

Summary
The correct segregation of DNA during cell division requires formation of a bipolar spindle, organized at each pole by a centrosome. The regulation of centrosome duplication such that each mitotic cell has exactly two centrosomes is therefore of central importance to cell division. Deregulation of centrosome duplication causes the appearance of supernumerary centrosomes, which are a hallmark of many cancer cells and can contribute to tumorigenesis. Overexpression of the kinase Plk4, which is required for centrosome duplication, causes the formation of extra centrosomes, and aberrant Plk4 expression levels are associated with cancer. Data from Drosophila and human cells show that Plk4 levels are regulated by the SCF ubiquitin ligase and proteasomal degradation. Recognition of Plk4 by the SCF complex is mediated by the F-box protein Slimb/βTrCP. We show that levels of the C. elegans Plk4 homolog ZYG-1 are elevated by impairing proteasome or SCF function, indicating that ZYG-1 is regulated by a conserved mechanism. In C. elegans, similar to Drosophila and humans, we find that the Slimb/βTrCP homolog LIN-23 regulates ZYG-1 levels. In addition, we show that a second F-box protein, SEL-10, also contributes to ZYG-1 regulation. Co-depletion of LIN-23 and SEL-10 suggests these proteins function cooperatively. Because SEL-10 is the homolog of human FBW7, which is frequently mutated in cancer, our findings have implications for understanding tumorigenesis.
Key words: F-box, ZYG-1, Centrosome
Introduction
The centrosome plays a key role in cell division by organizing the microtubules of the spindle, enabling the correct segregation of chromosomes during mitosis. In normal cells the presence of two centrosomes promotes the formation of a bipolar spindle. In cancer cells, increased numbers of centrosomes are frequently observed. This centrosome amplification is thought to contribute to tumorigenesis in two ways (Nigg and Raff, 2009). First, supernumerary centrosomes often cause the formation of transient multipolar spindle intermediates that are ultimately resolved into a bipolar spindle. Incorrect microtubule-chromosome attachments can however persist, and result in chromosome missegregation and aneuploidy (Ganem et al., 2009). Second, formation of transient multipolar spindle intermediates can perturb polarity cues, and may disrupt stem cell divisions leading to uncontrolled tissue growth (Basto et al., 2008). Therefore, ensuring that mitotic cells have exactly two centrosomes at mitosis is crucial to the fidelity of cell division. Maintenance of appropriate centrosome numbers is ensured by strict control of centrosome duplication.
The centrosome is composed of a pair of centrioles and associated pericentriolar material (PCM). Centrosome duplication proceeds by assembly of a new daughter centriole in close association with each existing centriole, once per cell cycle. The pathway leading to centriole duplication was first elucidated in C. elegans and requires the sequential recruitment of a set of evolutionarily-conserved modulators to the centrosome (Delattre et al., 2006). The most upstream factor is SPD-2, which is required for the recruitment of the kinase ZYG-1 to the centriole. Localization of SAS-5 and SAS-6 follows, and phosphorylation of SAS-6 by ZYG-1 promotes its maintenance at the centriole (Kitagawa et al., 2009). Lastly SAS-4 recruitment is required for addition of the centriolar microtubules to complete centriole duplication (Pelletier et al., 2006). The pathway is largely conserved in other species. Homologs of SPD-2 (Zhu et al., 2008), SAS-6 (Leidel et al., 2005) and SAS-4 (Basto et al., 2006) have been identified through sequence comparisons, and their functions in centrosome duplication have been confirmed. In Drosophila and human cells, the kinase Plk4 plays an equivalent role to ZYG-1 in promoting centrosome duplication (Strnad and Gönczy, 2008), yet their protein sequences lack obvious homology, particularly outside of the kinase domain. It is unclear, therefore, whether ZYG-1 is a true ortholog of Plk4 or is a product of convergent evolution (Carvalho-Santos et al., 2010; Hodges et al., 2010).
One of the early steps in centrosome duplication is recruitment of ZYG-1/Plk4 to the centrosome. Elevation of the levels of Drosophila Plk4 (dPlk4; also known as SAK) or human Plk4 (hPlk4) results in the formation of supernumerary centrosomes owing to dysregulated centrosome duplication (Basto et al., 2008; Habedanck et al., 2005; Kleylein-Sohn et al., 2007; Peel et al., 2007). Aberrant Plk4 expression levels are associated with cancer (Korzeniewski et al., 2011; Torres et al., 2011). It is therefore essential that cellular levels of Plk4 proteins are tightly controlled. Recent work in Drosophila and human cells demonstrated that appropriate levels of both dPlk4 and hPlk4 are maintained by SCF-mediated proteasomal degradation (Cunha-Ferreira et al., 2008; Guderian et al., 2010; Holland et al., 2010; Rogers et al., 2009; Sillibourne et al., 2010). The SCF complex has ubiquitin ligase activity and is composed of three core proteins: Skp1, Cullin 1/3, Roc1/Rbx1; and an interchangeable F-box protein that provides substrate specificity. The SCF complex mediates ubiquitination of many substrates, targeting them for degradation. In Drosophila and human cells the homologous F-box proteins Slimb and βTrCP facilitate recognition of their respective substrates, dPlk4 and hPlk4, by the SCF complex, leading to their subsequent degradation. Although it is clear that βTrCP contributes to Plk4 degradation in human cells, mutation of the βTrCP recognition motif in Plk4 neither prevents its ubiquitination, nor degradation. This evidence suggests that additional, as yet unidentified, factors regulate Plk4 stability in human cells (Holland et al., 2010).
Although the cascade of events that promotes centrosome duplication downstream of ZYG-1 is increasingly well understood, the regulation of ZYG-1 itself has not yet been investigated. Determining the upstream regulation of ZYG-1 will aid our understanding of how centrosome duplication is coupled to the cell cycle and may also provide insight into regulation of human Plk4. Furthermore, C. elegans provides the ideal opportunity to analyse the regulation of centrosome duplication in vivo, allowing us to monitor the regulation of endogenous proteins. The evolutionary origins of ZYG-1 remain controversial, and determining whether its degradation, like that of Plk4, is mediated by the SCF complex, may provide important insights. We therefore undertook a study to determine how the abundance of ZYG-1 is regulated in the C. elegans embryo. We find that ZYG-1 levels are regulated by proteasomal degradation, mediated by the SCF complex. Similar to Drosophila and human cells, the F-box protein LIN-23 (homolog of Slimb/βTrCP) contributes to the regulation of ZYG-1 levels. We additionally present the novel finding that a second F-box protein SEL-10, the C. elegans homolog of FBW7, regulates ZYG-1 levels.
Results
ZYG-1 levels are regulated by proteasomal degradation
It has previously been reported that ZYG-1 levels at the centrosome are dynamic and change in a cell-cycle-regulated manner (Delattre et al., 2006; O'Connell et al., 2001; Song et al., 2008). We first established the normal range of ZYG-1 levels by undertaking a quantitative analysis of changes in ZYG-1 intensity at the centrosome throughout the cell cycle, a well-established method for the analysis of centrosome protein levels (Cunha-Ferreira et al., 2008; Dammermann et al., 2008; Greenan et al., 2010; Guderian et al., 2010). During the first cell cycle in one-cell embryos, ZYG-1 levels increase 3-fold from prophase to anaphase (Fig. 1A). In agreement with previous work, we find ZYG-1 levels at the centrosome increase during mitosis, reaching a maximum in anaphase (O'Connell et al., 2001). To analyze the dynamic changes in ZYG-1 levels we used a GFP reporter in which GFP was fused to the C-terminal (non-kinase) portion of ZYG-1 (hereafter GFP::cZYG-1) (supplementary material Fig. S2) (Peters et al., 2010). The localization and cell cycle-dependent levels of GFP::cZYG-1 correlated with endogenous ZYG-1 (Fig. 1B), remaining low in prophase and increasing at the centrosome during mitosis, to reach maximal levels at anaphase.
Fig. 1.
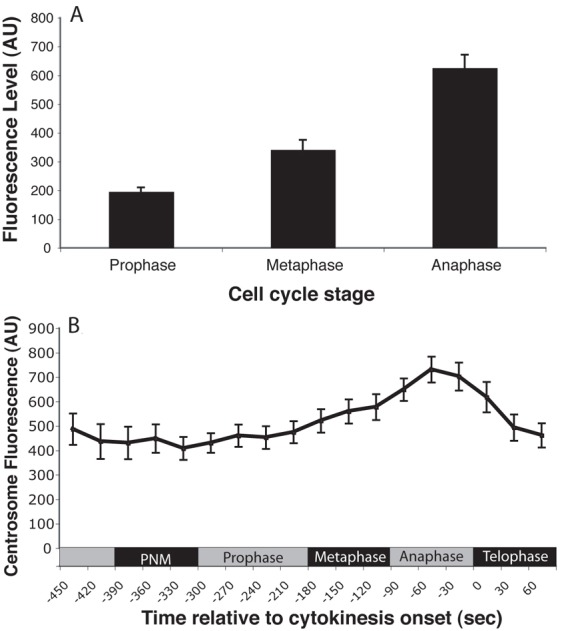
ZYG-1 levels show dynamic changes during the cell cycle. (A) Average levels of endogenous ZYG-1 at the centrosome in the one-cell embryo determined by quantification of fluorescence after immunostaining. Error bars denote s.e.m. (B) Average levels of GFP::cZYG-1 at the centrosome during the first cell cycle. n = 16; PNM, pronuclear meeting. Error bars denote s.e.m. AU, arbitrary units.
In Drosophila and human cells the abundance of the ZYG-1 homologs dPlk4 and hPlk4 is regulated by proteasomal degradation. In C. elegans the function of the proteasome can be disrupted by RNAi depletion of the proteasome component RPT-4 (Song et al., 2011). To test whether ZYG-1 levels are regulated in a similar way in C. elegans, we measured the levels of ZYG-1 at the centrosome following disruption of the proteasome. Because proteasome function is required for the completion of meiosis in C. elegans we calibrated our RNAi condition to partially deplete RPT-4 function, allowing us to avoid meiotic arrest. Our RNAi conditions largely led to an arrest during the first mitotic division. Reduced proteasome function in the rpt-4(RNAi) embryos led to increased levels of ZYG-1 at the centrosome at most stages of the cell cycle compared with controls (Fig. 2A,B). The effect was most pronounced during metaphase, when centrosomal ZYG-1 reached levels similar to those normally seen in anaphase. These results suggest that ZYG-1 is degraded by the proteasome and that degradation is most active during the early stages of mitosis. Centrosome accumulation of ZYG-1 is typically maximal during anaphase and this level is not affected by disruption of the proteasome.
Fig. 2.
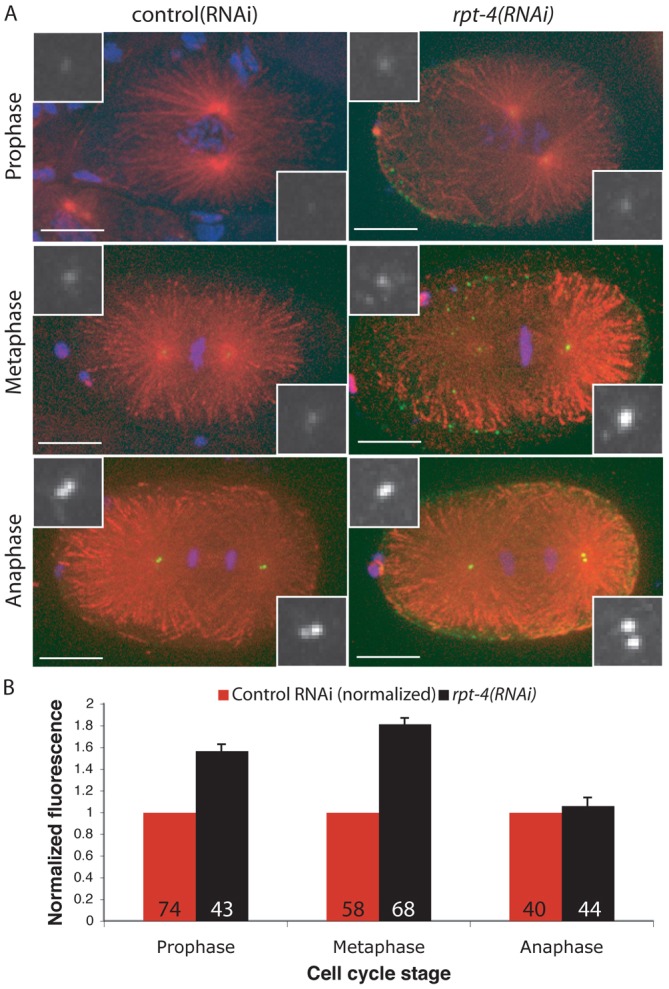
ZYG-1 levels are regulated by proteasomal degradation. (A) Representative control (RNAi) and rpt-4(RNAi) embryos stained for microtubules (red), ZYG-1 (green) and DNA (blue). Insets are magnified images of ZYG-1 at the centrosome. Scale bars: 10 µm. (B) Average levels of endogenous ZYG-1 at the centrosome in the one-cell embryo after RPT-4 depletion were quantitated and normalized to control RNAi, which is set at 1 for each stage. The number of centrosomes analysed is indicated. Error bars denote s.e.m.
SCF components SKR-1/2 and CUL-1 regulate ZYG-1 levels
We next determined which ubiquitin ligase complex is responsible for targeting ZYG-1 for degradation by the proteasome. In both Drosophila and human cells the SCF complex is a key regulator of dPlk4 and hPlk4 levels. We therefore investigated whether the SCF complex is required for ZYG-1 degradation. C. elegans possesses two closely related Skp1 homologs, SKR-1 and SKR-2, which play essential roles in cell division and are required for embryonic viability (Nayak et al., 2002; Yamanaka et al., 2002). These proteins are 83% identical (Yamanaka et al., 2002) and play overlapping roles in SCF function. We used an RNAi construct that depletes both skr-1 and skr-2 (henceforth referred to as skr-1/2) (supplementary material Fig. S1), and separately we carried out RNAi against the cullin homolog CUL-1. To test the effect of the depletion of SCF components on ZYG-1, we used embryos carrying the hypomorphic zyg-1(it25) mutation. These embryos fail to duplicate their centrosomes in the one-cell embryo, leading to the formation of monopolar spindles at the 2-cell stage (Fig. 3A) (Kemp et al., 2007). Importantly, this phenotype can be alleviated by increasing ZYG-1 levels at the centrosome (Song et al., 2008). Thus if the SCF promotes ZYG-1 degradation, perturbing SCF function would be expected to elevate ZYG-1 levels and suppress the zyg-1(it25) phenotype. We found that depletion of SKR-1/2 or CUL-1 restored centrosome duplication in zyg-1(it25) one-cell embryos (Fig. 3B,C), consistent with a role for SCF in regulating ZYG-1 degradation. The level of centrosome duplication was restored to around 30% for both skr-1/2 and cul-1 RNAi embryos (n = 28 and n = 32, respectively) (Fig. 3D). This is a significant increase over control RNAi where no duplication events were recorded (n>50). The majority of embryos underwent duplication of just one centrosome when treated with RNAi to deplete SCF components (Fig. 3B,C), although a smaller proportion showed duplication of both centrosomes. When centrosome duplication occurred we frequently found that one centrosome showed a reduced size, a phenotype previously observed in embryos with a partial block in centrosome duplication (Delattre et al., 2004). Nonetheless, these observations suggest that the SCF normally acts to inhibit centrosome duplication by suppressing levels of ZYG-1.
Fig. 3.

The SCF components SKR-1/2 and CUL-1 regulate ZYG-1. zyg-1(it25); gfp::h2b;gfp::spd-2 embryos were RNAi treated as indicated and grown at 24°C. Images from a series are presented in (A) Control(RNAi), (B) skr-1/2(RNAi) and (C) cul-1(RNAi), with time (in min:sec) relative to the onset of cytokinesis displayed in the bottom right of the panel. Arrowheads indicate centrosomes. Scale bars: 10 µm. (D) Quantification of the percentage of centrosomes that duplicated during the first cell cycle for each RNAi condition. Control RNAi, n = 50; Skr-1/2(RNAi), n = 28; cul-1(RNAi), n = 32. (E) Worms were treated as indicated and representative images of ZYG-1 levels at the centrosome are presented. Centrosomes from two embryos (total four centrosomes) are shown for each cell cycle stage. (F) Average levels of endogenous ZYG-1 at the centrosome in the one-cell embryo after SKR-1/2 RNAi were quantified and normalized to control RNAi. The average normalized fluorescence from multiple experiments is shown, error bars indicate s.e.m. The number of centrosomes analysed is indicated. (G) Average levels of GFP::cZYG-1 at the centrosome during the first cell cycle were calculated and normalized to control. Control, n = 16; skr-1/2(RNAi), n = 16. PNM, pronuclear meeting.
The ability of skr-1/2 and cul-1 RNAi to restore centrosome duplication in zyg-1(it25) embryos suggests that ZYG-1 levels become elevated when SCF function is impaired. We tested this directly by measuring the level of endogenous centrosome-associated ZYG-1 after depletion of SKR-1/2 in one-cell embryos (Fig. 3E,F). As expected, the levels of ZYG-1 were elevated upon depletion of SKR-1/2. As a second measure of ZYG-1 levels, we looked at GFP::cZYG-1 levels after skr-1/2(RNAi) (Fig. 3G). Levels of GFP::cZYG-1 were elevated after SKR-1/2 depletion, again suggesting that the SCF complex promotes ZYG-1 degradation.
The F-box protein LIN-23 regulates ZYG-1 levels
Our results indicated that the SCF complex components SKR-1/2 and CUL-1 regulate ZYG-1 levels. We next investigated which F-box protein is required in the SCF complex for ZYG-1 substrate recognition. In Drosophila and human cells the homologous F-box proteins Slimb/βTrCP are required for recognition of the ZYG-1 homolog, Plk4. We therefore performed RNAi against LIN-23, the C. elegans homolog of Slimb/βTrCP, to investigate its role in regulating ZYG-1 degradation. Depletion of LIN-23 by RNAi failed to rescue the centrosome duplication phenotype of zyg-1(it25) embryos (Fig. 4B, n = 26). We recombined the zyg-1(it25) and lin-23(ot1) alleles onto the same chromosome (see Materials and Methods for details), but found that the lin-23(ot1) hypomorph did not suppress the zyg-1(it25) phenotype (Fig. 4B, n = 20) (Hebeisen and Roy, 2008). However, we found that combining the lin-23(ot1) allele and lin-23(RNAi) produced a significant rescue of the zyg-1(it25) phenotype (Fig. 4A,B, n = 20). Combining a hypomophic allele with RNAi of the same gene is an effective method for maximizing reduction of gene function and our results indicate that substantial depletion of LIN-23 is only achieved by combining the lin-23(ot1) allele and RNAi. Rescue of the zyg-1(it25) phenotype by lin-23(ot1, RNAi) suggested that reducing LIN-23 availability was able to elevate ZYG-1 levels. We directly tested this by quantifying endogenous centrosome-associated ZYG-1. Although we did not see any increase in endogenous ZYG-1 levels at the centrosome in either lin-23(RNAi) embryos, or embryos carrying the hypomorphic lin-23(ot1) allele (Fig. 4D) when the lin-23(ot1) allele and RNAi were combined ZYG-1 levels were substantially elevated over controls (Fig. 4C,D). These results suggest that LIN-23 contributes to ZYG-1 degradation.
Fig. 4.

The F-box protein LIN-23 regulates ZYG-1 levels. (A) zyg-1(it25) lin-23(ot1); mcherry::h2b;gfp::spd-2 embryos were treated with lin-23 RNAi and grown at 24°C. Images from a series are presented with time (in min:sec) relative to the onset of cytokinesis displayed in the bottom right of the panel. Arrowheads indicate centrosomes. Scale bar: 10 µm. (B) Quantification of the percentage of centrosomes that duplicated during the first cell cycle for indicated genotypes. zyg-1(it25), n = 40 ; zyg-1(it25) lin-23(RNAi), n = 36; zyg-1(it25) lin-23(ot1), n = 20; zyg-1(it25) lin-23(ot1,RNAi), n = 20. (C) Representative images of ZYG-1 levels at the centrosome from control and lin-23(ot1,RNAi) treated embryos. (D) Average levels of endogenous ZYG-1 at the centrosome in one-cell lin-23(RNAi), lin-23(ot1) and lin-23(ot1,RNAi) embryos were quantified and normalized to controls. The average normalized fluorescence from multiple experiments is shown. The number of centrosomes analysed is indicated. Error bars indicate s.e.m.
The ZYG-1 protein contains a consensus sequence for Slimb-binding (Peters et al., 2010). However, when we compared ZYG-1 sequences among various nematode species we noticed that the Slimb-binding motif is not conserved in other Caenorhabditis species including the nearest relative C. briggsae (supplementary material Fig. S2A). This suggested that, like the situation in human cells, there may be additional regulatory inputs required to maintain appropriate ZYG-1 levels. To investigate this further we looked at the contribution of additional F-box proteins to ZYG-1 regulation.
The F-box protein SEL-10 regulates ZYG-1 levels
In contrast to other metazoans, C. elegans possesses a large number of F-box proteins (i.e. 326 compared to 68 in humans) (Kipreos and Pagano, 2000). C. elegans however possesses only three WD repeat-containing F-box proteins: the closely related LIN-23 and SEL-10 (BLAST E value = 1e–42) and the distantly related MEC-15 (BLAST E value = 0.044). Given the challenge involved in conducting an exhaustive analysis of all C. elegans F-box proteins, we chose to focus our efforts on SEL-10, the F-box protein most closely related to LIN-23. SEL-10 is a conserved F-box protein orthologous to human FBW7 and yeast Cdc4p (Hubbard et al., 1997).
We found that RNAi depletion of SEL-10 led to a robust rescue of the centrosome duplication failure associated with the zyg-1(it25) allele (Fig. 5A). Centrosome duplication was restored in approximately 50% of cases (n = 30) (Fig. 5B). To confirm that this was due to an increase in ZYG-1 levels we quantified ZYG-1 at the centrosome. We detected elevated levels of GFP::cZYG-1 at the centrosome in sel-10(RNAi) embryos (Fig. 5E), similar to that observed following SKR-1/2 depletion (Fig. 3E). In addition we confirmed that loss of SEL-10 increases endogenous ZYG-1 levels at the centrosome using the sel-10(ok1632) loss-of-function allele (Fig. 5D). This allele contains a 901bp deletion beginning 54bp after the ATG. The deletion removes the F-box and all eight WD40 repeats, similar to previously described null alleles (Jäger et al., 2004). In these sel-10(ok1632) worms ZYG-1 levels were substantially elevated at the centrosome throughout the first mitosis (Fig. 5C,D).
Fig. 5.

The F-box protein SEL-10 regulates ZYG-1 levels. (A) zyg-1(it25); gfp::h2b;gfp::spd-2 embryos were treated with SEL-10 RNAi and grown at 24°C. Images from a series are presented with time (in min:sec) relative to the onset of cytokinesis displayed in the bottom right of the panel. Arrowheads indicate centrosomes. Scale bar: 10 µm. (B) Quantification of the percentage of centrosomes that duplicated during the first cell cycle. Control RNAi, n = 20; sel-10(RNAi), n = 30. (C) Representative images of ZYG-1 levels at the centrosome in wild-type and sel-10(ok1632) embryos. (D) Average levels of endogenous ZYG-1 at the centrosome in one-cell sel-10(ok1632) embryos were normalized to controls. The average normalized fluorescence from multiple experiments is shown. The number of centrosomes analysed is indicated. Error bars indicate s.e.m. (E) Average levels of GFP::cZYG-1 at the centrosome during the first cell cycle of sel-10(RNAi) embryos were calculated and normalized to control. Control, n = 16; sel-10(RNAi), n = 18. PNM, pronuclear meeting. (F) Feminized worms of the indicated genotypes were mated with zyg-1(it25) males and centrosome duplication monitored in the resultant embryos using GFP::SPD-2. zyg-1(it25), n = 30; zyg-1(it25); sel-10(ok1632), n = 35; zyg-1(it25) lin-23(ot1, RNAi), n = 20; zyg-1(it25) lin-23(ot1); sel-10(ok1632), n = 24; zyg-1(it25) lin-23(ot1, RNAi); sel-10(ok1632), n = 16.
Although the sel-10(ok1632) allele reproducibly elevates ZYG-1 levels, self-fertilizing zyg-1(it25); sel-10(ok1632) hermaphrodites showed a substantially reduced rescue of centrosome duplication (15% suppression, n = 12), when compared with sel-10(RNAi) (Fig. 5B; 50% suppression, n = 30). It has been noted previously that in C. elegans spermatocyte-expressed genes are not efficiently silenced by RNAi (Reinke et al., 2004). This decreased suppression was therefore consistent with SEL-10 having two roles in centrosome duplication: a negative role in the embryo [seen after sel-10(RNAi)], and a positive role during spermatogenesis which is only revealed by the sel-10(ok1632) allele. By using feminized zyg-1(it25); sel-10(ok1632) worms (which do not produce sperm) fertilized by zyg-1(it25) males we were able to restore paternal SEL-10 activity. This allowed us to confirm that loss of SEL-10 during spermatogenesis was responsible for the relatively high levels of centrosome duplication failure in the embryos of self-fertilizing zyg-1(it25); sel-10(ok1632) hermaphrodites. This situation is reminiscent of class II mutations in zyg-1 which show differential effects in spermatogenesis and the embryo (Peters, 2010). After restoring paternal SEL-10 activity, the sel-10(ok1632) allele robustly rescues centrosome duplication failure (60% duplication, n = 35; Fig. 5F), similar to sel-10(RNAi) (Fig. 5B). As a control we used feminized zyg-1(it25) worms fertilized by zyg-1(it25) males and found a complete failure of centrosome duplication (0% duplication, n = 30; Fig. 5F). Thus our results indicate that reducing SEL-10 activity in the embryo, either using the sel-10(ok1632) allele or by sel-10(RNAi), rescues the zyg-1(it25) phenotype by elevating ZYG-1 levels. Our investigation was focused on understanding the regulation of ZYG-1 in the embryo, therefore to separate the embryonic phenotype of the zyg-1(it25); sel-10(ok1632) worms from the spermatogenesis phenotype all further work with the zyg-1(it25); sel-10(ok1632) double mutant was carried out using feminized worms mated with zyg-1(it25) males. The function of SEL-10 in spermatocytes is beyond the scope of the current study and will be reported elsewhere.
LIN-23 and SEL-10 cooperatively regulate ZYG-1 abundance
Taken together, our results indicated that the F-box proteins LIN-23 and SEL-10 both contribute to regulating ZYG-1 levels. To address whether these proteins may play redundant roles in the regulation of centrosome duplication we determined whether their co-depletion increased centrosome duplication in one-cell zyg-1(it25) embryos. Depleting either LIN-23 or SEL-10 function alone rescued centrosome duplication failure in slightly over 50% of cases (Fig. 5F). Using feminized zyg-1(it25); lin-23(ot1,RNAi); sel-10(ok1632) worms we assessed the effect of disrupting both F-box proteins in combination. Interestingly, co-depletion of LIN-23 and SEL-10 strongly suppressed the centrosome duplication failure associated with the zyg-1(it25) allele (Fig. 5F). In this background, centrosome duplication in the one-cell embryos occurs at close to wild-type levels. The synergistic effect of co-depletion on centrosome duplication suggests that LIN-23 and SEL-10 play redundant roles in the regulation of ZYG-1.
Suppression of zyg-1(it25) by reducing SEL-10 activity does not require Cyclin E
SEL-10 is a highly conserved protein with homologs in Drosophila (archiplelago: ago) and humans (FBW7). Both ago and FBW7 have been implicated in degradation of cyclin E (Koepp et al., 2001; Moberg et al., 2001). In vertebrate cells cyclin E is required during centriole duplication for the growth of the new centriole (Tsou and Stearns, 2006). In C. elegans, despite a requirement for Cyclin E (CYE-1) in the initial stages of centrosome maturation, CYE-1 does not function in centriole duplication (Cowan and Hyman, 2006). Nevertheless, we wanted to test whether reducing SEL-10 availability elevated CYE-1 levels leading to the observed rescue of centrosome duplication failure in zyg-1(it25) mutant embryos. We began by determining if CYE-1 levels are elevated by loss of SEL-10. Analysis of CYE-1 by western blot showed that levels were comparable in extracts from wild-type and sel-10(ok1632) embryos (Fig. 6A) and in control(RNAi) and sel-10(RNAi) embryo extracts (data not shown), indicating that embryonic CYE-1 levels are not elevated after loss of SEL-10. To further test a requirement for CYE-1 in suppression of the zyg-1(it25) phenotype we RNAi depleted CYE-1 in feminized zyg-1(it25); sel-10(ok1632) embryos and looked for rescue of centrosome duplication (Fig. 6B,C). Our RNAi conditions were able to successfully deplete CYE-1 (Fig. 6A). Despite this significant reduction in CYE-1, rescue of centrosome duplication was observed at the same frequency as in controls (Fig. 6C), indicating that cyclin E is not required for zyg-1(it25) rescue by the loss of SEL-10.
Fig. 6.

Elevated cyclin E activity is not required for suppression of zyg-1(it25) by SEL-10 depletion. (A) Western blot comparing CYE-1 levels between control and sel-10(ok1632) embryos and showing successful depletion of CYE-1 under our RNAi conditions. (B) A representative embryo from zyg-1(it25) fertilized, feminized zyg-1(it25); gfp::spd-2; fem-1(hc17); sel-10(ok1632); cye-1(RNAi) worms grown at 24°C. Arrowheads indicate centrosomes. (C) Quantification of the percentage of centrosomes that duplicated during the first cell cycle in control and CYE-1 depleted embryos. Control, n = 35; cye-1(RNAi), n = 17.
ZYG-1 and SEL-10 physically interact
Our data indicate that the ability of reduced SEL-10 activity to rescue of the zyg-1(it25) phenotype does not act through up-regulation of CYE-1. Moreover, we find that LIN-23 and SEL-10 cooperatively regulate ZYG-1 levels. We therefore hypothesized that SEL-10 may normally directly interact with ZYG-1 to target it for degradation. The loss of SEL-10 could thereby account for the elevated ZYG-1 levels (Fig. 5C,D,E) and the rescue of zyg-1(it25) (Fig. 5A,B). We therefore tested for a direct interaction between ZYG-1 and SEL-10. To examine the association between ZYG-1 and SEL-10, we cotransfected constructs encoding FLAG::ZYG-1 and Myc::SEL-10 into HEK293T cells and performed an immunoprecipitation using FLAG antibodies, a technique previously used to show association of SEL-10 with its substrates (Jäger et al., 2004; Wu et al., 1998). We found that Myc::SEL-10 was coimmunoprecipitated with FLAG::ZYG-1, and not by our FLAG::GST control, indicating a direct physical association between ZYG-1 and SEL-10 (Fig. 7), consistent with SEL-10 regulating ZYG-1 degradation.
Fig. 7.
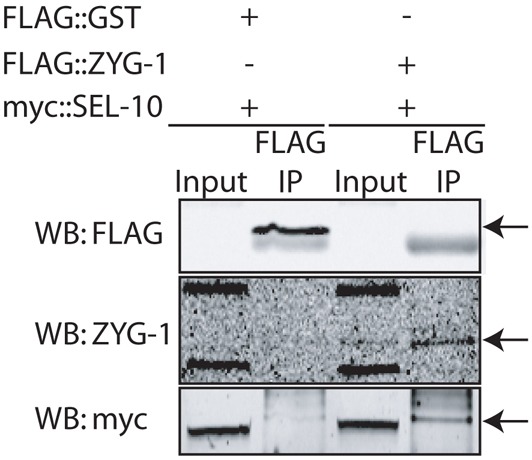
ZYG-1 and SEL-10 directly interact. Immunoprecipitation from HEK293T cells coexpressing FLAG::GST and Myc::SEL-10 (control) or FLAG::ZYG-1 and Myc::SEL-10. FLAG::ZYG-1 expression is low and cannot be detected in the extract. Proteins were immunoprecipitated with anti-FLAG beads and probed with antibodies against FLAG (to detect FLAG::GST), ZYG-1 (to detect FLAG::ZYG-1) and Myc. The apparent size difference of Myc::SEL-10 between input and immunoprecipitate lanes is probably due to differences in lysis and washing buffer compositions, because we see a similar shift when Myc::SEL-10 is immunoprecipitated using antibodies against Myc (data not shown).
Analysis of ZYG-1 protein sequences revealed the presence of multiple conserved consensus-recognition motifs for SEL-10 [known as cdc4 phosphodegron: CPD (Nash et al., 2001)](supplementary material Fig. S2), suggesting that SEL-10 can directly recognize ZYG-1 to target it for degradation. We mutated eight of these consensus sites in ZYG-1 (including the putative Slimb-box which also matches the CPD consensus) to try to define a SEL-10 interaction motif. After testing for an interaction between these mutant FLAG::ZYG-1 constructs and SEL-10 we found that none of the mutations were sufficient to disrupt the ZYG-1–SEL-10 interaction (supplementary material Fig. S2). We speculate that the interaction between SEL-10 and ZYG-1 does not rely on a single interaction motif, but instead uses multiple motifs, a situation reminiscent of the recognition of Myc by ago (Moberg et al., 2004).
Discussion
We have shown that the abundance of ZYG-1, a key regulator of centrosome duplication in C. elegans, is regulated by proteasomal degradation and the SCF ubiquitin ligase complex. This regulation is similar to that reported for the ZYG-1 homologs dPlk4 and hPlk4, which are also subject to SCF-dependent proteasomal degradation (Cunha-Ferreira et al., 2008; Guderian et al., 2010; Holland et al., 2010; Rogers et al., 2009; Sillibourne et al., 2010). Similar to previous studies in Drosophila and human cells, which implicate the F-box protein Slimb/βTrCP in targeting the SCF complex to Plk4, we find that regulation of ZYG-1 levels uses the homologous F-box protein, LIN-23. In addition, a second F-box protein, SEL-10, binds to and regulates ZYG-1 levels. Moreover, we present evidence for co-operativity between LIN-23 and SEL-10. Taken together, our data suggest that ZYG-1 levels are regulated by SCF-mediated targeting for proteasomal degradation using redundant F-box proteins. Although we cannot rule out an indirect effect, our data showing a direct interaction between SEL-10 and ZYG-1 suggests a direct role for the SCF in mediating ZYG-1 degradation.
The core centrosome duplication pathway was first elucidated in C. elegans, and subsequent work has found it to be conserved in other metazoans. In this hierarchy ZYG-1 is widely recognized to be the functional ortholog of Plk4 (Strnad and Gönczy, 2008). Recent reports have, however, cast doubt on whether ZYG-1 represents a true dPlk4/hPlk4 ortholog and imply it has instead emerged as a result of convergent evolution (Carvalho-Santos et al., 2010; Hodges et al., 2010). Our results show that ZYG-1 levels are regulated by the same pathways as dPlk-4/hPlk4, suggesting a conservation of the regulatory inputs on these homologous proteins. Although our results cannot answer the question of evolutionary origin of ZYG-1 they do show that the upstream regulatory pathways are conserved between C. elegans and other organisms. The fact that both upstream regulators and downstream effectors of ZYG-1 are conserved underscores the continued utility of studying centrosome duplication in C. elegans.
Loss of SCF, LIN-23 or SEL-10 activities lead to elevated levels of ZYG-1. Although the increase was sufficient to rescue the centrosome duplication defect of the zyg-1(it25) allele, we have never observed the formation of supernumerary centrosomes. Reducing SKR-1/2, CUL-1 or LIN-23 activities has pleiotrophic effects frequently resulting in chromosome mis-segregation, precluding our ability to assess the effect of elevated ZYG-1 beyond the early embryo. Null alleles of SEL-10 are, however, viable and fertile. In these embryos elevated ZYG-1 levels cause neither centrosome amplification in the embryo, nor overt adverse effects on later cell divisions. Since the formation of supernumerary centrosomes has not been reported in the C. elegans embryo our findings are consistent with previous studies. Clearly, although loss of SCFSEL-10 increases the accumulation of ZYG-1, these elevated levels are not sufficient to breach the normal controls of centrosome duplication, perhaps because ZYG-1 is not the sole limiting factor for centrosome duplication in the wild-type C. elegans embryo.
We found that in C. elegans in addition to the Slimb/βTrCP ortholog, LIN- 23, the FBW7 homolog, SEL-10, is required for ZYG-1 regulation. In C. elegans SEL-10 has been implicated in regulating sex determination, notch signaling and selective synapse elimination (Ding et al., 2007; Hubbard et al., 1997; Jäger et al., 2004). A role in the regulation of centrosome duplication has not previously been reported. A screen of F-box proteins in Drosophila cells found that depletion of the SEL-10 homolog, archipelago (ago), neither elevates dPlk4 levels (Cunha-Ferreira et al., 2008) nor increases centrosome number (Rogers et al., 2009). One explanation for this difference is evolutionary divergence leading to C. elegans using additional regulatory mechanisms, compared to Drosophila. Alternatively, the disparity may reflect a difference in the developmental status of the systems used. All previous work in Drosophila has used somatic cells whereas our work was carried out in the early embryo. It is therefore possible that the differences we have observed reflect a differential regulation of centrosome duplication in the embryo compared with the soma. In fact, tissue-specific adaptations of the regulation of centrosome duplication have been observed previously in Drosophila and in C. elegans (Peel et al., 2007; Peters et al., 2010).
Work in mammalian systems has confirmed that the Slimb homolog βTrCP plays a conserved role in degradation of mammalian Plk4; however, an exhaustive analysis of the involvement of additional F-box proteins has not been carried out. Depletion of βTrCP leads to increased Plk4 levels and the formation of supernumerary centrosomes in human cells (Guderian et al., 2010; Holland et al., 2010; Sillibourne et al., 2010). Significantly, deletion of the βTrCP recognition motif in Plk4 did not completely block Plk4 ubiquitination or degradation, suggesting the existence of additional regulatory mechanisms (Holland et al., 2010). Stabilization of Plk4 is instead only achieved by deletion of a 24 amino acid (aa) stretch of the protein from aa282-305 (Plk4Δ24), a region that includes the βTrCP degron and is rich in phosphorylatable residues. This has been taken to indicate that the 24aa region acts as a multidegron and, although βTrCP binding is important, additional substrate recognition elements also play an important role in Plk4 degradation. These additional regulators of Plk4 degradation have not yet been identified. Given our results showing redundancy between LIN-23 and SEL-10 in the regulation of ZYG-1 levels, we propose that the SEL-10 homolog FBW7 is a strong candidate for fulfilling this role in Plk4 regulation. Indeed the multidegron deleted in Plk4Δ24 contains multiple FBW7 consensus recognition motifs that could contribute to Plk4 degradation. We therefore suggest that Plk4 levels in human cells may be regulated by the combined action of FBW7 and βTrCP similar to the situation we describe here for C. elegans. FBW7 is a proto-oncogene that is frequently mutated in cancer, and reduced FBW7 activity results in supernumerary centrosomes and chromosome instability (Cizmecioglu et al., 2012; Rajagopalan et al., 2004). The formation of these extra centrosomes is thought to result from elevated cyclin E levels, however in light of our results it will be interesting to investigate whether elevated levels of Plk4 contribute to this phenotype. We have found SEL-10 function in C. elegans centrosome duplication to be independent of cyclin E. Similarly in Drosophila centrosome amplification caused by a loss of SCF function is also independent of cyclin E (Murphy, 2003). FBW7 regulates degradation of many oncogenes including Myc, notch and cyclin E and when FBW7 activity is lost, elevated levels of these substrates are thought to be key drivers of tumorigenesis (Welcker and Clurman, 2008). It will be important to determine whether Plk4 is an additional protein whose upregulation contributes to cellular transformation when FBW7 is mutated.
Throughout this study we have found that while abrogating SCF function has a substantial effect on metaphase ZYG-1 levels, it has little effect in anaphase. This suggests that degradation of ZYG-1 is cell cycle-regulated. How is this regulated degradation of ZYG-1 achieved? Recent evidence suggests that the abundance of FBW7 is cell cycle-regulated (Cizmecioglu et al., 2012). Similar regulation of SEL-10 could contribute to the cell cycle differences in ZYG-1 levels. In addition, for a substrate to be recognized by the SCF it must contain a motif for F-box binding and this motif must be phosphorylated. Regulating the phosphorylation of the F-box binding motif during the cell cycle can therefore control the timing of substrate degradation. The degradation of Plk4 in human cells is promoted by self-phosphorylation of its multidegron, although the action of an additional kinase has not been ruled out (Guderian et al., 2010; Holland et al., 2010; Sillibourne et al., 2010). It is possible that the timing of ZYG-1 degradation is achieved through cell-cycle-regulated phosphorylation of multiple degrons. Determining which kinase is responsible for ZYG-1 phosphorylation is of interest because it forms a key link between the cell cycle and centrosome duplication. It has previously been shown that ZYG-1 is capable of self-phosphorylation (O'Connell et al., 2001) and thus ZYG-1, like hPlk4, may regulate its own stability. In addition to kinase activity the action of a phosphatase may also impinge upon ZYG-1 regulation. Indeed, recent work has shown that loss of the phosphatase PP2A leads to a decrease in ZYG-1 levels (Song et al., 2011). An intriguing possibility is that PP2A dephosphorylates ZYG-1 to ‘rescue’ it from degradation similar to the regulation of Plk4 in Drosophila (Brownlee et al., 2011).
In summary, our work shows that ZYG-1 abundance in the C. elegans embryo is regulated by SCF-mediated degradation involving two F-box proteins, LIN-23 and SEL-10. This data is especially interesting since work in human cells suggests that in addition to βTrCP, further regulators contribute to the degradation of vertebrate Plk4. Regulated protein degradation by the SCF complex appears to be a common theme in controlling centrosome duplication. In addition to SCFβTrCP regulating Plk4, the degradation of human CP110, a protein implicated in centriole length control, is regulated by the SCFCyclinF complex (D'Angiolella et al., 2010) and degradation of human SAS-6 is promoted by SCFFBXW5 (Puklowski et al., 2011). The general picture that is emerging is that the SCF associates with a series of F-box proteins, and is required for the regulated degradation of multiple centrosome duplication factors in a temporally-controlled manner. In agreement with this, we show that in C. elegans centrosome duplication is also regulated by SCF-mediated protein degradation.
Materials and Methods
Worm strains and manipulation
Worm strains are listed in supplementary material Table S1. All worms were maintained at 20°C on MYOB plates seeded with E. coli OP50. Suppression of the zyg-1(it25) allele was tested at 24°C. All RNAi experiments were carried out by feeding bacteria expressing dsRNA as previously described (Kamath et al., 2003). For control RNAi we used an smd-1 feeding vector. The number of hours and age from which RNAi was carried out were as follows; skr-1/2, 36 h from L4; cul-1, 36 h from L4; sel-10, 48 h from L1; lin-23, 48 h from L1; rpt-4, 12–24 h from L4; cye-1 36 h from L1.
The zyg-1 and lin-23 loci are found at II:−0.99 and II:−0.38, respectively. To make the zyg-1(it25) lin-23(ot1) double mutant we carried out recombination between the dpy-25(e817) lin-23(ot1) and zyg-1(it25) unc-4(e120) chromosomes. We selected worms that had lost the dpy(e817) and unc(e120) markers and confirmed the presence of zyg-1(it25) and lin-23(ot1) by sequencing.
To distinguish the different effects of the sel-10(ok1632) allele on zyg-1(it25) in the sperm and embryo we used the temperature sensitive fem-1(hc17) allele. We feminized zyg-1(it25); gfp::spd-2; fem-1(hc17); sel-10(ok1632) worms by shifting L1 to 24°C. Feminized L4 were mated with zyg-1(it25) males and centrosome duplication observed. This was compared with centrosome duplication in zyg-1(it25); gfp::spd-2; sel-10(ok1632) self-fertilizing hermaphrodites. All subsequent work assessing the ability of the sel-10(ok1632) allele to suppress zyg-1(it25) utilized worms feminized as described using the fem-1(hc17) allele, and fertilized by zyg-1(it25) males.
Imaging
Fixation and staining of embryos was carried out as described previously (Kemp et al., 2007). For immunostaining, the following antibodies were used at 1/1000 dilution: DM1A (Sigma, St Louis, MO), ZYG-1 (Kemp et al., 2007). For live imaging we used a spinning disk confocal microscope which has been previously described (Peters et al., 2010). Levels of ZYG-1 at the centrosome were determined by quantification of average pixel intensity. Maximal projections of the centrosome were used for quantification of fluorescence in ImageJ 1.40g and background fluorescence was subtracted. For quantification of endogenous ZYG-1 we determined the average centrosome fluorescence and normalized to controls. We display normalized fluorescence averaged from at least two independent experiments. For GFP::cZYG-1 each experiment analyzed a minimum of eight test samples and eight controls. For each experiment the average fluorescence of test samples was normalized to that of wild-type. Average normalized fluorescence is displayed.
Antibodies and immunoblotting
Embryonic extracts were prepared and analyzed as in Song et al. (Song et al., 2011). The SKR-1/2 antibody was raised against peptide sequences that are present in both SKR-1 and SKR-2. The peptides MADQKKVSEAAKEREIKIS and MADQKEASEAAKNREIKIS were used to raise antibodies in rabbits and antibodies were affinity purified (Yenzym Antibodies LLC, San Fransisco, CA). The CYE-1 antibody was a gift from E. Kipreos (UGA, Athens, GA) and used at a 1/200 dilution. The TBB-2 antibody was raised against the peptide DVDGYAEGEAC and used at 1/500 dilution.
Immunoprecipitations
Point mutations in the putative CPD sites were made in FLAG::ZYG-1 using a Quick Change Lightning Site-Directed Mutagenesis kit (Agilent Technologies, Santa Clara, CA). The FLAG::GST fusion was made amplifying the GST open reading frame using HindIII and XbaI compatible primers and inserting the fragment into the pFLAG-CMV-2 vector (Sigma).
HEK293T cells were transfected with Myc::SEL-10 (Ding et al., 2007) and FLAG::ZYG-1 (Kitagawa et al., 2009) or FLAG::GST expression vectors, using Lipofectamine 2000 (Invitrogen, Carlsbad, CA). Cells were harvested after 40 h and lysed in buffer (50 mM Tris-HCl, pH 7.5, 150 mM NaCl, 5 mM EDTA, 1% Triton X-100, 1 mM DTT, 2 mM PMSF) in the presence of protease inhibitors. Lysates were incubated with anti-FLAG beads (Sigma, St Louis, MO) for 6 h, washed with PBS, 0.5% Triton X-100 and bound proteins eluted with LDS sample buffer. Western blots used the Myc 9E10 (Sigma, St Louis, MO) and FLAG (Sigma, St Louis, MO) antibodies at 1/1000 dilutions. The ZYG-1 antibody was used at 1/100 (Song et al., 2008).
Supplementary Material
Acknowledgments
We thank Edward Kipreos, Pierre Gonczy and Mei Ding for sharing reagents. We thank S. Bunting, A. Golden and O. Cohen-Fix for critical reading of the manuscript. Some stocks were provided by the Caenorhabditis Genetics Center and the C. elegans Gene Knockout Consortium.
Footnotes
Funding
This work was supported by the Intramural Research Program of the National Institutes of Health and the National Institute of Diabetes and Digestive and Kidney Diseases. Deposited in PMC for release after 12 months.
Supplementary material available online at http://jcs.biologists.org/lookup/suppl/doi:10.1242/jcs.097105/-/DC1
References
- Basto R., Lau J., Vinogradova T., Gardiol A., Woods C. G., Khodjakov A., Raff J. W. (2006). Flies without centrioles. Cell 125, 1375–1386 10.1016/j.cell.2006.05.025 [DOI] [PubMed] [Google Scholar]
- Basto R., Brunk K., Vinadogrova T., Peel N., Franz A., Khodjakov A., Raff J. W. (2008). Centrosome amplification can initiate tumorigenesis in flies. Cell 133, 1032–1042 10.1016/j.cell.2008.05.039 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Brownlee C. W., Klebba J. E., Buster D. W., Rogers G. C. (2011). The Protein Phosphatase 2A regulatory subunit Twins stabilizes Plk4 to induce centriole amplification. J. Cell Biol. 195, 231–243 10.1083/jcb.201107086 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Carvalho–Santos Z., Machado P., Branco P., Tavares–Cadete F., Rodrigues–Martins A., Pereira–Leal J. B., Bettencourt–Dias M. (2010). Stepwise evolution of the centriole-assembly pathway. J. Cell Sci. 123, 1414–1426 10.1242/jcs.064931 [DOI] [PubMed] [Google Scholar]
- Cizmecioglu O., Krause A., Bahtz R., Ehret L., Malek N., Hoffmann I. (2012). Plk2 regulates centriole duplication through phosphorylation-mediated degradation of Fbxw7 (human Cdc4). J. Cell. Sci. 125, 981–992 [DOI] [PubMed] [Google Scholar]
- Cowan C. R., Hyman A. A. (2006). Cyclin E-Cdk2 temporally regulates centrosome assembly and establishment of polarity in Caenorhabditis elegans embryos. Nat. Cell Biol. 8, 1441–1447 10.1038/ncb1511 [DOI] [PubMed] [Google Scholar]
- Cunha–Ferreira I., Rodrigues–Martins A., Bento I., Riparbelli M., Zhang W., Laue E., Callaini G., Glover D., Bettencourt–Dias M. (2008). The SCF/Slimb Ubiquitin Ligase Limits Centrosome Amplification through Degradation of SAK/PLK4. Curr. Biol. 19, 43–49 [DOI] [PubMed] [Google Scholar]
- D'Angiolella V., Donato V., Vijayakumar S., Saraf A., Florens L., Washburn M. P., Dynlacht B., Pagano M. (2010). SCF(Cyclin F) controls centrosome homeostasis and mitotic fidelity through CP110 degradation. Nature 466, 138–142 10.1038/nature09140 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Dammermann A., Maddox P. S., Desai A., Oegema K. (2008). SAS-4 is recruited to a dynamic structure in newly forming centrioles that is stabilized by the gamma-tubulin-mediated addition of centriolar microtubules. J. Cell Biol. 180, 771–785 10.1083/jcb.200709102 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Delattre M., Leidel S., Wani K., Baumer K., Bamat J., Schnabel H., Feichtinger R., Schnabel R., Gönczy P. (2004). Centriolar SAS-5 is required for centrosome duplication in C. elegans. Nat. Cell Biol. 6, 656–664 10.1038/ncb1146 [DOI] [PubMed] [Google Scholar]
- Delattre M., Canard C., Gönczy P. (2006). Sequential protein recruitment in C. elegans centriole formation. Curr. Biol. 16, 1844–1849 10.1016/j.cub.2006.07.059 [DOI] [PubMed] [Google Scholar]
- Ding M., Chao D., Wang G., Shen K. (2007). Spatial regulation of an E3 ubiquitin ligase directs selective synapse elimination. Science 317, 947–951 10.1126/science.1145727 [DOI] [PubMed] [Google Scholar]
- Ganem N. J., Godinho S. A., Pellman D. (2009). A mechanism linking extra centrosomes to chromosomal instability. Nature 460, 278–282 10.1038/nature08136 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Greenan G., Brangwynne C. P., Jaensch S., Gharakhani J., Jülicher F., Hyman A. A. (2010). Centrosome size sets mitotic spindle length in Caenorhabditis elegans embryos. Curr. Biol. 20, 353–358 10.1016/j.cub.2009.12.050 [DOI] [PubMed] [Google Scholar]
- Guderian G., Westendorf J., Uldschmid A., Nigg E. A. (2010). Plk4 trans-autophosphorylation regulates centriole number by controlling betaTrCP-mediated degradation. J. Cell Sci. 123, 2163–2169 10.1242/jcs.068502 [DOI] [PubMed] [Google Scholar]
- Habedanck R., Stierhof Y-D., Wilkinson C. J., Nigg E. A. (2005). The Polo kinase Plk4 functions in centriole duplication. Nat. Cell Biol. 7, 1140–1146 10.1038/ncb1320 [DOI] [PubMed] [Google Scholar]
- Hebeisen M., Roy R.2008). CDC-25.1 stability is regulated by distinct domains to restrict cell division during embryogenesis in C. elegans. Development 1351259–1269 10.1242/dev.014969 [DOI] [PubMed] [Google Scholar]
- Hodges M. E., Scheumann N., Wickstead B., Langdale J. A., Gull K. (2010). Reconstructing the evolutionary history of the centriole from protein components. J. Cell Sci. 123, 1407–1413 10.1242/jcs.064873 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Holland A. J., Lan W., Niessen S., Hoover H., Cleveland D. W. (2010). Polo-like kinase 4 kinase activity limits centrosome overduplication by autoregulating its own stability. J. Cell Biol. 188, 191–198 10.1083/jcb.200911102 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hubbard E. J., Wu G., Kitajewski J., Greenwald I. (1997). sel-10, a negative regulator of lin-12 activity in Caenorhabditis elegans, encodes a member of the CDC4 family of proteins. Genes Dev. 11, 3182–3193 10.1101/gad.11.23.3182 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Jäger S., Schwartz H. T., Horvitz H. R., Conradt B. (2004). The Caenorhabditis elegans F-box protein SEL-10 promotes female development and may target FEM-1 and FEM-3 for degradation by the proteasome. Proc. Natl. Acad. Sci. USA 101, 12549–12554 10.1073/pnas.0405087101 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kamath R. S., Fraser A. G., Dong Y., Poulin G., Durbin R., Gotta M., Kanapin A., Le Bot N., Moreno S., Sohrmann M., et al. (2003). Systematic functional analysis of the Caenorhabditis elegans genome using RNAi. Nature 421, 231–237 10.1038/nature01278 [DOI] [PubMed] [Google Scholar]
- Kemp C. A., Song M. H., Addepalli M. K., Hunter G., O'Connell K. (2007). Suppressors of zyg-1 define regulators of centrosome duplication and nuclear association in Caenorhabditis elegans. Genetics 176, 95–113 10.1534/genetics.107.071803 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kipreos E. T., Pagano M. (2000). The F-box protein family. Genome Biol 1 doi:10.1186/gb–2000–1–5–reviews3002 10.1186/gb-2000-1-5-reviews3002 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kitagawa D., Busso C., Flückiger I., Gönczy P. (2009). Phosphorylation of SAS-6 by ZYG-1 is critical for centriole formation in C. elegans embryos. Dev. Cell 17, 900–907 10.1016/j.devcel.2009.11.002 [DOI] [PubMed] [Google Scholar]
- Kleylein–Sohn J., Westendorf J., Le Clech M., Habedanck R., Stierhof Y-D., Nigg E. A. (2007). Plk4-induced centriole biogenesis in human cells. Dev. Cell 13, 190–202 10.1016/j.devcel.2007.07.002 [DOI] [PubMed] [Google Scholar]
- Koepp D. M., Schaefer L. K., Ye X., Keyomarsi K., Chu C., Harper J. W., Elledge S. J. (2001). Phosphorylation-dependent ubiquitination of cyclin E by the SCFFbw7 ubiquitin ligase. Science 294, 173–177 10.1126/science.1065203 [DOI] [PubMed] [Google Scholar]
- Korzeniewski N., Spardy N., Duensing A., Duensing S. (2011). Genomic instability and cancer: lessons learned from human papillomaviruses. Cancer Lett. 305, 113–122 10.1016/j.canlet.2010.10.013 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Leidel S., Delattre M., Cerutti L., Baumer K., Gönczy P. (2005). SAS-6 defines a protein family required for centrosome duplication in C. elegans and in human cells. Nat. Cell Biol. 7, 115–125 10.1038/ncb1220 [DOI] [PubMed] [Google Scholar]
- Moberg K. H., Bell D. W., Wahrer D. C. R., Haber D. A., Hariharan I. K. (2001). Archipelago regulates Cyclin E levels in Drosophila and is mutated in human cancer cell lines. Nature 413, 311–316 10.1038/35095068 [DOI] [PubMed] [Google Scholar]
- Moberg K. H., Mukherjee A., Veraksa A., Artavanis–Tsakonas S., Hariharan I. K. (2004). The Drosophila F box protein archipelago regulates dMyc protein levels in vivo. Curr. Biol. 14, 965–974 10.1016/j.cub.2004.04.040 [DOI] [PubMed] [Google Scholar]
- Murphy T. D. (2003). Drosophila skpA, a component of SCF ubiquitin ligases, regulates centrosome duplication independently of cyclin E accumulation. J. Cell Sci. 116, 2321–2332 10.1242/jcs.00463 [DOI] [PubMed] [Google Scholar]
- Nash P., Tang X., Orlicky S., Chen Q., Gertler F. B., Mendenhall M. D., Sicheri F., Pawson T., Tyers M. (2001). Multisite phosphorylation of a CDK inhibitor sets a threshold for the onset of DNA replication. Nature 414, 514–521 10.1038/35107009 [DOI] [PubMed] [Google Scholar]
- Nayak S., Santiago F. E., Jin H., Lin D., Schedl T., Kipreos E. T. (2002). The Caenorhabditis elegans Skp1-related gene family: diverse functions in cell proliferation, morphogenesis, and meiosis. Curr. Biol. 12, 277–287 10.1016/S0960-9822(02)00682-6 [DOI] [PubMed] [Google Scholar]
- Nigg E. A., Raff J. W. (2009). Centrioles, centrosomes, and cilia in health and disease. Cell 139, 663–678 10.1016/j.cell.2009.10.036 [DOI] [PubMed] [Google Scholar]
- O'Connell K. F., Caron C., Kopish K. R., Hurd D. D., Kemphues K. J., Li Y., White J. G. (2001). The C. elegans zyg-1 gene encodes a regulator of centrosome duplication with distinct maternal and paternal roles in the embryo. Cell 105, 547–558 10.1016/S0092-8674(01)00338-5 [DOI] [PubMed] [Google Scholar]
- Peel N., Stevens N. R., Basto R., Raff J. W. (2007). Overexpressing centriole-replication proteins in vivo induces centriole overduplication and de novo formation. Curr. Biol. 17, 834–843 10.1016/j.cub.2007.04.036 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Pelletier L., O'Toole E., Schwager A., Hyman A. A., Müller–Reichert T. (2006). Centriole assembly in Caenorhabditis elegans. Nature 444, 619–623 10.1038/nature05318 [DOI] [PubMed] [Google Scholar]
- Peters N., Perez D. E., Song M. H., Liu Y., Müller–Reichert T., Caron C., Kemphues K. J., O'Connell K. F. (2010). Control of mitotic and meiotic centriole duplication by the Plk4-related kinase ZYG-1. J. Cell Sci. 123, 795–805 10.1242/jcs.050682 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Puklowski A., Homsi Y., Keller D., May M., Chauhan S., Kossatz U., Grünwald V., Kubicka S., Pich A., Manns M. P., et al. (2011). The SCF-FBXW5 E3-ubiquitin ligase is regulated by PLK4 and targets HsSAS-6 to control centrosome duplication. Nat. Cell Biol. 13, 1004–1009 10.1038/ncb2282 [DOI] [PubMed] [Google Scholar]
- Rajagopalan H., Jallepalli P. V., Rago C., Velculescu V. E., Kinzler K. W., Vogelstein B., Lengauer C. (2004). Inactivation of hCDC4 can cause chromosomal instability. Nature 428, 77–81 10.1038/nature02313 [DOI] [PubMed] [Google Scholar]
- Reinke V., Gil I. S., Ward S., Kazmer K. (2004). Genome-wide germline-enriched and sex-biased expression profiles in Caenorhabditis elegans. Development 131, 311–323 10.1242/dev.00914 [DOI] [PubMed] [Google Scholar]
- Rogers G. C., Rusan N. M., Roberts D. M., Peifer M., Rogers S. L. (2009). The SCF Slimb ubiquitin ligase regulates Plk4/Sak levels to block centriole reduplication. J. Cell Biol. 184, 225–239 10.1083/jcb.200808049 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sillibourne J. E., Tack F., Vloemans N., Boeckx A., Thambirajah S., Bonnet P., Ramaekers F. C. S., Bornens M., Grand–Perret T. (2010). Autophosphorylation of polo-like kinase 4 and its role in centriole duplication. Mol. Biol. Cell 21, 547–561 10.1091/mbc.E09-06-0505 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Song M. H., Aravind L., Müller–Reichert T., O'Connell K. F. (2008). The conserved protein SZY-20 opposes the Plk4-related kinase ZYG-1 to limit centrosome size. Dev. Cell 15, 901–912 10.1016/j.devcel.2008.09.018 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Song M. H., Liu Y., Anderson D. E., Jahng W. J., O'Connell K. F. (2011). Protein phosphatase 2A-SUR-6/B55 regulates centriole duplication in C. elegans by controlling the levels of centriole assembly factors. Dev. Cell 20, 563–571 10.1016/j.devcel.2011.03.007 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Strnad P., Gönczy P. (2008). Mechanisms of procentriole formation. Trends Cell Biol. 18, 389–396 10.1016/j.tcb.2008.06.004 [DOI] [PubMed] [Google Scholar]
- Torres J. Z., Summers M. K., Peterson D., Brauer M. J., Lee J., Senese S., Gholkar A. A., Lo Y-C., Lei X., Jung K., et al. (2011). The STARD9/Kif16a kinesin associates with mitotic microtubules and regulates spindle pole assembly. Cell 147, 1309–1323 10.1016/j.cell.2011.11.020 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Tsou M-F. B., Stearns T. (2006). Mechanism limiting centrosome duplication to once per cell cycle. Nature 442, 947–951 10.1038/nature04985 [DOI] [PubMed] [Google Scholar]
- Welcker M., Clurman B. E. (2008). FBW7 ubiquitin ligase: a tumour suppressor at the crossroads of cell division, growth and differentiation. Nat. Rev. Cancer 8, 83–93 10.1038/nrc2290 [DOI] [PubMed] [Google Scholar]
- Wu G., Hubbard E. J., Kitajewski J. K., Greenwald I. (1998). Evidence for functional and physical association between Caenorhabditis elegans SEL-10, a Cdc4p-related protein, and SEL-12 presenilin. Proc. Natl. Acad. Sci. USA 95, 15787–15791 10.1073/pnas.95.26.15787 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Yamanaka A., Yada M., Imaki H., Koga M., Ohshima Y., Nakayama K-I. (2002). Multiple Skp1-related proteins in Caenorhabditis elegans: diverse patterns of interaction with Cullins and F-box proteins. Curr. Biol. 12, 267–275 10.1016/S0960-9822(02)00657-7 [DOI] [PubMed] [Google Scholar]
- Zhu F., Lawo S., Bird A., Pinchev D., Ralph A., Richter C., Müller–Reichert T., Kittler R., Hyman A. A., Pelletier L. (2008). The mammalian SPD-2 ortholog Cep192 regulates centrosome biogenesis. Curr. Biol. 18, 136–141 10.1016/j.cub.2007.12.055 [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.