, the distal colon, and the rectum should be considered as separate biologic organs; 4) results across all data sources should be considered in evaluating racial differences in cancer outcomes; and 5) formulation of testable models should yield valuable information on patient outcomes.
Moreover, to establish personalized medicine, research should focus on identification of individual differences based on tumor stage, tumor anatomic location, and race/ethnicity; on the discovery of molecules (genes, mRNA transcripts, and proteins) relevant to these differences; and on development of therapeutic approaches to target these molecules. Such strategies have the potential of reducing the personal and socioeconomic burden of all cancers, including CRN.
Table 7.
Racial differences in the distribution of CRCs and differences in p53 nuclear accumulation at UAB and VA Hospitals, Birmingham, Alabama
| CRC Location | African-American (n = 145)
|
Caucasian-American (n = 258)
|
||||||
|---|---|---|---|---|---|---|---|---|
| Total Cases | % of All Cases | p53+ Immuno- chemistry | % p53+ in Local Region | Total Cases | % of All Cases | p53+ Immuno- chemistry | % p53+ in Local Region | |
| Cecum | 46 | 32 | 20 | 43 | 51 | 20 | 23 | 45 |
| Ascending | 21 | 14 | 11 | 52 | 33 | 12 | 11 | 33 |
| 2/3 of Proximal Transverse | 18 | 13 | 10 | 56 | 25 | 10 | 14 | 56 |
| Total Proximal | 85 | 59 | 41 | 48 | 109 | 42 | 48 | 44 |
| 1/3 of Distal Transverse | None | None | None | None | None | None | None | None |
| Descending | 17 | 12 | 9 | 50 | 25 | 10 | 12 | 48 |
| Sigmoid | 25 | 17 | 8 | 32 | 65 | 25 | 38 | 58 |
| Total Distal Colon | 42 | 29 | 17 | 40 | 90 | 35 | 50 | 56 |
| Rectum | 18 | 12 | 10 | 56 | 59 | 23 | 34 | 59 |
| Total Distal Colon + Rectum | 60 | 41 | 27 | 44 | 149 | 58 | 84 | 56 |
Total no. cases = 403.
Acknowledgments
Supported in part by grants to Dr. U. Manne from the National Institutes of Health (U54CA118948, 2R01-CA98932 and R03-CA139629-01) and by a grant to William E. Grizzle for the Early Detection Reference Laboratory at UAB of the Early Detection Research Network (5U24CA086359-10). We thank Dr. Donald L. Hill, Division of Preventive Medicine, University of Alabama at Birmingham, for his critical review of this manuscript.
References
- 1.Jemal A, et al. Cancer statistics. CA Cancer J Clin. 2009;59(4):225–249. doi: 10.3322/caac.20006. [DOI] [PubMed] [Google Scholar]
- 2.Fearon ER, Vogelstein B. A genetic model for colorectal tumorigenesis. Cell. 1990;61(5):759–767. doi: 10.1016/0092-8674(90)90186-i. [DOI] [PubMed] [Google Scholar]
- 3.Huxley RR, et al. The impact of dietary and lifestyle risk factors on risk of colorectal cancer: a quantitative overview of the epidemiological evidence. Int J Cancer. 2009;125(1):171–180. doi: 10.1002/ijc.24343. [DOI] [PubMed] [Google Scholar]
- 4.Lin OS. Acquired risk factors for colorectal cancer. Methods Mol Biol. 2009;472:361–372. doi: 10.1007/978-1-60327-492-0_16. [DOI] [PubMed] [Google Scholar]
- 5.Fadden K, Hill MJ, Owen RW. Effect of fibre on bile acid metabolism by human faecal bacteria in batch and continuous culture. Eur J Cancer Prev. 1997;6(2):175–194. [PubMed] [Google Scholar]
- 6.Lim CC, Ferguson LR, Tannock GW. Dietary fibres as “prebiotics”: implications for colorectal cancer. Mol Nutr Food Res. 2005;49(6):609–619. doi: 10.1002/mnfr.200500015. [DOI] [PubMed] [Google Scholar]
- 7.Nagengast FM, Grubben MJ, van Munster IP. Role of bile acids in colorectal carcinogenesis. Eur J Cancer. 1995;31A(7–8):1067–1070. doi: 10.1016/0959-8049(95)00216-6. [DOI] [PubMed] [Google Scholar]
- 8.Visek WJ. Diet and cell growth modulation by ammonia. Am J Clin Nutr. 1978;31(10 Suppl):S216–S220. doi: 10.1093/ajcn/31.10.S216. [DOI] [PubMed] [Google Scholar]
- 9.Reeves HL, et al. Kruppel-like factor 6 (KLF6) is a tumor-suppressor gene frequently inactivated in colorectal cancer. Gastroenterology. 2004;126(4):1090–1103. doi: 10.1053/j.gastro.2004.01.005. [DOI] [PubMed] [Google Scholar]
- 10.Eaden JA, Abrams KR, Mayberry JF. The risk of colorectal cancer in ulcerative colitis: a meta-analysis. Gut. 2001;48(4):526–535. doi: 10.1136/gut.48.4.526. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Murata M, et al. Genotype difference of aldehyde dehydrogenase 2 gene in alcohol drinkers influences the incidence of Japanese colorectal cancer patients. Jpn J Cancer Res. 1999;90(7):711–719. doi: 10.1111/j.1349-7006.1999.tb00805.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Chen J, et al. A methylenetetrahydrofolate reductase polymorphism and the risk of colorectal cancer. Cancer Res. 1996;56(21):4862–4864. [PubMed] [Google Scholar]
- 13.Pereira P, et al. Loss of heterozygosity of methylenetetrahydrofolate reductase in colon carcinomas. Oncol Rep. 1999;6(3):597–599. doi: 10.3892/or.6.3.597. [DOI] [PubMed] [Google Scholar]
- 14.Shannon B, et al. A polymorphism in the methylenetetrahydrofolate reductase gene predisposes to colorectal cancers with microsatellite instability. Gut. 2002;50(4):520–524. doi: 10.1136/gut.50.4.520. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Chenevix-Trench G, et al. Glutathione S-transferase M1 and T1 polymorphisms: susceptibility to colon cancer and age of onset. Carcinogenesis. 1995;16(7):1655–1657. doi: 10.1093/carcin/16.7.1655. [DOI] [PubMed] [Google Scholar]
- 16.Zhong S, et al. Relationship between the GSTM1 genetic polymorphism and susceptibility to bladder, breast and colon cancer. Carcinogenesis. 1993;14(9):1821–1824. doi: 10.1093/carcin/14.9.1821. [DOI] [PubMed] [Google Scholar]
- 17.Kinzler KW, Vogelstein B. Lessons from hereditary colorectal cancer. Cell. 1996;87(2):159–170. doi: 10.1016/s0092-8674(00)81333-1. [DOI] [PubMed] [Google Scholar]
- 18.Boland CR. Clinical uses of microsatellite instability testing in colorectal cancer: an ongoing challenge. J Clin Oncol. 2007;25(7):754–756. doi: 10.1200/JCO.2006.09.4607. [DOI] [PubMed] [Google Scholar]
- 19.Boland CR, et al. A National Cancer Institute Workshop on Microsatellite Instability for cancer detection and familial predisposition: development of international criteria for the determination of microsatellite instability in colorectal cancer. Cancer Res. 1998;58(22):5248–5257. [PubMed] [Google Scholar]
- 20.Chao A, et al. Patient and tumor characteristics of colon cancers with microsatellite instability: a population-based study. Cancer Epidemiol Biomarkers Prev. 2000;9(6):539–544. [PubMed] [Google Scholar]
- 21.Dean PA. Hereditary intestinal polyposis syndromes. Rev Gastroenterol Mex. 1996;61(2):100–111. [PubMed] [Google Scholar]
- 22.Gomez Garcia EB, Knoers NV. Gardner’s syndrome (familial adenomatous polyposis): a cilia-related disorder. Lancet Oncol. 2009;10(7):727–735. doi: 10.1016/S1470-2045(09)70167-6. [DOI] [PubMed] [Google Scholar]
- 23.Hernegger GS, Moore HG, Guillem JG. Attenuated familial adenomatous polyposis: an evolving and poorly understood entity. Dis Colon Rectum. 2002;45(1):127–134. doi: 10.1007/s10350-004-6127-y. discussion 134–136. [DOI] [PubMed] [Google Scholar]
- 24.Galiatsatos P, Foulkes WD. Familial adenomatous polyposis. Am J Gastroenterol. 2006;101(2):385–398. doi: 10.1111/j.1572-0241.2006.00375.x. [DOI] [PubMed] [Google Scholar]
- 25.Aretz S, et al. High proportion of large genomic deletions and a genotype phenotype update in 80 unrelated families with juvenile polyposis syndrome. J Med Genet. 2007;44(11):702–709. doi: 10.1136/jmg.2007.052506. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.van Hattem WA, et al. Large genomic deletions of SMAD4, BMPR1A and PTEN in juvenile polyposis. Gut. 2008;57(5):623–627. doi: 10.1136/gut.2007.142927. [DOI] [PubMed] [Google Scholar]
- 27.Issa JP. Colon cancer: it’s CIN or CIMP. Clin Cancer Res. 2008;14(19):5939–5940. doi: 10.1158/1078-0432.CCR-08-1596. [DOI] [PubMed] [Google Scholar]
- 28.Rustgi AK. The genetics of hereditary colon cancer. Genes Dev. 2007;21(20):2525–2538. doi: 10.1101/gad.1593107. [DOI] [PubMed] [Google Scholar]
- 29.Pasche B, et al. TbetaR-I(6A) is a candidate tumor susceptibility allele. Cancer Res. 1999;59(22):5678–5682. [PubMed] [Google Scholar]
- 30.Bian Y, et al. TGFBR1*6A may contribute to hereditary colorectal cancer. J Clin Oncol. 2005;23(13):3074–3078. doi: 10.1200/JCO.2005.00.281. [DOI] [PubMed] [Google Scholar]
- 31.Pasche B, et al. Somatic acquisition and signaling of TGF-BR1*6A in cancer. Jama. 2005;294(13):1634–1646. doi: 10.1001/jama.294.13.1634. [DOI] [PubMed] [Google Scholar]
- 32.Valle L, et al. Germline allele-specific expression of TGF-BR1 confers an increased risk of colorectal cancer. Science. 2008;321(5894):1361–1365. doi: 10.1126/science.1159397. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.Zeng Q, et al. Tgfbr1 haploinsufficiency is a potent modifier of colorectal cancer development. Cancer Res. 2009;69(2):678–686. doi: 10.1158/0008-5472.CAN-08-3980. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.Kellokumpu I, Husa A. Colorectal adenomas: morphologic features and the risk of developing metachronous adenomas and carcinomas in the colorectum. Scand J Gastroenterol. 1987;22(7):833–841. doi: 10.3109/00365528708991923. [DOI] [PubMed] [Google Scholar]
- 35.Bird RP. Observation and quantification of aberrant crypts in the murine colon treated with a colon carcinogen: preliminary findings. Cancer Lett. 1987;37(2):147–151. doi: 10.1016/0304-3835(87)90157-1. [DOI] [PubMed] [Google Scholar]
- 36.Bird RP. Role of aberrant crypt foci in understanding the pathogenesis of colon cancer. Cancer Lett. 1995;93(1):55–71. doi: 10.1016/0304-3835(95)03788-X. [DOI] [PubMed] [Google Scholar]
- 37.Pretlow TP, et al. Aberrant crypts: putative preneoplastic foci in human colonic mucosa. Cancer Res. 1991;51(5):1564–1567. [PubMed] [Google Scholar]
- 38.Pretlow TP, et al. K-ras mutations in putative preneoplastic lesions in human colon. J Natl Cancer Inst. 1993;85(24):2004–2007. doi: 10.1093/jnci/85.24.2004. [DOI] [PubMed] [Google Scholar]
- 39.Pretlow TP, et al. Two types of putative preneoplastic lesions identified by hexosaminidase activity in whole-mounts of colons from F344 rats treated with carcinogen. Am J Pathol. 1993;142(6):1695–1700. [PMC free article] [PubMed] [Google Scholar]
- 40.Winawer SJ, et al. Prevention of colorectal cancer by colonoscopic polypectomy. The National Polyp Study Work-group. N Engl J Med. 1993;329(27):1977–1981. doi: 10.1056/NEJM199312303292701. [DOI] [PubMed] [Google Scholar]
- 41.Burgart LJ. Colorectal polyps and other precursor lesions. Need for an expanded view. Gastroenterol Clin North Am. 2002;31(4):959–970. doi: 10.1016/s0889-8553(02)00056-0. [DOI] [PubMed] [Google Scholar]
- 42.Rex DK. Colonoscopy: a review of its yield for cancers and adenomas by indication. Am J Gastroenterol. 1995;90(3):353–365. [PubMed] [Google Scholar]
- 43.Shanmugam C, et al. p53 Nuclear accumulation and Bcl-2 expression in contiguous adenomatous components of colorectal adenocarcinomas predict aggressive tumor behavior. J Histochem Cytochem. 2008;56(3):305–312. doi: 10.1369/jhc.7A7362.2007. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44.Richert MM, et al. Inhibition of CXCR4 by CTCE-9908 inhibits breast cancer metastasis to lung and bone. Oncol Rep. 2009;21(3):761–767. [PubMed] [Google Scholar]
- 45.Stafford LJ, Vaidya KS, Welch DR. Metastasis suppressors genes in cancer. Int J Biochem Cell Biol. 2008;40(5):874–891. doi: 10.1016/j.biocel.2007.12.016. [DOI] [PubMed] [Google Scholar]
- 46.Hurst DR, et al. Breast cancer metastasis suppressor 1 up-regulates miR-146, which suppresses breast cancer metastasis. Cancer Res. 2009;69(4):1279–1283. doi: 10.1158/0008-5472.CAN-08-3559. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 47.Norton L. Cancer stem cells, self-seeding, and decremented exponential growth: theoretical and clinical implications. Breast Dis. 2008;29:27–36. doi: 10.3233/bd-2008-29104. [DOI] [PubMed] [Google Scholar]
- 48.Kim MY, et al. Tumor self-seeding by circulating cancer cells. Cell. 2009;139(7):1315–1326. doi: 10.1016/j.cell.2009.11.025. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 49.Leung CT, Brugge JS. Tumor self-seeding: bidirectional flow of tumor cells. Cell. 2009;139(7):1226–1228. doi: 10.1016/j.cell.2009.12.013. [DOI] [PubMed] [Google Scholar]
- 50.Larson W. Human Embryology. Churchill Livingstone, Inc; New York, NY: 1993. pp. 205–234. [Google Scholar]
- 51.Grizzle WE, et al. Molecular and histopathologic changes in the development of colorectal neoplasia. In: Srivastava S, Henson DE, Gazdar A, editors. Molecular Pathology of Early Cancer. Chapter 10. IOS Press; Amsterdam: 1999. pp. 135–170. [Google Scholar]
- 52.Alexander D, et al. Postsurgical disparity in survival between African Americans and Caucasians with colonic adenocarcinoma. Cancer. 2004;101(1):66–76. doi: 10.1002/cncr.20337. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 53.Coates RJ, et al. Anatomic site distribution of colon cancer by race and other colon cancer risk factors. Dis Colon Rectum. 1995;38(1):42–50. doi: 10.1007/BF02053856. [DOI] [PubMed] [Google Scholar]
- 54.Delattre O, et al. Multiple genetic alterations in distal and proximal colorectal cancer. Lancet. 1989;2(8659):353–356. doi: 10.1016/s0140-6736(89)90537-0. [DOI] [PubMed] [Google Scholar]
- 55.Devesa SS, Chow WH. Variation in colorectal cancer incidence in the United States by subsite of origin. Cancer. 1993;71(12):3819–3826. doi: 10.1002/1097-0142(19930615)71:12<3819::aid-cncr2820711206>3.0.co;2-l. [DOI] [PubMed] [Google Scholar]
- 56.Freedman AN, et al. Familial and nutritional risk factors for p53 overexpression in colorectal cancer. Cancer Epidemiol Biomarkers Prev. 1996;5(4):285–291. [PubMed] [Google Scholar]
- 57.Ikeda Y, et al. Increased incidence of proximal colon cancer in the elderly. J Clin Gastroenterol. 1996;23(2):105–108. doi: 10.1097/00004836-199609000-00007. [DOI] [PubMed] [Google Scholar]
- 58.Jessup JM, Stewart AK, Menck HR. The National Cancer Data Base report on patterns of care for adenocarcinoma of the rectum, 1985–1995. Cancer. 1998;83(11):2408–2418. doi: 10.1002/(sici)1097-0142(19981201)83:11<2408::aid-cncr22>3.0.co;2-g. [DOI] [PubMed] [Google Scholar]
- 59.Kern SE, et al. Clinical and pathological associations with allelic loss in colorectal carcinoma [corrected] Jama. 1989;261(21):3099–3103. doi: 10.1001/jama.261.21.3099. [DOI] [PubMed] [Google Scholar]
- 60.Kern SE, et al. Oncogenic forms of p53 inhibit p53-regulated gene expression. Science. 1992;256(5058):827–830. doi: 10.1126/science.1589764. [DOI] [PubMed] [Google Scholar]
- 61.Scott N, et al. p53 in colorectal cancer: clinicopathological correlation and prognostic significance. Br J Cancer. 1991;63(2):317–319. doi: 10.1038/bjc.1991.74. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 62.Slattery ML, et al. A description of age, sex, and site distributions of colon carcinoma in three geographic areas. Cancer. 1996;78(8):1666–1670. doi: 10.1002/(sici)1097-0142(19961015)78:8<1666::aid-cncr5>3.0.co;2-c. [DOI] [PubMed] [Google Scholar]
- 63.Thibodeau SN, Bren G, Schaid D. Microsatellite instability in cancer of the proximal colon. Science. 1993;260(5109):816–819. doi: 10.1126/science.8484122. [DOI] [PubMed] [Google Scholar]
- 64.Shibata D, et al. Genomic instability in repeated sequences is an early somatic event in colorectal tumorigenesis that persists after transformation. Nat Genet. 1994;6(3):273–281. doi: 10.1038/ng0394-273. [DOI] [PubMed] [Google Scholar]
- 65.Grizzle WE, et al. Molecular characterization of colorectal neoplasia in translational research. Arch Pathol Lab Med. 2001;125(1):91–98. doi: 10.5858/2001-125-0091-MCOCNI. [DOI] [PubMed] [Google Scholar]
- 66.Manne U, et al. Nuclear accumulation of p53 in colorectal adenocarcinoma: prognostic importance differs with race and location of the tumor. Cancer. 1998;83(12):2456–2467. doi: 10.1002/(sici)1097-0142(19981215)83:12<2456::aid-cncr8>3.0.co;2-5. [DOI] [PubMed] [Google Scholar]
- 67.Sun XF, et al. Prognostic significance of p53 nuclear and cytoplasmic overexpression in right and left colorectal adenocarcinomas. Eur J Cancer. 1996;32A(11):1963–1967. doi: 10.1016/0959-8049(96)00205-5. [DOI] [PubMed] [Google Scholar]
- 68.Diez M, et al. Influence of tumor localization on the prognostic value of P53 protein in colorectal adenocarcinomas. Anticancer Res. 2000;20(5C):3907–3912. [PubMed] [Google Scholar]
- 69.Copeland MM. Introductory statement on the progress of The National Large Bowel Cancer Project: its goals, objectives, and programs. Cancer. 1977;40(5 Suppl):2406–2413. doi: 10.1002/1097-0142(197711)40:5+<2406::aid-cncr2820400903>3.0.co;2-b. [DOI] [PubMed] [Google Scholar]
- 70.American Cancer Society. Colon Cancer facts and figures for African Americans, 2009–2010. 2009. pp. 1–27. [Google Scholar]
- 71.Alexander DD, et al. African-American and Caucasian disparities in colorectal cancer mortality and survival by data source: an epidemiologic review. Cancer Biomark. 2007;3(6):301–313. doi: 10.3233/cbm-2007-3604. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 72.Anderson B, et al. Comparison of the predictive qualities of three prognostic models built on a molecular biomarker dataset of colorectal cancer. Front Biosci. doi: 10.2741/e146. In Press. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 73.Johnson H, Jr, Carstens R. Anatomical distribution of colonic carcinomas. Interracial differences in a community hospital population. Cancer. 1986;58(4):997–1000. doi: 10.1002/1097-0142(19860815)58:4<997::aid-cncr2820580435>3.0.co;2-2. [DOI] [PubMed] [Google Scholar]
- 74.Nelson RL, et al. The relation of age, race, and gender to the subsite location of colorectal carcinoma. Cancer. 1997;80(2):193–197. doi: 10.1002/(sici)1097-0142(19970715)80:2<193::aid-cncr4>3.0.co;2-v. [DOI] [PubMed] [Google Scholar]
- 75.Page WF, Kuntz AJ. Racial and socioeconomic factors in cancer survival. A comparison of Veterans Administration results with selected studies. Cancer. 1980;45(5):1029–1040. doi: 10.1002/1097-0142(19800301)45:5<1029::aid-cncr2820450533>3.0.co;2-3. [DOI] [PubMed] [Google Scholar]
- 76.Fitzgerald TL, et al. Gastrointestinal malignancies: when does race matter? J Am Coll Surg. 2009;209(5):645–652. doi: 10.1016/j.jamcollsurg.2009.08.007. [DOI] [PubMed] [Google Scholar]
- 77.Grizzle WE, et al. Providing human tissues for research: how to establish a program. Arch Pathol Lab Med. 1998;122(12):1065–1076. [PubMed] [Google Scholar]
- 78.Goli R, et al. Higher incidence of single nucleoptide polymorphisms at codon 72 of the p53 gene are associated with poor prognosis of African-American patients with high grade colorectal adenocarcinomas. Proceedings American Association for Cancer Research; 2006; Washington, DC. [Google Scholar]
- 79.Katkoori VR, et al. Prognostic Significance of p53 Codon 72 Polymorphism Differs with Race in Colorectal Adenocarcinoma. Clin Cancer Res. 2009;15(7):2406–2416. doi: 10.1158/1078-0432.CCR-08-1719. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 80.Burke HB, Henson DE. The American Joint Committee on Cancer. Criteria for prognostic factors and for an enhanced prognostic system. Cancer. 1993;72(10):3131–3135. doi: 10.1002/1097-0142(19931115)72:10<3131::aid-cncr2820721039>3.0.co;2-j. [DOI] [PubMed] [Google Scholar]
- 81.Grizzle WE, et al. Molecular staging of colorectal cancer in African-American and Caucasian patients using phenotypic expression of p53, Bcl-2, MUC-1 AND p27(kip-1) Int J Cancer. 2002;97(4):403–409. doi: 10.1002/ijc.1617. [DOI] [PubMed] [Google Scholar]
- 82.Grizzle WE, et al. Immunohistochemical evaluation of biomarkers in prostatic and colorectal neoplasia. In: Hanausek M, Walaszek Z, editors. John Walker’s Methods in Molecular Medicine-Tumor Marker Protocols. Humana Press; Totowa, NJ: 1998. pp. 143–160. [Google Scholar]
- 83.Manne U. Understanding racial differences in colorectal cancer aids in individualized medicine. Future Oncol. 2007;3(3):235–241. doi: 10.2217/14796694.3.3.235. [DOI] [PubMed] [Google Scholar]
- 84.McLeod HL, Murray GI. Tumour markers of prognosis in colorectal cancer. Br J Cancer. 1999;79(2):191–203. doi: 10.1038/sj.bjc.6690033. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 85.Manne U, et al. Prognostic significance of Bcl-2 expression and p53 nuclear accumulation in colorectal adenocarcinoma. Int J Cancer. 1997;74(3):346–358. doi: 10.1002/(sici)1097-0215(19970620)74:3<346::aid-ijc19>3.0.co;2-9. [DOI] [PubMed] [Google Scholar]
- 86.Ishioka C, Suzuki T, Kanamaru R. Mutations of p53 gene in human colorectal tumor in Japan: molecular epidemiological aspects. Tohoku J Exp Med. 1992;167(3):243–245. doi: 10.1620/tjem.167.243. [DOI] [PubMed] [Google Scholar]
- 87.Sayhan N, et al. P53 codon 72 genotypes in colon cancer. Association with human papillomavirus infection. Res Commun Mol Pathol Pharmacol. 2001;109(1–2):25–34. [PubMed] [Google Scholar]
- 88.Vos M, et al. Polymorphisms and mutations found in the regions flanking exons 5 to 8 of the TP53 gene in a population at high risk for esophageal cancer in South Africa. Cancer Genet Cytogenet. 2003;140(1):23–30. doi: 10.1016/s0165-4608(02)00638-6. [DOI] [PubMed] [Google Scholar]
- 89.Felley-Bosco E, et al. Functional studies of a germ-line polymorphism at codon 47 within the p53 gene. Am J Hum Genet. 1993;53(3):752–759. [PMC free article] [PubMed] [Google Scholar]
- 90.Matlashewski GJ, et al. Primary structure polymorphism at amino acid residue 72 of human p53. Mol Cell Biol. 1987;7(2):961–963. doi: 10.1128/mcb.7.2.961. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 91.Carbone D, Chiba I, Mitsudomi T. Polymorphism at codon 213 within the p53 gene. Oncogene. 1991;6(9):1691–1692. [PubMed] [Google Scholar]
- 92.Buchman VL, et al. A variation in the structure of the protein-coding region of the human p53 gene. Gene. 1988;70(2):245–252. doi: 10.1016/0378-1119(88)90196-5. [DOI] [PubMed] [Google Scholar]
- 93.Futreal PA, Barrett JC, Wiseman RW. An Alu polymorphism intragenic to the TP53 gene. Nucleic Acids Res. 1991;19(24):6977. doi: 10.1093/nar/19.24.6977. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 94.Oliva MR, et al. A new polymorphic site in intron 2 of TP53 characterizes LOH in human tumors by PCR-SSCP. Diagn Mol Pathol. 1995;4(1):54–58. doi: 10.1097/00019606-199503000-00010. [DOI] [PubMed] [Google Scholar]
- 95.Lazar V, et al. Simple sequence repeat polymorphism within the p53 gene. Oncogene. 1993;8(6):1703–1705. [PubMed] [Google Scholar]
- 96.Chumakov PM, Jenkins JR. BstNI/NciI polymorphism of the human p53 gene (TP53) Nucleic Acids Res. 1991;19(24):6969. doi: 10.1093/nar/19.24.6969-a. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 97.McDaniel T, et al. The MspI polymorphism in intron 6 of p53 (TP53) detected by digestion of PCR products. Nucleic Acids Res. 1991;19(17):4796. doi: 10.1093/nar/19.17.4796-a. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 98.Graf J, et al. Identification of novel polymorphisms in intron 7 of the human p53 gene in acute myeloid leukemia and healthy donors. Leuk Lymphoma. 2001;41(5–6):655–658. doi: 10.3109/10428190109060356. [DOI] [PubMed] [Google Scholar]
- 99.Prosser J, Condie A. Biallelic ApaI polymorphism of the human p53 gene (TP53) Nucleic Acids Res. 1991;19(17):4799. doi: 10.1093/nar/19.17.4799-a. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 100.Graziani D, et al. An Ava I polymorphism in the TP53 gene. Mol Cell Probes. 1999;13(5):393–395. doi: 10.1006/mcpr.1999.0256. [DOI] [PubMed] [Google Scholar]
- 101.Murata M, et al. Analysis of a germ line polymorphism of the p53 gene in lung cancer patients; discrete results with smoking history. Carcinogenesis. 1996;17(2):261–264. doi: 10.1093/carcin/17.2.261. [DOI] [PubMed] [Google Scholar]
- 102.Sjalander A, et al. P53 germ line haplotypes associated with increased risk for colorectal cancer. Carcinogenesis. 1995;16(7):1461–1464. doi: 10.1093/carcin/16.7.1461. [DOI] [PubMed] [Google Scholar]
- 103.Lung FW, et al. p53 codon 72 polymorphism and susceptibility malignancy of colorectal cancer in Taiwan. J Cancer Res Clin Oncol. 2004;130(12):728–732. doi: 10.1007/s00432-004-0605-4. [DOI] [PubMed] [Google Scholar]
- 104.Hainaut P, Hollstein M. p53 and human cancer: the first ten thousand mutations. Adv Cancer Res. 2000;77:81–137. doi: 10.1016/s0065-230x(08)60785-x. [DOI] [PubMed] [Google Scholar]
- 105.Dumont P, et al. The codon 72 polymorphic variants of p53 have markedly different apoptotic potential. Nat Genet. 2003;33(3):357–365. doi: 10.1038/ng1093. [DOI] [PubMed] [Google Scholar]
- 106.Baretton GB, et al. Apoptosis and immunohistochemical bcl-2 expression in colorectal adenomas and carcinomas. Aspects of carcinogenesis and prognostic significance. Cancer. 1996;77(2):255–264. doi: 10.1002/(SICI)1097-0142(19960115)77:2<255::AID-CNCR6>3.0.CO;2-L. [DOI] [PubMed] [Google Scholar]
- 107.Bosari S, et al. bcl-2 oncoprotein in colorectal hyperplastic polyps, adenomas, and adenocarcinomas. Hum Pathol. 1995;26(5):534–540. doi: 10.1016/0046-8177(95)90250-3. [DOI] [PubMed] [Google Scholar]
- 108.Hague A, et al. BCL-2 expression in human colorectal adenomas and carcinomas. Oncogene. 1994;9(11):3367–3370. [PubMed] [Google Scholar]
- 109.Hao XP, Ilyas M, Talbot IC. Expression of Bcl-2 and p53 in the colorectal adenomacarcinoma sequence. Pathobiology. 1997;65(3):140–145. doi: 10.1159/000164115. [DOI] [PubMed] [Google Scholar]
- 110.Sinicrope FA, et al. bcl-2 and p53 oncoprotein expression during colorectal tumorigenesis. Cancer Res. 1995;55(2):237–241. [PubMed] [Google Scholar]
- 111.Yang HB, et al. The role of bcl-2 in the progression of the colorectal adenomacarcinoma sequence. Anticancer Res. 1999;19(1B):727–730. [PubMed] [Google Scholar]
- 112.Giatromanolaki A, et al. Combined role of tumor angiogenesis, bcl-2, and p53 expression in the prognosis of patients with colorectal carcinoma. Cancer. 1999;86(8):1421–1430. doi: 10.1002/(sici)1097-0142(19991015)86:8<1421::aid-cncr6>3.0.co;2-x. [DOI] [PubMed] [Google Scholar]
- 113.Ilyas M, et al. Loss of Bcl-2 expression correlates with tumour recurrence in colorectal cancer. Gut. 1998;43(3):383–387. doi: 10.1136/gut.43.3.383. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 114.Ishijima N, et al. The immunohistochemical expression of BCL-2 oncoprotein in colorectal adenocarcinoma. Surg Today. 1999;29(7):682–684. doi: 10.1007/BF02483002. [DOI] [PubMed] [Google Scholar]
- 115.Chatla C, et al. Recurrence and survival predictive value of phenotypic expression of Bcl-2 varies with tumor stage of colorectal adenocarcinoma. Cancer Biomark. 2005;1(4–5):241–250. doi: 10.3233/cbm-2005-14-507. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 116.Ofner D, et al. Immunohistochemically detectable bcl-2 expression in colorectal carcinoma: correlation with tumour stage and patient survival. Br J Cancer. 1995;72(4):981–985. doi: 10.1038/bjc.1995.446. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 117.Sinicrope FA, et al. Prognostic value of bcl-2 oncoprotein expression in stage II colon carcinoma. Clin Cancer Res. 1995;1(10):1103–1110. [PubMed] [Google Scholar]
- 118.Schneider HJ, et al. Bcl-2 expression and response to chemotherapy in colorectal adenocarcinomas. Br J Cancer. 1997;75(3):427–431. doi: 10.1038/bjc.1997.70. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 119.Tollenaar RA, et al. Immunohistochemical detection of p53 and Bcl-2 in colorectal carcinoma: no evidence for prognostic significance. Br J Cancer. 1998;77(11):1842–1847. doi: 10.1038/bjc.1998.306. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 120.Bhatavdekar JM, et al. Coexpression of Bcl-2, c-Myc, and p53 oncoproteins as prognostic discriminants in patients with colorectal carcinoma. Dis Colon Rectum. 1997;40(7):785–790. doi: 10.1007/BF02055433. [DOI] [PubMed] [Google Scholar]
- 121.Jass JR, Roberton AM. Colorectal mucin histochemistry in health and disease: a critical review. Pathol Int. 1994;44(7):487–504. doi: 10.1111/j.1440-1827.1994.tb02599.x. [DOI] [PubMed] [Google Scholar]
- 122.Hanski C, et al. Altered glycosylation of the MUC-1 protein core contributes to the colon carcinoma-associated increase of mucin-bound sialyl-Lewis(x) expression. Cancer Res. 1993;53(17):4082–4088. [PubMed] [Google Scholar]
- 123.Ho SB, et al. Heterogeneity of mucin gene expression in normal and neoplastic tissues. Cancer Res. 1993;53(3):641–651. [PubMed] [Google Scholar]
- 124.Aoki R, et al. MUC-1 expression as a predictor of the curative endoscopic treatment of submucosally invasive colorectal carcinoma. Dis Colon Rectum. 1998;41(10):1262–1272. doi: 10.1007/BF02258227. [DOI] [PubMed] [Google Scholar]
- 125.Hiraga Y, et al. Immunoreactive MUC1 expression at the deepest invasive portion correlates with prognosis of colorectal cancer. Oncology. 1998;55(4):307–319. doi: 10.1159/000011868. [DOI] [PubMed] [Google Scholar]
- 126.Manne U, Weiss HL, Grizzle WE. Racial differences in the prognostic usefulness of MUC1 and MUC2 in colorectal adenocarcinomas. Clin Cancer Res. 2000;6(10):4017–4025. [PubMed] [Google Scholar]
- 127.Jessup JM, et al. Multivariate analysis of tissue-based prognostic markers in stage II–III colorectal carcinoma. Proc Annu Meet Am Soc Clin Oncol. 1997 [Google Scholar]
- 128.Loda M, et al. Increased proteasome-dependent degradation of the cyclin-dependent kinase inhibitor p27 in aggressive colorectal carcinomas. Nat Med. 1997;3(2):231–234. doi: 10.1038/nm0297-231. [DOI] [PubMed] [Google Scholar]
- 129.Tenjo T, et al. Prognostic significance of p27(kip1) protein expression and spontaneous apoptosis in patients with colorectal adenocarcinomas. Oncology. 2000;58(1):45–51. doi: 10.1159/000012078. [DOI] [PubMed] [Google Scholar]
- 130.Liu DF, et al. p27 cell-cycle inhibitor is inversely correlated with lymph node metastases in right-sided colon cancer. J Clin Lab Anal. 1999;13(6):291–295. doi: 10.1002/(SICI)1098-2825(1999)13:6<291::AID-JCLA7>3.0.CO;2-K. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 131.Manne U, et al. Prognostic significance of p27(kip-1) expression in colorectal adenocarcinomas is associated with tumor stage. Clin Cancer Res. 2004;10(5):1743–1752. doi: 10.1158/1078-0432.ccr-03-0037. [DOI] [PubMed] [Google Scholar]
- 132.Boland CR, et al. A National Cancer Institute Workshop on Microsatellite Instability for cancer detection and familial predisposition: development of international criteria for the determination of microsatellite instability in colorectal cancer. Cancer Res. 1998;58:5248–5257. [PubMed] [Google Scholar]
- 133.Chao A, et al. Patient and tumor characteristics of colon cancers with microsatellite instability: a population-based study. Cancer Epidemiol Biomarkers Prev. 2000;9(6):539–544. [PubMed] [Google Scholar]
- 134.Slattery ML, et al. Associations between cigarette smoking, lifestyle factors, and microsatellite instability in colon tumors. J Natl Cancer Inst. 2000;92(22):1831–1836. doi: 10.1093/jnci/92.22.1831. [DOI] [PubMed] [Google Scholar]
- 135.Grady WM. Genomic instability and colon cancer. Cancer Metastasis Rev. 2004;23(1–2):11–27. doi: 10.1023/a:1025861527711. [DOI] [PubMed] [Google Scholar]
- 136.Ashktorab H, et al. Clinicopathological features and microsatellite instability (MSI) in colorectal cancers from African Americans. Int J Cancer. 2005;116(6):914–919. doi: 10.1002/ijc.21062. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 137.Jass JR, et al. Morphology of sporadic colorectal cancer with DNA replication errors. Gut. 1998;42(5):673–679. doi: 10.1136/gut.42.5.673. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 138.Kim H, et al. Clinical and pathological characteristics of sporadic colorectal carcinomas with DNA replication errors in microsatellite sequences. Am J Pathol. 1994;145(1):148–156. [PMC free article] [PubMed] [Google Scholar]
- 139.Lothe RA, et al. Genomic instability in colorectal cancer: relationship to clinicopathological variables and family history. Cancer Res. 1993;53(24):5849–5852. [PubMed] [Google Scholar]
- 140.Ashktorab H, et al. High incidence of microsatellite instability in colorectal cancer from African Americans. Clin Cancer Res. 2003;9(3):1112–1117. [PubMed] [Google Scholar]
- 141.Kumar K, et al. Distinct BRAF (V600E) and KRAS mutations in high microsatellite instability sporadic colorectal cancer in African Americans. Clin Cancer Res. 2009;15(4):1155–1161. doi: 10.1158/1078-0432.CCR-08-1029. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 142.Ashktorab H, et al. Sporadic colon cancer: mismatch repair immunohistochemistry and microsatellite instability in Omani subjects. Dig Dis Sci. 2008;53(10):2723–2731. doi: 10.1007/s10620-007-0189-3. [DOI] [PubMed] [Google Scholar]
- 143.Kinzler KW, et al. Identification of a gene located at chromosome 5q21 that is mutated in colorectal cancers [see comments] Science. 1991;251(4999):1366–1370. doi: 10.1126/science.1848370. [DOI] [PubMed] [Google Scholar]
- 144.Kohonen-Corish MR, et al. Promoter methylation of the mutated in colorectal cancer gene is a frequent early event in colorectal cancer. Oncogene. 2007;26(30):4435–4441. doi: 10.1038/sj.onc.1210210. [DOI] [PubMed] [Google Scholar]
- 145.Nishisho I, et al. Mutations of chromosome 5q21 genes in FAP and colorectal cancer patients. Science. 1991;253(5020):665–669. doi: 10.1126/science.1651563. [DOI] [PubMed] [Google Scholar]
- 146.Miki Y, et al. Frequent loss of heterozygosity at the MCC locus on chromosome 5q21-22 in sporadic colorectal carcinomas. Jpn J Cancer Res. 1991;82(9):1003–1007. doi: 10.1111/j.1349-7006.1991.tb01935.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 147.Joslyn G, et al. Identification of deletion mutations and three new genes at the familial polyposis locus. Cell. 1991;66(3):601–613. doi: 10.1016/0092-8674(81)90022-2. [DOI] [PubMed] [Google Scholar]
- 148.Fukuyama R, et al. Mutated in colorectal cancer, a putative tumor suppressor for serrated colorectal cancer, selectively represses beta-catenin-dependent transcription. Oncogene. 2008;27(46):6044–6055. doi: 10.1038/onc.2008.204. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 149.Fearon ER, et al. Identification of a chromosome 18q gene that is altered in colorectal cancers. Science. 1990;247(4938):49–56. doi: 10.1126/science.2294591. [DOI] [PubMed] [Google Scholar]
- 150.Vogelstein B, et al. Genetic alterations during colorectal-tumor development. N Engl J Med. 1988;319(9):525–532. doi: 10.1056/NEJM198809013190901. [DOI] [PubMed] [Google Scholar]
- 151.Okamoto M, et al. Loss of constitutional heterozygosity in colon carcinoma from patients with familial polyposis coli. Nature. 1988;331(6153):273–277. doi: 10.1038/331273a0. [DOI] [PubMed] [Google Scholar]
- 152.Sasaki M, et al. Loss of constitutional heterozygosity in colorectal tumors from patients with familial polyposis coli and those with nonpolyposis colorectal carcinoma. Cancer Res. 1989;49(16):4402–4406. [PubMed] [Google Scholar]
- 153.Turley H, et al. The distribution of the deleted in colon cancer (DCC) protein in human tissues. Cancer Res. 1995;55(23):5628–5631. [PubMed] [Google Scholar]
- 154.Kataoka M, Okabayashi T, Orita K. Decreased expression of DCC mRNA in gastric and colorectal cancer. Surg Today. 1995;25(12):1001–1007. doi: 10.1007/BF00311682. [DOI] [PubMed] [Google Scholar]
- 155.Fearon ER, Hamilton SR, Vogelstein B. Clonal analysis of human colorectal tumors. Science. 1987;238(4824):193–197. doi: 10.1126/science.2889267. [DOI] [PubMed] [Google Scholar]
- 156.Kato M, et al. Detection of DCC and Kiras gene alterations in colorectal carcinoma tissue as prognostic markers for liver metastatic recurrence. Cancer. 1996;77(8 Suppl):1729–1735. doi: 10.1002/(SICI)1097-0142(19960415)77:8<1729::AID-CNCR47>3.0.CO;2-Z. [DOI] [PubMed] [Google Scholar]
- 157.Hedrick L, et al. Cold Spring Harbor Symp. on Quant. Biol. 1992;LVII:345–351. doi: 10.1101/sqb.1992.057.01.039. [DOI] [PubMed] [Google Scholar]
- 158.Kikuchi-Yanoshita R, et al. Loss of expression of the DCC gene during progression of colorectal carcinomas in familial adenomatous polyposis and non-familial adenomatous polyposis patients. Cancer Res. 1992;52(13):3801–3803. [PubMed] [Google Scholar]
- 159.Iino H, et al. Molecular genetics for clinical management of colorectal carcinoma. 17p, 18q, and 22q loss of heterozygosity and decreased DCC expression are correlated with the metastatic potential. Cancer. 1994;73(5):1324–1331. doi: 10.1002/1097-0142(19940301)73:5<1324::aid-cncr2820730503>3.0.co;2-w. [DOI] [PubMed] [Google Scholar]
- 160.Ookawa K, et al. Concordant p53 and DCC alterations and allelic losses on chromosomes 13q and 14q associated with liver metastases of colorectal carcinoma. Int J Cancer. 1993;53(3):382–387. doi: 10.1002/ijc.2910530307. [DOI] [PubMed] [Google Scholar]
- 161.Fazeli A, et al. Phenotype of mice lacking functional Deleted in colorectal cancer (Dcc) gene. Nature. 1997;386(6627):796–804. doi: 10.1038/386796a0. [DOI] [PubMed] [Google Scholar]
- 162.Gotley DC, et al. Alternatively spliced variants of the cell adhesion molecule CD44 and tumour progression in colorectal cancer. Br J Cancer. 1996;74(3):342–351. doi: 10.1038/bjc.1996.364. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 163.Cho KR, et al. The DCC gene: structural analysis and mutations in colorectal carcinomas. Genomics. 1994;19(3):525–531. doi: 10.1006/geno.1994.1102. [DOI] [PubMed] [Google Scholar]
- 164.Klingelhutz AJ, et al. The DCC gene suppresses the malignant phenotype of transformed human epithelial cells. Oncogene. 1995;10(8):1581–1586. [PubMed] [Google Scholar]
- 165.Tanaka S, et al. Immunoreactive transforming growth factor alpha is commonly present in colorectal neoplasia. Am J Pathol. 1991;139(1):123–129. [PMC free article] [PubMed] [Google Scholar]
- 166.Chan SS, et al. UNC-40, a C. elegans homolog of DCC (Deleted in Colorectal Cancer), is required in motile cells responding to UNC-6 netrin cues. Cell. 1996;87(2):187–195. doi: 10.1016/s0092-8674(00)81337-9. [DOI] [PubMed] [Google Scholar]
- 167.Keino-Masu K, et al. Deleted in Colorectal Cancer (DCC) encodes a netrin receptor. Cell. 1996;87(2):175–185. doi: 10.1016/s0092-8674(00)81336-7. [DOI] [PubMed] [Google Scholar]
- 168.Kolodziej PA, et al. frazzled encodes a Drosophila member of the DCC immunoglobulin subfamily and is required for CNS and motor axon guidance. Cell. 1996;87(2):197–204. doi: 10.1016/s0092-8674(00)81338-0. [DOI] [PubMed] [Google Scholar]
- 169.Gao X, et al. Frequent loss of expression and loss of heterozygosity of the putative tumor suppressor gene DCC in prostatic carcinomas. Cancer Res. 1993;53(12):2723–2727. [PubMed] [Google Scholar]
- 170.Murty VV, et al. Frequent allelic deletions and loss of expression characterize the DCC gene in male germ cell tumors. Oncogene. 1994;9(11):3227–3231. [PubMed] [Google Scholar]
- 171.Hohne MW, et al. Frequent loss of expression of the potential tumor suppressor gene DCC in ductal pancreatic adenocarcinoma. Cancer Res. 1992;52(9):2616–2619. [PubMed] [Google Scholar]
- 172.Shibata D, et al. Somatic microsatellite mutations as molecular tumor clocks. Nat Med. 1996;2(6):676–681. doi: 10.1038/nm0696-676. [DOI] [PubMed] [Google Scholar]
- 173.Jen J, et al. Allelic loss of chromosome 18q and prognosis in colorectal cancer. N Engl J Med. 1994;331(4):213–221. doi: 10.1056/NEJM199407283310401. [DOI] [PubMed] [Google Scholar]
- 174.Bernet A, et al. Inactivation of the UNC5C Netrin-1 receptor is associated with tumor progression in colorectal malignancies. Gastroenterology. 2007;133(6):1840–1848. doi: 10.1053/j.gastro.2007.08.009. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 175.Shin SK, et al. Epigenetic and genetic alterations in Netrin-1 receptors UNC5C and DCC in human colon cancer. Gastroenterology. 2007;133(6):1849–1857. doi: 10.1053/j.gastro.2007.08.074. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 176.Brand T, Schneider MD. Transforming growth factor-beta signal transduction. Circ Res. 1996;78(2):173–179. doi: 10.1161/01.res.78.2.173. [DOI] [PubMed] [Google Scholar]
- 177.Massague J. TGFbeta signaling: receptors, transducers, and Mad proteins. Cell. 1996;85(7):947–950. doi: 10.1016/s0092-8674(00)81296-9. [DOI] [PubMed] [Google Scholar]
- 178.Akiyama Y, et al. Mutations of the transforming growth factor-beta type II receptor gene are strongly related to sporadic proximal colon carcinomas with microsatellite instability. Cancer. 1996;78(12):2478–2484. doi: 10.1002/(sici)1097-0142(19961215)78:12<2478::aid-cncr5>3.0.co;2-g. [DOI] [PubMed] [Google Scholar]
- 179.Wang A, et al. Different expression patterns of cyclins A, D1 and E in human colorectal cancer. J Cancer Res Clin Oncol. 1996;122(2):122–126. doi: 10.1007/BF01226270. [DOI] [PubMed] [Google Scholar]
- 180.Eppert K, et al. MADR2 maps to 18q21 and encodes a TGFbeta-regulated MAD-related protein that is functionally mutated in colorectal carcinoma. Cell. 1996;86(4):543–552. doi: 10.1016/s0092-8674(00)80128-2. [DOI] [PubMed] [Google Scholar]
- 181.Friedman E, et al. High levels of transforming growth factor beta 1 correlate with disease progression in human colon cancer. Cancer Epidemiol Biomarkers Prev. 1995;4(5):549–554. [PubMed] [Google Scholar]
- 182.Robson H, et al. Transforming growth factor beta 1 expression in human colorectal tumours: an independent prognostic marker in a subgroup of poor prognosis patients. Br J Cancer. 1996;74(5):753–758. doi: 10.1038/bjc.1996.432. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 183.Tsushima H, et al. High levels of transforming growth factor beta 1 in patients with colorectal cancer: association with disease progression. Gastroenterology. 1996;110(2):375–382. doi: 10.1053/gast.1996.v110.pm8566583. [DOI] [PubMed] [Google Scholar]
- 184.Hsu S, Huang F, Friedman E. Platelet-derived growth factor-B increases colon cancer cell growth in vivo by a paracrine effect. J Cell Physiol. 1995;165(2):239–245. doi: 10.1002/jcp.1041650204. [DOI] [PubMed] [Google Scholar]
- 185.Huang S, Chakrabarty S. Expression of antisense 3-bronectin RNA in human colon carcinoma cells disrupts the regulation of carcinoembryonic antigen by transforming growth factor beta 1. J Biol Chem. 1994;269(46):28764–28768. [PubMed] [Google Scholar]
- 186.Shimizu S, et al. Involvement of transforming growth factor beta1 in autocrine enhancement of gelatinase B secretion by murine metastatic colon carcinoma cells. Cancer Res. 1996;56(14):3366–3370. [PubMed] [Google Scholar]
- 187.Fang F, et al. TGFB1 509 C/T polymorphism and colorectal cancer risk: a meta-analysis. Med Oncol. 2010;27(4):1324–1328. doi: 10.1007/s12032-009-9383-9. [DOI] [PubMed] [Google Scholar]
- 188.Forsti A, et al. Polymorphisms in the transforming growth factor beta 1 pathway in relation to colorectal cancer progression. Genes Chromosomes Cancer. 49(3):270–281. doi: 10.1002/gcc.20738. [DOI] [PubMed] [Google Scholar]
- 189.Wong SC, et al. Prognostic and diagnostic significance of beta-catenin nuclear immunostaining in colorectal cancer. Clin Cancer Res. 2004;10(4):1401–1408. doi: 10.1158/1078-0432.ccr-0157-03. [DOI] [PubMed] [Google Scholar]
- 190.Kobayashi M, et al. Nuclear translocation of beta-catenin in colorectal cancer. Br J Cancer. 2000;82(10):1689–1693. doi: 10.1054/bjoc.1999.1112. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 191.Miyamoto S, et al. Nuclear beta-catenin accumulation as a prognostic factor in Dukes’ D human colorectal cancers. Oncol Rep. 2004;12(2):245–251. [PubMed] [Google Scholar]
- 192.Pancione M, et al. Prognostic role of beta-catenin and p53 expression in the metastatic progression of sporadic colorectal cancer. Hum Pathol. 2010;41(6):867–876. doi: 10.1016/j.humpath.2009.09.019. [DOI] [PubMed] [Google Scholar]
- 193.Derynck R, Akhurst RJ, Balmain A. TGF-beta signaling in tumor suppression and cancer progression. Nat Genet. 2001;29(2):117–129. doi: 10.1038/ng1001-117. [DOI] [PubMed] [Google Scholar]
- 194.Hahn SA, et al. DPC4, a candidate tumor suppressor gene at human chromosome 18q21.1. Science. 1996;271(5247):350–353. doi: 10.1126/science.271.5247.350. [DOI] [PubMed] [Google Scholar]
- 195.Massague J, Wotton D. Transcriptional control by the TGF-beta/Smad signaling system. EMBO J. 2000;19(8):1745–1754. doi: 10.1093/emboj/19.8.1745. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 196.Arai T, et al. Allelotype analysis of early colorectal cancers with lymph node metastasis. Int J Cancer. 1998;79(4):418–423. doi: 10.1002/(sici)1097-0215(19980821)79:4<418::aid-ijc18>3.0.co;2-0. [DOI] [PubMed] [Google Scholar]
- 197.Mitelman F, Mertens F, Johansson B. A breakpoint map of recurrent chromosomal rearrangements in human neoplasia. Nat Genet. 1997;15(Spec No):417–474. doi: 10.1038/ng0497supp-417. [DOI] [PubMed] [Google Scholar]
- 198.Cohn KH, et al. The significance of allelic deletions and aneuploidy in colorectal carcinoma. Results of a 5-year follow-up study. Cancer. 1997;79(2):233–244. [PubMed] [Google Scholar]
- 199.Lanza G, et al. Chromosome 18q allelic loss and prognosis in stage II and III colon cancer. Int J Cancer. 1998;79(4):390–395. doi: 10.1002/(sici)1097-0215(19980821)79:4<390::aid-ijc14>3.0.co;2-9. [DOI] [PubMed] [Google Scholar]
- 200.Watanabe T, et al. Molecular predictors of survival after adjuvant chemotherapy for colon cancer. N Engl J Med. 2001;344(16):1196–1206. doi: 10.1056/NEJM200104193441603. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 201.Koyama M, et al. Inactivation of both alleles of the DPC4/SMAD4 gene in advanced colorectal cancers: identification of seven novel somatic mutations in tumors from Japanese patients. Mutat Res. 1999;406(2–4):71–77. doi: 10.1016/s1383-5726(99)00003-5. [DOI] [PubMed] [Google Scholar]
- 202.Miyaki M, et al. Higher frequency of Smad4 gene mutation in human colorectal cancer with distant metastasis. Oncogene. 1999;18(20):3098–3103. doi: 10.1038/sj.onc.1202642. [DOI] [PubMed] [Google Scholar]
- 203.Alazzouzi H, et al. SMAD4 as a prognostic marker in colorectal cancer. Clin Cancer Res. 2005;11(7):2606–2611. doi: 10.1158/1078-0432.CCR-04-1458. [DOI] [PubMed] [Google Scholar]
- 204.Seshimo I, et al. Expression and mutation of SMAD4 in poorly differentiated carcinoma and signet-ring cell carcinoma of the colorectum. J Exp Clin Cancer Res. 2006;25(3):433–442. [PubMed] [Google Scholar]
- 205.Shan YF, et al. Inhibitory effects of tanshinone II-A on invasion and metastasis of human colon carcinoma cells. Acta Pharmacol Sin. 2009;30(11):1537–1542. doi: 10.1038/aps.2009.139. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 206.Takaku K, et al. Intestinal tumorigenesis in compound mutant mice of both Dpc4 (Smad4) and Apc genes. Cell. 1998;92(5):645–656. doi: 10.1016/s0092-8674(00)81132-0. [DOI] [PubMed] [Google Scholar]
- 207.Mikami T, et al. KAI1, CAR, and Smad4 expression in the progression of colorectal tumor. J Gastroenterol. 2001;36(7):465–469. doi: 10.1007/s005350170069. [DOI] [PubMed] [Google Scholar]
- 208.Isaksson-Mettavainio M, et al. SMAD4/DPC4 expression and prognosis in human colorectal cancer. Anticancer Res. 2006;26(1B):507–510. [PubMed] [Google Scholar]
- 209.Kouvidou C, et al. Expression of Smad4 and TGF-beta2 in colorectal carcinoma. Anticancer Res. 2006;26(4B):2901–2907. [PubMed] [Google Scholar]
- 210.Maitra A, et al. Loss of Dpc4 expression in colonic adenocarcinomas correlates with the presence of metastatic disease. Am J Pathol. 2000;157(4):1105–1111. doi: 10.1016/S0002-9440(10)64625-1. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 211.Reinacher-Schick A, et al. Loss of Smad4 correlates with loss of the invasion suppressor E-cadherin in advanced colorectal carcinomas. J Pathol. 2004;202(4):412–420. doi: 10.1002/path.1516. [DOI] [PubMed] [Google Scholar]
- 212.Xie W, et al. Loss of Smad signaling in human colorectal cancer is associated with advanced disease and poor prognosis. Cancer J. 2003;9(4):302–312. doi: 10.1097/00130404-200307000-00013. [DOI] [PubMed] [Google Scholar]
- 213.Zhang B, et al. Antimetastatic Role of Smad4 Signaling in Colorectal Cancer. Gastroenterology. 2010;138(3):969–980. e1–3. doi: 10.1053/j.gastro.2009.11.004. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 214.Shanmugam C, et al. Prognostic Value of MUC4 Expression in Colorectal Adenocarcinomas. Cancer. 2010;116(15):3577–3586. doi: 10.1002/cncr.25095. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 215.Kotake K, et al. Noc2, a putative zinc finger protein involved in exocytosis in endocrine cells. J Biol Chem. 1997;272(47):29407–29410. doi: 10.1074/jbc.272.47.29407. [DOI] [PubMed] [Google Scholar]
- 216.Kato M, et al. Physical and functional interaction of rabphilin-3A with alpha-actinin. J Biol Chem. 1996;271(50):31775–31778. doi: 10.1074/jbc.271.50.31775. [DOI] [PubMed] [Google Scholar]
- 217.Shanmugam C, et al. Immunohistochemical expression of rabphilin-3A-like (Noc2) in normal and tumor tissues of human endocrine pancreas. Biotech Histochem. 2009;84(2):39–45. doi: 10.1080/10520290902738878. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 218.Katkoori VR, et al. Rabphillin-3A-Like gene is a candidate tumor suppressor in colorectal adenocarcinoma. Proc Am Assoc Cancer Res. 2007:Abstract # 3650. [Google Scholar]
- 219.Katkoori VR, et al. Down regulation of the Rabphillin-3A-Like gene, located on 17p 13.3, is associated with the metastasis of colorectal adenocarcinoma. Proc Am Assoc Cancer Res. 2008:Abstract # 334. [Google Scholar]
- 220.Goi T, et al. Mutations of rabphillin-3A-like gene in colorectal cancers. Oncol Rep. 2002;9(6):1189–1192. [PubMed] [Google Scholar]
- 221.Katkoori VR, et al. Clinical significance of a novel single nucleotide polymorphism in the 5′ untranslated region of the Rabphillin-3A-Like gene in colorectal adenocarcinoma. Front Biosci. 2008;13:1050–1061. doi: 10.2741/2742. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 222.Faber C, Kirchner T, Hlubek F. The impact of microRNAs on colorectal cancer. Virchows Arch. 2009;454(4):359–367. doi: 10.1007/s00428-009-0751-9. [DOI] [PubMed] [Google Scholar]
- 223.Calin GA, et al. A MicroRNA signature associated with prognosis and progression in chronic lymphocytic leukemia. N Engl J Med. 2005;353(17):1793–1801. doi: 10.1056/NEJMoa050995. [DOI] [PubMed] [Google Scholar]
- 224.Siena S, et al. Biomarkers predicting clinical outcome of epidermal growth factor receptor-targeted therapy in metastatic colorectal cancer. J Natl Cancer Inst. 2009;101(19):1308–1324. doi: 10.1093/jnci/djp280. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 225.Nagel R, et al. Regulation of the adenomatous polyposis coli gene by the miR-135 family in colorectal cancer. Cancer Res. 2008;68(14):5795–5802. doi: 10.1158/0008-5472.CAN-08-0951. [DOI] [PubMed] [Google Scholar]
- 226.Johnson SM, et al. RAS is regulated by the let-7 microRNA family. Cell. 2005;120(5):635–647. doi: 10.1016/j.cell.2005.01.014. [DOI] [PubMed] [Google Scholar]
- 227.Chen X, et al. Role of miR-143 targeting KRAS in colorectal tumorigenesis. Oncogene. 2009;28(10):1385–1392. doi: 10.1038/onc.2008.474. [DOI] [PubMed] [Google Scholar]
- 228.Guo C, et al. The noncoding RNA, miR-126, suppresses the growth of neoplastic cells by targeting phosphatidylinositol 3-kinase signaling and is frequently lost in colon cancers. Genes Chromosomes Cancer. 2008;47(11):939–946. doi: 10.1002/gcc.20596. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 229.Krichevsky AM, Gabriely G. miR-21: a small multi-faceted RNA. J Cell Mol Med. 2009;13(1):39–53. doi: 10.1111/j.1582-4934.2008.00556.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 230.Schetter AJ, et al. MicroRNA expression profiles associated with prognosis and therapeutic outcome in colon adenocarcinoma. J Am Med Assoc. 2008;299(4):425–436. doi: 10.1001/jama.299.4.425. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 231.Slaby O, et al. Altered expression of miR-21, miR-31, miR-143 and miR-145 is related to clinicopathologic features of colorectal cancer. Oncology. 2007;72(5–6):397–402. doi: 10.1159/000113489. [DOI] [PubMed] [Google Scholar]
- 232.Hermeking H. p53 enters the microRNA world. Cancer Cell. 2007;12(5):414–418. doi: 10.1016/j.ccr.2007.10.028. [DOI] [PubMed] [Google Scholar]
- 233.Chang TC, et al. Transactivation of miR-34a by p53 broadly influences gene expression and promotes apoptosis. Mol Cell. 2007;26(5):745–752. doi: 10.1016/j.molcel.2007.05.010. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 234.Gabriely G, et al. MicroRNA 21 promotes glioma invasion by targeting matrix metalloproteinase regulators. Mol Cell Biol. 2008;28(17):5369–5380. doi: 10.1128/MCB.00479-08. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 235.Asangani IA, et al. MicroRNA-21 (miR-21) post-transcriptionally downregulates tumor suppressor Pdcd4 and stimulates invasion, intravasation and metastasis in colorectal cancer. Oncogene. 2008;27(15):2128–2136. doi: 10.1038/sj.onc.1210856. [DOI] [PubMed] [Google Scholar]
- 236.Dews M, et al. Augmentation of tumor angiogenesis by a Myc-activated microRNA cluster. Nat Genet. 2006;38(9):1060–1065. doi: 10.1038/ng1855. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 237.Strillacci A, et al. MiR-101 downregulation is involved in cyclooxygenase-2 overexpression in human colon cancer cells. Exp Cell Res. 2009;315(8):1439–1447. doi: 10.1016/j.yexcr.2008.12.010. [DOI] [PubMed] [Google Scholar]
- 238.Chen K, et al. Polymorphisms in microRNA targets: a gold mine for molecular epidemiology. Carcinogenesis. 2008;29(7):1306–1311. doi: 10.1093/carcin/bgn116. [DOI] [PubMed] [Google Scholar]
- 239.Landi D, et al. Polymorphisms within micro-RNA-binding sites and risk of sporadic colorectal cancer. Carcinogenesis. 2008;29(3):579–584. doi: 10.1093/carcin/bgm304. [DOI] [PubMed] [Google Scholar]
- 240.Chen X, et al. Characterization of microRNAs in serum: a novel class of biomarkers for diagnosis of cancer and other diseases. Cell Res. 2008;18(10):997–1006. doi: 10.1038/cr.2008.282. [DOI] [PubMed] [Google Scholar]
- 241.Xi Y, et al. Prognostic Values of microRNAs in Colorectal Cancer. Biomark Insights. 2006;2:113–121. [PMC free article] [PubMed] [Google Scholar]
- 242.Bovell L, et al. MicroRNA profiles of colorectal adenocarcinomas: Racial disparity. Proc Am Assoc Cancer Res. 2009:Abstract # 549. [Google Scholar]
- 243.Schepeler T, et al. Diagnostic and prognostic microRNAs in stage II colon cancer. Cancer Res. 2008;68(15):6416–6424. doi: 10.1158/0008-5472.CAN-07-6110. [DOI] [PubMed] [Google Scholar]
- 244.Moertel CG, et al. Fluorouracil plus levamisole as effective adjuvant therapy after resection of stage III colon carcinoma: a final report. Ann Intern Med. 1995;122(5):321–326. doi: 10.7326/0003-4819-122-5-199503010-00001. [DOI] [PubMed] [Google Scholar]
- 245.Saltz LB, et al. Irinotecan plus fluorouracil and leucovorin for metastatic colorectal cancer. Irinotecan Study Group [see comments] New Eng J Med. 2000;343(13):905–914. doi: 10.1056/NEJM200009283431302. [DOI] [PubMed] [Google Scholar]
- 246.Douillard JY, et al. Irinotecan combined with fluorouracil compared with fluorouracil alone as first-line treatment for metastatic colorectal cancer: a multicentre randomised trial. Lancet. 2000;355(9209):1041–1047. doi: 10.1016/s0140-6736(00)02034-1. [DOI] [PubMed] [Google Scholar]
- 247.de Gramont A, et al. Oxaliplatin with high-dose leucovorin and 5-fluorouracil 48-hour continuous infusion in pretreated metastatic colorectal cancer. Eur J Cancer. 1997;33(2):214–219. doi: 10.1016/s0959-8049(96)00370-x. [DOI] [PubMed] [Google Scholar]
- 248.Andre T, et al. Current issues in adjuvant treatment of stage II colon cancer. Ann Surg Oncol. 2006;13(6):887–898. doi: 10.1245/ASO.2006.07.003. [DOI] [PubMed] [Google Scholar]
- 249.Andre T, et al. Adjuvant treatment of colon cancer MOSAIC study’s main results. Bull Cancer. 2006;93(Suppl 1):S5–S9. [PubMed] [Google Scholar]
- 250.Sun W, Haller DG. Adjuvant therapy for colon cancer. Curr Oncol Rep. 2005;7(3):181–185. doi: 10.1007/s11912-005-0071-4. [DOI] [PubMed] [Google Scholar]
- 251.Enker WE, et al. Abdominoperineal resection via total mesorectal excision and autonomic nerve preservation for low rectal cancer. World J Surg. 1997;21(7):715–720. doi: 10.1007/s002689900296. [DOI] [PubMed] [Google Scholar]
- 252.Martling AL, et al. Effect of a surgical training programme on outcome of rectal cancer in the County of Stockholm. Stockholm Colorectal Cancer Study Group, Basingstoke Bowel Cancer Research Project. Lancet. 2000;356(9224):93–96. doi: 10.1016/s0140-6736(00)02469-7. [DOI] [PubMed] [Google Scholar]
- 253.Martling A, et al. The Stockholm II trial on preoperative radiotherapy in rectal carcinoma: long-term follow-up of a population-based study. Cancer. 2001;92(4):896–902. doi: 10.1002/1097-0142(20010815)92:4<896::aid-cncr1398>3.0.co;2-r. [DOI] [PubMed] [Google Scholar]
- 254.Martling A, et al. The surgeon as a prognostic factor after the introduction of total mesorectal excision in the treatment of rectal cancer. Br J Surg. 2002;89(8):1008–1013. doi: 10.1046/j.1365-2168.2002.02151.x. [DOI] [PubMed] [Google Scholar]
- 255.Kapiteijn E, et al. Preoperative radiotherapy combined with total mesorectal excision for resectable rectal cancer. N Engl J Med. 2001;345(9):638–646. doi: 10.1056/NEJMoa010580. [DOI] [PubMed] [Google Scholar]
- 256.Minsky BD, et al. Preoperative 5-FU, low-dose leucovorin, and radiation therapy for locally advanced and unresectable rectal cancer. Int J Radiat Oncol Biol Phys. 1997;37(2):289–295. doi: 10.1016/s0360-3016(96)00487-7. [DOI] [PubMed] [Google Scholar]
- 257.Tepper JE, et al. Adjuvant postoperative fluorouracil-modulated chemotherapy combined with pelvic radiation therapy for rectal cancer: initial results of intergroup 0114. J Clin Oncol. 1997;15(5):2030–2039. doi: 10.1200/JCO.1997.15.5.2030. [DOI] [PubMed] [Google Scholar]
- 258.Crane CH, et al. Response to preoperative chemoradiation increases the use of sphincter-preserving surgery in patients with locally advanced low rectal carcinoma. Cancer. 2003;97(2):517–524. doi: 10.1002/cncr.11075. [DOI] [PubMed] [Google Scholar]
- 259.Crane CH, et al. The addition of continuous infusion 5-FU to preoperative radiation therapy increases tumor response, leading to increased sphincter preservation in locally advanced rectal cancer. Int J Radiat Oncol Biol Phys. 2003;57(1):84–89. doi: 10.1016/s0360-3016(03)00532-7. [DOI] [PubMed] [Google Scholar]
- 260.Gill S, Goldberg RM. New developments in therapy for colorectal cancer. Curr Oncol Rep. 2003;5(3):183–191. doi: 10.1007/s11912-003-0109-4. [DOI] [PubMed] [Google Scholar]
- 261.Gill S, Thomas RR, Goldberg RM. New targeted therapies in gastrointestinal cancers. Curr Treat Options Oncol. 2003;4(5):393–403. doi: 10.1007/s11864-003-0040-9. [DOI] [PubMed] [Google Scholar]
- 262.Gill S, Thomas RR, Goldberg RM. Colorectal cancer chemotherapy. Aliment Pharmacol Ther. 2003;18(7):683–692. doi: 10.1046/j.1365-2036.2003.01735.x. [DOI] [PubMed] [Google Scholar]
- 263.Rowinsky EK, Windle JJ, Von Hoff DD. Ras protein farnesyltransferase: A strategic target for anticancer therapeutic development. J Clin Oncol. 1999;17(11):3631–3652. doi: 10.1200/JCO.1999.17.11.3631. [DOI] [PubMed] [Google Scholar]
- 264.Hamid O, et al. Phase II trial of intravenous CI-1042 in patients with metastatic colorectal cancer. J Clin Oncol. 2003;21(8):1498–1504. doi: 10.1200/JCO.2003.09.114. [DOI] [PubMed] [Google Scholar]
- 265.Midgley RS, Kerr DJ. Immunotherapy for colorectal cancer. Expert Rev Anticancer Ther. 2003;3(1):63–78. doi: 10.1586/14737140.3.1.63. [DOI] [PubMed] [Google Scholar]
- 266.Kerr D. Clinical development of gene therapy for colorectal cancer. Nat Rev Cancer. 2003;3(8):615–622. doi: 10.1038/nrc1147. [DOI] [PubMed] [Google Scholar]
- 267.Nicum S, Midgley R, Kerr DJ. Colorectal cancer. Acta Oncol. 2003;42(4):263–275. doi: 10.1080/02841860310000412. [DOI] [PubMed] [Google Scholar]
- 268.Elsaleh H, et al. Association of tumour site and sex with survival benefit from adjuvant chemotherapy in colorectal cancer. Lancet. 2000;355(9217):1745–1750. doi: 10.1016/S0140-6736(00)02261-3. [DOI] [PubMed] [Google Scholar]
- 269.Stoehlmacher J, et al. Association between glutathione S-transferase P1, T1, and M1 genetic polymorphism and survival of patients with metastatic colorectal cancer. J Natl Cancer Inst. 2002;94(12):936–942. doi: 10.1093/jnci/94.12.936. [DOI] [PubMed] [Google Scholar]
- 270.Ribic CM, et al. Tumor microsatellite-instability status as a predictor of benefit from fluorouracil-based adjuvant chemotherapy for colon cancer. N Engl J Med. 2003;349(3):247–257. doi: 10.1056/NEJMoa022289. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 271.Midgley R, Kerr DJ. Adjuvant chemotherapy for stage II colorectal cancer: the time is right! Nat Clin Pract Oncol. 2005;2(7):364–369. doi: 10.1038/ncponc0228. [DOI] [PubMed] [Google Scholar]
- 272.Lowe SW, et al. p53-dependent apoptosis modulates the cytotoxicity of anticancer agents. Cell. 1993;74(6):957–967. doi: 10.1016/0092-8674(93)90719-7. [DOI] [PubMed] [Google Scholar]
- 273.McCurrach ME, et al. bax-deficiency promotes drug resistance and oncogenic transformation by attenuating p53-dependent apoptosis. Proc Natl Acad Sci U S A. 1997;94(6):2345–2349. doi: 10.1073/pnas.94.6.2345. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 274.Sturm I, et al. Analysis of the p53/BAX pathway in colorectal cancer: low BAX is a negative prognostic factor in patients with resected liver metastases. J Clin Oncol. 1999;17(5):1364–1374. doi: 10.1200/JCO.1999.17.5.1364. [DOI] [PubMed] [Google Scholar]
- 275.Manne U, et al. Nuclear accumulation of p53 in colorectal adenocarcinoma: prognostic importance differs with race and location of the tumor. Cancer. 1998;83:2456–2467. doi: 10.1002/(sici)1097-0142(19981215)83:12<2456::aid-cncr8>3.0.co;2-5. [DOI] [PubMed] [Google Scholar]
- 276.Katkoori V, et al. Mutations in the L3 loop of zinc-binding domain of p53 are poor prognostic indicators of Caucasians with colorectal adenocarcinomas. In. American Association of Cancer Research. 2005:Abstract #2548. [Google Scholar]
- 277.Manne U, et al. Prognostic significance of Bcl-2 expression and p53 nuclear accumulation in colorectal adenocarcinoma. Int J Cancer. 1997;74(3):346–358. doi: 10.1002/(sici)1097-0215(19970620)74:3<346::aid-ijc19>3.0.co;2-9. [DOI] [PubMed] [Google Scholar]
- 278.Manne U, Weiss HL, Grizzle WE. Bcl-2 expression is associated with improved prognosis in patients with distal colorectal adenocarcinomas. Int J Cancer. 2000;89(5):423–430. doi: 10.1002/1097-0215(20000920)89:5<423::aid-ijc5>3.0.co;2-t. [DOI] [PubMed] [Google Scholar]
- 279.Suarez-Cuervo C, et al. Predictive value of molecular markers in clinical response of colorectal carcinoma to 5-FU treatment. Proc Am Assoc Cancer Res. 2004:Abstract # 1145. [Google Scholar]
- 280.Manne U, et al. Phenotypic expression of Bax is a predictive marker of 5-fluorouracil treatment in colorectal cancer. Proc Am Soc Clin Oncol. 2006:Abstract # 3605. [Google Scholar]
- 281.Manne U, et al. Altered subcellular localization of suppressin, a novel inhibitor of cell-cycle entry, is an independent prognostic factor in colorectal adenocarcinomas. Clin Cancer Res. 2001;7(11):3495–3503. [PubMed] [Google Scholar]
- 282.Manne U, et al. p53 nuclear accumulation (p53NAC), Bcl-2 expression and clinicopathological characteristics of primary colorectal adenocarcinomas (CAC) with microsatellite instability (MSI) Proc Am Assoc Cancer Res. 1997:Abstract #1003. [Google Scholar]
- 283.Gandham M, et al. Analysis of microsatellite instability in colorectal adenocarcinomas of African-American and Caucasian patients. Proc Am Assoc Cancer Res. 2004:Abstract # 918. [Google Scholar]
- 284.Katkoori V, et al. A novel single nucleotide polymorphism in the 5′untranslated region of the Rabphillin-3A-like gene is associated with nodal metastasis, early recurrence and poor survival of patients with colorectal adenocarcinomas. Cancer Res. (in revision) [Google Scholar]
- 285.Katkoori V, et al. A mutation in the 5′UTR of Rabphillin-3A-Like (RPH3AL) gene is associated with nodal metastasis in colorectal adenocarcinoma. Proc Am Assoc Cancer Res. 2005:Abstract # 2816. [Google Scholar]
- 286.Katkoori V, et al. A novel single nucleotide polymorphism in the 5′-untranslated region of the Rabphillin-3A-like gene is associated with nodal metastasis, early recurrence and poor survival of patients with colorectal adenocarcinoma. Proc Am Assoc Cancer Res. 2006:Abstract # 5728. [Google Scholar]
- 287.Ahnen DJ, et al. Kiras mutation and p53 overexpression predict the clinical behavior of colorectal cancer: a Southwest Oncology Group study. Cancer Res. 1998;58(6):1149–1158. [PubMed] [Google Scholar]
- 288.Elsaleh H, et al. P53 alteration and microsatellite instability have predictive value for survival benefit from chemotherapy in stage III colorectal carcinoma. Clin Cancer Res. 2001;7(5):1343–1349. [PubMed] [Google Scholar]
- 289.Sinicrope FA, et al. Reduced expression of cyclooxygenase 2 proteins in hereditary nonpolyposis colorectal cancers relative to sporadic cancers. Gastroenterology. 1999;117(2):350–358. doi: 10.1053/gast.1999.0029900350. [DOI] [PubMed] [Google Scholar]
- 290.Sinicrope FA, et al. Prognostic value of bcl-2 oncoprotein expression in stage II colon carcinoma. Clin Cancer Res. 1995;1(10):1103–1110. [PubMed] [Google Scholar]
- 291.McLeod HL, Murray GI. Tumour markers of prognosis in colorectal cancer. Br J Cancer. 1999;79(2):191–203. doi: 10.1038/sj.bjc.6690033. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 292.Paradiso A, et al. Expression of apoptosis-related markers and clinical outcome in patients with advanced colorectal cancer. Br J Cancer. 2001;84(5):651–658. doi: 10.1054/bjoc.2000.1658. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 293.Oltvai ZN, Milliman CL, Korsmeyer SJ. Bcl-2 heterodimerizes in vivo with a conserved homolog, Bax, that accelerates programmed cell death. Cell. 1993;74(4):609–619. doi: 10.1016/0092-8674(93)90509-o. [DOI] [PubMed] [Google Scholar]
- 294.Evertsson S, et al. Apoptosis in relation to proliferating cell nuclear antigen and Dukes’ stage in colorectal adenocarcinoma. Int J Oncol. 1999;15(1):53–58. doi: 10.3892/ijo.15.1.53. [DOI] [PubMed] [Google Scholar]
- 295.Zhang L, et al. Role of BAX in the apoptotic response to anticancer agents. Science. 2000;290(5493):989–992. doi: 10.1126/science.290.5493.989. [DOI] [PubMed] [Google Scholar]
- 296.Xiong Y, et al. p21 is a universal inhibitor of cyclin kinases [see comments] Nature. 1993;366(6456):701–704. doi: 10.1038/366701a0. [DOI] [PubMed] [Google Scholar]
- 297.Harper JW, et al. The p21 Cdk-interacting protein Cip1 is a potent inhibitor of G1 cyclin-dependent kinases. Cell. 1993;75(4):805–816. doi: 10.1016/0092-8674(93)90499-g. [DOI] [PubMed] [Google Scholar]
- 298.el-Deiry WS, et al. WAF1/CIP1 is induced in p53-mediated G1 arrest and apoptosis. Cancer Res. 1994;54(5):1169–1174. [PubMed] [Google Scholar]
- 299.Waldman T, Kinzler KW, Vogelstein B. p21 is necessary for the p53-mediated G1 arrest in human cancer cells. Cancer Res. 1995;55(22):5187–5190. [PubMed] [Google Scholar]
- 300.Suzuki H, Matsumoto K, Terabe M. Ki-67 antibody labeling index in colorectal carcinoma. J Clin Gastroenterol. 1992;15(4):317–320. doi: 10.1097/00004836-199212000-00010. [DOI] [PubMed] [Google Scholar]
- 301.Zirbes TK, et al. Prognostic impact of p21/waf1/cip1 in colorectal cancer. Int J Cancer. 2000;89(1):14–18. doi: 10.1002/(sici)1097-0215(20000120)89:1<14::aid-ijc3>3.0.co;2-l. [DOI] [PubMed] [Google Scholar]
- 302.Slebos RJ, et al. Clinical and pathological associations with p53 tumour-suppressor gene mutations and expression of p21WAF1/Cip1 in colorectal carcinoma. Br J Cancer. 1996;74(2):165–171. doi: 10.1038/bjc.1996.333. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 303.Han EK, et al. Increased expression of cyclin D1 in a murine mammary epithelial cell line induces p27Kip1, inhibits growth, and enhances apoptosis. Cell Growth Different. 1996;7(6):699–710. [PubMed] [Google Scholar]
- 304.Uchida H, et al. 5-Fluorouracil efficiently enhanced apoptosis induced by adenovirus-mediated transfer of caspase-8 in DLD-1 colon cancer cells. J Gene Med. 2003;5(4):287–299. doi: 10.1002/jgm.339. [DOI] [PubMed] [Google Scholar]
- 305.Manne U, et al. Prognostic significance of p27kip–1 expression in colorectal adenocarcinomas is associated with tumor stage. Clin Cancer Res. doi: 10.1158/1078-0432.ccr-03-0037. In press. [DOI] [PubMed] [Google Scholar]
- 306.Tenjo T, et al. Prognostic significance of p27Kip1 protein expression and spontaneous apoptosis in patients with colorectal adenocarcinomas. Oncology. 2000;58(1):45–51. doi: 10.1159/000012078. [DOI] [PubMed] [Google Scholar]
- 307.Liu DF, et al. p27 cell-cycle inhibitor is inversely correlated with lymph node metastases in right-sided colon cancer. J Clin Lab Analysis. 1999;13(6):291–295. doi: 10.1002/(SICI)1098-2825(1999)13:6<291::AID-JCLA7>3.0.CO;2-K. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 308.Buglioni S, et al. Evaluation of multiple bio-pathological factors in colorectal adenocarcinomas: independent prognostic role of p53 and bcl-2. Int J Cancer. 1999;84(6):545–552. doi: 10.1002/(sici)1097-0215(19991222)84:6<545::aid-ijc1>3.0.co;2-2. [DOI] [PubMed] [Google Scholar]
- 309.Farrugia DC, et al. Thymidylate synthase expression in advanced colorectal cancer predicts for response to raltitrexed. Clin Cancer Res. 2003;9(2):792–801. [PubMed] [Google Scholar]
- 310.Metzger R, et al. High basal level gene expression of thymidine phosphorylase (platelet-derived endothelial cell growth factor) in colorectal tumors is associated with nonresponse to 5-fluorouacil. Clin Cancer Res. 1998;4(10):2371–2376. [PubMed] [Google Scholar]
- 311.Paradiso A, et al. Thymidilate synthase and p53 primary tumour expression as predictive factors for advanced colorectal cancer patients. Br J Cancer. 2000;82(3):560–567. doi: 10.1054/bjoc.1999.0964. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 312.Johnston PG, et al. The role of thymidylate synthase expression in prognosis and outcome of adjuvant chemotherapy in patients with rectal cancer. J Clin Oncol. 1994;12(12):2640–2647. doi: 10.1200/JCO.1994.12.12.2640. [DOI] [PubMed] [Google Scholar]
- 313.Lenz HJ, et al. p53 and thymidylate synthase expression in untreated stage II colon cancer: associations with recurrence, survival, and site. Clin Cancer Res. 1998;4:1227–1234. [PubMed] [Google Scholar]
- 314.Lincoln DW, Hrushesky WJ, 2nd, Wood PA. Circadian organization of thymidylate synthase activity in normal tissues: a possible basis for 5-fluorouracil chronotherapeutic advantage. Int J Cancer. 2000;88(3):479–485. [PubMed] [Google Scholar]
- 315.Salonga D, et al. Colorectal tumors responding to 5-Fluorouracil have low gene expression levels of dihydropyrimidine dehydrogenase, thymidylate synthase, and thymidine phosphorylase. Clin Cancer Res. 2000;6:1322–1327. [PubMed] [Google Scholar]
- 316.Diasio RB, Harris BE. Clinical pharmacology of 5-fluorouracil. Clin Pharmacokinet. 1989;16(4):215–237. doi: 10.2165/00003088-198916040-00002. [DOI] [PubMed] [Google Scholar]
- 317.Johnson MR, et al. Life-threatening toxicity in a dihydropyrimidine dehydrogenase-deficient patient after treatment with topical 5-fluorouracil. Clin Cancer Res. 1999;5(8):2006–2011. [PubMed] [Google Scholar]
- 318.Yoshinare K, et al. Gene expression in colorectal cancer and in vitro chemosensitivity to 5-fluorouracil: A study of 88 surgical specimens. Cancer Sci. 2003;94(7):633–638. doi: 10.1111/j.1349-7006.2003.tb01495.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 319.Yamashita K, et al. Gender differences in the dihydropyrimidine dehydrogenase expression of colorectal cancers. Cancer Lett. 2002;188(1–2):231–236. doi: 10.1016/s0304-3835(02)00435-4. [DOI] [PubMed] [Google Scholar]
- 320.Khalil MY, Grandis JR, Shin DM. Targeting epidermal growth factor receptor: novel therapeutics in the management of cancer. Expert Rev Anticancer Ther. 2003;3(3):367–380. doi: 10.1586/14737140.3.3.367. [DOI] [PubMed] [Google Scholar]
- 321.Herbst RS, Shin DM. Monoclonal antibodies to target epidermal growth factor receptor-positive tumors: a new paradigm for cancer therapy. Cancer. 2002;94(5):1593–1611. doi: 10.1002/cncr.10372. [DOI] [PubMed] [Google Scholar]
- 322.Shin DM, et al. Epidermal growth factor receptor-targeted therapy with C225 and cisplatin in patients with head and neck cancer. Clin Cancer Res. 2001;7(5):1204–1213. [PubMed] [Google Scholar]
- 323.Lango MN, Shin DM, Grandis JR. Targeting growth factor receptors: integration of novel therapeutics in the management of head and neck cancer. Curr Opin Oncol. 2001;13(3):168–175. doi: 10.1097/00001622-200105000-00007. [DOI] [PubMed] [Google Scholar]
- 324.Cascinu S, et al. Expression of vascular endothelial growth factor can predict event-free survival in stage II colon cancer. Clin Cancer Res. 2000;6(7):2803–2807. [PubMed] [Google Scholar]
- 325.Kabbinavar F, et al. Phase II, randomized trial comparing bevacizumab plus fluorouracil (FU)/leucovorin (LV) with FU/LV alone in patients with metastatic colorectal cancer. J Clin Oncol. 2003;21(1):60–65. doi: 10.1200/JCO.2003.10.066. [DOI] [PubMed] [Google Scholar]
- 326.Aoki R, et al. MUC-1 expression as a predictor of the curative endoscopic treatment of submucosally invasive colorectal carcinoma. Dis Colon Rectum. 1998;41(10):1262–1272. doi: 10.1007/BF02258227. [DOI] [PubMed] [Google Scholar]
- 327.Karanikas V, et al. Mannan Mucin-1 Peptide Immunization: Influence of Cyclophosphamide and the Route of Injection. J Immunother. 2001;24(2):172–183. [PubMed] [Google Scholar]
- 328.Stintzing S, et al. The treatment of colorectal carcinoma with monoclonal antibodies: the importance of KRAS mutation analysis and EGFR status. Dtsch Arztebl Int. 2009;106(12):202–206. doi: 10.3238/arztebl.2009.0202. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 329.Di Nicolantonio F, et al. Wild-type BRAF is required for response to panitumumab or cetuximab in metastatic colorectal cancer. J Clin Oncol. 2008;26(35):5705–5712. doi: 10.1200/JCO.2008.18.0786. [DOI] [PubMed] [Google Scholar]
- 330.Zlobec I, et al. Prognostic and predictive value of TOPK stratified by KRAS and BRAF gene alterations in sporadic, hereditary and metastatic colorectal cancer patients. Br J Cancer. 102(1):151–161. doi: 10.1038/sj.bjc.6605452. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 331.Jass JR, et al. Characterization of a subtype of colorectal cancer combining features of the suppressor and mild mutator pathways. J Clin Pathol. 1999;52(6):455–460. doi: 10.1136/jcp.52.6.455. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 332.Mori Y, et al. The impact of microsatellite instability on the molecular phenotype of colorectal tumors. Cancer Res. 2003;63(15):4577–4582. [PubMed] [Google Scholar]
- 333.Kim GP, et al. Predictive value of microsatellite instability-high remains controversial. J Clin Oncol. 2007;25(30):4857. 4857–4858. doi: 10.1200/JCO.2007.13.2019. author reply. [DOI] [PubMed] [Google Scholar]
- 334.Kim GP, et al. Prognostic and predictive roles of high-degree microsatellite instability in colon cancer: a National Cancer Institute-National Surgical Adjuvant Breast and Bowel Project Collaborative Study. J Clin Oncol. 2007;25(7):767–772. doi: 10.1200/JCO.2006.05.8172. [DOI] [PubMed] [Google Scholar]
- 335.Gruber SB, et al. Genetic variation in 8q24 associated with risk of colorectal cancer. Cancer Biol Ther. 2007;6(7):1143–1147. doi: 10.4161/cbt.6.7.4704. [DOI] [PubMed] [Google Scholar]
- 336.Wijnen JT, et al. Chromosome 8q23.3 and 11q23.1 variants modify colorectal cancer risk in Lynch syndrome. Gastroenterology. 2009;136(1):131–137. doi: 10.1053/j.gastro.2008.09.033. [DOI] [PubMed] [Google Scholar]
- 337.Nakajima G, et al. Non-coding MicroRNAs hsa-let-7g and hsa-miR-181b are Associated with Chemoresponse to S-1 in Colon Cancer. Cancer Genomics Proteomics. 2006;3(5):317–324. [PMC free article] [PubMed] [Google Scholar]
- 338.Marsoni S. International Multicenter Pooled Analysis of Colon Cancer Trials Investigators. Efficacy of adjuvant fluorouracil and leucovorin in Stage B2 and C colon cancer. Semin Oncol 2001. 2001;28(1):14–19. doi: 10.1053/sonc.2001.19723. [DOI] [PubMed] [Google Scholar]
- 339.el-Deiry WS, et al. WAF1, a potential mediator of p53 tumor suppression. Cell. 1993;75(4):817–825. doi: 10.1016/0092-8674(93)90500-p. [DOI] [PubMed] [Google Scholar]
- 340.Zirbes TK, et al. Prognostic impact of p21/waf1/cip1 in colorectal cancer. Int J Cancer. 2000;89(1):14–18. doi: 10.1002/(sici)1097-0215(20000120)89:1<14::aid-ijc3>3.0.co;2-l. [DOI] [PubMed] [Google Scholar]
- 341.Sherr CJ. Mammalian G1 cyclins. Cell. 1993;73(6):1059–1065. doi: 10.1016/0092-8674(93)90636-5. [DOI] [PubMed] [Google Scholar]
- 342.Zuo L, et al. Germline mutations in the p16INK4a binding domain of CDK4 in familial melanoma. Nat Genet. 1996;12(1):97–99. doi: 10.1038/ng0196-97. [DOI] [PubMed] [Google Scholar]
- 343.Fornace AJ., Jr Mammalian genes induced by radiation; activation of genes associated with growth control. Annu Rev Genet. 1992;26:507–526. doi: 10.1146/annurev.ge.26.120192.002451. [DOI] [PubMed] [Google Scholar]
- 344.Kastan MB, et al. A mammalian cell cycle checkpoint pathway utilizing p53 and GADD45 is defective in ataxia-telangiectasia. Cell. 1992;71(4):587–597. doi: 10.1016/0092-8674(92)90593-2. [DOI] [PubMed] [Google Scholar]
- 345.Fornace AJ, Jr, et al. Genotoxic-stress-response genes and growth-arrest genes. gadd, MyD, and other genes induced by treatments eliciting growth arrest. Ann N Y Acad Sci. 1992;663:139–153. doi: 10.1111/j.1749-6632.1992.tb38657.x. [DOI] [PubMed] [Google Scholar]
- 346.Carrier F, et al. Characterization of human Gadd45, a p53-regulated protein. J Biol Chem. 1994;269(51):32672–36277. [PubMed] [Google Scholar]
- 347.Kearsey JM, et al. Gadd45 is a nuclear cell cycle regulated protein which interacts with p21Cip1. Oncogene. 1995;11(9):1675–1683. [PubMed] [Google Scholar]
- 348.Cox LS, Lane DP. Tumour suppressors, kinases and clamps: how p53 regulates the cell cycle in response to DNA damage. Bioessays. 1995;17(6):501–508. doi: 10.1002/bies.950170606. [DOI] [PubMed] [Google Scholar]
- 349.Wang XW, et al. GADD45 induction of a G2/M cell cycle checkpoint. Proc Natl Acad Sci U S A. 1999;96(7):3706–3711. doi: 10.1073/pnas.96.7.3706. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 350.Campomenosi P, Hall PA. Gadd45 mutations are uncommon in human tumour cell lines. Cell Prolif. 2000;33(5):301–306. doi: 10.1046/j.1365-2184.2000.00187.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 351.Sensi E, et al. Clinicopathological Significance of GADD45 Gene Alterations in Human Familial Breast Carcinoma. Breast Cancer Res Treat. 2004;87(2):197–201. doi: 10.1023/B:BREA.0000041625.60280.4a. [DOI] [PubMed] [Google Scholar]
- 352.Yamasawa K, et al. Clinicopathological significance of abnormalities in Gadd45 expression and its relationship to p53 in human pancreatic cancer. Clin Cancer Res. 2002;8(8):2563–2569. [PubMed] [Google Scholar]
- 353.Contente A, et al. A polymorphic microsatellite that mediates induction of PIG3 by p53. Nat Genet. 2002;30(3):315–320. doi: 10.1038/ng836. [DOI] [PubMed] [Google Scholar]
- 354.Polyak K, et al. A model for p53-induced apoptosis. Nature. 1997;389(6648):300–305. doi: 10.1038/38525. [DOI] [PubMed] [Google Scholar]
- 355.Wozney JM, et al. Novel regulators of bone formation: molecular clones and activities. Science. 1988;242(4885):1528–1534. doi: 10.1126/science.3201241. [DOI] [PubMed] [Google Scholar]
- 356.Montesano R, Sarkozi R, Schramek H. Bone morphogenetic protein-4 strongly potentiates growth factor-induced proliferation of mammary epithelial cells. Biochem Biophys Res Commun. 2008;374(1):164–168. doi: 10.1016/j.bbrc.2008.07.007. [DOI] [PubMed] [Google Scholar]
- 357.Shepherd TG, Theriault BL, Nachtigal MW. Autocrine BMP4 signalling regulates ID3 protooncogene expression in human ovarian cancer cells. Gene. 2008;414(1–2):95–105. doi: 10.1016/j.gene.2008.02.015. [DOI] [PubMed] [Google Scholar]
- 358.Ogata S, et al. TGF-beta signaling-mediated morphogenesis: modulation of cell adhesion via cadherin endocytosis. Genes Dev. 2007;21(14):1817–1831. doi: 10.1101/gad.1541807. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 359.Haines BP, et al. Regulated expression of FLRT genes implies a functional role in the regulation of FGF signalling during mouse development. Dev Biol. 2006;297(1):14–25. doi: 10.1016/j.ydbio.2006.04.004. [DOI] [PubMed] [Google Scholar]
- 360.Packer LM, et al. Gene expression profiling in melanoma identifies novel downstream effectors of p14ARF. Int J Cancer. 2007;121(4):784–790. doi: 10.1002/ijc.22725. [DOI] [PubMed] [Google Scholar]
- 361.Frenzel A, et al. Bcl2 family proteins in carcinogenesis and the treatment of cancer. Apoptosis. 2009;14(4):584–596. doi: 10.1007/s10495-008-0300-z. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 362.Miled C, et al. A genomic map of p53 binding sites identifies novel p53 targets involved in an apoptotic network. Cancer Res. 2005;65(12):5096–5104. doi: 10.1158/0008-5472.CAN-04-4232. [DOI] [PubMed] [Google Scholar]
- 363.Inbal B, et al. Death-associated protein kinase-related protein 1, a novel serine/threonine kinase involved in apoptosis. Mol Cell Biol. 2000;20(3):1044–1054. doi: 10.1128/mcb.20.3.1044-1054.2000. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 364.Kawai T, et al. Death-associated protein kinase 2 is a new calcium/calmodulin-dependent protein kinase that signals apoptosis through its catalytic activity. Oncogene. 1999;18(23):3471–3480. doi: 10.1038/sj.onc.1202701. [DOI] [PubMed] [Google Scholar]
- 365.Thirumoorthy N, et al. Metallothionein: an overview. World J Gastroenterol. 2007;13(7):993–996. doi: 10.3748/wjg.v13.i7.993. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 366.Dziegiel P. Expression of metallothioneins in tumor cells. Pol J Pathol. 2004;55(1):3–12. [PubMed] [Google Scholar]
- 367.Jin R, et al. Clinicopathological significance of metallothioneins in breast cancer. Pathol Oncol Res. 2004;10(2):74–79. doi: 10.1007/BF02893459. [DOI] [PubMed] [Google Scholar]
- 368.Wehder L, et al. Annexin A5 is involved in migration and invasion of oral carcinoma. Cell Cycle. 2009;8(10) doi: 10.4161/cc.8.10.8404. [DOI] [PubMed] [Google Scholar]
- 369.Rimkus C, et al. Expression of the mitotic checkpoint gene MAD2L2 has prognostic significance in colon cancer. Int J Cancer. 2007;120(1):207–211. doi: 10.1002/ijc.22155. [DOI] [PubMed] [Google Scholar]
- 370.Pinto M, et al. Expression changes of the MAD mitotic checkpoint gene family in renal cell carcinomas characterized by numerical chromosome changes. Virchows Arch. 2007;450(4):379–385. doi: 10.1007/s00428-007-0386-7. [DOI] [PubMed] [Google Scholar]
- 371.McMahan CJ, et al. A novel IL-1 receptor, cloned from B cells by mammalian expression, is expressed in many cell types. Embo J. 1991;10(10):2821–2832. doi: 10.1002/j.1460-2075.1991.tb07831.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 372.Wu J, et al. Translocation and phosphorylation of calcyclin binding protein during retinoic acid-induced neuronal differentiation of neuroblastoma SH-SY5Y cells. J Biochem Mol Biol. 2003;36(4):354–358. doi: 10.5483/bmbrep.2003.36.4.354. [DOI] [PubMed] [Google Scholar]
- 373.Sun S, et al. Overexpressed CacyBP/SIP leads to the suppression of growth in renal cell carcinoma. Biochem Biophys Res Commun. 2007;356(4):864–871. doi: 10.1016/j.bbrc.2007.03.080. [DOI] [PubMed] [Google Scholar]
- 374.Zhai H, et al. Expression of calcyclin-binding protein/Siah-1 interacting protein in normal and malignant human tissues: an immunohistochemical survey. J Histochem Cytochem. 2008;56(8):765–772. doi: 10.1369/jhc.2008.950519. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 375.Hayflick JS, Kilgannon P, Gallatin WM. The inter-cellular adhesion molecule (ICAM) family of proteins. New members and novel functions. Immunol Res. 1998;17(3):313–327. doi: 10.1007/BF02786454. [DOI] [PubMed] [Google Scholar]
- 376.Simmons DL. The role of ICAM expression in immunity and disease. Cancer Surv. 1995;24:141–155. [PubMed] [Google Scholar]
- 377.Brown EJ, Frazier WA. Integrin-associated protein (CD47) and its ligands. Trends Cell Biol. 2001;11(3):130–135. doi: 10.1016/s0962-8924(00)01906-1. [DOI] [PubMed] [Google Scholar]
- 378.Kim MJ, et al. Association of CD47 with natural killer cell-mediated cytotoxicity of head-and-neck squamous cell carcinoma lines. Tumour Biol. 2008;29(1):28–34. doi: 10.1159/000132568. [DOI] [PubMed] [Google Scholar]
- 379.Mateo V, et al. CD47 ligation induces caspase-independent cell death in chronic lymphocytic leukemia. Nat Med. 1999;5(11):1277–1284. doi: 10.1038/15233. [DOI] [PubMed] [Google Scholar]
- 380.Fielding LP, Henson DE. Multiple prognostic factors and outcome analysis in patients with cancer. Communication from the American Joint Committee on Cancer. Cancer. 1993;71(7):2426–2429. doi: 10.1002/1097-0142(19930401)71:7<2426::aid-cncr2820710742>3.0.co;2-l. [DOI] [PubMed] [Google Scholar]
- 381.Simon R, Altman DG. Statistical aspects of prognostic factor studies in oncology. Br J Cancer. 1994;69(6):979–985. doi: 10.1038/bjc.1994.192. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 382.Grizzle WE, Srivastava S, Manne U. The biology of incipient, pre-invasive or intraepithelial neoplasia. Cancer Biomark. 2011;9:21–39. doi: 10.3233/CBM-2011-0172. [DOI] [PMC free article] [PubMed] [Google Scholar]