

Abstract
9-Alkyladenine derivatives and ribose-modified N6-benzyladenosine derivatives were synthesized in an effort to identify selective ligands for the rat A3 adenosine receptor and leads for the development of antagonists. The derivatives contained structural features previously determined to be important for A3 selectivity in adenosine derivatives, such as an N6-(3-iodobenzyl) moiety, and were further substituted at the 2-position with halo, amino, or thio groups. Affinity was determined in radioligand binding assays at rat brain A3 receptors stably expressed in Chinese hamster ovary (CHO) cells, using [125I]AB-MECA (N6-(4-amino-3-iodobenzyl)adenosine-5′-(N-methyluronamide)), and at rat brain A1 and A2a receptors using [3H]-N6-PIA ((R)-N6-phenylisopropyladenosine) and [3H]CGS 21680 (2-[[[4-(2-carboxyethyl)-phenyl]ethyl]amino]-5′-(N-ethylcarbamoyl)adenosine), respectively. A series of N6-(3-iodobenzyl) 2-amino derivatives indicated that a small 2-alkylamino group, e.g., methylamino, was favored at A3 receptors. N6-(3-Iodobenzyl)-9-methyl-2-(methylthio)adenine was 61-fold more potent than the corresponding 2-methoxy ether at A3 receptors and of comparable affinity at A1 and A2a receptors, resulting in a 3–6-fold selectivity for A3 receptors. A pair of chiral N6-(3-iodobenzyl) 9-(2,3-dihydroxypropyl) derivatives showed stereoselectivity, with the R-enantiomer favored at A3 receptors by 5.7-fold. 2-Chloro-9-(β-d-erythrofuranosyl)-N6-(3-iodobenzyl)adenine had a Ki value at A3 receptors of 0.28 µM. 2-Chloro-9-[2-amino-2,3-dideoxy-β-d-5-(methylcarbamoyl)-arabinofuranosyl]-N6-(3-iodobenzyl)adenine was moderately selective for A1 and A3 vs A2a receptors. A 3′-deoxy analogue of a highly A3-selective adenosine derivative retained selectivity in binding and was a full agonist in the inhibition of adenylyl cyclase mediated via cloned rat A3 receptors expressed in CHO cells. The 3′-OH and 4′-CH2OH groups of adenosine are not required for activation at A3 receptors. A number of 2′,3′-dideoxyadenosines and 9-acyclic-substituted adenines appear to inhibit adenylyl cyclase at the allosteric “P” site.
Introduction
Adenosine is a ubiquitous chemical messenger or “local hormone” involved in regulation of many physiological functions.1 There are three classes of adenosine receptors: A1, A2, and A3. Tremendous advances have been made in recent years in the synthesis of selective agents acting at subtypes of adenosine receptors.2 Selective adenosine antagonists are under development for use in cognitive diseases (A1),3,4 renal failure (A1),5 Parkinson’s and Huntington’s diseases (A2),6 and cardiac arrhythmias (A1)7 Adenosine agonists (A1 and A3) are likewise of potential therapeutic interest as cerebroprotective agents, antiepileptic drugs, etc.4
The A3 receptor was only recently discovered,8 with its cloning from a rat brain library. When expressed in Chinese hamster ovary (CHO) cells, rat A3 receptors were found to inhibit adenylyl cyclase. A3 receptors are also present in the RBL-2H3 (rat basophilic leukemia) cell line, where adenosine activates phospholipase C.9 Fozard and Carruthers10 have attributed to A3 receptor activation a component of the hypotensive effects of adenosine agonists in rats that is not antagonized by xanthines. Activation of A3 receptors has been suggested by Downey and colleagues11 to be involved in the cardioprotective effects of preconditioning by adenosine agonists in rabbits. The occurrence of A3 receptors in the testes and brain12–14 also suggests that it may be important in regulation of reproduction and CNS function. It has been suggested that A3-selective antagonists might have anti-inflammatory properties.15 Recently, the A3 receptor was found to be localized on eosinophils in the human lung, and tissue from patients with pulmonary disease showed differential occurrence of A3 receptor expression.16 MacKenzie et al.29 reported evidence that A3 receptor activation inhibits the adhesion of killer lymphocytes to adenocarcinoma cells.
We have studied in detail the structure–activity relationships (SAR) for N6- and 5′-substituted adenosine derivatives17,18 as agonists at rat A3 receptors and for alkylxanthines as antagonists.19 We recently reported that an adenosine derivative, N6-(3-iodobenzyl)-5′-(N-methylcarbamoyl)adenosine (IB-MECA, 1; Figure 1), is a 50-fold selective agonist for rat brain A3 vs A1 or A2a receptors and is selective in in vivo behavioral experiments.14 Additional structure–activity probing led us to the highly A3-selective agonist N6-(3-iodobenzyl)-2-chloro-5′-(N-methylcarbamoyl)adenosine(Cl-IB-MECA, 2).
Figure 1.

Structures of adenosine (1 and 2) and adenine (3 and 4) derivatives studied as adenosine receptor A3 agonists and A1/A2 antagonists, respectively.
Rat A3 receptors are unlike A1 and A2 receptors in lack of antagonism by the usual high-affinity xanthine ligands, such as the xanthine amine congener, XAC. An A3 antagonist that is selective in rodents is lacking. Many xanthines that are potent antagonists at A1 and A2 receptors in the rat, rabbit, and human only weakly displaced the binding of radioligand from cloned rat A3 receptors.20 Linden et al.12 found that certain xanthines do bind appreciably to cloned sheep A3 receptors but generally with less affinity than at A1 and A2 receptors in a variety of species. The human A3 receptor was recently cloned,13 and its phamacological profile was found to resemble that of the sheep A3 receptor, i.e., many potent xanthines bind in the submicromolar range. We have studied the unusually large species dependence of affinity at A3 receptors.21 Xanthines that are generally A3-selective across species are needed as pharmacological and biochemical probes, in order to define more clearly the physiological role, distribution, and regulation of A3 adenosine receptors.
A class of 9-alkyladenine derivatives was reported to act as antagonists at A1 or A2a receptors.22–24 There are parallels in the structural determinants of affinity among adenine derivatives (antagonists) and those of the corresponding 9-ribosides (agonists) at A1 receptors. These structural features include cycloalkyl groups, such as the N6-cyclopentyl group, leading to selectivity for A1 receptors.23 The N6-cycloalkyladenine derivative (R,S)-N-0861, 3 (Figure 1), is 610-fold selective for A1 receptors.7 A similar attempt to introduce parallel A2a selectivity in 9-methyladenine derivatives, using 2-sub-stitution known to favor that subtype when present in adenosine analogues, was less successful.24 2-[(Phenyl-ethyl)oxy]-9-methyladenine, 4, for example, distinguishes between subtypes of A2 receptors and appears to be selective for the A2a subtype in the coronary vasculature but is nonselective between A2a and A1 receptors.24 In the present study we have applied to the 9-alkyladenines the structural features we have determined to be important for A3 selectivity when occurring in adenosine derivatives, including both N6-and 2-substituents.17,18
Results
Chemical Synthesis
Adenine analogues modified with the N6-(3-iodobenzyl) group were synthesized (chemical characterization in Table 1, structures and biological properties in Tables 2 and 3). Scheme 1 outlines the synthesis of 9-methyl derivatives of adenine. The synthesis of analogues with non-methyl substitution at the 9-position is shown in Scheme 2. The N6-(3-iodobenzyl) substituent in adenine derivatives is likely to be well suited for A3 affinity, on the basis of an assumed parallel in structure–activity relationship with adenosine derivatives. N6-(3-Iodobenzyl)adenosine is the only singly substituted adenosine derivative reported to be selective for A3 receptors in rat brain.18 Additional modifications were made at the 9-position, using groups other than methyl, and by substituting at the 2-position. Compounds 8–24 contain acyclic sub-stituents at the 9-position of adenine, and compounds 25–40 contain cyclic substituents. Adenine nucleoside analogues, containing erythrose (Scheme 3), modified 3′-deoxy- (35) or 2′,3′-dideoxyribose (29), 2′-substituted 2′,3′-dideoxyarabinose (30–32), arabinose (39), or talose (40) sugars, were included. Procedures for synthesis of 3′-deoxy and 2′,3′-dideoxy analogues are outlined in Schemes 4–6.
Table 1.
Characterization of 9-Alkyladenine and Ribose-Modified Adenosine Derivatives
| compd no. | mp (°C) | MS | formula | anal. |
|---|---|---|---|---|
| 8 | 159–161 | 366 (CI) | C13H12N5I1·0.3EtOAc | C, H, N |
| 9 | 185–187 | C14H14IN5O | C, H, N | |
| 10 | 125–128 | C15H16IN5O2·1H2O | C, H, N | |
| 11 | 126–127 | C15H16IN5O2 | C, H, N | |
| 12 | 160 dec | C14H12IN5O2·0.5H2O | C, H, N | |
| 13 | oil | 418 (EI) | C16H15IN6·1.5H2O | C, H; Nb |
| 14 | 192–193 | 400 (CI) | C13H11N5Cl1I1 | C, H, N |
| 15 | 203–205 | 381 (CI) | C13H13N6I1 | a |
| 16 | 202–203 | 396 (CI) | C13H14N7I1·0.2C6H14 | C, H, N |
| 17 | 185–186 | 395 (CI) | C14H15N6I1 | a |
| 18 | 190–191 | 409 (CI) | C15H17N6I1·0.6MeOH | C, H, N |
| 19 | 134–135 | 423 (CI) | C16H19N6I1 | C, H, N |
| 20 | 138 | 465 (CI) | C19H25N6I1·0.35C6H14 | C, H, N |
| 21 | 159 | 396 (CI) | C14H14N5O1I1·0.2C6H14·0.5MeOH | C, H, N |
| 22 | 160–161 | 412 (CI) | C14H13N5S1I1·0.35C6H14 | C, H, N |
| 23 | 199 dec | 474 (CI) | C18H15N6S1I1 | a |
| 24 | 130 | C16H18IN5O2S1 | a | |
| 25 | 145–147 | C16H15N5O3Cl1I1 | C, H, N | |
| 26 | 158–161 | C17H19N6O3I1 | a | |
| 27 | 180–182 | 456 (CI) | C16H15N5O1Cl1I1 | a |
| 29 | 130 | 387(CI) | C18H19N6O2C1 | a |
| 30 | 184 | 553 (EI) | C18H17N9O2Cl1I1 | a |
| 31 | 98 | 528 (CI) | C18H19N7O2Cl1I1 | a |
| 32 | 119–129 | C18H17N6O2Cl1I1F1·2H2O | C, H, N | |
| 33 | foam | 571 (CI) | C20H20N6O4Cl1I1 | a |
| 34 | foam | 607 (CI) | C19H20N6O5Cl1I1S1 | C, H, N |
| 35 | 162 | 528 (EI) | C18H18N6O3Cl1I1 | C, H, N |
| 36 | foam | 759 (EI) | C29H43N5O5Cl1I1Si1 | a |
| 37 | 120 dec | C19H16N6O4Cl1I1S1 | C, H, N | |
| 38 | foam | 753 (CI) | C32H26N6O6Cl1I1 | a |
High-resolution mass in FAB+ mode m / z determined to be within acceptable limits. 15: calcd, 381.0325; found, 381.0335. 17: calcd, 395.0481; found, 395.0463. 23: calcd, 475.0202; found, 475.0201. 27: calcd, 456.0078; found, 456.0077. 29: calcd, 386.1258; found, 386.1249. 30: calcd, 553.0239; found, 553.0226. 31: calcd, 527.0333; found, 527.0318. 33: calcd, 571.0358; found, 571.0361. 36: calcd, 760.1615; found, 760.1614. 38: calcd, 753.0725; found, 753.0745.
N: calcd, 18.87; found, 17.65.
Table 2.
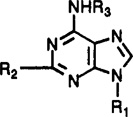
Affinities of 9-Alkyladenine and Ribose-Modified Adenosine Derivatives in Radioligand Binding Assays at Rat Brain A1, A2a, and A3 Receptorsa–c
 | ||||||||
|---|---|---|---|---|---|---|---|---|
| Ki (µM) or % inhibition | ||||||||
| Compound | R1 | R2 | R3 | Ki(A1)a | Ki(A2a)b | Ki(A3)c | A1/A3 | A2a/A3 |
| 1 | 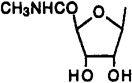 |
H | 3-I-Bz | 0.054 | 0.056 | 0.0011 | 49 | 51 |
| 2 | 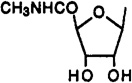 |
Cl | 3-I-Bz | 0.820 | 0.470 | 0.00033 | 2500 | 1400 |
| 5d | CH3 | H | cyclopentyl | 0.54 | 11 | 1330±390 | 0.00041 | 0.0083 |
| 6d,e | CH3CH2 | H | cyclopentyl | 0.44 | 17 | 30% (10−4) | <<1 | <1 |
| 7e | 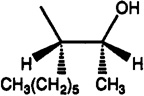 |
H | H | 0.455 | 60% (10−4) | 57% (10−4) | <<1 | - |
| 8 | CH3 | H | 3-I-Bz | 5.73±1.88 | 2.23±1.33 | 48.3±6.0 | 0.12 | 0.046 |
| 9 | HO(CH2)2 | H | 3-I-Bz | 22.9±3.7 | 15.1±1.6 | 62.5±14.5 | 0.37 | 0.24 |
| 10 | R-HOCH2-CHOH-CH2- | H | 3-I-Bz | 13.8±2.2 | 18.9±1.9 | 24.9±10.7 | 0.55 | 0.76 |
| 11 | S-HOCH2-CHOH-CH2- | H | 3-I-Bz | 19.1±2.2 | 41.8±12.5 | 142±13 | 0.13 | 0.29 |
| 12 | HO2C-CH2 | H | 3-I-Bz | 17% (10−4) | 9% (10−4) | 225±17 | - | - |
| 13 | NC(CH2)3 | H | 3-I-Bz | 6.03±1.37 | 18.7±5.8 | 185±17 | 0.032 | 0.10 |
| 14 | CH3 | CI | 3-I-Bz | 0.45±0.11 | 2.7±0.56 | 51.0±10.0 | 0.0088 | 0.053 |
| 15 | CH3 | NH2 | 3-I-Bz | 5.57±1.32 | 3.22±1.52 | 40.1±5.7 | 0.14 | 0.080 |
| 16 | CH3 | NH2NH | 3-I-Bz | 5.44±0.05 | 19.6±7.8 | 109±9 | 0.050 | 0.18 |
| 17 | CH3 | CH3NH | 3-I-Bz | 0.648±0.102 | 3.56±0.84 | 0.974±0.340 | 0.67 | 3.7 |
| 18 | CH3 | (CH3)2N | 3-I-Bz | 1.48±0.12 | 9.89±3.01 | 15.0±0.9 | 0.099 | 0.66 |
| 19f | CH3 | CH3(CH2)2NH | 3-I-Bz | 0.33±0.08 | 1.72±0.70 | 20% (30 µM) | <<1 | <<1 |
| 20f | CH3 | CH3(CH2)5NH | 3-I-Bz | 4.48±0.82 | 11±4% (10−5) | 19% (30 µM) | <1 | - |
| 21 | CH3 | CH3O | 3-I-Bz | 0.50±0.21 | 1.24±0.11 | 18.3±12.9 | 0.027 | 0.068 |
| 22 | CH3 | CH3S | 3-I-Bz | 1.89±0.59 | 1.64±0.39 | 0.299±0.074 | 6.3 | 5.5 |
| 23 | CH3 | 4-pyridyl-S | 3-I-Bz | 0.84±0.19 | 11.6±4.0 | 166±57 | 0.0051 | 0.070 |
| 24 | R-HOCH2-CHOH-CH2- | CH3S | 3-I-Bz | 1.34±0.09 | 78.9±23.5 | 8.59±4.29 | 0.16 | 9.2 |
| 25 | 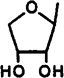 |
Cl | 3-I-Bz | 0.811±0.123 | 2.89±1.00 | 0.276±0.110 | 2.9 | 10 |
| 26 | 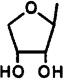 |
CH3NH | 3-I-Bz | 0.660±0.010 | 3.39±0.29 | 73.1±11.3 | 0.0090 | 0.046 |
| 27f | 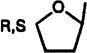 |
Cl | 3-I-Bz | 0.174±0.017 | 4.12±0.18 | 3.47±0.58 | 0.050 | 1.2 |
| 28 | 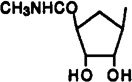 |
H | 3-I-Bz | 35.9±8.3 | 28±5% (10−4) | 19.5±4.7 | 1.8 | >1 |
| 29 | 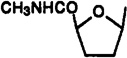 |
Cl | Bz | 11.5±1.3 | 220±65 | 30.9±1.3 | 0.37 | 7.1 |
| 30 | 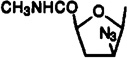 |
Cl | 3-I-Bz | 0.401±0.041 | 28.1±3.2 | 6.01±0.63 | 0.067 | 4.7 |
| 31 | 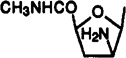 |
Cl | 3-I-Bz | 6.69±0.74 | 2% (10−4) | 3.40±0.79 | 2.0 | >50 |
| 32 | 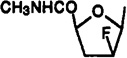 |
Cl | 3-I-Bz | 1.42±0.27 | 98.0±9.7 | 17.8±2.4 | 0.080 | 5.5 |
| 33 | 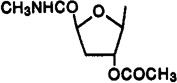 |
Cl | 3-I-Bz | 0.778±0.044 | 15.9±3.7 | 0.0625±0.0310 | 12 | 250 |
| 34 | 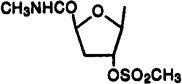 |
Cl | 3-I-Bz | 1.29±0.08 | 41.9±6.2 | 7.27±1.19 | 0.18 | 5.8 |
| 35 | 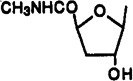 |
Cl | 3-I-Bz | 1.03±0.15 | 4.66±0.74 | 0.0329±0.0078 | 31 | 140 |
| 36 | 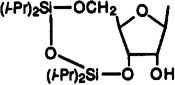 |
Cl | 3-I-Bz | 66.3±19.8 | 18±2% (10−4) | 13.1±3.5 | 5.1 | >7 |
| 37 | 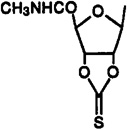 |
Cl | 3-I-Bz | 0.179±0.024 | 0.871±0.219 | 0.0122±0.0013 | 15 | 71 |
| 38 | 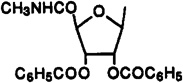 |
Cl | 3-I-Bz | 21% (10−4) | 7% (10−4) | 55±2% (10−4) | - | - |
| 39 | 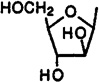 |
Cl | H | 24.2±7.9 | 90.0±12.7 | 14%(10−5) | - | - |
| 40 | 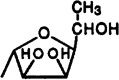 |
H | H | 150±28 | 54.7±3.1 | 6%(10−4) | <1 | <1 |
Displacement of specific [3H]PIA binding, unless noted, in rat brain membranes expressed as Ki ± SEM (µM) or percent inhibition at the indicated molar concentration (n = 3–6).
Displacement of specific [3H]CGS 21680 binding, unless noted, in rat striatal membranes expressed as Ki ± SEM (µM) or percent inhibition at the indicated molar concentration (n = 3–6).
Displacement of specific binding of [125I]-N6-(4-amino-3-iodobenzyl)adenosine-5′-(N-methyluronamide)25 from membranes of CHO cells stably transfected with the rat A3–cDNA expressed as Ki ± SEM (µM) or percent inhibition at the indicated molar concentration (n = 3–7).
Values at A1 and A2a receptors are from Thompson et al.23
Values are from van Galen et al.20 A3 affinity was measured by displacement of specific binding of [125I]APNEA in membranes of CHO cells stably transfected with the rat A3–cDNA.8 Ki values at A1 receptors are vs specific binding of [3H]-N6-cyclohexyladenosine or [3H]R-PIA. Ki values at A2a receptors are vs specific binding of [3H]NECA in the presence of 50 nM CPA or vs specific binding of [3H]CGS 21680 in rat striatal membranes.
Low aqueous solubility.
Table 3.
Effects on Adenylyl Cyclase in CHO Cells, Either Stably Transfected with Rat A3 Adenosine Receptors or Untransfecteda
| % inhibition of a. cyclase | effect on IB-MECA dose–resp curve |
||||
|---|---|---|---|---|---|
| compd | conc (µM) | ratio conc/Ki(A3) | CHO (A3) | CHO (cntrl) | |
| 1 | 100 | 9.1 × 104 | 44.4 ± 1.0 | nd | c |
| 0.1 | 91 | 22.0 ± 0.9 | nd | c | |
| 7 | 40 | 5.9 | nd | c | |
| 22 | 100 | 330 | 19.5 | nd | c |
| 24 | 40 | 4.6 | 5.5 ± 2.5 | 7.3 ± 3.3 | c |
| 25 | 100 | 360 | 28.8 ± 4.0 | 14.9 ± 1.4 | nd |
| 29 | 40 | 1.3 | 7.5 ± 3.8 | 10.8 ± 4.4 | c |
| 30 | 20 | 3.3 | 25.7 ± 3.6 | 18.0 ± 3.5 | nd |
| 100 | 17 | 27.8 | nd | nd | |
| 32 | 100 | 5.6 | 11.2 | nd | nd |
| 33 | 20 | 320 | 12.7 ± 1.0 | nd | nd |
| 35 | 40 | 1200 | 35.2 ± 7.8 | 5.2 ± 2.6 | nd |
| 100 | 3000 | 49.2 ± 3.7 | nd | nd | |
| 36 | 100 | 7.6 | 8.7 | nd | nd |
| 37 | 100 | 8200 | 63.4 ± 5.8 | 4.9 ± 2.7 | nd |
| 39 | 40 | 16.1 | nd | nd | |
| 40 | 40 | 0 | nd | c | |
| 5′-MeSAdo | 40 | 28 | 19.7 ± 2.7 | nd | nd |
In the presence of 1 µM forskolin.
Average ± SEM for three determinations or a single value.
No antagonism. nd, not determined.
Scheme 1a.

aReagents: (a) 3-iodobenzylamine-HCl, triethylamine, EtOH, rt; (b) CH3I, K2CO3, DMF; (c) NH2NH2; (d) NH2CH3/THF; (e) DMF, triethylamine, CH3O2CCH2NH2-HCl; (f) CH3(CH2)2NH2; (g) CH3(CH2)5NH2; (h) NaOCH3, MeOH; (i) NaSCH3, DMF–DME; (j) NaSH, pyridine.
Scheme 2a.

a Reagents: (a) R’I (9, 12), R’Br (13), or (2,2-dimethyl-1,3-dioxolan-4-yl)methyl p-toluenesulfonate, R (10, 47) or S (ll), and K2CO3, DMF; (b) 1 N HCl, 90 °C, 1 h; (c) NaSCH3, DMF.
Scheme 3a.

a Reaction conditions: (a) Ac2O, pyridine, rt, 24 h; (b) SnCl4, MeCN, N6-(3-iodobenyl)-2-chloroadenine,50 °C; (c) conc NH4OH, reflux.
Scheme 4a.

a Reagents: (a) i. CS2, NaH, MeI, THF, ii. Bu3SnH, Et3B, benzene; (b) NH3/MeOH; (c) RuO2, NaIO4, CHCl3:CH3CN:H2O (2:2:3); (d) MeOH, EDAC, DMAP; (e) CH3NH2/THF; (f) H2SO4, Ac2O, AcOH.
Scheme 6a.

a Reagents: (a) methanesulfonyl chloride, pyridine, CH2Cl2; (b) NaN3, DMF, 100 °C; (c) PPh3, NH4OH, THF–MeOH; (d) DAST.
Scheme 1 shows the route used to synthesize 9-methyladenine derivatives. The synthesis of the 2-unsub-stituted adenine derivative was carried out by substitution of 6-chloropurine, 41, using 3-iodobenzylamine, to provide N6-(3-iodobenzyl)adenine, 44. This was followed by alkylation at the 9-position, resulting in the 9-methyl analogue 8. Alternately, 2-substitution was introduced at the first synthetic stage with 2,6-dichlo-ropurine, 42, or 2-amino-6-chloropurine, 43, carried through the same sequence, leading to compound 14 or 15, respectively. 2-Chloro-N6-(iodobenzyl)adenine, 45a, was prepared as reported,18 except that the reaction condition used was at 50 °C for 3 h followed by stirring overnight at room temperature, resulting in an improved yield (70%). The 2-chloro group was readily replaced at elevated temperature by various nucleo-philes, such as amines (leading to compounds 16–20) or alkoxides (leading to compounds 21 and 22). Compound 23 was the unanticipated product of the reaction of 14 with sodium hydrosulfide in the presence of pyridine. The expected product, the corresponding 2-thiol, was not detected.
Combinations of 2- and 6-modifications with 9-sub-stituents larger than methyl were made according to Scheme 2. A 9-(2,3-dihydroxypropyl) substituent was introduced as the isopropylidene-protected form, and the protecting group was later cleaved in acid. Replacement of 2-chloro with the methylthio group was carried out as the final step, leading to compound 24.
The synthesis of a 9-erythrose derivative, 25, is shown in Scheme 3. Only the β-isomer was isolated from the condensation of N6-(3-iodobenzyl)-2-chloroadenine, 45b, with triacetylerythrose, 49. The synthesis of compound 27, a tetrahydrofuran derivative, was based on a similar procedure by Olsson and co-worker.23
Synthesis of ribose- and arabinose-modified analogues began with 5-O-benzoyl-1,2-O-isopropylidene-α-d-xylo-furanoside, 51 (Scheme 4). Following conversion of the 3-hydroxyl group to the 3-xanthate in situ, the material was deoxygenated by the action of tributyltin hydride and triethylborane, to give compound 52. Debenzoylation of the 5-position and oxidation of resulting alcohol 53 yielded acid 54 in good yields. The methylamide at the 5-position of compound 56 was introduced by esterification of the carboxylic acid to yield compound 55 followed by displacement with methylamine in a sealed bottle. The 1,2-isopropylidene group of compound 56 was cleaved, and the diol was acetylated in one pot by conventional methods to give compound 57. This sugar intermediate was condensed with the silylated adenine base 45b by a modified Vorbrüggen method37 to produce compound 33, which was deprotected in methanolic ammonia to yield 3′-deoxy-2-chloro-IB-MECA, 35. Deoxygenation of compound 35 via intermediate 61 produced the deiodinated 2′,3′-dideoxy compound 29. The β-2′-azide of 30 was introduced by displacement of the mesylate group of 34 with sodium azide. Furthermore, the 2′-azide could be reduced using triphenylphosphine/ammonium hydroxide in THF–methanol38 to give the β-2′-amino derivative 31. The β-2′-fluoro compound 32 was synthesized by reaction of compound 35 with DAST ((diethylamino)sulfur trifluoride).
In an attempt to synthesize 2-chloro-2′-deoxy-N6-(3-iodobenzyl)adenosine, the 3′- and 5′-hydroxyl groups of 2-chloro-N6-(3-iodobenzyl)adenosine18 were protected with 1,1,3,3,-tetraisopropyldisiloxyl protective group to yield compound 36. However, attempted deoxygenation of 36 using tributyltin hydride and AIBN (2,2′-azobis-(2-methylpropionitrile)) in toluene39 was sluggish and did not give the desired product.
Biological Activity
The analogues were tested in radioligand binding assays (Table 2) using rat cortical A1 receptors or striatal A2a receptors or in CHO cells stably transfected with rat brain A3 receptors.8,20 Radioligands for A1 and A2a receptors were the selective agonists [3H]-N6-(R)-phenylisopropyladenosine26 and [3H]CGS 21680, respectively.27 The radioligand used for binding to A3 receptors was the recently reported high-affinity agonist [125I]AB-MECA (N6-(4-amino-3-iodobenzyl)adenosine-5′-(N-methyluronamide)).25
Compound 5 (N-0840)23 and the corresponding 9-ethyl derivative 6 were similar in their binding profiles at adenosine receptors. The previously reported high selectivity of 5 for A1 vs A2a receptors was even greater vs A3 receptors. The structure of EHNA, 7, an inhibitor of adenosine deaminase, corresponded to removal of the cyclopentyl group of 5 and lengthening and hydroxylation of the 9-alkyl chain. The affinity of EHNA was comparable to that of 5 and 6; thus it bound well at A1 receptors and only weakly at A2a and A3 receptors.
The inclusion of the 3-iodobenzyl group at the N6-amine position of 9-methyladenine resulted in compound 8. The Ki value of this analogue at A3 receptors was 48 µM: very weak, yet more potent than the cyclopentyl analogues 5 and 6. At A1 receptors compound 8 was 1 order of magnitude less potent than the corresponding N6-cyclopentyl analogue 5. Thus, although perhaps not optimized for A3 selectivity in the adenine series, the 3-iodobenzyl group had properties favorable toward such selectivity. Consequently, it was included in additional analogues.
With N6-substitution constant, the 9-alkyl substituent was varied in compounds 9–13. An anionic alkyl group, as in the carboxylic acid derivative 12, led to diminished affinity at all receptor subtypes. Hydroxylic alkyl groups at the 9-position (compounds 9–11) offered no advantage in affinity at A3 receptors vs 9-Me. The hydroxyethyl derivative 9 was nearly identical in A3 affinity to the corresponding methyl analogue 4 yet was 4–6-fold less potent at both A1 and A2a receptors. A pair of chiral dihydroxy analogues, 10 and 11, demonstrated moderate stereoselectivity of binding favoring the R-configuration β to the 9-nitrogen. The R-isomer 10 was 5.7-fold more potent at A3 receptors than the corresponding S-isomer 11. No selectivity was observed at A1 receptors, and at A2a receptors the enantiomers differed in affinity by only 2-fold. Compound 10 was slightly more potent at A1 and A3 receptors than the monohydroxy derivative 9. The 9-(2,3-dihydroxypropyl)-adenines also appeared to have favorable water solubility. The maximum aqueous solubility of compound 10 was found to be 0.6 mM.
Substitution at the 2-position was probed in N6-(iodobenzyl)-9-methyladenine derivatives 14–24. Such substitutions had major effects on the affinity at A3, and to a lesser degree A1 and A2a, receptors. Chloro (14), amino (15), alkylamino (16–20), methyl ether (21), and methylthio ether (22–24) groups were included at this position. The 2-chloro analogue 14 was moderately A1-selective, by 110-fold vs A3 receptors but only by 6-fold vs A2a receptors. Affinity of 15 at A1 receptors was 13-fold greater than that found for the corresponding 2-unsubstituted derivative 8, while affinities at A2a and A3 receptors were unchanged.
Among 2-amino derivatives (15–20), Ki values at A3 receptors ranged from 1 to roughly 100 µM. At A1 receptors the range was more narrow, with the most potent displaying a Ki value of 0.33 µM (2-n-propylamino, 19) and the least potent 8.6 µM (2-n-hexylamino, 20). The primary amine 15 was identical in A1 affinity to the 2-unsubstituted derivative 8. Substitution on the 2-amino group indicated that a small alkyl group, as in the 2-methylamino analogue 17, was favored at rat A3 receptors. Lengthening of the chain (compounds 19 and 20) or formation of the corresponding hydrazine derivative 16 greatly diminished affinity at A3 receptors while maintaining affinity at A1 receptors. Thus, the 2-hydrazino compound 16 was 20-fold selective for A1 vs A3 receptors. In addition to having diminished potency at A3 receptors, the longer chain 2-amino analogues 19 and 20 proved to be of low water solubility, which interfered during the binding assay. 2-Dialkylamino, 18, vs monoalkylamino, 17, substitution was less well tolerated at rat A3 receptors than at A1 and A2a receptors.
The affinities of 2-thio and 2-oxo ethers were compared. The most dramatic difference between the 2-methoxy (21) and 2-methylthio (22) ethers was found at A3 receptors, at which the 2-methylthio analogue was 61-fold more potent. Thus, compound 22 proved to be only slightly selective (5–6-fold) for A3 vs either A1 or A2a receptors. At A1 receptors 21 was somewhat more potent (4-fold) than 22, while at A2a receptors there was no difference in affinity with Ki values of approximately 1 µM. The affinity of compound 23 indicates that the bulky pyridyl ring is tolerated at the 2-position well at A1 and poorly at A3 receptors. The combination of selectivity enhancing features at the 2- and 9-positions in compound 24 failed to achieve an additive effect on A3 selectivity; instead the compound was 9-fold A1-selective.
There is evidence that at A1 receptors 2′,3′-dideoxy-adenosines and other truncated ribose analogues act as antagonists or partial agonists.31–34 Thus, in an effort to identify leads for selective antagonists, derivatives of adenosine, i.e., based on 9-ribosides and other cyclic groups, were also included (compounds 25–38). Selectivity for A3 vs A2a receptors was observed for adenosine analogues 25 and 29–36. Omission of the 5′-hydroxy-methyl group in the erythrose derivative 25 provided slight A3 vs A1 selectivity with a Ki value of 0.28 µM. Combination of favorable N6- and 2-substitution, as in compound 26, maintained roughly micromolar potency at A1 and A2a receptors but was not tolerated at A3 receptors. A tetrahydrofuran derivative, compound 27, had a Ki value of 3.5 µM at A3 receptors. Compound 28, the carbocyclic analogue of IB-MECA, 1, was reported previously31 to be slightly selective for A3 receptors.
Compounds 29–35 contain 3′-deoxy or 2′,3′-dideoxy modifications of ribose-5′-(N-methylamide). The β-2′-azido derivative 30 was slightly more potent than the corresponding fluoro derivative 32 at all the receptor subtypes. A β-2′-amino derivative, 31, was 2-fold selective for A3 vs A1 receptors and inactive at A2a receptors. The 3′-deoxy analogue of IB-MECA, compound 35, was moderately A3-selective (31-fold vs A1 receptors) in the binding assays.
In compounds 33, 34, and 36–38, the ribose hydroxyl groups have been blocked by acylation or silylation. It is possible that some of the binding displacement observed resulted from lability of the blocking group, in which case these derivatives would constitute pro-drugs. Although they were all found to be stable in aqueous medium, the consequences of incubation with membranes remain untested. The potential use of these relatively hydrophobic yet biologically active adenosine analogues for in vivo therapeutics is under investigation.
Two other adenine glycosides, i.e., compounds 39 and 40, derivatives of arabinose and talose, respectively, having free 2′,3′-dihydroxy groups have been included in this study. These derivatives displayed only weak affinity at A1 and A2a receptors and no selectivity.
We examined the agonist and antagonist properties of 9-alkyladenine and adenosine derivatives in an adenylyl cyclase assay in A3 receptor-transfected CHO cells (Table 3). As in previous studies,18 adenylyl cyclase was inhibited by IB-MECA, 1, with an IC50 value of ~10−7 M in A3-transfected CHO cells (Figure 2), with a maximal degree of inhibition of 40–50%. The corresponding 2-chloro-3′-deoxyadenosine derivative, compound 35, proved to be a full agonist in the A3-mediated inhibition of adenylyl cyclase (Figure 2). 5′-Deoxy-5′-(methylthio)adenosine (Table 3) gave a robust agonist response at A3 receptors. This compound was reported to be an agonist at A1 receptors, a low-efficacy agonist at A2a receptors,35 and an antagonist at A2b receptors.36
Figure 2.

Agonist-elicited inhibition of adenylyl cyclase via rat A3 receptors in transfected CHO cells: circles, NECA; squares, Cl-IB-MECA triangles, compound 35.
Although the novel 9-methyladenine derivatives were designed to act as adenosine antagonists, we were unable to detect antagonism of A3 agonist-elicited inhibition of adenylyl cyclase in the transfected CHO cells. Compound 22, the 9-methyl 2-methylthio analogue, displaced radioligand binding with at Ki value <1 µM (Table 2), but at concentrations as high as 50 µM it failed to reverse the agonist-induced inhibition of adenylyl cyclase. Curiously, compound 22 alone inhibited adenylyl cyclase in A3-transfected CHO cells by 19%. Other ribose-truncated adenosine derivatives, such as the 9-acyclic compound 24, the erythrose derivative 25, and the 2′,3′-dideoxyadenosine derivative 29, similarly were found to inhibit adenylyl cyclase in A3-transfected CHO cell membranes (Table 3).
We investigated the possibility that the observed inhibition of adenylyl cyclase resulted from action at a site other than A3 receptors. For example, certain adenosine analogues with extensively modified ribose moieties, e.g., 9-(tetrahydrofur-2-yl)adenine,40 have been found to inhibit adenylyl cyclase by acting directly on the catalytic subunit at the allosteric “P” site. The control experiments, in which selected adenine and adenosine derivatives were tested for effects on adenylyl cyclase in untransfected CHO cells, were carried out. Several of the agents, such as 35 and 37, showed little inhibition, relative to that observed with the transfected cells (Table 3). Thus, for 35 and 37, activation of A3 receptors is still the most plausible explanation for the biological activity. However, for some of the derivatives, e.g., compounds 24 and 29, degrees of inhibition of adenylyl cyclase in control and transfected CHO cells were comparable. Therefore the “P” site may account for the cyclase inhibition seen for these adenine derivatives. Compounds 24 and 29 were roughly equipotent in inhibiting adenylyl cyclase directly, and the 2′,3′-dideoxy 2′-azido derivative 30 was somewhat more potent, with 18% inhibition at a concentration of 20 µM. Compounds 25 and 30 appeared to have mixed A3 agonist and “P” site inhibitory properties, since the inhibition in transfected CHO cells was significantly greater than in control CHO cells.
Another possible explanation for apparent inhibition of adenylyl cyclase through a non-receptor-mediated mechanism is that some of the compounds might be inhibiting adenosine deaminase and thereby raising the levels of endogenous agonist, which becomes available to activate A3 receptors. EHNA, 7, is known as an effective inhibitor of this enzyme and indeed at 40 µM inhibited adenylyl cyclase in the transfected CHO cells by 6%. However, with respect to most of the analogues synthesized in this study, it is known that the N6-substitution precludes potent interaction with adenosine deaminase, either as substrate or inhibitor, at adenosine deaminase.41
Discussion
In this study 9-alkyladenine and truncated adenosine derivatives were examined for selectivity for rat A3 receptors. Among the compounds studied, 22, 25, 28, 31, 33, and 35–38 were somewhat A3-selective in binding to rat adenosine receptors. Several of the compounds, 17, 24, 29, 30, 32, and 34, were A3-selective vs A2a but not A1 receptors. Ki values determined at A3 receptors were at best in the 10−7–10−6 M range. Due to the well-documented large species dependence among adenosine antagonists, specifically xanthines, at A3 receptors,8,12,13,21 it will be essential to examine compounds from this series for affinity at A3 receptors in other species. Even some of the nonselective adenines in this study may turn out to be selective ligands at human or sheep A3 receptors. It is still undetermined whether these species differences represent distinct receptor subtypes.
A comparison of the structural features of A3-selective agonists17,18 with the present results is useful. Due to the high selectivity of N6-(3-iodobenzyl)adenosine derivatives for A3 receptors, the same N6-substituent was included in most of the present adenine and adenosine derivatives. Although the N6-iodobenzyl group was found to be preferred over the N6-cyclopentyl group at A3 receptors (with a 28-fold increase in affinity in 8 vs 5), the SAR at this position must be explored in greater detail in order to draw general conclusions concerning adenosine/adenine parallels at this position. The 2-methylamino and 2-methylthio groups were favorable for affinity at A3 receptors in both the adenosine18 and adenine series, yet the 2-chloro group, which resulted in A1 selectivity in this series, was present in the highly A3 selective agonist N6-(3-iodobenzyl)-2-chloroadenosine-5′-(methyluronamide).18 Thus, in the series of N6-(3-iodobenzyl)adenosine-5′-(methyluronamide)s, A3 affinity varied in the order: 2-Cl > 2-CH3S > 2-CH3NH. In the current series of 9-methyladenines, the order was 2-CH3S > 2-CH3NH ≫ 2-Cl. Effects on affinity of substitution at 9- and 2-positions were highly interdependent; the groups were simply not additive. 2-Methylthio and 2-methylamino groups did not maintain micromolar affinity at A3 receptors when combined with 9-substituents larger than methyl, i.e., dihydroxypropyl (24) and erythrose (26), respectively. The lack of additivity in the structure–activity relationships of the adenine derivatives possibly indicates that those analogues having large 9-substituents and those with small 9-substituents have different binding modes. Another possible explanation for the low affinity of 26 could be that the methylamino group is increasing the basicity of the 6-amino group; a positive charge on the compound at neutral pH would render it much less active.
Increasing the number of hydroxyl groups on the 9-substituent (cf. 8–11) enhanced water solubility and had only minor effects on A3 affinity. The pair of chiral N6-(3-iodobenzyl) 9-(2,3-dihydroxypropyl) derivatives showed stereoselectivity only at A3 receptors, with the S-isomer more potent. If the 2′- and 3′-hydroxyl groups of the adenine derivatives correspond spatially at the receptor binding site to the 2′- and 3′-hydroxyls of receptor-bound adenosine, then it is the S-isomer that would more closely resemble adenosine. Thus, this slight stereoselectivity is without explanation.
Bruns30 reported that certain analogues of adenosine that were missing portions of the ribose moiety, such as the erythrose derivative, were low-efficacy activators or even antagonists of human A2b receptors. Bruns also was the first to detect adenosine antagonism by 9-methyladenine.42 On the basis of these and later finding,31–34 we have prepared various deoxy and other analogues of known A3-selective agonists, in an effort to identify antagonists. Included in this study are derivatives of 9-alkyladenine and 9-erythrose adenine.
The apparent inhibition of adenylyl cyclase by the dihydroxypropyl derivative 24 and the 2′,3′-dideoxy analogue 29 was shown to be due not to A3 receptor activation but to action at the allosteric “P” site on adenylate cyclase. It is likely that the adenylyl cyclase inhibition exhibited by the 9-methyladenine derivative 22 and the 2′,3′-dideoxy 2′-fluoro derivative 32 was also due to “P” site inhibition. The erythrose derivative 25 inhibited adenylyl cyclase as both a “P” site ligand and an A3 agonist in roughly equal proportions.
In the case of agonists there is a good correlation between potency in inhibiting cyclase in the rat A3-transfected cell membranes and the relative Ki values obtained in binding experiments at A3 receptors.18 However, there is a discrepancy for antagonists at rat A3 receptors. We have shown that theophylline, which has a Ki value of 85 µM at rat A3 receptors,21 and other xanthines lack functional antagonistic properties vs A3 agonist-elicited adenylyl cyclase inhibition in A3-transfected CHO cell membranes.20 So far, none of the ligands we have examined, including xanthines in a previous study,19 are useful antagonists at A3 receptors (even nonselective). Although some of these adenine derivatives, such as 22, were considerably more potent in A3 binding than theophylline, functional antagonism in this assay was not seen, perhaps because of inhibition of adenylate cyclase through the “P” site. It will be instructive to test the adenine derivatives at A3 receptors in other species, in which a possible gain in receptor affinity may avoid the “P” site complication.
Structural requirements for activation at A3 receptors do not include either the 3′-hydroxyl group (see 35) or the 4′-CH2OH group (see 25). The 3′-deoxy-IB-MECA derivative, 35, elicited a full inhibitory effect on adenylyl cyclase (Figure 2), and 37, the 2′,3′-thiocarbonyl derivative of Cl-IB-MECA, caused a >60% inhibition at high concentrations. Thus both gave full agonist responses. Similarly, it has been shown that the 3′-deoxy analogue of R-PIA was a full agonist at A1 receptors.33 Additional pharmacological studies are needed to clearly distinguish full and partial agonism of the other adenosine derivatives, such as the erythrose derivative 25, the azido derivative 30, and the 2′-acetoxy 3′-deoxy derivative 33. At A2b receptors, adenosine-9-β-d-erythrofuranoside, the parent structure related to 25, acted as a competitive antagonist.30
In conclusion, we have demonstrated the feasibility of developing A3 receptor-selective ligands based on substituted adenine derivatives, although optimization of selectivity remains a challenge. But the expectation of antagonizing rat A3 receptors in an adenylyl cyclase functional assay by modifying the ribose sugar has not been realized. It is possible that antagonism of second-messenger effects by adenine derivatives, that in our study weakly inhibited adenylyl cyclase, or xanthines, that bind to A3 receptors but have no effect on agonist-elicited inhibition of adenylyl cyclase, would be observed under different conditions. Such conditions might include higher concentrations of the agents, use of a different species in which higher affinity is attained,21 or another functional assay, such as phospholipase C.9 It is also possible that non-purine antagonists will provide leads for A3 selectivity, although a screen of five such A1 antagonists of diverse structure indicated negligible affinity at rat A3 receptors.20
Experimental Section
Chemistry
New compounds were characterized (and resonances assigned) by 300 MHz proton nuclear magnetic resonance spectroscopy using a Varian GEMINI-300 FT-NMR spectrometer. Unless noted, chemical shifts are expressed as ppm downfield from tetramethylsilane. Synthetic intermediates were characterized by chemical ionization mass spectrometry (NH3) on a JEOL SX102 mass spectrometer. In the EI mode accurate mass was determined using a VG7070F mass spectrometer. C, H, and N analyses (Table 1) were carried out by Atlantic Microlabs (Norcross, GA), and ±0.4% was acceptable. All adenine derivatives were judged to be homogeneous using thin layer chromatography (silica gel, 0.25 mm, glass backed; Alltech Assoc., Deerfield, IL) following final purification. Compound 5, 2-chloroadenosine, and EHNA were obtained from Research Biochemicals International (Natick, MA). Analytical TLC plates and silica gel (230–400 mesh) were purchased from VWR (Bridgeport, NJ). Compound 6 was kindly provided by Prof. Ray A. Olsson (University of South Florida). The solubility of 10 was measured by boiling the solid in water and cooling followed by measurement of the concentration by UV. The ε270 (λmax) value for compound 3 in methanol was found to be 19 000. IB-MECA, Cl-IB-MECA, and compound 38 were prepared as described.17,18 Compounds 39 and 40 were obtained from Dr. John W. Daly (NIDDK).
N6-(3-Iodobenzyl)-9-methyladenine (8)
A mixture of 6-chloropurine (41; 100 mg, 0.65 mmol), 3-iodobenzylamine hydrochloride (192 mg, 0.71 mmol), and triethylamine (0.27 mL, 1.94 mmol) in absolute ethanol (2 mL) was heated for 24 h at 80 °C in a sealed tube. After cooling, a solid was collected by suction filtration, washed with ethyl alcohol, and dried to give compound 44 (191.3 mg, 84.0%): 1H NMR (DMSO-d6) δ 4.67 (br s, 2 H, CH2), 7.11 (pseudo t, J = 7.6 and 7.5 Hz, 1 H, H-16), 7.37 (d, J = 7.9 Hz, 1 H, H-17), 7.58 (d, J = 7.6 Hz, 1 H, H-15), 7.73 (s, 1 H, H-13), 8.12 and 8.17 (each s, 1 H, H-8 and H-2), 8.25 (br s, 1 H, exchangeable with D2O, N6H), 12.95 (br s, 1 H, exchangeable with D2O, N9H).
To a solution of compound 44 (100 mg, 0.28 mmol) in dry DMF (4 mL) were added anhydrous potassium carbonate (78.7 mg, 0.57 mmol) and methyl iodide (0.365 mL, 5.7 mmol). The reaction mixture was stirred for 1 h and 40 min at room temperature. The solid was removed by suction, and the residue was purified by preparative TLC (chloroform–methanol, 10:1) to give compound 8 [Rf = 0.51 (chloroform–methanol, 10:1); 25 mg, 24.0%]: 1H NMR (DMSO-d6) δ 3.73 (s, 3 H, CH3), 4.67 (br s, 2 H, CH2), 7.10 (pseudo t, J = 7.9 and 7.6 Hz, 1 H, H-16), 7.36 (d, J = 7.5 Hz, 1 H, H-17), 7.58 (d, J = 7.7 Hz, 1 H, H-15), 7.71 (s, 1 H, H-13), 8.12 and 8.21 (each s, 1 H, H-8 and H-2), 8.29 (br s, 1 H, exchangeable with D2O, N6H).
9-(2-Hydroxyethyl)-N6-(3-iodobenzyl)adenine (9)
To a solution of compound 44 (20 mg, 0.056 mmol) and iodoethanol (100 µL) in dry DMF (0.5 mL) was added anhydrous K2CO3 (50 mg). The mixture was stirred at room temperature for 10 h and filtered to remove inorganic solids. The filtrate was evaporated to dryness and the residue purified by preparative TLC (CH2Cl2-MeOH, 10:1) to give compound 9 (Rf = 0.42), 27 mg (80%): 1H NMR (DMSO-d6) δ 3.20 (br s, 1 H, exchangeable with D2O, OH), 3.75 (t, J = 7 Hz, 2 H, CH2), 4.21 (t, J = 7 Hz, 2 H, CH2), 4.67 (br s, 2 H, CH2), 7.10 (pseudo t, J = 7.9 and 7.6 Hz, 1 H, H-16), 7.40 (d, J = 7.5 Hz, 1 H, H-17), 7.57 (d, J = 7.7 Hz, 1 H, H-15), 7.71 (s, 1 H, H-13), 8.12 and 8.20 (each s, 1 H, H-8 and H-2), 8.31 (br s, 1 H, exchangeable with D2O, N6H).
(R)-9-(2,3-Dihydroxypropyl)-N6-(3-iodobenzyl)adenine (10)
To a solution of compound 44 (60 mg, 0.267 mmol) and (R)-(−)-(2,2-dimethyl-1,3-dioxolan-4-yl)methyl p-toluene-sulfonate (100 mg, 0.35 mmol) in dry DMF (2 mL) was added anhydrous K2CO3 (200 mg). The reaction mixture was heated at 50 °C for 20 h. After cooling to room temperature, the reaction mixture was filtered and the filtrate evaporated to dryness. The residue was dissolved in 1 N HCl (10 mL) and heated at 80 °C for 1 h. With cooling in ice, the reaction mixture was neutralized by dropwise addition of concentrated NH4OH and evaporated to dryness. The residue was purified by preparative TLC (CH2Cl2-MeOH, 9:1, Rf = 0.35) to give 10, 80 mg (70%): 1H NMR (DMSO-d6) δ 3.46 (m, 2 H, CH2), 3.68 (m, 2 H, CH2), 4.05 (m, 1 H, CH), 4.67 (br s, 2 H, CH2), 4.85 (t, 1 H, exchangeable with D2O, OH), 5.12 (d, 1 H, exchangeable with D2O, OH), 7.13 (pseudo t, J = 7.9 and 7.6 Hz, 1 H, H-16), 7.36 (d, J = 7.5 Hz, 1 H, H-17), 7.52 (d, J = 7.7 Hz, 1 H, H-15), 7.64 (s, 1 H, H-13), 8.07 and 8.19 (each s, 1 H, H-8 and H-2), 8.33 (br s, 1 H, exchangeable with D2O, N6H).
(S)-9-(2,3-Dihydroxypropyl)-N6-(3-iodobenzyl)adenine (11)
Compound 11 was synthesized as described for 10 (same scale) from (S)-(+)-(2,2-dimethyl-1,3-dioxolan-4-yl)methyl p-toluenesulfonate. Yield of the purified product 11 was 69%. The 1H NMR in DMSO-d6 was similar to that of compound 10.
2-[N6-(3-Iodobenzyl)adenin-9-yl]acetic Acid (12)
This compound was prepared by a similar procedure as described for 9, starting with compound 44 (0.056 mmol), iodoacetic acid (100 mg), and K2CO3 (50 mg) in dry DMF (0.5 mL). The reaction mixture was neutralized with glacial acetic acid and evaporated to dryness. The yield of 12 after preparative TLC purification (CH2Cl2-MeOH, 9:1, Rf = 0.25) was 31 mg (85%): 1H NMR (DMSO-d6) δ 4.55 (s, 2 H, CH2), 4.78 (s, 2 H, CH2), 7.16 (pseudo t, J = 7.9 and 7.6 Hz, 1 H, H-16), 7.42 (d, J = 7.5 Hz, 1 H, H-17), 7.61 (d, J = 7.7 Hz, 1 H, H-15), 7.79 (s, 1 H, H-13), 8.40 and 8.45 (each s, 1 H, H-8 and H-2), 8.90 (br s, 1 H, exchangeable with D2O, N6H), 12.90 (br s, 1 H, CO2H).
9-(3-Cyanopropyl)-N6-(3-iodobenzyl)adenine (13)
A solution of N6-(3-iodobenzyl)adenine (44; 50 mg, 140 µmol), 4-bromobutyronitrile (300 mg, 2.0 mmol), and anhydrous potassium carbonate (150 mg, 1.1 mmol) in DMF (2 mL) was stirred for 12 h at 80 °C. Following addition of 10 mL of half-saturated sodium chloride, an oil separated. The oil was chromatographed on a preparative silica gel TLC plate (chloroform–methanol, 95:5, Rf = 0.31) to give compound 13 (40 mg, 66%): MS (EI) m / z 418 (M+), 350, 291, 232, 187.
2-Chloro-N6-(3-iodobenzyl)-9-methyladenine (14)
A solution of 2,6-dichloropurine (42; 2 g, 10.6 mmol), 3-iodoben-zylamine hydrochloride (3.14 g, 11.6 mmol), and triethylamine (4.42 mL, 31.7 mmol) in ethanol (20 mL) was stirred for 5 days at room temperature. A solid was collected by suction, washed with a small amount of ethanol, and dried to give compound 45a (2.32 g, 57.0%) which was recrystallized from methanol: 1H NMR (DMSO-d6) δ 4.59 (br s, 2 H, CH2), 7.13 (pseudo t, J = 8.2 and 7.5 Hz, 1 H, H-16), 7.36 (d, J = 7.5 Hz, 1 H, H-17), 7.61 (d, J = 7.5 Hz, 1 H, H-15), 7.74 (s, 1 H, H-13), 8.14 (s, 1 H, H-8), 8.75 (br s, 1 H, exchangeable with D2O, N6H), 13.14 (br s, 1 H, exchangeable with D2O, N9H); MS (CI, NH3) m/z 386 (M+ + 1).
A mixture of compound 45a (356 mg, 0.92 mmol), methyl iodide (2.08 mL, 32.4 mmol), and potassium carbonate (256 mg, 1.85 mmol) in DMF (12 mL) was stirred for 1 h and 40 min at room temperature. After filtration of potassium carbonate, the filtrate was mixed with water (100 mL) and chloroform (30 mL). During evaporation of the organic solvent, a slightly yellow solid formed. It was collected by suction and dried to yield compound 14 (303 mg, 82.0%): 1H NMR (DMSO-d6) δ 3.70 (s, 3 H, CH3), 4.60 (br s, 2 H, CH2), 7.13 (t, J = 7.6 Hz, 1 H, H-16), 7.36 (d, J = 7.7 Hz, 1 H, H-17), 7.60 (d, J = 7.7 Hz, 1 H, H-15), 7.73 (s, 1 H, H-13), 8.14 (s, 1 H, H-8), 8.80 (br s, 1 H, exchangeable with D2O, N6H).
2-Amino-N6-(3-iodobenzyl)-9-methyladenine (15)
A mixture of 6-chloroguanine (43; 100 mg, 0.59 mmol), 3-iodobenzylamine hydrochloride (175 mg, 0.65 mmol), and triethylamine (0.25 mL, 1.79 mmol) in ethanol (2 mL) was heated for 94 h at 80 °C. The solution was cooled and crystallized by addition of water. A colorless solid was collected by suction and dried to give compound 46 (161 mg, 75.0%): 1H NMR (DMSO-d6) δ 4.62 (br s, 2 H, CH2), 5.70 (br s, 2 H, exchangeable with D2O, NH2), 7.11 (pseudo t, J = 7.9 and 7.7 Hz, 1 H, H-16), 7.37 (d, J = 7.6 Hz, 1 H, H-17), 7.57 (d, J = 7.9 Hz, 1 H, H-15), 7.71 (s, 1 H, H-13), 7.66 (s, 1 H, H-8), 12.09 (br s, 1 H, exchangeable with D2O, N6H).
A mixture of compound 46 (100 mg, 0.27 mmol), methyl iodide (0.35 mL, 5.46 mmol), and anhydrous potassium carbonate (75 mg, 0.54 mmol) in dry DMF (4 mL) was stirred for 1.1 h at room temperature. A solid was removed by suction filtration, and the filtrate was concentrated and purified by preparative TLC (chloroform–methanol, 10:1) to give compound 15 [Rf = 0.46 (chloroform-methanol, 10:1); 3 mg, 2.9%]: 1H NMR (DMSO-d6) δ 3.54 (s, 3 H, CH3), 4.61 (br s, 2 H, CH2), 5.86 (br s, 2 H, exchangeable with D2O, NH2), 7.10 (t, J = 7.7 and 7.6 Hz, 1 H, H-16), 7.27 (d, J = 7.3 Hz, 1 H, H-17), 7.36 (d, J = 7.5 Hz, 1 H, H-15), 7.55 and 7.58 (each s, 1 H, H-13 and H-8), 7.75 (br s, 1 H, exchangeable with D2O, N6H).
2-Hydrazino-N6-(3-iodobenzyl)-9-methyladenine (16)
A solution of compound 14 (25 mg, 0.06 mmol) in hydrazine hydrate (1 mL) was heated for 17 h at 82 °C in a sealed bottle. Water (3 mL) was added, and a colorless solid was separated by suction and dried to yield compound 16 (19.9 mg, 80.6%): 1H NMR (DMSO-d6) δ 3.59 (s, 3 H, 9-CH3), 4.08 (br s, 2 H, exchangeable with D2O, NH2), 4.61 (br s, J = 5.3 Hz, 2 H, CH2), 7.10 (t, J = 7.6 Hz, 1 H, H-16), 7.35 (s, 1 H, exchangeable with D2O, NH), 7.39 (d, J = 7.6 Hz, 1 H, H-17), 7.57 (d, J = 7.6 Hz, 1 H, H-15), 7.73 (s, 2 H, H-13 and H-8), 7.92 (br s, 1 H, exchangeable with D2O, N6H).
N6-(3-Iodobenzyl)-2-(methylamino)-9-methyladenine (17)
A mixture of compound 14 (25 mg, 0.06 mmol), 2 M methylamine in THF (1 mL), and 40% methylamine in water (1 mL) was stirred for 14 h at 85 °C in a sealed bottle. After removal of volatiles in vacuo, the residue was triturated with methanol–water and a solid was collected by suction, washed with water (10 mL), and dried to give compound 17 (22 mg, 89.0%): 1H NMR (DMSO-d6) δ 2.76 (d, J = 4.6 Hz, 3 H, NHCH3), 3.55 (s, 3 H, 9-CH3), 4.59 (br s, 2 H, CH2), 6.28 (br s, 1 H, exchangeable with D2O, NHCH3), 7.10 (pseudo t, J = 7.9 and 7.6 Hz, 1 H, H-16), 7.38 (d, J = 7.6 Hz, 1 H, H-17), 7.57 (d, J = 7.6 Hz, 1 H, H-15), 7.67 (s, 1 H, H-13), 7.35 (s, 1 H, H-8), 7.83 (br s, 1 H, exchangeable with D2O, N6H).
2-(Dimethylamino)-N6-(3-iodobenzyl)-9-methyladenine (18)
A mixture of compound 14 (40 mg, 0.1 mmol), glycine methyl ester hydrochloride (310 mg, 2.47 mmol), and triethylamine (0.7 mL, 5.0 mmol) in DMF (2 mL) was heated for 22 h at room temperature in a sealed bottle. After cooling, the mixture was concentrated to dryness and purified using silica gel column chromatography (chloroform–methanol, 20:1) to give compound 18 (25 mg, 53.5%) as a colorless solid: 1H NMR (DMSO-d6) δ 3.06 (s, 6 H, N(CH3)2), 3.58 (s, 3 H, 9-CH3), 4.55 (br s, 2 H, CH2), 7.10 (pseudo t, J = 8.0 and 7.6 Hz, 1 H, H-16), 7.38 (d, J = 7.7 Hz, 1 H, H-17), 7.56 (d, J = 8.0 Hz, 1 H, H-15), 7.70 (s, 1 H, H-13), 7.77 (s, 1 H, H-8), 7.92 (br s, 1 H, exchangeable with D2O, N6H).
N6-(3-Iodobenzyl)-9-methyl-2-(n-propylamino)adenine (19)
A mixture of compound 14 (22.5 mg, 0.056 mmol) and n-propylamine (2 mL) was stirred at 85 °C for 36 h in a sealed bottle. After evaporation of volatiles, the residue was purified on preparative TLC (chloroform–methanol, 20:1) to give compound 19 (17.3 mg, 72.8%) as a slightly yellow solid: 1H NMR (DMSO-d6) δ 0.85 (pseudo t, J = 7.5 and 7.3 Hz, 3 H, CH3), 1.47 (sixtet, J = 7.2 Hz, 2 H, CH2), 3.30 (m, 2 H, CH2), 3.54 (s, 3 H, 9-CH3), 4.58 (br s, 2 H, CH2), 6.33 (br s, 1 H, exchangeable with D2O, NH), 7.10 (pseudo t, J = 8.0 and 7.7 Hz, 1 H, H-16), 7.36 (d, J = 7.7 Hz, 1 H, H-17), 7.57 (d, J = 8.2 Hz, 1 H, H-15), 7.66 (s, 1 H, H-13), 7.72 (s, 1 H, H-8), 7.80 (br s, 1 H, exchangeable with D2O, N6H).
2-(n-Hexylamino)-N6-(3-iodobenzyl)-9-methyladenine (20)
A mixture of compound 14 (23.5 mg, 0.059 mmol) and n-hexylamine (1 mL) was heated for 4.5 days at 80 °C in a sealed bottle. After evaporation of volatiles, the residue was purified on preparative TLC (chloroform–methanol, 20:1) to give compound 20 (23.5 mg, 86.0%): 1H NMR (DMSO-d6) δ 0.84 (m, 3 H, CH3), 1.25 (m, 6 H, CH2), 1.45 (m, 2 H, CHd), 3.17 (m, 2 H, CH2), 3.54 (s, 3 H, 9-CH3), 4.58 (br s, 2 H, CH2), 6.32 (br s, 1 H, exchangeable with D2O, NH), 7.09 (pseudo t, J = 7.8 and 7.6 Hz, 1 H, H-16), 7.35 (d, J = 7.8 Hz, 1 H, H-17), 7.57 (d, J = 7.7 Hz, 1 H, H-15), 7.66 (s, 1 H, H-13), 7.71 (s, 1 H, H-8), 7.82 (br s, 1 H, exchangeable with D2O, N6H).
N6-(3-Iodobenzyl)-2-methoxy-9-methyladenine (21)
A mixture of compound 14 (21 mg, 0.052 mmol) and sodium methoxide (1.5 mg of Na) was heated for 14 h at 85 °C in a sealed bottle. The reaction mixture was concentrated to dryness, and the residue was crystallized from methanol–water to give compound 21 (19 mg, 86.0%): 1H NMR (DMSO-d6) δ 3.64 (s, 3 H, 9-CH3), 3.81 (s, 3 H, OCH3), 4.59 (br s, 2 H, CH2), 7.11 (t, J = 7.6 Hz, 1 H, H-16), 7.37 (d, J = 7.6 Hz, 1 H, H-17), 7.59 (d, J = 7.6 Hz, 1 H, H-15), 7.74 (s, 1 H, H-13), 7.92 (s, 1 H, H-8), 8.37 (br s, 1 H, exchangeable with D2O, N6H).
N6-(3-Iodobenzyl)-9-methyl-2-(methylthio)adenine(22)
A mixture of compound 14 (24.4 mg, 0.061 mmol) and sodium thiomethoxide (8 mg, 0.1 mmol) in DMF–DME (1:1, 1.5 mL) was heated for 22 h at 110 °C in a sealed bottle. After cooling, the reaction mixture was concentrated to dryness and the residue was purified using silica gel column chromatography (chloroform–methanol, 20:1) to give compound 22 (13 mg, 52.0%) as a colorless solid: 1H NMR (DMSO-d6) δ 2.45 (s, 3 H, SCH3), 3.67 (s, 3 H, 9-CH3), 4.60 (br s, 2 H, CH2), 7.11 (pseudo t, J = 7.9 and 7.6 Hz, 1 H, H-16), 7.36 (d, J = 7.0 Hz, 1 H, H-17), 7.58 (d, J = 8.0 Hz, 1 H, H-15), 7.74 (s, 1 H, H-13), 7.99 (s, 1 H, H-8), 8.43 (br s, 1 H, exchangeable with D2O, N6H).
N6-(3-Iodobenzyl)-9-methyl-2-(4-pyridylthio)adenine (23)
A mixture of compound 14 (20.4 mg, 0.051 mmol) and sodium hydrosulfide hydrate (11 mg, 0.2 mmol) in pyridine (1.5 mL) was heated for 5 days at 100 °C in a sealed bottle. After cooling, the reaction mixture was concentrated to dryness and the residue was purified using preparative TLC (chloroform–methanol, 20:1) to give compound 23 (6.5 mg, 27.4%) as a yellow solid: 1H NMR (DMSO-d6) δ 3.78 (s, 3 H, CH3), 4.70 (br s, 2 H, CH2), 7.13 (pseudo t, J = 7.6 and 7.5 Hz, 1 H, H-16), 7.29 (d, J = 7.2 Hz, 2 H, pyr), 7.45 (d, J = 7.2 Hz, 1 H, H-17), 7.60 (d, J = 8.2 Hz, 1 H, H-15), 7.86 (s, 1 H, H-13), 8.22 (s, 1 H, H-8), 8.73 (d, J = 7.2 Hz, 2 H, pyr), 9.03 (br s, 1 H, exchangeable with D2O, N6H).
2-Chloro-9-(β-d-erythrofuranosyl)-N6-(3-iodobenzyl)-adenine (25)
To a solution of d-erythrOSe 1,2,3-triacetate (49; 0.5 g, 2.03 mmol, prepared from erythrose and acetic anhydride/pyridine) in dry acetonitrile (10 mL), cooled to 0 °C, were added 45a (0.8 g, 2.08 mmol) and SnCl4 (0.8 mg, 3.07 mmol). After warming to room temperature, the reaction mixture was heated at 70 °C for 20 h. Solvent was removed in vacuo, and the residue was dissolved in concentrated NH4OH. This solution was refluxed for 1 h. After evaporation of volatiles, the residue was purified using preparative TLC (eluent CH2-Cl2-MeOH, 9.5:0.5, Rf = 0.45) to give 25 (150 mg, 15%): 1H NMR (DMSO-d6) δ 3.93 (m, 2 H, CH2), 4.27 (m, 1 H, H-3′), 4.43 (m, 1 H, H-2′), 4.60 (br s, 2 H, CH2), 5.31 (d, J = 4.5 Hz, 1 H, exchangeable with D2O, OH), 4.50 (d, J = 4.5 Hz, exchangeable with D2O, OH), 6.13 (d, J = 5.9 Hz, 1 H, H-1′), 7.14 (pseudo t, J = 7.9 and 7.6 Hz, 1 H, H-16), 7.34 (d, J = 7.5 Hz, 1 H, H-17), 7.60 (d, J = 7.8 Hz, 1 H, H-15), 7.62 (s, 1 H, H-13), 8.37 (s, 1 H, H-8), 8.85 (br s, 1 H, exchangeable with D2O, N6H).
9-(β-d-Erythrofuranosyl)-2-(methylamino)-N6-(3-iodobenzyl)adenine (26)
A solution of 25 (10 mg, 0.021 µmol) in MeOH (1 mL) and 40% aqueous methylamine (1 mL) was heated in a sealed vessel at 100 °C for 5 days. After cooling to room temperature, the volatiles were evaporated and the residue was purified using preparative TLC (eluent CH2Cl2–MeOH, 9.5:0.5) to give 26 as a white solid (9.6 mg, 98%): 1H NMR (DMSO-d6) δ 2.80 (s, 3 H, NHMe), 3.86 (m, 2 H, CH2), 4.40 (m, 1 H, H-2′), 4.60 (s, 2 H, CH2), 5.29 (d, J = 4.5 Hz, 1 H, exchangeable with D2O, OH), 4.98 (d, J = 4.5 Hz, exchangeable with D2O, OH), 6.13 (d, J = 5.9 Hz, 1 H, H-1′), 7.14 (pseudo t, J = 7.9 and 7.6 Hz, 1 H, H-16”), 7.34 (d, J = 7.5 Hz, 1 H, H-17), 7.59 (d, J = 7.8 Hz, 1 H, H-15), 7.59 (s, 1 H, H-8), 8.60 (br s, 1 H, exchangeable with D2O, N6H).
2-Chloro-[(3-iodobenzyl)amino]-9-(2-tetrahydrofuryl)-9H-purine (27)
A solution of 45a (350 mg, 0.91 mmol), 2,3-dihydrofuran (0.38 g, 5.42 mmol), and 6 drops of ethanesulfonic acid in 30 mL of dry ethyl acetate was heated for 20 h at 50 °C. After cooling to the room temperature, volatiles were removed by rotary evaporation and the residue was purified on preparative silica gel TLC plates (eluent CH2Cl2-MeOH, 10:1). After recrystallization from MeOH, 27 (53 mg, 13%) was obtained as a white solid: 1H NMR (DMSO-d6) δ 2.15 (m, 2 H, H-3′), 2.45 (q, J = 7.38 Hz, 2 H, H-2′), 3.92 (q, J = 7.38 Hz, 1 H, H-4′), 4.14 (q, J = 7.49 Hz, 1 H, H-4′), 4.60 (d, J = 5.65 Hz, 2 H, CH2-Ph), 6.21 (t, J = 5.1 Hz, 1 H, H-1), 7.14 (pseudo t, J = 7.9 and 7.6 Hz, 1 H, H-16), 7.35 (d, J = 7.5 Hz, 1 H, H-17), 7.60 (d, J = 7.8 Hz, 1 H, H-15), 7.62 (d, J = 7.8 Hz, 1 H, H-13), 7.74 (s, 1 H, H-8), 8.87 (br s, 1 H, exchangeable with D2O, N6H).
N6-Benzyl-2-chloro-9-[2,3-dideoxy-5-(methylcarbamoyl)-β-d-ribofuranosyl]adenine (29)
A mixture of compound 35 (58.55 mg, 0.11 mmol), (phenoxythio)carbonyl chloride (0.027 mL, 0.19 mmol), and DMAP (35.7 mg, 0.29 mmol) in dry acetonitrile (1.5 mL) was stirred for 6.5 h at room temperature. The reaction mixture was concentrated to dryness, and the residue was purified using preparative TLC (chloroform–methanol, 20:1) to give compound 58 as a glassy solid: 1H NMR (CDCl3) δ 2.75 (m, 1 H, H-3′a), 2.89 (d, J = 4.7 Hz, 21 H, NH-CH3), 3.05 (m, 1 H, H-3′b), 4.75 (m, 3 H, H-4′ and CH2), 5.81 (m, 1 H, H-2′), 6.12 (s, 1 H, H-1′), 7.00–7.80 (m, 10 H, Ar).
A mixture of compound 58, l.0 M triethylborane in hexanes (0.28 mL, 0.28 mmol), and tributyltin hydride (0.074 mL, 0.28 mmol) in benzene was stirred for 2.5 h at room temperature. The reaction mixture was concentrated to dryness, and the residue was purified using preparative TLC (chloroform–methanol, 20:1) to yield compound 29 (11 mg, 23%) as a colorless solid: 1H NMR (CDCl3) δ 2.24–2.60 (m, 4 H, H-2′ and H-3′), 2.89 (d, J = 4.8 Hz, 3 H, NHCH3), 4.53 (dd, J = 8.5 and 4.8 Hz, 1 H, H-4′), 4.76 (br s, 2 H, CHz), 6.01 (pseudo t, J = 6.8 and 5.9 Hz; 1 H, H-1′), 6.24 (br s, 1 H, NH), 7.23–7.31 (m, 4 H, H-13,15,16,17), 7.66 (s, 1 H, H-8), 7.83 (br s, 1 H, N6H).
9-[2-Azido-2,3-dideoxy-5-(methylcarbamoyl)-β-d-arabinofuranosyl]-2-chloro-N6-(3-iodobenzyl)adenine (30)
A mixture of compound 34 (56.6 mg, 0.12 mmol) and sodium azide (83 mg, 1.26 mmol) in anhydrous DMF (2.5 mL) was heated for 41 h at 100 °C. Diethyl ether (30 mL) and water (25 mL) were added, and two layers were separated after shaking. The aqueous layer was extracted with ether (3 × 30 mL), and the combined organic layer and extracts were washed with brine (30 mL), dried over anhydrous MgSO4, filtered, and concentrated to dryness. The residue was purified using preparative TLC (chloroform–methanol, 20:1) to give compound 30 [Rf(chloroform–methanol, 20:1) = 0.29; 22 mg, 34%] as a colorless solid: 1H NMR (CDCl3) δ 2.61–2.87 (m, 2 H, H-3′), 2.89 (d, J = 5.0 Hz, 3 H, NHCH3), 4.37 (dd, J = 11.1 and 5.2 Hz, 1 H, H-2′), 4.56 (dd, J = 8.5 and 6.1 Hz, 1 H, H-4′), 4.71 (br s, 2 H, CH2), 6.18 (br s, 1 H, NH), 6.19 (d, J = 4.9 Hz, 1 H, H-1′), 7.05 (t, J = 7.7 Hz, 1 H, H-16), 7.30 (d, J = 6.8 Hz, 1 H, H-17), 7.43 (br s, 1 H, N6H), 7.59 (d, J = 7.6 Hz, 1 H, H-15), 7.68 (s, 1 H, H-13), 7.81 (s, 1 H, H-8).
9-[2-Amino-2,3-dideoxy-5-(methylcarbamoyl)-β-d-arabinofuranosyl]-2-chloro-N6-(3-iodobenzyl)adenine (31)
A solution of compound 30 (15 mg, 0.027 mmol) and triphenylphosphine (78 mg, 0.3 mmol) in dry THF (2 mL) was stirred for 3 days at room temperature. Water (0.5 mL) and methanolic ammonia (5 mL) were added, and the reaction mixture was stirred for 21 h at room temperature. The reaction mixture was concentrated to dryness, and the residue was purified using preparative TLC (chloroform–methanol, 10:1) to give compound 31 (6 mg, 43%) as a colorless solid: 1H NMR (DMSO-d6) δ 2.01 (m, 1 H, H-3′a), 2.45 (m, 1 H, H-3′b), 2.64 (d, J = 4.7 Hz, 3 H, NHCH3), 3.80 (m, 1 H, H-2′), 4.40 (pseudo t, J = 8.7 and 7.5 Hz, 1 H, H-4′), 4.63 (br s, 2 H, CH2), 6.09 (d, J = 5.7 Hz, 1 H, H-1′), 7.13 (pseudo t, J = 8.2 and 7.7 Hz, 1 H, H-16), 7.37 (d, J = 7.5 Hz, 1 H, H-17), 7.61 (d, J = 8.0 Hz, 1 H, H-15), 7.75 (s, 1 H, H-13), 8.11 (br s, 2 H, NH2, exchangeable with D2O), 8.52 (s, 1 H, H-8), 8.88 (br s, 1 H, exchangeable with D2O, N6H).
2-Chloro-9-[2,3-dideoxy-2-fluoro-5-(methylcarbamoyl)-β-d-arabinofuranosyl]-N6-(3-iodobenzyl)adenine(32)
To a −78 °C solution of 3′-deoxy-Cl-IB-MECA (35; 20 mg, 0.04 mmol) in dry dichloromethane (0.5 mL) was added 50 µL of DAST. After stirring at −78 °C for 2 h, the reaction mixture was warmed to room temperature over a period of 1 h and the reaction quenched by adding methanol (0.5 mL) and solid K2-CO3 (2 mg). The solvent was removed by evaporation, and the residue was purified using preparative TLC (CH2Cl2–MeOH, 9.5:0.5, Rf = 0.3) to give 32, 10 mg (50%): 1H NMR (DMSO-d6) δ 2.75 (m, 2 H, H-3′), 3.31 (s, 3 H, NHMe), 4.65 (br s, 2 H, CH2), 4.75 (m, 1 H, H-2′), 5.51 (d, J = 3.6 Hz, H-4′), 6.21 (d, J = 4.0 Hz, 1 H, H-1′), 7.13 (pseudo t, J = 7.9 and 7.6 Hz, 1 H, H-16), 7.36 (d, J = 7.5 Hz, 1 H, H-17), 7.61 (d, J = 7.8 Hz, 1 H, H-15), 7.60 (s, 1 H, H-13), 8.35 (s, 1 H, H-8), 8.90 (br s, 1 H, exchangeable with D2O, N6H).
9-[2-Acetyl-3-deoxy-5-(methylcarbamoyl)-β-d-ribofuranosyl]-2-chloro-N6-(3-iodobenzyl)adenine (33)
A mixture of 2-chloro-N6-(3-iodobenzyl)adenine (45a; 163 mg, 0.42 mmol), ammonium sulfate (catalytic amount), and HMDS (10 mL) was refluxed for 2 h under N2. The reaction mixture was concentrated to dryness in vacuo with exclusion of moisture. The resulting white solid 45b was dissolved in dry 1,2-dichloroethane (1 mL), and a solution of compound 57 (75 mg, 0.3 mmol) in dry 1,2-dichloroethane (2 mL) and TMS triflate (0.082 mL, 0.42 mmol) were added. The reaction solution under N2 was stirred for 1.5 h at room temperature and then refluxed for 17 h at 90 °C. Saturated NaHCO3 (10 mL) and methylene chloride (10 mL) were added, and the mixture was stirred for 15 min. Two layers separated, and the aqueous layer was extracted with methylene chloride (3 × 30 mL). The combined organic layer and extracts were washed with brine, dried over anhydrous MgSO4, filtered, and concentrated to dryness. The residue was separated on preparative TLC (chloroform–methanol, 20:1) to give compound 33 (71 mg, 42%): 1H NMR (CDCl3) δ 2.06 (s, 3 H, OAc), 2.50 and 2.75 (each m, 1 H, H-3′), 2.89 (d, J = 4.7 Hz, 3 H, NHCH3), 4.70 (m, 3 H, H-4′ and CH2), 5.31 (m, 1 H, H-2′), 5.85 (d, J = 3.2 Hz, 1 H, H-1′), 6.31 (br s, 1 H, NH), 7.02 (pseudo t, J = 7.8 and 7.6 Hz, 1 H, H-16), 7.29 (d, J = 7.6 Hz, 1 H, H-17), 7.58 (d, J = 7.8 Hz, 1 H, H-15), 7.67 and 7.72 (each s, 1 H, H-8 and H-13), 7.84 (br s, 1 H, exchangeable with D2O, N6H); UV (MeOH) λmax 271.5 nm.
2-Chloro-9-[3-deoxy-2-(methylsulfonyl)-5-(methylcarbamoyl)-β-d-ribofuranosyl]-N6-(3-iodobenzyl)adenine(34)
Compound 35 (100 mg, 0.18 mmol) was dissolved in an equivolume mixture of dry pyridine and methylene chloride (4 mL), and methanesulfonyl chloride (0.05 mL, 0.65 mmol) was added. The reaction mixture was stirred for 1.5 h at room temperature, and the solvents were removed using rotary evaporation. The residue was purified using silica gel column chromatography (chloroform–methanol, 20:1) to give compound 34 (87.5 mg, 78%) as a colorless foam: 1H NMR (CDCl3) δ 2.66 (ddd, J = 11.1, 7.5, and 3.9 Hz, 1 H, H-3′a), 2.86 (m, 1 H, H-3′b), 2.89 (d, J = 5.0 Hz, 3 H, NHCH3), 3.03 (s, 3 H, OSO2CH3), 4.72 (m, 3 H, H-4′ and CH2), 5.39 (m, 1 H, H-2′), 6.05 (d, J = 3.1 Hz, 1 H, H-1′), 6.31 (br s, 1 H, NH), 7.05 (pseudo t, J = 7.8 and 7.6 Hz, 1 H, H-16), 7.28 (d, J = 7.7 Hz, 1 H, H-17), 7.58 (d, J = 7.7 Hz, 1 H, H-15), 7.67 and 7.77 (each s, 1 H, H-8 and H-13), 8.65 (br s, 1 H, exchangeable with D2O, N6H).
2-Chloro.9-[3-deoxy-5-(methylcarbamoyl)-β-d-ribofuranosyl]-N6-(3-iodobenzyl)adenine (35)
A mixture of compound 33 (15 mg, 0.027 mmol) and NH3/MeOH (1.5 mL) was stirred for 18 h at room temperature. The reaction mixture was concentrated to dryness, and the residue was purified using silica gel column chromatography (chloroform–methanol, 20:1) to give compound 35 (6.22 mg, 43%) as a slightly yellow solid: 1H NMR (DMSO-d6) δ 2.15–2.23 and 2.26–2.35 (each m, 1 H, H-3′), 2.65 (d, J = 4.3 Hz, 3 H, NHCH3), 4.55–4.68 (m, 4 H, H-2′, H-4′, CH2), 5.83 (d, J = 3.9 Hz, 1 H, OH, exchangeable with D2O), 5.90 (s, 1 H, H-1′), 7.13 (t, J = 7.6 Hz, 1 H, H-16), 7.37 (d, J = 7.6 Hz, 1 H, H-17), 7.61 (d, J = 7.7 Hz, 1 H, H-15), 7.75 (s, 1 H, H-13), 8.14 (br s, 1 H, exchangeable with D2O, NHCH3), 8.59 (s, 1 H, H-8), 8.95 (br t, J = 5.7 Hz, 1 H, exchangeable with D2O, N6H).
2-Chloro-N6-(3-iodobenzyl)-9-[3,5-O-(1,1,3,3-tetraiso-propyldisiloxyl)-β-d-ribofuranosyl]adenine (36)
To a solution of 2-chloro-N6-(3-iodobenzyl)adenosine18 (300 mg, 0.58 mmol) in dry pyridine (9 mL) was added 1,3-dichloro-1,1,3,3-tetraisopropyldisiloxane (0.41 mL, 1.28 mmol) at room temperature, and the reaction mixture was stirred for 2.5 h at room temperature. After workup as described,39 the residue was purified via silica gel column chromatography (chloroform–methanol, 100:1) to give compound 36 (375 mg, 91%) as a colorless foam: 1H NMR (DMSO-d6) δ 0.91–1.18 (m, 28 H, isopropyl), 3.17 and 3.49 (each s, 1 H), 4.03 (m, 3 H), 4.52 (d, J = 5.3 Hz, 1 H), 4.70 (br s, 2 H), 5.01 (m, 1 H), 5.83 (s, 1 H), 6.15 (br s, 1 H), 7.01 (t, J = 7.6 Hz, 1 H, H-16), 7.28 (d, J = 7.6 Hz, 1 H, H-17), 7.55 (d, J = 7.7 Hz, 1 H, H-15), 7.66 (s, 1 H, H-13), 7.78 (s, 1 H, H-8).
2-Chloro-N6-(3-iodobenzyl)-9-[5-(methylcarbamoyl)-2,3-O-(thiocarbonyl)-β-d-ribofuranosyl]adenine (37)
To a solution of Cl-IB-MECA (2; 10 mg, 0.02 mmol) in dry DMF (0.5 mL) were added 1,l-thiocarbonyldiimidazole (30 mg, 0.17 mmol) and DMAP (2 mg). The resulting mixture was stirred overnight at room temperature. After removal of DMF using rotary evaporation under high vacuum, the residue was purified using preparative TLC (CH2Cl2-MeOH, 9.5:0.5, Rf = 0.6) to give 37, 8.6 mg (80%): 1H NMR (DMSO-d6) δ 2.73 (d, J = 4.3 Hz, 3 H, NHMe), 4.21 (m, 1 H, H-3′), 4.62 (br s, 2 H, CH2), 5.09 (s, 1 H, H-4′), 5.95 (m, 1 H, H-2′), 6.31 (d, J = 7.3 Hz, 1 H, H-1′), 7.14 (pseudo t, J = 7.9 and 7.6 Hz, 1 H, H-16), 7.40 (d, J = 7.6 Hz, 1 H, H-17), 7.60 (d, J = 7.8 Hz, 1 H, H-15), 7.76 (s, 1 H, H-13), 8.27 (br d, J = 4.3 Hz, 1 H, exchangeable with D2O, NH), 8.49 (s, 1 H, H-8), 9.02 (br t, J = 6.2 and 5.7 Hz, 1 H, exchangeable with D2O, N6H).
5-O-Benzoyl-3-deoxy-1,2-isopropylidene-α-d-ribofuranose (52)
A solution of 5-O-benzoyl-1,2-isopropylidene-α-d-xylofuranose (5.9 g, 0.02 mol) and carbon disulfide (6.03 mL, 0.1 mol) in anhydrous THF (60 mL) was immersed in an ice bath under N2 atmosphere. Sodium hydride in mineral oil (60%, 1.6 g, 0.04 mol) was added all at once. The reaction mixture was stirred for 50 min at 0 °C, and methyl iodide (25.7 mL, 0.4 mol) was added. After stirring for 1 h at 0 °C, the reaction mixture was neutralized with glacial acetic acid until the precipitate dissolved. The mixture was concentrated to dryness in vacuo. The residue was dissolved in ethyl acetate and filtered through a short silica gel column (hexanes–ethyl acetate, 10:1) to give the xanthate as a brown thick syrup.
A mixture of the xanthate, tributyltin hydride (7.6 mL, 0.029 mol), and triethylborane (28.6 mL, 0.029 mol) in benzene was stirred for 4 h at room temperature. The reaction mixture was concentrated to dryness, and the residue was purified using silica gel column chromatography (hexanes–ethyl acetate, 100:1 → 10:1 → 3:1) to give compound 52 (1.67 g, 30%): 1H NMR (CDCl3) δ 1.27 (s, 3 H, isopropylidene), 1.47 (s, 3 H, isopropylidene), 1.69 (td, J = 13.1 and 4.8 Hz, 1 H, H-3b), 2.12 (dd, J = 13.3 and 4.2 Hz, 1 H, H-3a), 4.29 (dd, J = 12.1 and 6.0 Hz, 1 H, H-5b), 4.50 (m, 2 H, H-4 and H-5a), 4.72 (t, J = 4.2 Hz, 1 H, H-2), 5.81 (d, J = 3.7 Hz, 1 H, H-1), 7.35–8.01 (m, 5 H, Bz).
3-Deoxy-1,2-isopropylidene-α-d-ribofuranose (53)
A mixture of 52 (1.67 g, 6 mmol) and methanolic ammonia (50 mL, saturated at 0 °C) was stirred for 5 days at room temperature. The reaction mixture was concentrated to dryness, and the residue was purified using silica gel column chromatography (hexanes–ethyl acetate, 100:1 → 1:1) to give compound 53 (0.83 g, 79%) as a colorless solid: 1H NMR (CDCl3) δ 1.26 (s, 3 H, isopropylidene), 1.45 (s, 3 H, isopropylidene), 1.66–1.83 (m, 1 H, H-3b), 1.95 (dd, J = 13.4 and 4.6 Hz, 1 H, H-3a), 3.50 (m, 1 H, H-5b), 3.83 (1 H, H-5a), 4.28 (1 H, H-4), 4.70 (pseudo t, J = 4.2 and 4.1 Hz, 1 H, H-2), 5.76 (d, J = 3.6 Hz, 1 H, H-1).
3-Deoxy-1,2-isopropylidene-α-d-5-ribofuronic Acid (54)
A mixture of compound 53 (0.503 g, 2.89 mmol), ruthenium oxide (38 mg), and sodium periodate (2.47 g, 11.6 mmol) in acetonitrile:chloroform:water (2:2:3, 14 mL) was stirred vigorously for 4 h at room temperature. After separation of the two layers, the aqueous layer was extracted with chloroform (3 × 50 mL). The combined organic layer and extracts were washed with brine, dried over anhydrous MgSO4, filtered, concentrated to dryness, and dried in vacuo to give compound 54 (0.537 g, 98%) as a solid: 1H NMR (CDCl3) δ 1.28 (s, 3 H, isopropylidene), 1.46 (s, 3 H, isopropylidene), 1.91 (td, J = 12.3 and 4.3 Hz, 1 H, H-3b), 2.48 (dd, J = 13.6 and 5.2 Hz, 1 H, H-3a), 4.70 (m, 2 H, H-2 and H-4), 5.89 (d, J = 3.3 Hz, 1 H, H-1).
Methyl 3-Deoxy-1,2-isopropylidene-α-d-ribofuronamide (56)
The mixture of compound 54 (0.48 g, 2.55 mmol), EDAC (1.226 g, 6.42 mmol), and DMAP (0.031 g, 0.25 mmol) in anhydrous methanol (10 mL) was stirred for 24 h at room temperature. The reaction mixture was concentrated to dryness, and the residue was dissolved in chloroform (30 mL) and water (20 mL). Two layers separated, and the aqueous layer was extracted with chloroform (3 × 30 mL). The combined organic layer and extracts were washed with brine, dried over anhydrous MgSO4, filtered, and concentrated to dryness. The residue was purified using silica gel column chromatography (chloroform–methanol, 20:1) to give compound 55 (0.217 g, 42%): 1H NMR (CDCl3) δ 1.33 and 1.55 (each s, 3 H, isopropylidene), 1.90–2.00 (m, 3 H, H-3a), 2.39–2.45 (dd, J = 13.5 and 4.9 Hz, 1 H, H-3b), 3.78 (s, 3H, OCH3), 4.71 (dd, J = 10.9 and 5.0 Hz, 1 H, H-2 or -4), 4.77 (t, J = 4.2 Hz, 1 H, H-2 or -4), 5.95 (d, J = 3.4 Hz, 1 H, H-1).
A solution of compound 55 (217 mg, 1.07 mmol) and 2 M methylamine in THF (5 mL) was heated for 24 h at 55 °C in a sealed tube. The reaction mixture was concentrated to dryness, and the residue was dried in vacuo to give compound 56 (216 mg, 99%) as needles: 1H NMR (CDCl3) δ 1.27 (s, 3 H, isopropylidene), 1.44 (s, 3 H, isopropylidene), 1.69–1.78 (m, 1 H, H-3b), 2.53 (dd, J = 13.7 and 5.2 Hz, 1 H, H-3a), 2.77 (d, J = 4.9 Hz, 3 H, NHCH3), 4.59 (dd, J = 11.1 and 5.2 Hz, 1 H, H-4), 4.69 (t, J = 4.0 Hz, 1 H, H-2), 5.81 (d, J = 3.5 Hz, 1 H, H-1), 6.42 (br s, 1 H, exchangeable with D2O, N6H). Anal. Calcd for C9H15N1O4: C, 53.72; H, 7.51; N, 6.96. Found: C, 53.97; H, 7.65; N, 6.93.
Methyl 3-Deoxy-1,2-diacetyl-β-d-ribofuronamide (57)
A mixture of compound 56 (189 mg, 0.94 mmol), concentrated sulfuric acid (0.276 mL, 5.18 mmol), and acetic anhydride (0.93 mL, 9.86 mmol) in glacial acetic acid (4.68 mL) was stirred for 18 h at room temperature. Following cooling in an ice bath, saturated NaHCO3 solution (10 mL) and methylene chloride (10 mL) were added slowly, and the mixture was stirred for 10 min. After separation of the two layers, the aqueous layer was extracted with methylene chloride (3 × 30 mL). The organic layer and extracts were combined, washed with saturated NaHCO3 and brine, dried over anhydrous MgSO4, filtered, concentrated to dryness, and dried in vacuo to yield crude compound 57 (184 mg, 80%) as a yellow syrup: 1H NMR (CDCl3) δ 2.00 and 2.03 (each s, 3 H, OAc), 2.25–2.35 (m, 1 H, H-3b), 2.40–2.47 (m, 1 H, H-3a), 2.77 (d, J = 5.0 Hz, 3 H, NHCH3), 4.68 (m, 1 H, H-4), 5.12 (d, J = 4.8 Hz, 1 H, H-2), 6.12 (s, 1 H, H-1), 6.35 (br s, 1 H, exchangeable with D2O, N6H). Anal. Calcd for C10H15N1O6: C, 48.98; H, 6.17; N, 5.71. Found: C, 58.94; H, 6.06; N, 5.42.
Biological Methods. Receptor Binding. Materials
F-12 (Ham’s) medium, fetal bovine serum (FBS), and penicillin/streptomycin were from Gibco BRL (Gaithersburg, MD). [125I]-AB-MECA was prepared as described.25 [3H]R-PIA was from Amersham (Arlington Heights, IL), and [3H]CGS 21680 was from DuPont NEN (Boston, MA). Adenosine deaminase (ADA) was from Boehringer Mannheim (Indianapolis, IN). Composition of lysis buffer: 10 mM Tris/5 mM EDTA, pH 7.4 at 5 °C. The incubation buffer for A3 competition experiments consisted of 50 mM Tris, 10 mM MgCl2, 1 mM EDTA, pH 8.26 at 5 °C. All other materials were from standard local sources and of the highest grade commercially available.
CHO cells stably expressing the A3 receptol.8 were grown, and cell membranes were prepared by homogenization and centrifugation, as previously described.17,20 The preparation was stored at −70 °C and retained its A3 radioligand binding properties for at least 1 month.
Binding of [125I]-N6-(4-amino-3-iodobenzyl)adenosine-5′-(N-methyluronamide) ([125I]AB-MECA) to membranes from CHO cells stably transfected with the A3 receptor clone was performed essentially as described.20,25 Assays were performed in 50/10/1 buffer in glass tubes and contained 100 µL of the membrane suspension, 50 µL of [125I]AB-MECA (final concentration 0.3 nM), and 50 µL of inhibitor. Inhibitors were routinely dissolved in DMSO and then diluted with buffer; final DMSO concentrations never exceeded 2%. Incubations were carried out in duplicate for 1 h at 37 °C and terminated by rapid filtration over Whatman GF/B filters, using a Bran-dell cell harvester (Brandell, Gaithersburg, MD). Nonspecific binding was determined in the presence of 200 µM NECA.
Binding of [3H]PIA to A1 receptors from rat brain membranes and binding of [3H]CGS 21680 to A2 receptors from rat striatal membranes were performed as described previously.20,26,27 Rat cerebral cortical membranes and striatal membranes were prepared and treated with adenosine deaminase (2 U/mL) for 30 min at 37 °C prior to storage at −70 °C. Nonspecific binding was determined in the presence of 2-chlo-roadenosine at a concentration of 10 µM for A1 receptors and 200 µM for A2a receptors.
For all radioligand binding assays, IC50 values were computer-generated using a nonlinear regression formula using the InPlot program (GraphPad Software, San Diego, CA) and converted to apparent Ki values using Kd values of 1.0 and 14 nM for [3H]PIA and [3H]CGS 21680 binding, respectively, and the Cheng–Prusoff equation.28 The Kd for [125I]AB-MECA was assumed to be 1.48 nM as found previously at cloned rat A3 receptors in CHO cells.25 Adenylyl cyclase in transfected CHO cell membranes was measured as previously described.8,20
Scheme 5a.

a Reagents: (a) TMSOTf, Cl(CH)2Cl; (b) NH3MeOH; (c) PhOC(S)Cl, DMAP, AcCN; (d) n-Bu3SnH, Et3B, benzene.
Acknowledgment
We thank Mary Pound for technical assistance. G.L.S. is supported by NHLBI Grant RO1HL35134 from the National Institutes of Health. We thank Gilead Sciences (Foster City, CA) for support.
Footnotes
Abbreviations: [125I]AB-MECA, N6-(4-amino-3-iodobenzyl)adenosine-5′-(N-methyluronamide); ADA, adenosine deaminase; AIBN, 2,2′-azobis(2-methylpropionitrile); CGS 21680, 2-[[[4-(2-carboxyethyl)-phenyl]ethyl]amino]-5′-(N-ethylcarbamoyl)adenosine; CHO, Chinese hamster ovary; CNS, central nervous system; DAST, (diethylamino)-sulfur trifluoride; DMAP, 4-(dimethylamino)pyridine; DMF, N,N-dimethylformamide; DMSO, dimethyl sulfoxide; EHNA, erythro-9-(2-hydroxy-3-nonyl)adenine; EDTA, ethylenediaminetetraacetic acid; FBS, fetal bovine serum; HMDS, 1,1,1,3,3,3-hexamethyldisilazane; IB-MECA, N6-(3-iodobenzyl)adenosine-5′-(N-methyluronamide); Ki, inhibition constant; NECA, 5′-(N-ethylcarbamoyl)adenosine; PIA, (R)-N6-phenylisopropyladenosine; THF, tetrahydrofuran; Tris, tris(hydroxy-methyl)aminomethane; XAC, 8-[4-[[[[(2-aminoethyl)amino]carbonyl]-methyl]oxy]phenyl]-1,3-dipropylxanthine.
References
- 1.Jacobson KA, van Galen PJM, Williams M. Perspective. Adenosine receptors: pharmacology, structure-activity relationships and therapeutic potential. J. Med. Chem. 1992;35:407–422. doi: 10.1021/jm00081a001. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2.Daly JW, Jacobson KA. Adenosine receptors: Selective agonists and antagonists. In: Bellardinelli L, Pelleg A, editors. Adenosine and adenine nucleotides: From molecular biology to integrative physiology. Norwell, MA: Kluwer; 1995. pp. 157–166. [Google Scholar]
- 3.Suzuki F, Shimada J, Shiozaki S, Ichikawa S, Ishii A, Nakamura J, Nonaka H, Kobayashi H, Fuse E. Adenosine A1 antagonists. 3. Structure activity relationships on amelioration against scopolamine- or N6-[(R)-phenylisopropyl]adenosine-induced cognitive disturbance. J. Med. Chem. 1993;36:2508–2518. doi: 10.1021/jm00069a009. [DOI] [PubMed] [Google Scholar]
- 4.von Lubitz DKJE, Jacobson KA. Behavioral effects of adenosine receptor stimulation. In: Bellardinelli L, Pelleg A, editors. Adenosine and adenine nucleotides: From molecular biology to integrative physiology. Norwell, MA: Kluwer; 1995. pp. 489–498. [Google Scholar]
- 5.Suzuki F, Shimada J, Mizumoto H, Karasawa A, Kubo K, Nonaka H, Ishii A, Kawakita T. Adenosine-A1 antagonists. 2. Structure-activity relationships on diuretic activities and protective effects against acute renal failure. J. Med. Chem. 1992;35:3066–3075. doi: 10.1021/jm00094a022. [DOI] [PubMed] [Google Scholar]
- 6.Schiffman SN, Halleux P, Menu R, Vanderhaeghen J-J. Adenosine A2a receptor expression in striatal neurons: implications for basal ganglia pathophysiology. Drug Dev. Res. 1993;28:381–385. [Google Scholar]
- 7.Hill JA, Bertholet B, Franco E, Kerensky R, Nichols W, Cowley S, Miller J, Bellardinelli L. Attenuation of adenosine A1 receptor mediated effects by N-0861 (N6-endonorboran-2-yl-9-methyladenine), in humans. Drug. Dev. Res. 1994;31:278. [Google Scholar]
- 8.Zhou QY, Li CY, Olah ME, Johnson RA, Stiles GL, Civelli O. Molecular cloning and characterization of an adenosine receptor - the A3 adenosine receptor. Proc. Natl. Acad. Sci. U.S.A. 1992;89:7432–7436. doi: 10.1073/pnas.89.16.7432. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Ramkumar V, Stiles GL, Beaven MA, Ali H. The A3AR is the unique adenosine receptor which facilitates release of allergic mediators in mast cells. J. Biol. Chem. 1993;268:16887–16890. [PubMed] [Google Scholar]
- 10.Fozard JR, Carruthers AM. Adenosine A3 receptors mediate hypotension in the angiotensin II-supported circulation of the pithed rat. Br. J. Pharmacol. 1993;109:3–5. doi: 10.1111/j.1476-5381.1993.tb13522.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Liu GS, Richards RA, Olsson RA, Mullane K, Walsh RS, Downey JM. Evidence that the adenosine A3 receptor may mediate the protection afforded by preconditioning in the isolated rabbit heart. Cardiovasc. Res. 1994;28:1057–1061. doi: 10.1093/cvr/28.7.1057. [DOI] [PubMed] [Google Scholar]
- 12.Linden J, Taylor HE, Robeva AS, Tucker AL, Stehle JH, Rivkees SA, Fink JS, Reppert SM. Molecular cloning and functional expression of a sheep A3 adenosine receptor with widespread tissue distribution. Mol. Pharmacol. 1993;44:524–532. [PubMed] [Google Scholar]
- 13.Salvatore CA, Jacobson MA, Taylor HE, Linden J, Johnson RG. Molecular cloning and characterization of the human A3 adenosine receptor. Proc. Natl. Acad. Sci. U.S.A. 1993;90:10365–10369. doi: 10.1073/pnas.90.21.10365. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Jacobson KA, Nikodijević O, Shi D, Gallo-Rodriguez C, Olah ME, Stiles GL, Daly JW. A role for central A3-adenosine receptors: Mediation of behavioral depressant responses. FEBS Lett. 1993;336:57–60. doi: 10.1016/0014-5793(93)81608-3. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Beaven MA, Ramkumar V, Ali H. Adenosine A3 receptors in mast cells. Trends Pharmacol. Sci. 1994;15:13–14. doi: 10.1016/0165-6147(94)90124-4. [DOI] [PubMed] [Google Scholar]
- 16.Bai TR, Weir T, Walker BAM, Salvatore CA, Johnson RG, Jacobson MA. Comparison and localization of adeonsine A3 receptor expression in normal and asthmatic lung. Drug Dev. Res. 1994;31:244. [Google Scholar]
- 17.Gallo-Rodriguez C, Ji X-D, Melman N, Siegman BD, Sanders LH, Orlina J, Pu Q-L, Olah ME, van Galen PJM, Stiles GL, Jacobson KA. Structure-activity relationships at A3-adenosine receptors. J. Med. Chem. 1994;37:636–646. doi: 10.1021/jm00031a014. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Kim HO, Ji X-d, Siddiqi SM, Stiles GL, Jacobson KA. 2-Substitution of N6-benzyladenosine-5′-uronamides enhances selectivity for A3 receptors. J. Med. Chem. 1994;37:3614–3621. doi: 10.1021/jm00047a018. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Kim HO, Ji X-d, Melman N, Olah ME, Stiles GL, Jacobson KA. Structure activity relationships of 1,3-dialkylx-anthine derivatives at rat A3-adenosine receptors. J. Med. Chem. 1994;37:3373–3382. doi: 10.1021/jm00046a022. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.(a) van Galen PJM, van Bergen AH, Gallo-Rodriguez C, Melman N, Olah ME, IJzerman AP, Stiles GL, Jacobson KA. Mol. Pharmacol. 1994;45:1101–1111. [PMC free article] [PubMed] [Google Scholar]; (b) van Bergen A, van Galen PJM, Stiles GL, Jacobson KA. A3 receptors: Structure activity relationships and molecular modeling. ACS 206th National Meeting; Aug., 1993; Chicago, IL. Abstract MEDI217. [Google Scholar]
- 21.Ji X-d, von Lubitz D, Olah ME, Stiles GL, Jacobson KA. Species differences in ligand affinity at central A3-adenosine receptors. Drug Dev. Res. 1994;33:51–59. doi: 10.1002/ddr.430330109. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Ukena D, Padgett WL, Hong O, Daly JW, Daly DT, Olsson RA. N6-Substituted 9-methyladenines: a new class of adenosine receptor antagonists. FEBS Lett. 1987;215:203–208. doi: 10.1016/0014-5793(87)80146-1. [DOI] [PubMed] [Google Scholar]
- 23.Thompson RD, Secunda S, Daly JW, Olsson RA. N6,9-Disubstituted adenines - potent, selective antagonists at the A1-adenosine receptor. J. Med. Chem. 1991;34:2877–2882. doi: 10.1021/jm00113a029. [DOI] [PubMed] [Google Scholar]
- 24.Martin PL, Ueeda M, Olsson RA. 2-Phenylethoxy-9-methyl-adenine: an adenosine receptor antagonist that discriminates between A2 adenosine receptors in the atria and the coronary vessels from the guinea pig. J. Pharmacol. Exp. Ther. 1999;265:248–253. [PubMed] [Google Scholar]
- 25.Olah ME, Gallo-Rodriguez C, Jacobson KA, Stiles GL. [125I]AB-MECA, a high affinity radioligand for the rat A3 adenosine receptor. Mol. Pharmacol. 1994;45:978–982. [PMC free article] [PubMed] [Google Scholar]
- 26.Schwabe U, Trost T. Characterization of adenosine receptors in rat brain by (−)[3H]N6-phenylisopropyladenosine. Naunyn-Schmiedeberg’s Arch. Pharmacol. 1980;313:179–187. doi: 10.1007/BF00505731. [DOI] [PubMed] [Google Scholar]
- 27.Jarvis MF, Schulz R, Hutchison AJ, Do UH, Sills MA, Williams M. [3H]CGS 21680, a selective A2 adenosine receptor agonist directly labels A2 receptors in rat brain. J. Pharmacol. Exp. Ther. 1989;251:888–893. [PubMed] [Google Scholar]
- 28.Cheng YC, Prusoff WH. Relationship between the inhibition constant (Ki) and the concentration of inhibitor which causes 50 percent inhibition (IC50) of an enzyme reaction. Biochem. Pharmacol. 1973;22:3099–3108. doi: 10.1016/0006-2952(73)90196-2. [DOI] [PubMed] [Google Scholar]
- 29.MacKenzie WM, Hoskin DW, Blay J. Adenosine inhibits the adhesion of anti-CD3-activated killer lymphocytes to adeno-carcinoma cells through an A3 receptor. Cancer Res. 1994;54:3521–3526. [PubMed] [Google Scholar]
- 30.Bruns RF. Adenosine receptor activation in human fibroblasts: nucleoside agonists and antagonists. Can. J. Physiol. Pharmacol. 1980;58:673–691. doi: 10.1139/y80-110. [DOI] [PubMed] [Google Scholar]
- 31.Siddiqi SM, Jacobson KA, Esker JL, Olah ME, Ji X-d, Melman N, Tiwari KN, Secrist JA, Schneller SW, Cristalli G, Stiles GL, Johnson CR, IJzerman AP. Search for new purine- and ribose-modified adenosine analogues as selective agonists and antagonists at adenosine receptors. J. Med. Chem. 1995;38:1174–1188. doi: 10.1021/jm00007a014. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Lohse MJ, Klotz KN, Diekmann E, Friedrich K, Schwabe U. 2′,3′-Dideoxy-N6-cyclohexyladenosine: an adenosine derivative with antagonist properties at adenosine receptors. Eur. J. Pharmacol. 1988;156:157–160. doi: 10.1016/0014-2999(88)90158-6. [DOI] [PubMed] [Google Scholar]
- 33.Taylor MD, Moos WH, Hamilton HW, Szotek DS, Patt WC, Badger EW, Bristol JA, Bruns RF, Heffner TG, Mertz TE. Ribose-modified adenosine analogues as adenosine receptor agonists. J. Med. Chem. 1986;29:346–353. doi: 10.1021/jm00153a008. [DOI] [PubMed] [Google Scholar]
- 34.van der Wenden EM, von Frijtag Drabbe Künzel JK, Mathôt RAA, IJzerman AP. Ribose modified adenosine ananlogues as potential partial agonists for the adenosine receptor. Drug Dev. Res. 1994;31:330. doi: 10.1021/jm00020a014. [DOI] [PubMed] [Google Scholar]
- 35.Daly JW, Padgett WL. Agonist activity of 2- and 5′-substituted adenosine analogs and their N6-cycloalkyl derivatives at A1-adenosine and A2-adenosine receptors coupled to adenylyl cyclase. Biochem. Pharmacol. 1992;43:1089–1093. doi: 10.1016/0006-2952(92)90616-q. [DOI] [PubMed] [Google Scholar]
- 36.Munshi R, Clanachan AS, Baer HP. 5′-Deoxy-5′-methyl-thioadenosine: a nucleotide which differentiates between adenosine receptor types. Biochem. Pharmacol. 1988;37:2085–2089. doi: 10.1016/0006-2952(88)90560-6. [DOI] [PubMed] [Google Scholar]
- 37.Vorbrüggen H, Krolikiewicz K, Bennua B. Nucleoside synthesis with trimethylsilyl triflate and perchlorate as catalysts. Chem. Ber. 1981;114:1234–1255. [Google Scholar]
- 38.Mungall WS, Greene GL, Heavner GA, Letsinger RL. Use of the azido group in the synthesis of 5′ terminal aminode-oxythymdine oligonucleotides. J. Org. Chem. 1975;40:1659–1662. doi: 10.1021/jo00899a038. [DOI] [PubMed] [Google Scholar]
- 39.Tiwari KN, Secrist JA, Montgomery JA. Synthesis and biological activity of 4′-thionucleosides of 2-chloroadenine. Nucleosides Nucleotides. 1994;13:1819–1828. [Google Scholar]
- 40.Goldsmith BA, Abrams TW. Reversal of synaptic depression by serotonin at aplysia sensory neuron synapses involves activation of adenylyl cyclase. Proc. Natl. Acad. Sci. U.S.A. 1991;88:9021–9025. doi: 10.1073/pnas.88.20.9021. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41.Hampton A. Substrate binding of adenine nucleosides and nucleotides to certain enzymes. In: Townsend L, editor. Chemistry of Nucleosides and Nucleotides. Vol. 2. New York: Plenum; 1991. pp. 399–450. [Google Scholar]
- 42.Bruns RF. Adenosine antagonism by purines, pteridines, and benzopteridines in human fibroblasts. Biochem. Pharmacol. 1981;30:325–333. doi: 10.1016/0006-2952(81)90062-9. [DOI] [PubMed] [Google Scholar]