

Summary
Contact inhibition ubiquitously exists in non-transformed cells that are in contact with neighboring cells. This phenomenon explains the poor regenerative capacity of in vivo human corneal endothelial cells during aging, injury and surgery. This study demonstrated that the conventional approach of expanding human corneal endothelial cells by disrupting contact inhibition with EDTA followed by bFGF activated canonical Wnt signaling and lost the normal phenotype to endothelial–mesenchymal transition, especially if TGFβ1 was added. By contrast, siRNA against p120 catenin (CTNND1) also uniquely promoted proliferation of the endothelial cells by activating trafficking of p120 catenin to the nucleus, thus relieving repression by nuclear Kaiso. This nuclear p120-catenin–Kaiso signaling is associated with activation of RhoA–ROCK signaling, destabilization of microtubules and inhibition of Hippo signaling, but not with activation of Wnt–β-catenin signaling. Consequently, proliferating human corneal endothelial cells maintained a hexagonal shape, with junctional expression of N-cadherin, ZO-1 and Na+/K+-ATPase. Further expansion of human corneal endothelial monolayers with a normal phenotype and a higher density was possible by prolonging treatment with p120 catenin siRNA followed by its withdrawal. This new strategy of perturbing contact inhibition by selective activation of p120-catenin–Kaiso signaling without disrupting adherent junction could be used to engineer surgical grafts containing normal human corneal endothelial cells to meet a global corneal shortage and for endothelial keratoplasties.
Key words: Contact inhibition, Endothelial–mesenchymal transformation, Human corneal endothelium, Kaiso, Proliferation, p120 catenin
Introduction
The corneal endothelium forms a single monolayer of hexagonal cells lining the basement-membrane-containing Descemet membrane of the posterior cornea and facing the aqueous humor, which contains TGFβ2 (Chen et al., 1999). Through expression of ZO-1, a tight junction component, and Na+/K+-ATPase to exert barrier and pump functions, respectively, human corneal endothelial cells (HCECs) play a pivotal role in regulating corneal stromal hydration and hence transparency (reviewed by Bonanno, 2003). Unlike other species, such as mouse, rabbit and cow, HCECs are notorious for their limited proliferative capacity in vivo after injury, aging and surgery (Laing et al., 1984). The limited proliferative capacity of HCECs is caused by mitotic arrest at the G1 phase of the cell cycle (reviewed by Joyce, 2005). A similar mitotic block is also reported in cat (Petroll et al., 1998) and human (Senoo et al., 2000) corneal explants, as well as confluent rat corneal endothelial cultures (Joyce et al., 2002) as a result of ‘contact inhibition’. The conventional approach of expanding HCECs in vitro is to disrupt cell junctions by EDTA, followed by culture in medium supplemented with mitogens, such as bFGF (Engelmann et al., 1988). However, such a culture method could activate endothelial–mesenchymal transformation (EMT), a pathological process that can generate ‘retrocorneal fibrous membrane’, leading to corneal blindness (reviewed by Lee and Kay, 2006).
Intercellular junctions include gap junctions, adherent junctions (AJs), and tight junctions, among which AJs play an important role in controlling many cellular behaviors, including proliferation, differentiation and survival (reviewed by Perez-Moreno et al., 2003). Although not fully elucidated, the mechanism governing contact- inhibition-mediated mitotic block likely involves signaling transmitted from AJs to the nucleus (reviewed by Matter et al., 2005; Perez-Moreno et al., 2003). Conventionally, two signaling pathways could be elicted by β-catenin and p120 catenin (CTNND1, hereafter referred to as p120), respectively, when the AJ junction is disrupted. The former is known as the canonical Wnt pathway, in which β-catenin, if accumulated in the cytoplasm without prompt degradation by binding with β-TrCP, can be translocated into the nucleus, where it acts as a transcriptional coactivator through binding with the T-cell-factor/lymphoid enhancer factor (TCF/LEF) transcription factors (reviewed by Nelson and Nusse, 2004). The latter might trigger the p120–Kaiso pathway, in which nuclear-translocated p120 relieves the repressor activity of Kaiso, a member of the BTB/POZ-ZF transcription factor family (Kelly et al., 2004; Daniel, 2007). It has been known that p120 negatively regulates RhoA (Anastasiadis et al., 2000; Anastasiadis and Reynolds, 2001). However, it is unclear whether and how activation of RhoA, following the release of p120 inhibition, can be linked to p120 nuclear trafficking and subsequent signaling. Most recently, the Hippo pathway, an evolutionarily conserved protein kinase cascade, has also been identified to control in vivo organ size and in vitro contact inhibition by governing cell proliferation and apoptosis (Zeng and Hong, 2008). Cytoplasmic phosphorylated and nuclear non-phosphorylated Yes-associated protein (YAP) and transcriptional coactivator with PDZ-binding motif (TAZ) have been proposed as the prime mediators of the Hippo pathway (Zhao et al., 2010). The Hippo signaling can intercede Wnt–β-catenin signaling via an interaction between phosphorylated TAZ and CK1δ/3-mediated phosphorylation of Disheveled in the cytoplasm (Varelas et al., 2010) and between non-phosphorylated YAP and β-catenin in the nucleus (Heallen et al., 2011). It is unclear whether and how any of these three signaling pathways are linked to contact inhibition of HCEC monolayers.
To resolve these questions, we isolated HCECs using collagenase instead of trypsin-EDTA or dispase to avoid disrupting intercellular junctions and cell matrix (basement membrane) interaction (Li et al., 2007), and we demonstrated that such cultured HCEC monolayers also exhibit contact inhibition when AJs mature to an in vivo pattern (Zhu et al., 2008). Here, we demonstrate that the mitotic block unlocked by EDTA with bFGF, is mediated by canonical Wnt signaling and induces EMT, especially if TGFβ1 is added. By contrast, the mitotic block unlocked by treatment with siRNA against p120 is causatively related to activation of p120 trafficking to the nucleus, relief of repression by nuclear Kaiso, activation of RhoA–ROCK signaling, inhibition of Hippo signaling, but not activation of β-catenin–Wnt signaling. Consequently, HCECs retain a normal phenotype without EMT, despite addition of bFGF and TGFβ1. These results highlight the feasibility of deploying a novel strategy of tissue engineering of HCECs and other similar tissues by selective activation of p120–Kaiso signaling without the use of single cells, by disrupting adherent junctions.
Results
Mitotic block unlocked by EDTA-bFGF induces EMT, especially if TGFβ1 is added
As a first step of exploring the signaling involved in controlling contact inhibition, we isolated HCECs by collagenase, but not trypsin or dispase, to retain cell junctions and cell to basement membrane interaction (Li et al., 2007), and we demonstrated that in vitro HCEC monolayers exhibit contact inhibition when their AJ matures to an in vivo pattern (Zhu et al., 2008). Similar contact inhibition can be unlocked by scraping, followed by culturing with bFGF or bFGF plus TGFβ in cat corneal buttons (Petroll et al., 1998), or by disrupting cell junctions with EDTA in human corneal buttons pre-incubated in a medium with EGF and FGF (Senoo et al., 2000). Exposure of mitotically quiescent HCEC monolayers to 5 mM EDTA for 1 hour alone led to notable disruption of intercellular junction (Fig. 1A). Without bFGF, cells restored their pre-treatment monolayer morphology in 2 days (not shown). However, immediately following EDTA treatment, when 20 ng/ml bFGF was added for 2 days, 10 ng/ml TGFβ1 was added for 3 days, or 20 ng/ml bFGF was added for 2 days followed by 10 ng/ml TGFβ1 for 3 days, HCECs transformed into fibroblastic-like cells, with relocation of N-cadherin from the cell junction to the cytoplasm (Fig. 1A). Nuclear BrdU staining was negative when the cells were treated with PBS or EDTA alone (Fig. 1A). However, the BrdU labeling index was promoted to ∼19% by bFGF, but not TGFβ1 alone or bFGF plus TGFβ1 (Fig. 1A), suggesting that the mitotic block could only be unlocked by bFGF when EDTA was used to disrupt cell junctions. Immunostaining for S100A4, an early marker of epithelial–mesenchymal transition (Basile et al., 2011), showed the staining was negative in cells with or without EDTA treatment, but became positive in the cytoplasm and the nucleus after addition of bFGF or TGFβ1, and exclusively in the nucleus when both bFGF and TGFβ1 were added (Fig. 1A). In addition, α-smooth muscle actin (α-SMA), a marker indicative of myofibroblast differentiation (Gabbiani, 2003), became strongly positive only when TGFβ was added (Fig. 1A). The EMT caused by EDTA-bFGF was accompanied by the loss of the normal HCEC phenotype because junctional staining of N-cadherin, ZO-1, and Na+/K+-ATPase resembled the cytoplasmic staining pattern (Fig. 1B). Collectively, these results indicated that the contact inhibition unlocked by EDTA-bFGF initiated EMT with proliferation, whereas full-blown EMT without proliferation developed after TGFβ1 was added.
Fig. 1.

EMT with or without proliferation is caused by EDTA with or without bFGF and TGFβ1. (A) HCECs assume a fibroblastic shape after addition of bFGF and/or TGFβ. BrdU labeling is increased by 19% with EDTA-bFGF (n = 3, *P<0.05) but not with EDTA-TGFβ1 or EDTA-bFGF-TGFβ1. S100A4 staining was increased with EGTA-bFGF or TGFβ1, but became exclusively nuclear upon addition of EDTA-bFGF-TGFβ1. Cytoplasmic α-SMA staining was apparent with EDTA-TGFβ1 and was increased in EDTA-bFGF-TGFβ1-treated cells. In addition, N-cadherin switch from the membrane to the cytoplasm was apparent with EDTA-bFGF, EDTA-TGFβ1 or EDTA-bFGF-TGFβ1. (B) Membrane staining of N-cadherin, ZO-1 and Na+/K+-ATPase was disrupted in EDTA-bFGF-treated cells. Scale bars: 100 µm.
Mitotic block unlocked by EDTA-bFGF is mediated by Wnt signaling
Because S100A4 is a transcriptional target of canonical β-catenin–Wnt signaling (Sack and Stein, 2009), we wondered whether the Wnt signaling was activated to unlock the mitotic block of HCEC monolayers. Compared with the PBS control, EDTA alone did not change the expression of β-catenin and LEF1 mRNAs and proteins (not shown). However, EDTA-bFGF elevated the level of β-catenin and LEF1 mRNAs twofold and threefold, respectively (Fig. 2A, n = 3). Immunostaining confirmed the junctional localization of β-catenin and the absence of nuclear LEF1 in cells treated with PBS (Fig. 2B) or EDTA alone (not shown). By contrast, EDTA-bFGF reduced β-catenin in the cell junction, increased its accumulation in the cytoplasm and the nucleus, and notably enhanced the LEF1 nuclear staining (Fig. 2B). The aforementioned change of immunostaining was confirmed by semi-quantitative protein dot blotting using membranous, cytosolic and nuclear extracts. Based on the loading control, i.e. CN43, α-tubulin and histone, respectively, the β-catenin protein level was decreased fivefold in the membranous compartment, but increased three- and sixfold in cytosolic and nuclear compartments, respectively. At the same time, EDTA-bFGF increased the LEF1 level threefold in the nuclear compartment (Fig. 2C). These results collectively supported the notion that the mitotic block unlocked by EDTA-bFGF correlated with activation of the canonical Wnt signaling. Such activation of Wnt signaling was not observed when TGFβ1 was added (not shown).
Fig. 2.

EMT induced by EDTA-bFGF is mediated by Wnt signaling. (A) Real-time PCR shows two- and threefold upregulation of β-catenin and LEF1 transcripts with EDTA-bFGF, respectively (n = 3, *P<0.05). (B) EDTA-bFGF promotes translocalization of β-catenin from the membrane to the nucleus and nuclear accumulation of LEF1. (C) Dot protein blotting confirmed that EDTA-bFGF causes a shift of β-catenin from the membrane to the nucleus and promotes nuclear accumulation of LEF1 (n = 3, *P<0.05; CN43, α-tubulin and histone were used as the loading control for membranous, cytosolic and nuclear compartments, respectively). (D) EDTA-bFGF activates Wnt signaling by activating the TCF/LEF promoter activity sixfold (n = 3, P<0.05). Addition of a specific Wnt inhibitor, XAV 939, completely abolishes the activity of TCF/LEF promoter and BrdU labeling. (E) EMT caused by addition of EDTA-bFGF was prevented by XAV 939. After addition of XAV 939, the staining of β-catenin, S100A4, N-cadherin, ZO-1 and Na+/K+-ATPase was similar to that from normal HCEC monolayers (cf. Fig. 1B, Fig. 2B). (F) Overexpression of S33Y β-catenin in HCEC monolayers treated with EGTA without growth factors increases BrdU labeling (by 21-fold, n = 3, P<0.05), nuclear β-catenin, LEF-1, S100A4 and cytoplasmic α-SMA, N-cadherin. (G) The activation of Wnt signaling by S33Y β-catenin was confirmed by Western blotting, indicating that nuclear β-catenin and LEF-1 are activated. Scale bars: 100 µm.
To confirm that the above correlation was causative, we transfected HCEC monolayers with a plasmid containing a TCF/LEF promoter construct and simultaneously treated the cells with XAV939, an inhibitor of β-catenin-mediated signaling through inhibiting the poly-ADP-ribosylating enzymes tankyrase 1 and tankyrase 2 (Huang et al., 2009). As expected, the promoter activity was low in PBS- or EDTA-treated groups alone, but was elevated sixfold in the EDTA-bFGF-treated group (Fig. 2D). Such an elevated promoter activity was completely suppressed by XAV939 (Fig. 2D). As a result, the BrdU labeling reverted to baseline quiescence, whereas β-catenin remained in the junction and S-100A4 was negative without EMT (Fig. 2E). In fact, the junctional staining of N-cadherin, ZO-1, and Na+/K+-ATPase was retained (Fig. 2E). In addition, the notion that activation of Wnt signaling induced proliferation and EMT was confirmed by overexpression of stable S33Y β-catenin in contact-inhibited HCEC monolayers treated with EDTA, but without bFGF. In such cases, Wnt signaling was activated (Fig. 2G) whereas nuclear BrdU labeling, β-catenin, LEF-1 and S100A4 and cytoplasmic α-SMA and N-cadherin were significantly increased (Fig. 2F). In fact, overexpression of stable S33Y β-catenin also rescued proliferation and EMT inhibited by XAV939 (not shown).
HCEC mitotic block is uniquely unlocked by p120 siRNA without EMT
Because disruption of AJs by EDTA induced EMT by activating Wnt signaling (Figs 1,2), we took a different approach by perturbing intercellular junction components with siRNAs to downregulate p120, β-catenin, N-cadherin, or ZO-1. The knockdown efficiency of these siRNAs was verified by both real-time PCR and western blotting (supplementary material Fig. S1A,B). To our surprise, the BrdU labeling index was also promoted by ∼18% in HCEC monolayers when treated with p120 siRNA, but not with siRNA against β-catenin, N-cadherin or ZO-1 (Fig. 3A). Unlike EDTA treatment (Fig. 1A), addition of bFGF did not further increase BrdU labeling that was already increased by addition of p120 siRNA (Fig. 3B). The above result was confirmed using three other independent p120 siRNAs with target sequences 5′-GCCAGAGGTGGTTCGGATA-3′ (Davis et al., 2003), 5′-AACGAGGTTATCGCTGAGAAC-3′ (Xiao et al., 2003) and 5′-GCGATTGCTTCGAAAGGCTCGTGAT-3′ (designed by us, not shown). Most importantly, HCECs treated with p120 siRNA remained hexagonal and did not express cytoplasmic or nuclear S100A4 and cytoplasmic α-SMA, even with additional bFGF and TGFβ (Fig. 3B). These results collectively indicated that the mitotic block unlocked by p120 siRNA retained the normal HCEC phenotype without inducing EMT. By contrast, siRNA against p120 decreased HCEC proliferation sevenfold when the cells were dissociated into single cells using EDTA-trypsin (Fig. 3C).
Fig. 3.

Mitotic block is uniquely unlocked by p120 siRNA without EMT. (A) HCEC monolayers treated with siRNA against p120 but not β-catenin, N-cadherin, or ZO-1, significantly promoted nuclear BrdU labeling up to 18-fold (n = 3, *P<0.05). (B) The proliferation promoted by p120 siRNA was not further promoted by additional bFGF (n = 3, P>0.05). Addition of bFGF and/or TGFβ1 did not induce staining of S100A4 or α-SMA. However, addition of TGFβ1 abolished BrdU labeling promoted by p120 siRNA (n = 3, *P<0.05). (C) By contrast, the proliferation of HCEC monolayers decreased sevenfold (n = 3, P<0.05) upon addition of p120 siRNA when the cells were dissociated into single cells using EDTA-trypsin. Scale bars: 100 µm.
p120 siRNA activates p120–Kaiso but not β-catenin–Wnt signaling
Although p120 is usually found at the cell junction and undergoes nucleocytoplasmic shuttling (Kelly et al., 2004; Roczniak-Ferguson and Reynolds, 2003; van Hengel et al., 1999), the controversy of nuclear p120 in normal or tumor cells exists (Daniel, 2007). We suspected that such a controversy might stem from the difference in the fixative used for immunostaining. In fact, nuclear p120 staining was not apparent when HCEC monolayers were fixed with 4% paraformaldehyde as practiced in previous studies (Davis et al., 2003; Roczniak-Ferguson and Reynolds, 2003; Wildenberg et al., 2006). By contrast, nuclear p120 staining was apparent when cells were fixed using 25% acetic acid and 75% methanol (supplementary material Fig. S2). To determine whether nuclear accumulation of p120 triggered by p120 siRNA might cause the release of transcriptional repression of nuclear Kaiso as suggested previously (Daniel, 2007), we first examined the expression of p120 and Kaiso transcripts by real-time PCR. Compared with the control treated with scrambled RNA (scRNA), HCECs treated with p120 siRNA expressed a 20- and 3-fold decrease of p120 and Kaiso transcripts, respectively (Fig. 4A). By contrast, p120 siRNA did not alter the levels of β-catenin and LEF1 transcripts (Fig. 4A). Compared with scRNA, p120 siRNA markedly reduced p120 staining at the cell junction, but increased that in the nucleus, where it colocalized with increased BrdU nuclear staining (Fig. 4B). At the same time, nuclear staining of Kaiso was markedly reduced (Fig. 4B). As a comparison, junctional β-catenin staining and nuclear LEF1 staining were not changed by addition of p120 siRNA when compared with the controls (Fig. 4B). These immunostaining differences were confirmed by semi-quantitation of the protein levels by dot blotting using membranous, cytosolic and nuclear lysates. p120 siRNA indeed reduced the membranous p120 level fourfold, but increased the nuclear p120 level threefold, whereas the nuclear Kaiso level was reduced fourfold (Fig. 4C). As a comparison, the dot blotting showed that p120 siRNA did not change β-catenin and LEF1 levels in these three compartments (Fig. 4C). EDTA-bFGF, which activated Wnt signaling (Fig. 2), affected neither junctional p120 nor nuclear Kaiso staining, (Fig. 4D) nor did it reduce p120 and Kaiso protein levels in these three compartments (Fig. 4E). Collectively, these results showed that the mitotic block unlocked by p120 siRNA led to nuclear translocation of p120, which correlated well with the decrease of nuclear Kaiso, and that such an unlocking effect was not accompanied by changes of cytolocalization of β-catenin and LEF1 and the TCF/LEF promoter activity (not shown).
Fig. 4.

p120 siRNA triggers p120–Kaiso, but not Wnt signaling. (A) p120 siRNA downregulates expression of both p120 and Kaiso transcripts by 95% and 70%, respectively (n = 3, *P<0.05), but not β-catenin and LEF1 transcripts. (B) p120 siRNA induces nuclear translocalization of p120 (green), which colocalizes with nuclear BrdU labeling (red) (n = 3, *P<0.05) and decreases nuclear Kaiso staining, but does not affect membrane β-catenin or activate nuclear LEF1 staining. (C) Dot blotting confirmed that p120 siRNA increased the level of p120 and decreased that of Kaiso in the nuclear compartment, but did not alter that of β-catenin and LEF1. (D) In comparison, EDTA-bFGF did not alter the staining pattern of p120 and Kaiso. Scale bars: 100 µm. (E) Dot blotting confirmed that EDTA-bFGF did not affect p120 and Kaiso levels.
p120 nuclear translocation is directly responsible for releasing nuclear Kaiso to unlock HCEC mitotic block
Although p120 siRNA led to nuclear translocation of p120 and nuclear release of Kaiso (Fig. 4B,C), it also reduced the level of Kaiso transcript by 70% (Fig. 4A). To discern whether Kaiso nuclear release was directly influenced by p120 nuclear translocation or indirectly via downregulation of Kaiso transcription, we tested the effect of Kaiso siRNA knockdown, of which the efficiency was confirmed by a reduction of 85% in Kaiso transcripts (supplementary material Fig. S1). When HCEC monolayers were treated with Kaiso siRNA alone, the protein dot assay showed that nuclear Kaiso protein level (Fig. 5A) and Kaiso staining (Fig. 5B) were not altered when compared with the control treated with scRNA. Double immunostaining confirmed that junctional staining of p120 was not reduced, and nuclear BrdU labeling was not elevated (Fig. 5B). These results indicated that reduction of Kaiso transcription alone by Kaiso siRNA knockdown could not alter the nuclear Kaiso level and hence was not sufficient to unlock the mitotic block. Nonetheless, the nuclear Kaiso level was reduced fourfold when cells were treated with p120 siRNA alone, and reduced sixfold when cells were treated with both p120 and Kaiso siRNAs (Fig. 5A). Consequently, the BrdU labeling index was increased by 18% upon treatment with p120 siRNA alone, and increased by 26% upon treatment with both p120 and Kaiso siRNAs (Fig. 5B). These results further suggested that the release of nuclear Kaiso was crucial for unlocking the mitotic block of HCEC monolayers, and such an effect could not be achieved by Kaiso siRNA alone, unless p120 siRNA was added to trigger p120 nuclear translocation.
Fig. 5.

p120 nuclear translocation plays an important role in releasing nuclear Kaiso to unlock HCEC mitotic block. (A) The nuclear Kaiso level was significantly decreased by p120 siRNA and further by combined p120 and Kaiso siRNAs (n = 3, *P<0.05), but not by Kaiso siRNA (n = 3, P>0.05). By contrast, the nuclear p120 level was not affected by Kaiso siRNA (not shown, n = 3, P>0.05). (B) Double immunostaining of p120 and BrdU (left) showed that BrdU labeling (red) correlated with nuclear p120 staining (green) was promoted by p120 siRNA but not Kaiso siRNA. A synergistic effect was noted by combined treatment with both p120 and Kaiso siRNAs (n = 3, *P<0.05). (C) Nuclear dot blotting shows the nuclear p120 protein level promoted by p120 siRNA is further enhanced by nocodazole, but is decreased by taxol (n = 3, *P<0.05). (D) Dot blotting shows the nuclear Kaiso protein level decreased by p120 siRNA is further decreased by nocodazole, but is increased by taxol to the control level (n = 3, *P<0.05). (E) The extent of nuclear p120 and BrdU labeling is negatively correlated with nuclear Kaiso levels, which is decreased by nocodazole but increased by taxol (n = 3, *P<0.05). Scale bars: 100 µm.
To further prove that nuclear translocation of p120 was crucial for Kaiso nuclear release to unlock the mitotic block, we examined the effect of nocodazole or taxol (Roczniak-Ferguson and Reynolds, 2003), which are known to depolymerize and stabilize the microtubule network, respectively, to affect the cytoplasmic p120 pool, thus indirectly affecting p120 nuclear translocation (Yanagisawa et al., 2004). Compared with the control treated with scRNA, addition of nocodazole increased the nuclear p120 level from three- to fivefold when HCEC monolayers were treated with p120 siRNA (Fig. 5C). By contrast, addition of taxol reduced the p120 nuclear level to that of the control (Fig. 5C,E). Consequently, the nuclear Kaiso level was decreased twofold by nocodazole, but increased by taxol to the level of the scRNA control (Fig. 5D). The aforementioned changes of nuclear p120 and Kaiso levels were also reflected by the extent of nuclear p120 and Kaiso staining (Fig. 5E). Consequently, the BrdU labeling index was enhanced from 18% to 24% with nocodazole, but decreased to control levels with taxol (Fig. 5E). Collectively, these results indicated that the extent of p120 nuclear translocalization played a dominant role in reducing the nuclear level of Kaiso to unlock the mitotic block of HCEC monolayers.
Nuclear p120-mediated proliferation is controlled by RhoA–ROCK signaling
p120 is known to interact and stabilize microtubules (Franz and Ridley, 2004) and to inhibit RhoA (Anastasiadis et al., 2000; Anastasiadis and Reynolds, 2001). Activation of RhoA signaling can destabilize microtubules (Takesono et al., 2010). Because destabilization of microtubules by nocodazole facilitated p120–Kaiso signaling (Fig. 5), we wondered whether RhoA was activated by p120 siRNA in a similar manner to that seen with nocodazole, resulting in proliferation mediated by p120 nuclear translocation. Indeed, we noted that the level of active RhoA was increased threefold by p120 siRNA and fourfold with additional nocodazole (Fig. 6A). By contrast, the level of active RhoA promoted by p120 siRNA or p120 siRNA and nocodazole was suppressed by Taxol to the baseline level. Under this scenario, nuclear p120 staining and BrdU labeling promoted by p120 siRNA and nocodazole was abolished by taxol, CT-04 (a RhoA inhibitor) or Y27632 (a ROCK inhibitor) (Fig. 6B). These results suggest that activation of RhoA–ROCK signaling is correlated with destabilization of microtubules and p120 nuclear translocation and proliferation.
Fig. 6.
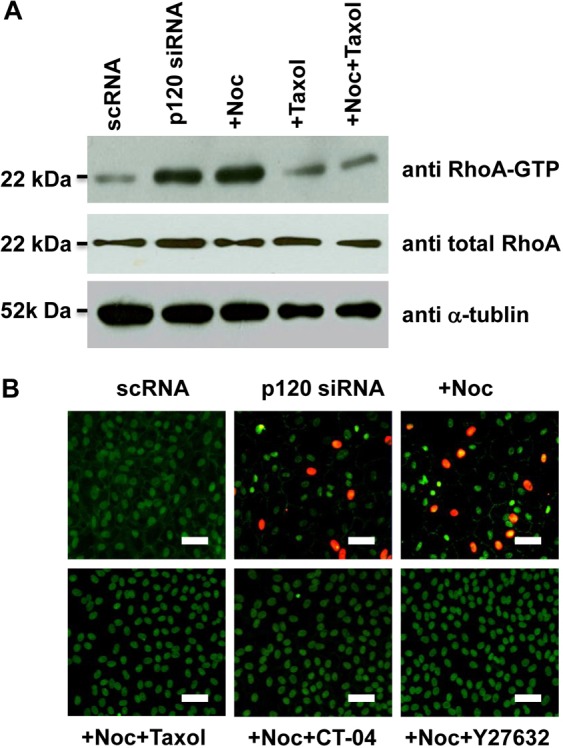
RhoA–ROCK signaling controls p120 nuclear translocation and its associated proliferation. (A) The level of active RhoA is promoted threefold by p120 siRNA and fourfold with addition of nocodazole. The level of active RhoA promoted by p120 siRNA or p120 siRNA+nocodazole is inhibited by taxol back to baseline levels. (B) Nuclear BrdU and p120 promoted by p120 siRNA+nocodazole was abolished by Taxol, CT-04 (RhoA inhibitor) or Y27632 (ROCK inhibitor). Scale bars: 100 µm.
Hippo signaling is inhibited by p120 siRNA, but not EDTA-bFGF
The Hippo pathway is a key protein kinase cascade involved in regulating in vivo organ size and in vitro contact inhibition by governing cell proliferation and apoptosis (Zeng and Hong, 2008). To determine whether Hippo signaling was also affected, we examined and quantified the cytolocalization of phosphorylated and non-phosphorylated YAP and TAZ, which are the prime mediators of the Hippo pathway (Zhao et al., 2010). Both immunostaining and western blotting showed that non-phosphorylated YAP and TAZ were accumulated in the nucleus after treatment of p120 siRNA, whereas cytoplasmic phosphorylated YAP and TAZ was dramatically reduced in the cytoplasm compared with levels in the control treated with scRNA (Fig. 7A,B). As a comparison, EDTA-bFGF failed to elicit any change (Fig. 7C,D). Because the pYAP (S127) antibody detected both phosphorylated YAP (pYAP) and phosphorylated TAZ (pTAZ) (Habbig et al., 2011; Lei et al., 2008), we performed western blotting to confirm the presence of cytoplasmic pTAZ and pYAP and nuclear TAZ and YAP. These results indicated that p120 siRNA, but not EDTA-bFGF, inhibited Hippo signaling by decreasing cytoplasmic pTAZ and pYAP, but increasing nuclear TAZ and YAP, further suggesting that selective activation of p120–Kaiso signaling also uniquely inhibited the Hippo signaling.
Fig. 7.
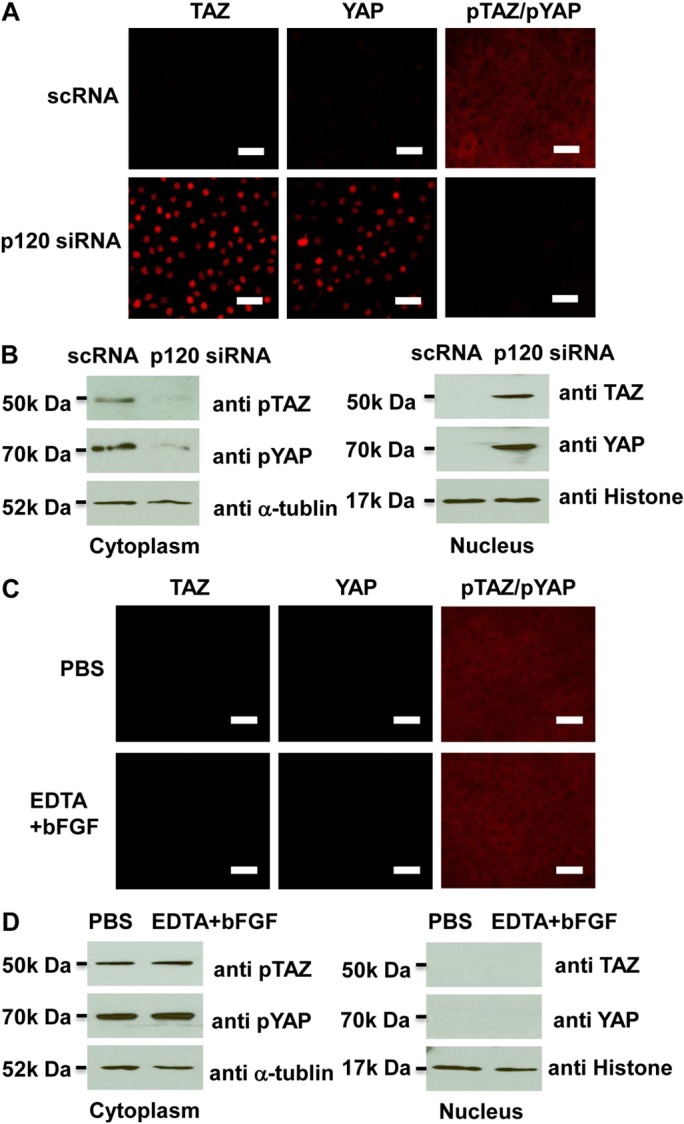
p120 knockdown but not EDTA-bFGF inhibits Hippo signaling. (A) Immunostaining shows that p120 siRNA causes nuclear accumulation of non-phosphorylated TAZ and YAP, and cytoplasmic deletion of phosphorylated TAZ and YAP when compared with addition of scRNA. (B) Because the same antibody detects both pYAP and pTAZ, Western blotting was used to confirm that both pTAZ and pYAP bands were decreased in the cytoplasm, whereas TAZ and YAP (detected by different antibodies) were increased in the nuclear extract. (C,D) By contrast, immunostaining (C) and western blotting (D) show that EDTA-bFGF fails to elicit such a change.
Prolonged treatment of p120 siRNA followed by withdrawal leads to further expansion of HCEC monolayers without EMT
The above experiments based on a short pulse of 100 nM p120 siRNA knockdown for 48 hours was capable of unlocking the HCEC mitotic block without inducing EMT by selective activation of p120–Kaiso and inhibition of Hippo signaling. It remained unclear whether proliferation could be sustained without EMT by prolonging treatment with p120 siRNA. A total of 18 HCEC monolayers derived from three separate donors with ages of 55 (Fig. 8A, d1 and D1), 58 (Fig. 8A, d2 and D1), and 76 (Fig. 8A, d3 and D3) were treated with 40 nM of either p120 siRNA or scRNA (n = 3 for each for each donor) weekly from Day 14 for 2 weeks followed by withdrawal on Day 28 for 10 days. The HCEC monolayers treated with scRNA virtually turned quiescent after 18 days of culturing (Fig. 8A, d1–d3). By contrast, the HCEC monolayers treated with p120 siRNA continued growth during the entire 27 days of culture (Fig. 8A, D1–D3). At Day 27, the monolayer size of the control treated with scRNA rapidly declined to an average of 1.7±0.4 mm2 (n = 9), whereas that of the p120 siRNA-treated counterpart continued to expand to 3.7±0.7 mm2 (n = 9).
Fig. 8.

Prolonged treatment with p120 siRNA causes further expansion of HCEC monolayers without EMT. (A) The HCEC monolayer surface area reached a plateau when treated with scRNA (d1, d2 and d3; 1.7±0.4 mm2; n = 9, P<0.05), but was continuously promoted by p120 siRNA (D1, D2 and D3) (P<0.05 on day 18 except the D3/d3 pair, and on Day 21, 24 and 27 for all three pairs; 3.7±0.7 mm2; n = 9, P<0.05) without cell enlargement in the center. (B) The HCEC density was 2241±104/mm2 when the Descemet membrane was stripped from the peripheral cornea (a). It increased to 2548±93/mm2 for HCEC monolayers cultured on Day 14 (b, n = 5, P<0.05). For the control treated with scRNA, the HCEC density dropped to 2083±86/mm2 on Day 28 (c, n = 5, P>0.05) and 1764±96 mm2 on Day 38 (d, n = 5, P<0.05), i.e. 10 days after withdrawal. By contrast, the HCEC density was maintained at 2316±79/mm2 on Day 28 (i.e. two weeks of p120 siRNA3 treatment) (e, n = 5, P>0.05), and 2289±113/mm2 on Day 38 (i.e. 10 days after withdrawal, f, n = 5, P>0.05). (C) Prolonged p120 siRNA treatment results in nuclear translocation of p120 and dissolution of F-actin without disturbing the junctional staining pattern of N-cadherin, ZO-1 and Na+/K+-ATPase. Ten days after withdrawal of p120 siRNA, the staining pattern of p120 and F-actin reverted to the normal pattern whereas that of the other proteins remained unchanged. Scale bars: 100 µm.
To ensure that the aforementioned change of monolayer size resulted from cell proliferation rather than cell enlargement, we measured the HCEC cell density. HCECs of the Descemet membrane stripped from the donor peripheral cornea showed a characteristic in vivo hexagonal pattern with an average density of 2241±104/mm2 (Fig. 8Ba, n = 5 donors). HCEC monolayers cultured in SHEM up to Day 14 also maintained a similar hexagonal pattern with a significant increase of cell density to 2548±93/mm2 (Fig. 8Bb, n = 5). For the control treated with scRNA, although the hexagonal pattern was maintained, the cell density was 2083±86/mm2 at Day 28 (after 2 weeks of treatment) (Fig. 8Bc, n = 5), and dropped to 1764±96/mm2 on Day 38 (10 days after withdrawal), indicative of cell enlargement (Fig. 8Bd, n = 5). By contrast, prolonged treatment with p120 siRNA maintained not only the hexagonal pattern but also the cell density at 2316±79/mm2 at Day 28 and 2289±113/mm2 at Day 38 (Fig. 8Be and 8Bf, respectively, both n = 5).
Immunostaining confirmed that the normal HCEC phenotype, judged by junctional staining of p120, N-cadherin, ZO-1 and Na+/K+-ATPase as well as by peri-membranous staining of F-actin, was maintained in the control treated with scRNA (Fig. 8C, top row). Prolonged treatment with p120 siRNA for 2 weeks resulted in a significant reduction of p120 junctional staining and an increase in p120 nuclear staining (Fig. 8C, middle row) similar to that shown in Fig. 4. Furthermore, it also reduced peri-junctional staining of F-actin, but only mildly reduced the junctional staining of N-cadherin, ZO-1 and Na+/K+-ATPase (Fig. 8C, middle row). Ten days after withdrawal of p120 siRNA, the immunostaining pattern was completely reverted to that of the control for all these five markers (Fig. 8C, bottom row). These results collectively indicated that prolonged treatment with p120 siRNA further expanded HCEC monolayer sizes while maintaining the normal hexagonal morphology at the same cell density without EMT (not shown). Following withdrawal of p120 siRNA, the normal HCEC phenotype was completely restored in 10 days.
Discussion
Disruption of cell junctions by EDTA (Senoo et al., 2000), scraping (Petroll et al., 1998) or freezing (Petroll et al., 1997) is a pre-requisite to unlock the mitotic block mediated by contact inhibition in corneal endothelial cells. In cat corneal endothelial explants (Petroll et al., 1998), scraping not only stimulates proliferation but also leads to EMT when cultured in a medium containing bFGF and/or TGFβ (Petroll et al., 1998). In cultured HCEC monolayers, contact inhibition disrupted by EDTA did not unlock the mitotic block even in EGF-containing SHEM until bFGF was added (Fig. 1). By that time HCECs had lost their normal phenotype expressing the junctional staining of N-cadherin, ZO-1 and Na+/K+-ATPase, changed to a slender shape and began to express cytoplasmic and nuclear staining of S100A4 (Fig. 1), a marker of epithelial–mesenchymal transition (Zeisberg et al., 2007). Such a phenotypic transition was reminiscent of the early phase of EMT elicited in confluent rabbit corneal endothelial cells when bFGF was added to promote proliferation in a serum-containing medium (Kay et al., 1993). Upon addition of TGFβ1 to EDTA-bFGF-treated cells, full-blown EMT ensued as evidenced by cytoplasmic α-SMA and heralded by cessation of BrdU labeling (Fig. 1). These results collectively let us conclude that conventional methods of expanding single EDTA or EDTA-trypsin-treated HCECs in a medium containing such growth factors as EGF, bFGF and TGFβ1 are potentially at risk of losing the HCEC important barrier and pump functions to EMT, which is a pathological process leading to corneal blindness by forming a retrocorneal membrane (reviewed in Lee and Kay, 2006).
We further demonstrated that the mitotic block of HCEC monolayers unlocked by EDTA-bFGF was caused by selective activation of canonical Wnt signaling. EDTA-bFGF led to nuclear translocation of β-catenin, a significant increase of nuclear β-catenin and LEF1 protein levels, and a significant increase of TCF/LEF promoter activity (Fig. 2). Furthermore, addition of a specific Wnt inhibitor XAV939 (Huang et al., 2009) abolished BrdU labeling, cytoplasmic and nuclear staining of S100A4, cytoplasmic α-SMA, and retained the junctional staining of N-cadherin, ZO-1 and Na+/K+-ATPase in EDTA-bFGF-treated HCECs even if TGFβ1 was added (Fig. 2). Such a change could be completely reverted by overexpression of stable S33Y β-catenin (Fig. 2). To our knowledge, the above finding is the first demonstrating a clear causative role played by canonical Wnt signaling in governing the transition from contact inhibition to EMT with proliferation in HCEC monolayers. This finding is similar to the known role of the Wnt signaling in epithelial–mesenchymal transition (Polette et al., 2007), where the canonical Wnt signaling cooperates with TGFβ-mediated Smad signaling in mouse epithelial cells (Eger et al., 2004) or proximal tubular epithelial cell lines (Masszi et al., 2004).
Realizing the aforementioned drawback of activating Wnt signaling by EDTA-bFGF in HCECs, we discovered that contact inhibition of HCEC monolayers could also be unlocked by p120 siRNA. Similar to EDTA-bFGF treatment, a high BrdU labeling index was uniquely promoted by knockdown of p120, but not β-catenin, N-cadherin or ZO-1 (Fig. 3). Unlike EDTA-bFGF, p120 siRNA, which downregulated p120 transcript and protein levels, actually perturbed neither transcription nor junctional staining of β-catenin and N-cadherin (not shown) nor transcription and nuclear protein level of LEF1 (Fig. 4). The above finding was surprising, given the fact that p120 is known to play a key role in stabilizing cadherins at the membrane when AJs mature in many types of cells (Anastasiadis and Reynolds, 2001; Rossman et al., 2005). In fact, p120 siRNA reportedly decreases the level of β-catenin (Davis et al., 2003) and E-cadherin in tumor epithelial cells (Davis et al., 2003; Ireton et al., 2002), VE-cadherin in vascular endothelial cells (Davis et al., 2003; Ferreri et al., 2008) and N-cadherin in cardiac myocytes (Davis et al., 2003) and vascular endothelial cells (Ferreri et al., 2008). But none of these studies showed increased proliferation after treatment with p120 siRNA. We attributed such a discrepancy to their use of EDTA-trypsin-dissociated cells, because p120 siRNA actually reduced cell proliferation when HCECs were dissociated into single cells by EDTA-trypsin (Fig. 3). These data highlight a unique role of p120 in governing contact inhibition in post-confluent cultures.
The mitotic block of contact-inhibited HCEC monolayers unlocked by p120 siRNA was mediated by selective activation of the p120–Kaiso signaling. Previous studies have shown that the intrinsic nucleocytoplasmic shuttling activity of p120 can be modulated by extrinsic factors, such as cadherin binding (Roczniak-Ferguson and Reynolds, 2003; van Hengel et al., 1999) and interactions with the microtubule network (Roczniak-Ferguson and Reynolds, 2003; Yanagisawa et al., 2004). However, the importance of nuclear p120 in eliciting downstream signaling has not been appropriately addressed, presumably because of the controversy surrounding nuclear p120 staining when cells were fixed with paraformaldehyde. We discovered that p120 nuclear staining was apparent in HCEC monolayers only when they were fixed using methanol and acetic acid (supplementary material Fig. S2). Accompanied by downregulation of p120 transcript and protein, p120 siRNA dramatically shifts p120 cytolocalization from the junction or membrane to the nucleus, where it was colocalized with BrdU labeling and correlated with the release of nuclear Kaiso (Fig. 4). Consistent with what has been noted in a number of cell lines (Roczniak-Ferguson and Reynolds, 2003), the extent of nuclear p120 level elicited by p120 siRNA could be augmented by nocodazole, a microtubule-disrupting agent, but diminished by taxol, a microtubule-stabilizing agent (Fig. 5), confirming that p120 shuttling from the cytoplasm to the nucleus could be facilitated by perturbing the microtubular network. Consequently, the nuclear p120 level manipulated by nocodazole or taxol, was inversely correlated with the nuclear level of Kaiso, strongly suggesting that the former dictates the latter. Similar to porcine pulmonary artery endothelial cells (Zhang et al., 2010), p120 knockdown in HCEC monolayers also downregulated Kaiso transcription (Fig. 4). Here, for the first time, we showed that Kaiso siRNA per se, which downregulated Kaiso transcript and protein (supplementary material Fig. S1), was incapable of reducing the nuclear Kaiso level unless nuclear p120 was elicited (Fig. 5). Such a finding suggests that the homeostasis of nuclear Kaiso is tightly regulated and cannot be influenced by its own transcription and translation in the short term. Nonetheless, Kaiso siRNA was synergistic with p120 siRNA in further lowering the nuclear Kaiso level (Fig. 5), a finding resembling that in H. pylori-mediated up-regulation of MMP-7 in MKN28 cells (Ogden et al., 2008). These data collectively support the notion that nuclear translocalization of p120 is a prerequisite to relieve nuclear Kaiso, in agreement with the data that the Kaiso binding domain for p120 is the same as that for DNA (Kelly et al., 2004).
The notion that p120–Kaiso signaling activated by p120 siRNA was facilitated by destabilization of microtubules is supported by the finding that p120 is capable of regulating microtubule dynamics in a cadherin-independent manner (Ichii and Takeichi, 2007). Consistent with the known action of p120 in downregulating RhoA (Anastasiadis et al., 2000; Anastasiadis and Reynolds, 2001), p120 siRNA activated RhoA–ROCK signaling (Fig. 6), which was associated with destabilization of microtubules. We discovered that p120 nuclear translocation induced by p120 siRNA was dependent on RhoA–ROCK signaling and destabilization of microtubules because inhibition of RhoA by CT-4 or ROCK by Y-27632 or stabilization of microtubules by Taxol abolished p120 nuclear translocation and BrdU labeling (Fig. 6). Although p120 signaling might modulate canonical and non-canonical Wnt signaling (Kim et al., 2004; Ruzov et al., 2009; Spring et al., 2005), p120 knockdown did not activate canonical Wnt signaling, leading to EMT in HCEC monolayers. The finding that p120 signaling did not activate Wnt signaling might partly be explained by our finding that p120 siRNA, but not EGTA-bFGF, elicited a dramatic decrease of cytoplasmic pYAP and pTAZ, but an increase in nuclear non-phosphorylated YAP and TAZ (Fig. 7). This was because Hippo signaling intercedes with Wnt signaling through the binding between pTAZ and CK1δ/3 to block phosphorylation of Disheveled in the cytoplasm (Varelas et al., 2010) and interaction between non-phosphorylated YAP and β-catenin in the nucleus (Heallen et al., 2011) and disruption of the cadherin–catenin complex (Kim et al., 2011). Such a contrast between p120 and β-catenin when the adherens junction is perturbed by EDTA and p120 knockdown, respectively, paves the way for future investigation of how Hippo signaling might be controlled by contact inhibition.
p120 is the prototypic member of the subfamily of p120 armadillo-related proteins (Hatzfeld, 2005) associated with cadherin-mediated adherens junctions (Davis et al., 2003; Ireton et al., 2002; Xiao et al., 2003). Several authors have reported that the cell proliferation was promoted in a p120 siRNA stable-transfected NIH3T3 cell line (Wildenberg et al., 2006), in the keratinocytes of p120 conditional-knockout mouse (Perez-Moreno et al., 2006; Perez-Moreno et al., 2008) by activation of RhoA signaling. Such an effect of p120 might be reversely linked to E-cadherin or isoform 1A of p120 at the cell membrane (Slorach et al., 2011; Soto et al., 2008). However, others have demonstrated that the growth of MDA-MB-231 cells was promoted by p120 (Soto et al., 2008) or the growth of single MDCK cells was inhibited by p120 knockdown (Dohn et al., 2009). Our results clearly indicate that relief of inhibition of RhoA activity by p120 siRNA might play a major role in promoting proliferation of contact-inhibited HCEC monolayers. Such an activation of RhoA might be due to the fact that p120 acts as a guanine nucleotide dissociation inhibitor (GDI) by binding to and preventing RhoA activity (Anastasiadis et al., 2000; Rossman et al., 2005). Interestingly, downregulation of p120 was not associated with a change of junctional N-cadherin. Such a finding is not in line with the finding that p120 siRNA resulted in downregulation of N-cadherin in bovine pulmonary artery endothelial cells (Ferreri et al., 2008). We attribute such a discrepancy to a different type of endothelial cell and different culture conditions.
To rule out the concern of carcinogenesis based on the finding that enhanced nuclear p120 levels are noted in several tumor cell lines (reviewed by Daniel, 2007), we prolonged p120 siRNA treatment for 2 weeks. Our results showed a wider spread of BrdU labeling from the periphery to the center of HCEC monolayers (not shown). Consequently, the mitotically arrested HCEC monolayers continued to expand its size from an average of 1.6 mm2 to 3.7 mm2 while maintaining the same cell density. Importantly, prolonged p120 knockdown did not alter the characteristic hexagonal shape and junctional expression of N-cadherin, ZO-1 and Na+/K+-ATPase, suggesting their barrier and pump functions were preserved (Fig. 8). Both p120 nuclear translocation and dissolution of peri-membranous F-actin cables were transient and reversed to the normal pattern following the withdrawal of p120 siRNA. Hence, it is plausible to deploy this novel strategy of selectively activating the p120–Kaiso signaling to expand HCEC monolayers using the donor scleral rim left after corneal transplantation. This new engineering strategy will not only obviate the use of enzymatic digestion to produce single HCECs but will also avoid the loss of their normal phenotype to EMT during ex vivo expansion of HCECs. If accomplished, this new technology will not only help solve the global shortage of human donor corneas but will also facilitate the current popular trend of transplanting only the corneal endothelium in procedures collectively termed ‘endothelial keratoplasty’ (reviewed by Terry, 2006). Our ongoing experiments show that this technology can also be applied to achieve the same goal in the human retinal pigment epithelium.
Materials and Methods
Reagents
Dulbecco’s modified Eagle’s medium (DMEM), Ham’s/F12 medium, human epidermal growth factor (hEGF), HEPES buffer, Hanks’ balanced salt solution (HBSS), phosphate-buffered saline (PBS), gentamicin, fetal bovine serum (FBS), Texas-Red-X phalloidin and Alexa-Fluor-conjugated secondary IgG were purchased from Invitrogen (Carlsbad, CA). Collagenase A was obtained from Roche Applied Science (Indianapolis, IN). Hydrocortisone, dimethyl sulfoxide, cholera toxin, insulin-transferrin-sodium selenite media supplement, bovine serum albumin, agarose, PCR marker, paraformaldehyde, methanol, Triton X-100, Hoechst 33342 dye, Y-27632, CT-04, XAV 939, nocodazole and taxol were purchased from Sigma-Aldrich (St Louis, MO) or Cytoskeleton (Denver, CO) or Calbiochem (La Jolla, CA). Specific monoclonal antibodies against α-tubulin, β-catenin, BrdU, Kaiso, Na+/K+-ATPase, N-cadherin as well as polyclonal antibodies against α-SMA, Histone, N-cadherin (type I), p120-catenin, RhoA, S100A4, ZO-1, YAP, TAZ and pYAP (S127) were purchased from Abcam (La Jolla, CA), Cell Signaling (Boston, MA), Chemicon (Billerica, MA), Upstate (Billerica, MA), Cytoskeleton, Santa Cruz Biotechnology (Santa Cruz, CA), Sigma and Zymed (Carlsbad, CA) (supplementary material Table S1). RNeasy Mini Kit was purchased from Qiagen (Valencia, CA). Dual luciferase assay system was purchased from Promega (Madison, WI). High Capacity Reverse Transcription Kit and TaqMan Universal PCR Master Mix were obtained from Applied Biosystems (Foster City, CA). Control scRNA and HP validated siRNAs to β-catenin, N-cadherin, ZO-1, and Kaiso (Qiagen, Catalog Number SI02662478, SI02663927, SI02655149 and S104165924, respectively), HiPerFect siRNA and SuperFect plasmid transfection reagents were obtained from Qiagen. p120 siRNA was designed by us and obtained from Invitrogen with the target sequence of 5′-CAGAGGTGATCGCCATGCTTGGATT-3′. The TCF/LEF reporter plasmid kit was from SABiosciences (Valencia, CA). Both control and stable S33Y β-catenin plasmids were a gift from Jan-Kan Chen (The Chang Gung University, Taiwan) (Chen et al., 1994).
HCEC isolation and culture
A total of 336 human corneas from individuals aged 20–81 years and maintained at 4°C in Optisol (Chiron Vision, Irvine, CA) for less than 5 days were obtained from the Florida Lions Eye Bank (Miami, FL) and handled according to the declaration of Helsinki. The isolation and culture of HCECs followed reported methods (Li et al., 2007; Zhu et al., 2008). In short, after central corneal buttons had been used for corneal transplantation, the remaining corneoscleral tissues were rinsed three times with DMEM containing 50 mg/ml gentamicin and 1.25 mg/ml amphotericin B. Under a dissecting microscope, the trabecular meshwork was cleaned, the rim was trephined within Schwalbe’s line, and Descemet’s membranes containing HCEC were stripped. After digestion at 37°C for 16 hours with 1 mg/ml collagenase A in SHEM, which was made of an equal volume of HEPES-buffered DMEM and Ham’s F12 supplemented with 5% FBS, 0.5% dimethyl sulfoxide, 2 ng/ml hEGF, 5 µg/ml insulin, 5 µg/ml transferrin, 5 ng/ml selenium, 0.5 µg/ml hydrocortisone, 1 nM cholera toxin, 50 µg/ml gentamicin, and 1.25 µg/ml amphotericin B, HCEC aggregates were collected by centrifugation at 2000 r.p.m. for 3 minutes to remove the digestion solution, and they were cultured in 24-well dishes coated with Collagen IV in SHEM. Cultures were monitored by phase contrast micrography, and the size of monolayer was determined by digitizing the surface area using ImageJ.
siRNA transfection and other treatments
HCEC monolayers were cultured to Day 14–19, when they exhibited contact inhibition coinciding with the maturation of AJs and ZO-1 staining resembling the in vivo pattern (Zhu et al., 2008). Such HCEC monolayers were treated with PBS with or without 5 mM EDTA at 37°C for 1 hour, followed by culturing in SHEM with or without addition of 20 ng/ml bFGF for 2 days, which was then followed with or without 10 ng/ml TGFβ1 for 3 additional days or TGFβ1 for 3 days. For the short pulse siRNA knockdown, parallel HCEC monolayers were subjected to scRNA or siRNA transfection by mixing 50 µl of serum-free, antibiotic-free SHEM with 1 µl of HiPerFect siRNA transfection reagent (final dilution, 1∶300) and 3 µl of 20 µM of scRNA or siRNA (final concentration, 100 nM) to p120, β-catenin, N-cadherin, ZO-1 or Kaiso drop-wise, followed by culturing in 250 µl of fresh SHEM at 37°C for 2 days. For prolonged p120 siRNA knockdown, HCEC monolayers were treated with 40 nM of scRNA or p120 siRNA added once a week on Day 14 for 2 weeks before switching to siRNA-free fresh SHEM for 10 days. BrdU was added at a final concentration of 10 µM in the culture medium for 24 hours before termination. Some cultures were treated with 1 µM XAV 939 in the culture medium for 48 hours immediately following 5 mM EDTA treatment (Senoo et al., 2000) or with 5 µg/ml nocodazole or 10 µM taxol or 5 µg/ml CT-04 or 20 µM Y27632 in the culture medium (Roczniak-Ferguson and Reynolds, 2003; Yanagisawa et al., 2004) during the entire period of p120 siRNA transfection. For cultures receiving transfection of the control plasmid or the S33Y β-catenin plasmid, 0.2 µg of the control plasmid or the S33Y β-catenin plasmid was mixed with 3 µl of SuperFect transfection reagent (Qiagen, Valencia, CA) and 50 µl of a serum-free DMEM/F12 medium for 30 minutes before being drop-wise added to the culture and incubated for 1 day.
TCF/LEF promoter assay
HCEC monolayers in 24-well dishes were cotransfected with 0.4% of the TCF/LEF construct that harbors TCF/LEF-binding sites and 0.01% of pRL-TK internal control plasmids with 1% SuperFect plasmid transfection reagent in SHEM. Twenty-four hours after transfection, the transfection medium was removed. HCEC monolayers were then treated with PBS or 5 mM EDTA for 1 hour and cultured in SHEM with or without bFGF or TGFβ1 or both or with or without addition of XAV 939 for another 48 hours. The samples collected were assayed for firefly luciferase and Renilla luciferase activities using a Dual-Luciferase Reporter Assay System (Promega, Madison, WI) and TD-20/20 luminometer (Turner BioSystems, Sunnyvale, CA). The ratio of firefly luciferase and Renilla luciferase activities was used to determine whether the promoters are activated.
RNA extraction, reverse transcription and real-time PCR
Total RNAs were extracted using RNeasy Mini Kit (Qiagen) and were reverse-transcribed using High Capacity Reverse Transcription Kit (Applied Biosystems). cDNA of each cell junction component was amplified by real-time RT-PCR using specific primer-probe mixtures and DNA polymerase in 7000 Real-time PCR System (Applied Biosystems). Real-time RT-PCR profile consisted of 10 minutes of initial activation at 95°C, followed by 40 cycles of 15 seconds denaturation at 95°C, and 1 minute annealing and extension at 60°C. The genuine identity of each PCR product was confirmed by the size determination using 2% agarose gels followed by ethidium bromide staining together with PCR marker according to EC3 Imaging System (BioImaging System, Upland, CA).
Immunostaining
HCEC monolayer cultures were air-dried and fixed in 4% formaldehyde, pH 7.0, for 15 minutes at room temperature, rehydrated in PBS, incubated with 0.2% Triton X-100 for 15 minutes, and rinsed three times with PBS for 5 minutes each. For double immunostaining to both BrdU and p120 or nuclear S100A4 or Kaiso, samples were fixed with 75% methanol plus 25% acetic acid for 15 minutes, denatured with 2 M HCl for 30 minutes at 37°C and neutralized by 0.1 M borate buffer, pH 8.5 for 5 minutes three times. After incubation with 2% BSA to block non-specific staining for 30 minutes, they were incubated with the desired first antibody (all at 1∶50 dilution) for 16 hours at 4°C. After three washes with PBS, they were incubated with corresponding Alexa-Fluor-conjugated secondary IgG for 60 minutes. The samples were then counterstained with Hoechst 33342 and analyzed with Zeiss LSM 700 confocal microscope (Thornhood, NY). Corresponding mouse and rabbit sera were used as negative controls for primary monoclonal and polyclonal antibodies, respectively.
RhoA activity assay
The assay of Rho activation was performed in 10–50 µg of protein of cell lysates using RhoA Activation Assay Biochem Kit (Cytoskeleton) to pull down the GTP-bound form of RhoA by a GST fusion protein containing rhotekin (7–89 residues) and RBD protein using brightly colored glutathione affinity beads. The amount of activated RhoA pulled down was quantitatively determined by western blot using anti-RhoA antibody.
Dot or western blotting
Cell lysates were prepared in RIPA buffer and resolved on 4–15% (w/v) gradient acrylamide gels under denaturing and reducing conditions for Western blotting. To prepare protein extracts from the membrane, the cytosol, and the nucleus, we followed Qproteome Cell Compartment protocol (Qiagen). Briefly, cells were first added with Extraction Buffer CE1, which selectively disrupts, but without solubilizing, the plasma membrane, followed by centrifugation at 100 g for 10 minutes to pellet plasma membranes and compartmentalized organelles, such as nuclei, mitochondria and the endoplasmic reticulum. The pellet was then resuspended in Extraction Buffer CE2, which solubilizes the plasma membrane as well as all organelle membranes except the nuclear membrane, followed by centrifugation at 6000 g for 10 minutes to pellet nuclei. The supernatant contains membrane proteins and proteins from the endoplasmic reticulum and mitochondria. Finally, the pellet containing nuclei was solubilized using Extraction Buffer CE3 and pelleted by centrifugation at 6800 g for 10 minutes. The protein extracts from the above three compartments were transferred to a nitrocellulose membrane, which was then blocked with 5% (w/v) fat-free milk in TBST (50 mM Tris-HCl, pH 7.5, 150 mM NaCl, 0.05% (v/v) Tween-20), followed by sequential incubation with specific primary antibodies against β-catenin, LEF1, p120, Kaiso, RhoA and their respective secondary antibodies using connexin (CN) 43, α-tubulin, and histone as the loading control for the membranous, cytosolic, and nuclear compartments, respectively. Immunoreactive proteins were detected with Western Lighting Chemiluminesence Reagent.
Statistics
All summary data were reported as means ± s.d. calculated for each group and compared using the Student’s unpaired t-test by Microsoft Excel (Microsoft, Redmont, WA). Test results were reported as two-tailed P values, where P<0.05 was considered statistically significant.
Supplementary Material
Acknowledgments
The innovations, methods for promoting cellular proliferation limited by contact inhibition because of adherent junctions in cells including HCECs, and for generating surgical grafts and tissues, were filed in an International PCT Patent Application (PCT/US07/79757) on September 27, 2007.
Footnotes
Funding
This study has been supported by the National Eye Institute, National Institutes of Health [grant number R43 EY 022502 to Y.-T.Z. and S.C.G.T.]; and in part by TissueTech, Miami, FL. Deposited in PMC for release after 12 months.
Supplementary material available online at http://jcs.biologists.org/lookup/suppl/doi:10.1242/jcs.103267/-/DC1
References
- Anastasiadis P. Z., Reynolds A. B. (2001). Regulation of Rho GTPases by p120-catenin. Curr. Opin. Cell Biol. 13, 604–610 10.1016/S0955-0674(00)00258-1 [DOI] [PubMed] [Google Scholar]
- Anastasiadis P. Z., Moon S. Y., Thoreson M. A., Mariner D. J., Crawford H. C., Zheng Y., Reynolds A. B. (2000). Inhibition of RhoA by p120 catenin. Nat. Cell Biol. 2, 637–644 10.1038/35023588 [DOI] [PubMed] [Google Scholar]
- Basile D. P., Friedrich J. L., Spahic J., Knipe N., Mang H., Leonard E. C., Changizi–Ashtiyani S., Bacallao R. L., Molitoris B. A., Sutton T. A. (2011). Impaired endothelial proliferation and mesenchymal transition contribute to vascular rarefaction following acute kidney injury. Am. J. Physiol. Renal Physiol. 300, F721–F733 10.1152/ajprenal.00546.2010 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bonanno J. A. (2003). Identity and regulation of ion transport mechanisms in the corneal endothelium. Prog. Retin. Eye Res. 22, 69–94 10.1016/S1350-9462(02)00059-9 [DOI] [PubMed] [Google Scholar]
- Chen J-K., Tsai R. J. F., Lin S-S. (1994). Fibroblasts isolated from human pterygia exhibit transformed cell charateristics. In Vitro Cell. Dev. Biol. Anim. 30, 243–248 10.1007/BF02632046 [DOI] [PubMed] [Google Scholar]
- Chen K. H., Harris D. L., Joyce N. C. (1999). TGF-beta2 in aqueous humor suppresses S-phase entry in cultured corneal endothelial cells. Invest. Ophthalmol. Vis. Sci. 40, 2513–2519 [PubMed] [Google Scholar]
- Daniel J. M. (2007). Dancing in and out of the nucleus: p120(ctn) and the transcription factor Kaiso. Biochim. Biophys. Acta 1773, 59–68 10.1016/j.bbamcr.2006.08.052 [DOI] [PubMed] [Google Scholar]
- Davis M. A., Ireton R. C., Reynolds A. B. (2003). A core function for p120-catenin in cadherin turnover. J. Cell Biol. 163, 525–534 10.1083/jcb.200307111 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Dohn M. R., Brown M. V., Reynolds A. B. (2009). An essential role for p120-catenin in Src- and Rac1-mediated anchorage-independent cell growth. J. Cell Biol. 184, 437–450 10.1083/jcb.200807096 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Eger A., Stockinger A., Park J., Langkopf E., Mikula M., Gotzmann J., Mikulits W., Beug H., Foisner R. (2004). beta-Catenin and TGFbeta signalling cooperate to maintain a mesenchymal phenotype after FosER-induced epithelial to mesenchymal transition. Oncogene 23, 2672–2680 10.1038/sj.onc.1207416 [DOI] [PubMed] [Google Scholar]
- Engelmann K., Böhnke M., Friedl P. (1988). Isolation and long-term cultivation of human corneal endothelial cells. Invest. Ophthalmol. Vis. Sci. 29, 1656–1662 [PubMed] [Google Scholar]
- Ferreri D. M., Minnear F. L., Yin T., Kowalczyk A. P., Vincent P. A. (2008). N-cadherin levels in endothelial cells are regulated by monolayer maturity and p120 availability. Cell Commun. Adhes. 15, 333–349 10.1080/15419060802440377 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Franz C. M., Ridley A. J. (2004). p120 catenin associates with microtubules: inverse relationship between microtubule binding and Rho GTPase regulation. J. Biol. Chem. 279, 6588–6594 10.1074/jbc.M312812200 [DOI] [PubMed] [Google Scholar]
- Gabbiani G. (2003). The myofibroblast in wound healing and fibrocontractive diseases. J. Pathol. 200, 500–503 10.1002/path.1427 [DOI] [PubMed] [Google Scholar]
- Habbig S., Bartram M. P., Müller R. U., Schwarz R., Andriopoulos N., Chen S., Sägmüller J. G., Hoehne M., Burst V., Liebau M. C., et al. (2011). NPHP4, a cilia-associated protein, negatively regulates the Hippo pathway. J. Cell Biol. 193, 633–642 10.1083/jcb.201009069 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hatzfeld M. (2005). The p120 family of cell adhesion molecules. Eur. J. Cell Biol. 84, 205–214 10.1016/j.ejcb.2004.12.016 [DOI] [PubMed] [Google Scholar]
- Heallen T., Zhang M., Wang J., Bonilla–Claudio M., Klysik E., Johnson R. L., Martin J. F. (2011). Hippo pathway inhibits Wnt signaling to restrain cardiomyocyte proliferation and heart size. Science 332, 458–461 10.1126/science.1199010 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Huang S. M., Mishina Y. M., Liu S., Cheung A., Stegmeier F., Michaud G. A., Charlat O., Wiellette E., Zhang Y., Wiessner S., et al. (2009). Tankyrase inhibition stabilizes axin and antagonizes Wnt signalling. Nature 461, 614–620 10.1038/nature08356 [DOI] [PubMed] [Google Scholar]
- Ichii T., Takeichi M. (2007). p120-catenin regulates microtubule dynamics and cell migration in a cadherin-independent manner. Genes Cells 12, 827–839 10.1111/j.1365-2443.2007.01095.x [DOI] [PubMed] [Google Scholar]
- Ireton R. C., Davis M. A., van Hengel J., Mariner D. J., Barnes K., Thoreson M. A., Anastasiadis P. Z., Matrisian L., Bundy L. M., Sealy L., et al. (2002). A novel role for p120 catenin in E-cadherin function. J. Cell Biol. 159, 465–476 10.1083/jcb.200205115 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Joyce N. C. (2005). Cell cycle status in human corneal endothelium. Exp. Eye Res. 81, 629–638 10.1016/j.exer.2005.06.012 [DOI] [PubMed] [Google Scholar]
- Joyce N. C., Harris D. L., Mello D. M. (2002). Mechanisms of mitotic inhibition in corneal endothelium: contact inhibition and TGF-beta2. Invest. Ophthalmol. Vis. Sci. 43, 2152–2159 [PubMed] [Google Scholar]
- Kay E. P., Gu X., Ninomiya Y., Smith R. E. (1993). Corneal endothelial modulation: a factor released by leukocytes induces basic fibroblast growth factor that modulates cell shape and collagen. Invest. Ophthalmol. Vis. Sci. 34, 663–672 [PubMed] [Google Scholar]
- Kelly K. F., Spring C. M., Otchere A. A., Daniel J. M. (2004). NLS-dependent nuclear localization of p120ctn is necessary to relieve Kaiso-mediated transcriptional repression. J. Cell Sci. 117, 2675–2686 10.1242/jcs.01101 [DOI] [PubMed] [Google Scholar]
- Kim N. G., Koh E., Chen X., Gumbiner B. M. (2011). E-cadherin mediates contact inhibition of proliferation through Hippo signaling-pathway components. Proc. Natl. Acad. Sci. USA 108, 11930–11935 10.1073/pnas.1103345108 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kim S. W., Park J. I., Spring C. M., Sater A. K., Ji H., Otchere A. A., Daniel J. M., McCrea P. D. (2004). Non-canonical Wnt signals are modulated by the Kaiso transcriptional repressor and p120-catenin. Nat. Cell Biol. 6, 1212–1220 10.1038/ncb1191 [DOI] [PubMed] [Google Scholar]
- Laing R. A., Neubauer L., Oak S. S., Kayne H. L., Leibowitz H. M. (1984). Evidence for mitosis in the adult corneal endothelium. Ophthalmology 91, 1129–1134 [DOI] [PubMed] [Google Scholar]
- Lee J. G., Kay E. P. (2006). FGF-2-mediated signal transduction during endothelial mesenchymal transformation in corneal endothelial cells. Exp. Eye Res. 83, 1309–1316 10.1016/j.exer.2006.04.007 [DOI] [PubMed] [Google Scholar]
- Lei Q. Y., Zhang H., Zhao B., Zha Z. Y., Bai F., Pei X. H., Zhao S., Xiong Y., Guan K. L. (2008). TAZ promotes cell proliferation and epithelial-mesenchymal transition and is inhibited by the hippo pathway. Mol. Cell. Biol. 28, 2426–2436 10.1128/MCB.01874-07 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Li W., Sabater A. L., Chen Y. T., Hayashida Y., Chen S. Y., He H., Tseng S. C. (2007). A novel method of isolation, preservation, and expansion of human corneal endothelial cells. Invest. Ophthalmol. Vis. Sci. 48, 614–620 10.1167/iovs.06-1126 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Masszi A., Fan L., Rosivall L., McCulloch C. A., Rotstein O. D., Mucsi I., Kapus A. (2004). Integrity of cell-cell contacts is a critical regulator of TGF-beta 1-induced epithelial-to-myofibroblast transition: role for beta-catenin. Am. J. Pathol. 165, 1955–1967 10.1016/S0002-9440(10)63247-6 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Matter K., Aijaz S., Tsapara A., Balda M. S. (2005). Mammalian tight junctions in the regulation of epithelial differentiation and proliferation. Curr. Opin. Cell Biol. 17, 453–458 10.1016/j.ceb.2005.08.003 [DOI] [PubMed] [Google Scholar]
- Nelson W. J., Nusse R. (2004). Convergence of Wnt, beta-catenin, and cadherin pathways. Science 303, 1483–1487 10.1126/science.1094291 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ogden S. R., Wroblewski L. E., Weydig C., Romero–Gallo J., O’Brien D. P., Israel D. A., Krishna U. S., Fingleton B., Reynolds A. B., Wessler S., et al. (2008). p120 and Kaiso regulate Helicobacter pylori-induced expression of matrix metalloproteinase-7. Mol. Biol. Cell 19, 4110–4121 10.1091/mbc.E08-03-0283 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Perez–Moreno M., Jamora C., Fuchs E. (2003). Sticky business: orchestrating cellular signals at adherens junctions. Cell 112, 535–548 10.1016/S0092-8674(03)00108-9 [DOI] [PubMed] [Google Scholar]
- Perez–Moreno M., Davis M. A., Wong E., Pasolli H. A., Reynolds A. B., Fuchs E. (2006). p120-catenin mediates inflammatory responses in the skin. Cell 124, 631–644 10.1016/j.cell.2005.11.043 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Perez–Moreno M., Song W., Pasolli H. A., Williams S. E., Fuchs E. (2008). Loss of p120 catenin and links to mitotic alterations, inflammation, and skin cancer. Proc. Natl. Acad. Sci. USA 105, 15399–15404 10.1073/pnas.0807301105 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Petroll W. M., Barry–Lane P. A., Cavanagh H. D., Jester J. V. (1997). ZO-1 reorganization and myofibroblast transformation of corneal endothelial cells after freeze injury in the cat. Exp. Eye Res. 64, 257–267 10.1006/exer.1996.0211 [DOI] [PubMed] [Google Scholar]
- Petroll W. M., Jester J. V., Bean J. J., Cavanagh H. D. (1998). Myofibroblast transformation of cat corneal endothelium by transforming growth factor-beta1, -beta2, and -beta3. Invest. Ophthalmol. Vis. Sci. 39, 2018–2032 [PubMed] [Google Scholar]
- Polette M., Mestdagt M., Bindels S., Nawrocki–Raby B., Hunziker W., Foidart J. M., Birembaut P., Gilles C. (2007). Beta-catenin and ZO-1: shuttle molecules involved in tumor invasion-associated epithelial-mesenchymal transition processes. Cells Tissues Organs 185, 61–65 10.1159/000101304 [DOI] [PubMed] [Google Scholar]
- Roczniak–Ferguson A., Reynolds A. B. (2003). Regulation of p120-catenin nucleocytoplasmic shuttling activity. J. Cell Sci. 116, 4201–4212 10.1242/jcs.00724 [DOI] [PubMed] [Google Scholar]
- Rossman K. L., Der C. J., Sondek J. (2005). GEF means go: turning on RHO GTPases with guanine nucleotide-exchange factors. Nat. Rev. Mol. Cell Biol. 6, 167–180 10.1038/nrm1587 [DOI] [PubMed] [Google Scholar]
- Ruzov A., Hackett J. A., Prokhortchouk A., Reddington J. P., Madej M. J., Dunican D. S., Prokhortchouk E., Pennings S., Meehan R. R. (2009). The interaction of xKaiso with xTcf3: a revised model for integration of epigenetic and Wnt signalling pathways. Development 136, 723–727 10.1242/dev.025577 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sack U., Stein U. (2009). Wnt up your mind – intervention strategies for S100A4-induced metastasis in colon cancer. Gen. Physiol. Biophys. 28, F55–F64 [PubMed] [Google Scholar]
- Senoo T., Obara Y., Joyce N. C. (2000). EDTA: a promoter of proliferation in human corneal endothelium. Invest. Ophthalmol. Vis. Sci. 41, 2930–2935 [PubMed] [Google Scholar]
- Slorach E. M., Chou J., Werb Z. (2011). Zeppo1 is a novel metastasis promoter that represses E-cadherin expression and regulates p120-catenin isoform expression and localization. Genes Dev. 25, 471–484 10.1101/gad.1998111 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Soto E., Yanagisawa M., Marlow L. A., Copland J. A., Perez E. A., Anastasiadis P. Z. (2008). p120 catenin induces opposing effects on tumor cell growth depending on E-cadherin expression. J. Cell Biol. 183, 737–749 10.1083/jcb.200805113 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Spring C. M., Kelly K. F., O’Kelly I., Graham M., Crawford H. C., Daniel J. M. (2005). The catenin p120ctn inhibits Kaiso-mediated transcriptional repression of the beta-catenin/TCF target gene matrilysin. Exp. Cell Res. 305, 253–265 10.1016/j.yexcr.2005.01.007 [DOI] [PubMed] [Google Scholar]
- Takesono A., Heasman S. J., Wojciak–Stothard B., Garg R., Ridley A. J. (2010). Microtubules regulate migratory polarity through Rho/ROCK signaling in T cells. PLoS ONE 5, e8774 10.1371/journal.pone.0008774 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Terry M. A. (2006). Endothelial keratoplasty: history, current state, and future directions. Cornea 25, 873–878 10.1097/01.ico.0000244869.54761.50 [DOI] [PubMed] [Google Scholar]
- van Hengel J., Vanhoenacker P., Staes K., van Roy F. (1999). Nuclear localization of the p120(ctn) Armadillo-like catenin is counteracted by a nuclear export signal and by E-cadherin expression. Proc. Natl. Acad. Sci. USA 96, 7980–7985 10.1073/pnas.96.14.7980 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Varelas X., Miller B. W., Sopko R., Song S., Gregorieff A., Fellouse F. A., Sakuma R., Pawson T., Hunziker W., McNeill H., et al. (2010). The Hippo pathway regulates Wnt/beta-catenin signaling. Dev. Cell 18, 579–591 10.1016/j.devcel.2010.03.007 [DOI] [PubMed] [Google Scholar]
- Wildenberg G. A., Dohn M. R., Carnahan R. H., Davis M. A., Lobdell N. A., Settleman J., Reynolds A. B. (2006). p120-catenin and p190RhoGAP regulate cell-cell adhesion by coordinating antagonism between Rac and Rho. Cell 127, 1027–1039 10.1016/j.cell.2006.09.046 [DOI] [PubMed] [Google Scholar]
- Xiao K., Allison D. F., Buckley K. M., Kottke M. D., Vincent P. A., Faundez V., Kowalczyk A. P. (2003). Cellular levels of p120 catenin function as a set point for cadherin expression levels in microvascular endothelial cells. J. Cell Biol. 163, 535–545 10.1083/jcb.200306001 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Yanagisawa M., Kaverina I. N., Wang A., Fujita Y., Reynolds A. B., Anastasiadis P. Z. (2004). A novel interaction between kinesin and p120 modulates p120 localization and function. J. Biol. Chem. 279, 9512–9521 10.1074/jbc.M310895200 [DOI] [PubMed] [Google Scholar]
- Zeisberg E. M., Tarnavski O., Zeisberg M., Dorfman A. L., McMullen J. R., Gustafsson E., Chandraker A., Yuan X., Pu W. T., Roberts A. B., et al. (2007). Endothelial-to-mesenchymal transition contributes to cardiac fibrosis. Nat. Med. 13, 952–961 10.1038/nm1613 [DOI] [PubMed] [Google Scholar]
- Zeng Q., Hong W. (2008). The emerging role of the hippo pathway in cell contact inhibition, organ size control, and cancer development in mammals. Cancer Cell 13, 188–192 10.1016/j.ccr.2008.02.011 [DOI] [PubMed] [Google Scholar]
- Zhang J., O’Donnell J. J., 3rd, Holian O., Vincent P. A., Kim K. S., Lum H. (2010). P120 catenin represses transcriptional activity through Kaiso in endothelial cells. Microvasc. Res. 80, 233–239 10.1016/j.mvr.2010.04.001 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zhao B., Li L., Lei Q., Guan K. L. (2010). The Hippo-YAP pathway in organ size control and tumorigenesis: an updated version. Genes Dev. 24, 862–874 10.1101/gad.1909210 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zhu Y. T., Hayashida Y., Kheirkhah A., He H., Chen S. Y., Tseng S. C. (2008). Characterization and comparison of intercellular adherent junctions expressed by human corneal endothelial cells in vivo and in vitro. Invest. Ophthalmol. Vis. Sci. 49, 3879–3886 10.1167/iovs.08-1693 [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.