

Abstract
Tumor metastases and epithelial to mesenchymal transition (EMT) involve tumor cell invasion and migration through the dense collagen-rich extracellular matrix surrounding the tumor. Little is known about the mechanobiological mechanisms involved in this process, nor the role of the mechanical forces generated by the cells in their effort to invade and migrate through the stroma. In this paper we propose a new fundamental mechanobiological mechanism involved in cancer growth and metastasis, which can be both protective or destructive depending on the magnitude of the forces generated by the cells. This new mechanobiological mechanism directly challenges current paradigms that are focused mainly on biological and biochemical mechanisms associated with tumor metastasis. Our new mechanobiological mechanism describes how tumor expansion generates mechanical forces within the stroma to not only resist tumor expansion but also inhibit or enhance tumor invasion by, respectively, inhibiting or enhancing matrix metalloproteinase (MMP) degradation of the tensed interstitial collagen. While this mechanobiological mechanism has not been previously applied to the study of tumor metastasis and EMT, it may have the potential to broaden our understanding of the tumor invasive process and assist in developing new strategies for preventing or treating cancer metastasis.
Keywords: Tumor, stroma, metastasis, invasion, biomechanical forces, collagen
Introduction
Breast cancer is the most commonly diagnosed malignancy and leading cause of cancer mortality among women in the United States [1]. Ninety percent of all cancer deaths arise from tumor metastases, the process in which abnormal tumor cells proliferate, expand, reorganize, degrade and migrate through the surrounding stroma’s microenvironment (extracellular matrix (ECM)) into the circulation to invade other tissues. Triple negative breast cancer (TNBC), an aggressive subtype of invasive breast cancer that responds less well to current chemotherapeutic agents, is associated with an increased risk of tumor relapse, metastasis, and a poorer overall patient prognosis [2]. A better understanding of the tumor invasive process is essential to developing more effective and targeted therapies to both treat and prevent metastatic breast cancer, specifically the TNBC subtype.
The delamination and spread of tumor cells are prerequisites for metastatic disease. To develop new strategies for the prevention and treatment of cancer metastasis, including TNBC, it is important to understand the basic molecular mechanisms that control how cells from a primary tumor leave their neighboring cells, break into the vascular bed, migrate into the blood or lymph stream, reattach to a distant site and develop into a metastatic tumor. The early steps of the metastatic process are characterized by migration and invasion of the tumor cells into and through the stroma to enter the circulation through a process known as epithelial to mesenchymal transition (EMT) [3]. An ever expanding literature is now evolving to describe the mechanisms controlling this process [4]. During EMT, cells acquire a more spindle-like fibroblastic morphology, lose their adhesive properties, gain enhanced motility, increase their stroma interactions and invasive behavior in a process that involves increased cell traction forces to reorganize and degrade the extracellular matrix [4-7]. To increase their traction force on the ECM, the epithelial cells must incorporate α-smooth muscle actin (α-SMA) into stress fibers by synthesizing TGFβ-1 or activating it from the surrounding ECM [5]. As the cells increase their traction force on the ECM in response to increasing ECM stiffness as the tumor expands, the epithelial cells will transition from a more fibroblastic phenotype to a differentiated mesenchymal/myofibroblastic phenotype. Degradation of the ECM is a necessity for invasion and dispersion of cells and has been thoroughly studied during the last decade. Matrix metalloproteinases (MMPs) have been identified as key enzymes in this process as they are capable of degrading the ECM components, specifically proteoglycan, basement membrane, fibronectin and collagen [8, 9].
The importance and role of biomechanical forces within the tumor and stroma in the EMT process have not been extensively studied. We introduce a new paradigm for how biomechanical forces can modify the ECM’s molecular conformation during tumor growth and how these forces can be both protective and destructive depending on the stage of tumor growth. We describe how tumor expansion generates mechanical forces (tensile and compressive) within the stroma to resist tumor expansion and how these same forces can inhibit or enhance tumor invasion of the stroma. We also propose a possible biomechanical trigger for EMT based on the magnitude of traction force exerted by the tumor and stroma cells to deform and unwind the collagen molecule for enzyme cleavage. These conceptual force-based mechanisms have not been previously described, and we believe they will have application to the study of the tumor invasive process. In this paper we focused on breast cancer and the cells and tissues involved in the metastatic process. Our paradigm was developed in the context of breast cancer because the mechanics of tumor expansion, ECM deformation, and cellular traction forces were easiest to conceptualize. However the proposed biomechanical force mechanism is intrinsic to the collagen molecule itself, as well as other ECM components, and as such is applicable to many different cell types, tissues and disease processes. In the broader sense, we are proposing a new biomechanical mechanism for ECM degradation based on previous literature and our own research, with the hope that this will provide new and useful insights into not only cancer biology but many other disease processes.
Tumor Metastasis and Biomechanical Forces
Biomechanical forces within the microenvironment at the tumor-stroma interface may be a key mechanism driving EMT, and ultimately lead to metastatic spread to target organs. In this section we will explore the role and importance of mechanobiological (cell-matrix) and mechanochemical (cell-enzyme) mechanisms for eliciting EMT and subsequent degradation of the surrounding stroma. We will discuss possible biomechanical triggers that may be involved in tumor expansion in the early stages of the metastatic process, such as tumor and stroma cell responses to collagen conformational changes and increased tension and compression in the ECM of the stroma. We take a multiscale approach to describe possible mechanisms present at the tumor-stroma interface, ranging from the atomic level (molecule conformation) through the molecular level (collagen, enzymes), cellular level (tumor and stroma cells), and finally to the structural level (ECM) [10]]. Key players in this multiscale approach are the tumor and stroma cells, the surrounding stroma composed primarily of collagen, specific enzymes secreted by these cells which cleave collagen (MMPs such as collagenases) [7-9], and the role of transforming growth factor beta (TGFβ) in transforming benign epithelial cell morphology (cuboidal-shape) to an invasive myofibroblastic/mesenchymal cell morphology (spindle-shaped) [5], the later capable of reorganizing, degrading and migrating through the stroma [11].
It is now well recognized that cells can exert high and continuous tensile traction forces on their ECM via the cell’s integrin attachments (for instance, smooth muscle cells attached to fibronectin-coated posts exerted ~50-60 nN for 20 minutes) [12]. Likewise the ECM can transmit mechanical forces to the cell through these same integrin attachments [13-15]. The mechanotransduction of signals (forces) between the cell and its surrounding ECM are fundamental mechanisms during tissue and organ embryonic development, maturation, aging and in response to disease and injury, such as occurs during wound healing and tumor growth and metastasis [16-19]. The biomechanical forces exerted within the ECM and sensed by both the tumor and stroma cells will change as the tumor cells proliferate and increase the tumor’s size, concomitantly pressing on, expanding and compacting the ECM in the surrounding stroma [4, 16, 20, 21]. Tumor and most other cells have been shown to respond differentially to the biomechanical properties of their microenvironment, such as happens when cells are in contact with a soft or hard ECM [5, 22-25]. The cell’s ability to apply and vary the traction force it can apply to the ECM is generated by the cell’s internal contractile actin cytoskeleton through external focal adhesions (integrin clusters) attached to the ECM [5, 26]. Fibroblasts within the microenvironment of the stroma are normally unable to generate sufficiently large forces to deform and reorganize the collagen in the ECM. However, in the presence of cancerous/transformed epithelial cells or with the addition of the cytokine TGF-β1, the fibroblasts transition into myofibroblasts by the expression α-smooth muscle actin (α-SMA) [5, 27, 28], and thus are able to deform, contract and reorganize the ECM [4, 6, 20]. The ability of tumor and stroma cells to exert tensile traction forces to deform the ECM, in particular the collagen, is a key mechanism for tumor invasion into the surrounding stroma, not only to reorient the collagen for cell migration but possibly also to unwind the collagen for the degradation by enzymes. We will explore this new paradigm after describing the basic concepts associated with mechanical forces and material properties.
Mechanical Forces and Properties of Soft Tissues
Loads or forces are commonly exerted and sensed by cells attached to the ECM (Fig. 1). External cellular forces are generated by the extracellular matrix at the cell’s ECM contact sites (focal adhesions) between the cell’s integrins and their ECM ligands (e.g., α2β1 and α5β1 and collagen and fibronectin, respectively). On the other hand, internal cellular forces are intracellularly generated, through cytoskeletal proteins or filaments (e.g., actin filaments, microtubles). These forces act as mechanical signals to stimulate the cell’s metabolism (outside-in transduction) and allow the cell to apply traction forces to deform the ECM (inside-out transduction), respectively. A delicate balance exists between the external and internal forces which is necessary for cell-ECM stability (maintenance and repair).
Figure 1.
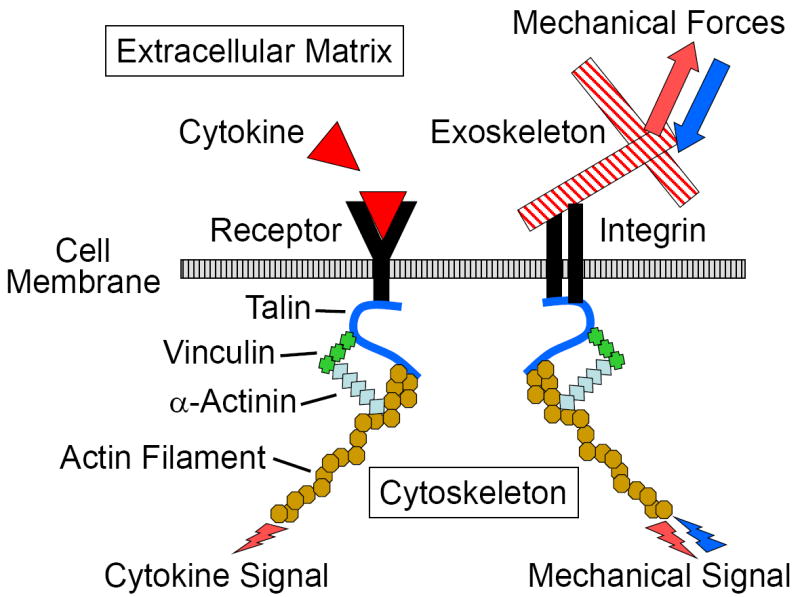
External mechanical forces are transmitted to the cell by the extracellular matrix through the cell’s integrins, and the cells can apply a force to the extracellular matrix through the same integrins. The external mechanical forces are internally transmitted or applied through the cell’s cytoskeletal proteins or filaments. Thus mechanical forces can be transmitted in both directions (outside-in and inside-out), while the cell’s response to cytokines, transmitted through the cell’s receptors, are one directional (inside-out).
The study of how these forces affect cellular function is termed mechanobiology while how the forces affect ECM function is termed biomechanics. Both are based on the principles of engineering mechanics, the study of how forces affect stability and motion. When cell-ECM forces are balanced the cell and ECM will be in static equilibrium or at rest, but when the forces are unbalanced there will be cell and ECM dynamic movement. In addition the study of how the external and internal forces can change the shape or deform the cell and ECM is termed mechanics of materials or biomaterials. In mechanics the study of the mechanical behavior of an ideal elastic material is termed the theory of elasticity, where the deformation of the material is proportional to the applied load and the deformation will persist (remain constant) until the load is removed. For man-made materials this proportionality holds for deformations of ~15%, while for biological materials it can be as high as 25-30%. If the deformation of the material depends on the rate of loading, the material is described as being deformation or strain-rate dependent, and the mechanical behavior termed viscoelastic. Finally, if the material flows, such as with viscous liquids, the mechanical behavior is more complex as it must describe both the deformation and flow of the material. This behavior is characterized by material’s rheological properties.
The fields of mechanobiology, biomechanics, mechanics of materials, viscoelasticity and rheology are too broad to cover adequately in this review. Only the basic concepts of elasticity will be described in order to provide a consistent biomechanical and biophysical-based terminology in which to discuss cell-ECM interactions. The reader is referred to other papers which will provide a broader and more comprehensive description of these different mechanical theories as applied to biological cells and tissues (see for instance [29-31].
The basic units of mechanics are length (L), mass (M), and time (t). Force is defined as the action of one object or body on another body, which can be generated with direct or indirect contact, for example, magnetic, electrostatic, and gravitational actions. Force is a vector quantity, and to correctly define a force four properties must be reported - its magnitude, line of action, direction or sense, and point of application (Fig. 2). The magnitude describes the amount of force, the line of action defines its orientation, the direction defines its effect, such as tensile or compressive, and the location is the point where the force is applied. Changing any one of these four properties will change the definition of the force. Since a force can be applied to the cell and ECM in any orientation and direction, it can deform the cell and ECM in pure tension, compression and shear, or any combination of these deformations (Fig. 3). When the force acts on a known surface area of an object, the distribution of the force over that area is termed a stress and equal to the force per unit area, or stress = force/area. Likewise, the deformation of the object resulting from the force can be normalized to the initial shape of the object, which is termed the strain and usually equal to the change in shape (length, area or volume) per initial shape (length, area or volume). This is illustrated in Figure 4 for a tensile force applied to a typical cylindrical or rectangular shaped specimen (e.g., a beam or tendon). As the magnitude of the tensile force increases the specimen will be stretched (elongated), and concomitantly the cross-sectional area will contract or “neck down” (A<Ao). In compression the cross-sectional area will expand or “bulge” (A>Ao). The contraction or expansion in the plane transverse to the applied force direction (axial) is termed the Poisson effect, and the ratio of the transverse to axial deformations is termed the Poisson’s ratio.
Figure 2.
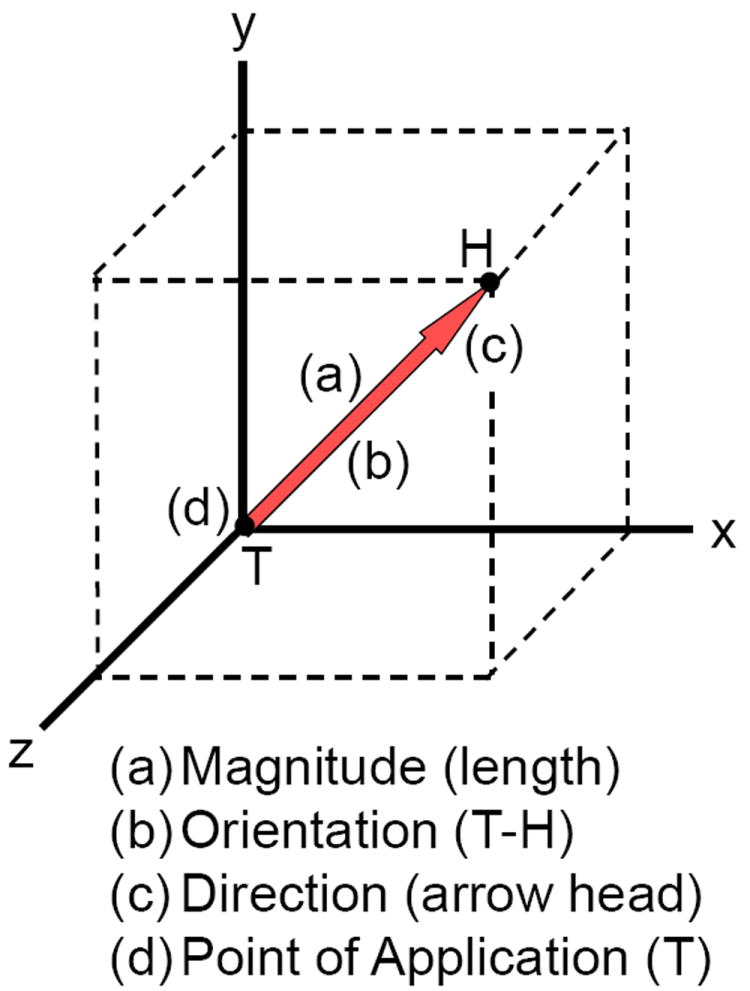
Force is a vector quantity and described as having a (a) magnitude (amount of force or length of arrow), (b) orientation or line of action (T-to-H), (c) direction or sense (arrow head), and (d) point of application (T). The magnitude (a) describes the amount of force (Newtons), the line of action (b) defines its orientation (xyz coordinates), the direction (c) defines its effect on the point of application (T), such as tensile or compressive, and the location (d) is the point (T) where the force is applied, such as to a cell’s integrin or to the extracellular matrix.
Figure 3.
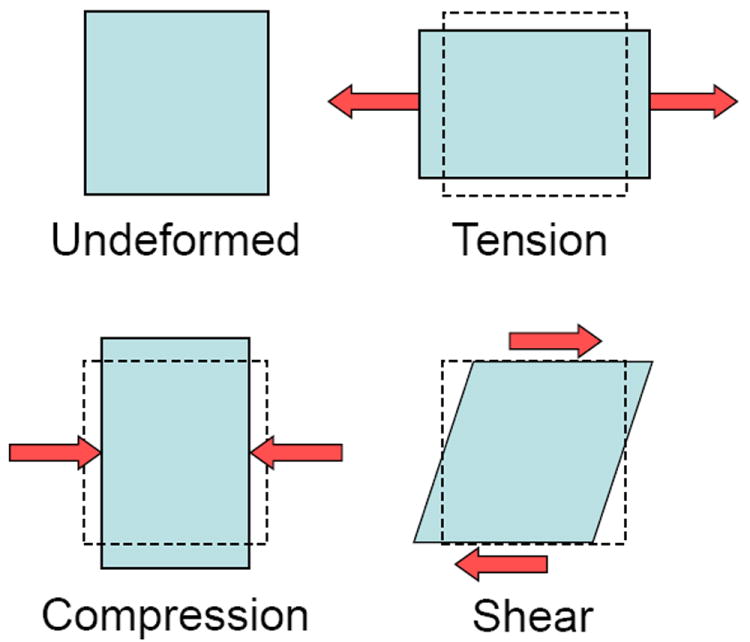
A force applied to an undeformed object will deform the object, the final shape of the object dependent on the characteristics of the force (magnitude, orientation, direction and point of application) and the material composition of the object.
Figure 4.
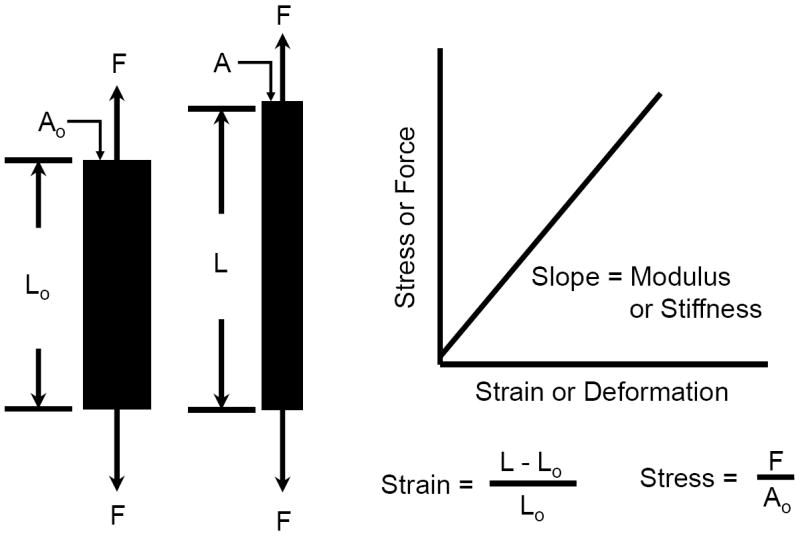
A tensile force applied to a specimen will increase its length (L>Lo) and decrease its cross-sectional area (A<Ao). The ratio of the transverse to axial deformation is termed the Poisson’s ratio. The slope of the force-deformation response is termed the specimen’s stiffness, a structural property. When the force and deformation are normalized to the specimen’s original length (Lo) and cross-sectional area (Ao), the slope of the stress-strain response is termed the specimen’s modulus, a material property.
Most isotropic and homogeneous materials have a linear force-deformation (stress-strain) response as shown in Figure 4, which may or may not be the same if the specimen is loaded in tension, compression or shear. The slope of the force-deformation response is a measure of the mechanical property of a structure, termed the stiffness, while the slope of the stress-strain response is a measure of the mechanical property of the material making up the structure, termed the modulus. An important distinction between the stiffness and modulus is that the stiffness is a function of both the geometry and composition (material) of the structure, for example a whole bone (femur), while the modulus is a function of only the material itself, such as for cortical or trabecula regions of the bone. However, most biological materials are composed of several different components, are inhomogeneous and anisotropic, and exhibit unique spatial morphology, such that at different length scales (macro, micro, nano and atomic) they can be considered as a structure with different stiffness and modulus when loaded in different directions and locations within the tissue.
While most engineered materials (e.g., rubber, aluminum, steel) have a linear mechanical response, almost all biological tissues have a nonlinear mechanical response (Fig. 5). In addition, most engineered materials and biological tissues are elastic, in that they will regain or recover their original shape once the deforming force is removed, providing the force has not caused any internal permanent damage to the tissue. The recovery may take seconds or even days. If the material does not regain its original shape, it is termed an inelastic material, which is an intrinsic property of the material and not a result of internal damage (e.g., cross-link rupture). The nonlinear mechanical response of most biological tissues (see Fig. 6) is characterized by an initial nonlinear, low-load and high-deformation response (concave curve), termed the toe region, where the tissue’s internal structure begins to reorient, straighten, bend or unwind (e.g., collagen crimp). This is followed by a linear region where the linear stiffness or modulus can be determined from the slope. As the load and deformation increase the response again becomes nonlinear (convex), where internal permanent damage begins, termed the yield point. Thereafter the tissue continues to deform until it eventually fails (failure point). Each of these regions and points represent a unique mechanical property of the structure and material response.
Figure 5.
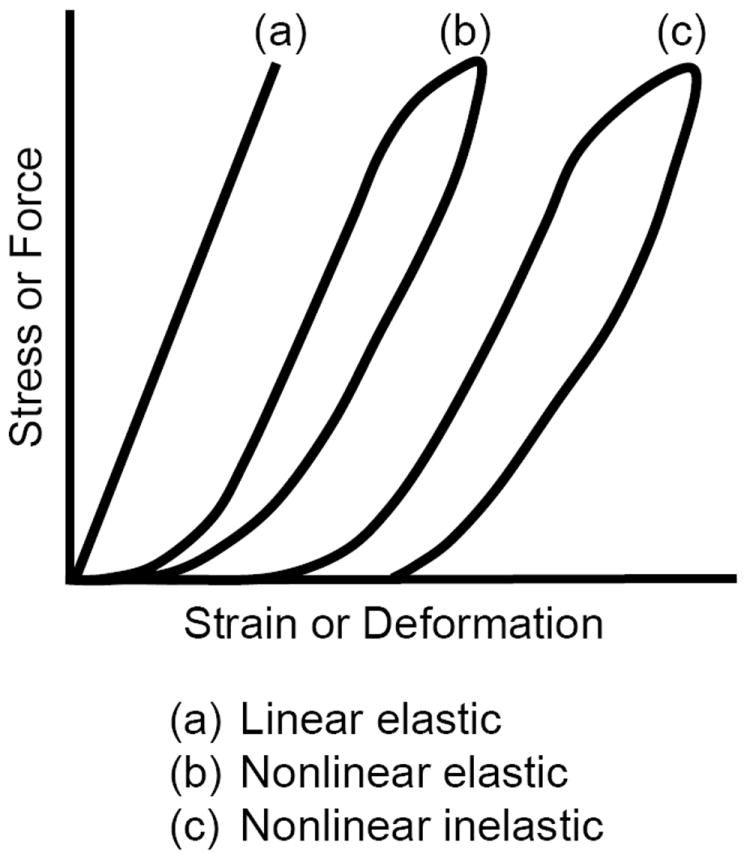
Typical mechanical responses of three different materials. (a) Elastic linear response. (b) Nonlinear elastic response (returns to its initial shape). (c) Nonlinear inelastic response (does not return to its initial shape).
Figure 6.
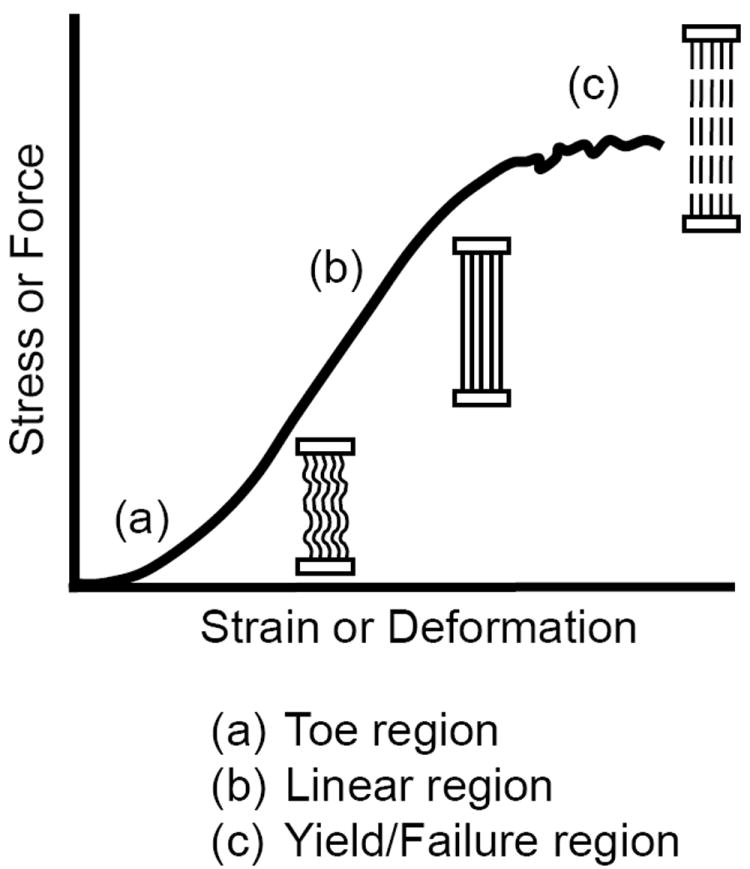
Typical nonlinear mechanical response of most biological tissues containing collagen. (a) The toe region is characterized by a nonlinear response at low-loads (concave curve) where the tissue straightens or unwinds to remove any crimp or slack in the structure. (b) In the linear region the tissue is stretched and the stiffness or modulus can be determined from the slope. (c) At higher forces the response again becomes nonlinear (convex curve) when tissue damage begins (yield point) until it eventually fails (failure point).
Listed in Table 1 is the elastic modulus, E, for various biological and synthetic materials having a reversible (elastic) mechanical response (note: the elastic modulus is often referred to as the Young’s modulus). These are representative values for the linear region of the mechanical response, keeping in mind that there is wide variability within and between different tissue types and species. For instance, the mechanical tensile response for whole bovine Achilles tendon is nonlinear, with an elastic modulus of ~400 MPa (Fig. 7). However on the macro scale, the collagen fibril’s tensile response is almost linear with a modulus of ~430 MPa, while on the nano scale the collagen molecule (monomer triple helix) is linear with a modulus ~750 MPa.
Table 1.
Elastic Modulus for Various Materials (Pa=N/m2)
| Cells | 0.3-0.6 kPa |
| Breast tissues [32] | 0.1-2 |
| Adipose tissue [33] | 1.9 |
| Eye sclera [57] | 2.9 |
| Ductal Carcinoma [33] | 12 |
| Muscle, Lung, Liver [32] | 9-25 |
| Skin (foot) [58] | 593 |
| Skin (foot diabetic) [58] | 1,147 |
| Rubber | 4 MPa |
| Cartilage (OA) | 2 |
| Cartilage (normal) | 12 |
| Ligament | 111 |
| Polyethylene | 240 |
| Tendon | 400 |
| Nylon fiber | 700 |
| Bone cement | 2.1 GPa |
| Polystyrene culture dish [32] | 3.6 |
| Douglas fir (soft wood) | 4.1 |
| Hickory (hard wood) | 13.8 |
| Bone | 15.0 |
| Titanium | 125 |
| 316 Stainless steel | 180 |
| Cobalt chrome | 225 |
Figure 7.
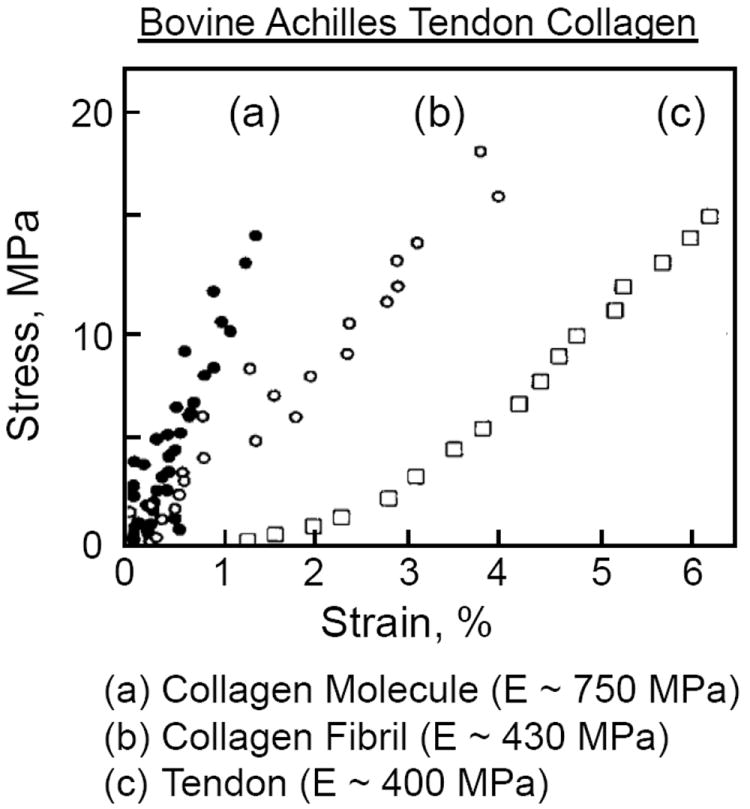
Mechanical tensile response for (a) collagen molecule (triple helix), (b) collagen fibril, and (c) whole bovine Achilles tendon. Adapted from Sasaki and Odajima [59].
Limited information exists on the mechanical properties of breast tissues (mammary gland and stroma) and cells (epithelial, myoepithelial, fibroblast and adipocytes). Because the breast tissues and cells are difficult to test in tension due to size limitations, mechanical tests are most often performed in compression using a small custom built indentation apparatus or an atomic force microscope. The measured elastic compressive modulus for normal breast tissue ranges between 2.2 kPa and 0.1 MPa [17, 32]. The compressive mechanical response for breast adipose and tumor tissues are both nonlinear elastic (Fig. 8), with elastic compressive modulus of approximately 1.9 kPa and 12.0 kPa, respectively [17, 33]. The compressive mechanical response of a fibroblast is also nonlinear elastic. However the cell’s stiffness and modulus will depend on the elastic stiffness and modulus of the substrate to which the cell is attached [22]. For fibroblasts the average compressive modulus ranges from 1kPa to 8 kPa as the cell’s surface area increases threefold, and the substrate’s modulus increases from 0.8 kPa to 4 kPa, the cell’s modulus being slightly lower then the substrate modulus.
Figure 8.
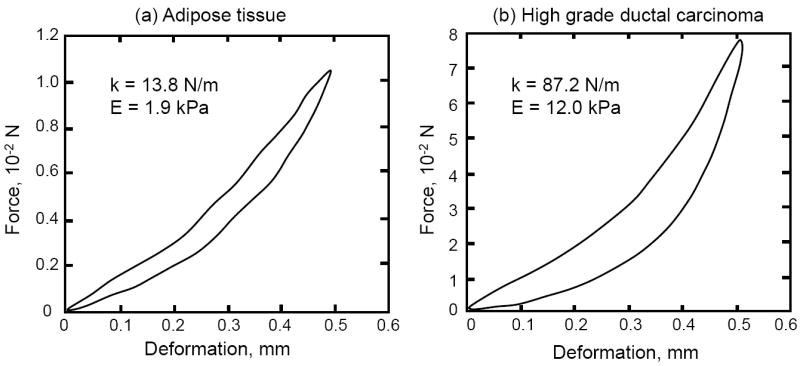
Compressive mechanical response for (a) breast adipose tissue and (b) high grade ductal carcinoma The compressive modulus E for each tissue is approximately 1.9 kPa and 12.0 kPa, while the stiffness k is 13.8 N/m and 87.2 N/m, respectively. Modified from Samani et al. [33].
Finally, the term pressure is often used (incorrectly) to describe stress. Pressure is defined as the force per unit area acting perpendicular to the area’s surface. However pressure is a scalar quantity, magnitude (Pa), whereas stress is a vector (tensor) quantity. Pressure is normally used to describe the stress exerted by fluids and gases within a confining container, such as with fluid hydrostatic pressure which always acts perpendicular to a container’s surfaces. However when a fluid flows obliquely to a surface, it can generate shear, compressive and tensile stresses depending on the friction between the fluid and surface. In the tumor-stroma microenvironment, tumor cell proliferation and growth can exert these different stress types on the surrounding stroma, including interstitial fluid pressure (IFP) [21]. Interstitial fluid pressures (hydrostatic) have been measured in breast tumors. In patients with invasive ductal carcinomas the IFP was 3.87±0.40 kPa, significantly higher than in individuals with normal breast parenchyma (-0.04±0.01 kPa), benign tumors (0.48±0.11 kPa), noninvasive carcinomas (0.04±0.03 kPa), and other benign breast conditions (0.05±0.05 kPa) [34]. In patients having high-grade osteosacoma tumors, the IFP was 4.5±2.3 kPa, significantly higher compared to normal tissue IFP of 0.39±0.76 kPa [35]. Interstitial fluid pressures in tumors have also been found to increase with tumor size and vascularity [34, 35].
Tumor Invasion and Migration through the ECM
Degradation of the ECM by tumor and stroma cells is necessary for tumor invasion of the stroma, and the interaction between both cell types and with their surrounding ECM are recognized as primary factors in the EMT process [9]. The most significant ECM components that inhibit cell motility through the ECM are collagen, fibronectin (FN) and proteoglycan (PG). Two primary cellular mechanisms exist for tumor invasion and migration through the ECM, cellular catabolism of the ECM by enzymatic cleavage of collagen, FN and PG [7, 36-39], and cellular physical rearrangement or reorientation of the collagen by traction forces generated by the cells [4, 6, 14, 15, 20]. How the cells do this, and whether both the tumor and stroma cells are involved, is as yet not fully understood.
It is well known that mechanically collagen resists tensile forces while the FN and PG resist compressive forces. These later two components are rather easily digested by most MMPs [40] [41]. However, the collagen component is the largest, strongest, and most difficult to penetrate by the tumor or stroma cells. This is due to the collagen’s limited degradation by only a few MMPs, specifically the collagenases (MMPs-1, 8 and 13) and membrane type1-MMP (MT1-MMP or MMP-14), and the ability of the tumor cells to exert sufficient traction forces to reorient the collagen fibrils, especially when they are tensed due to tumor expansion. Cleavage of the collagen triple helix occurs at the 3/4-1/4 site, known as the “hinge region,” where the MMP squeezes into the “hole” between the α-chains to cleave the molecule [42, 43]. It has also shown that MMP-2 (gelatinase A) can aid in the cleavage process by a mechanism termed triple helicase activity, in which MMP-2 attaches to and unwinds (opens) the collagen α-chains at the hinge region for easier MMP access (Fig. 9). This mechanism may also be present at the tumor invasion front where MMP-2 has been shown to be expressed at high level and involved in cell migration through the ECM by its interaction with the collagen [41]. What is still unknown is how the increased tension in the collagen from the expanding ECM will affect the ability of MMPs to open, unwind and enter the hinge region for helices cleavage, and how the increased tension affects the tumor and stroma cells’ ability to apply sufficient traction forces on the collagen triple helix to rearrange the collagen for migration.
Figure 9.
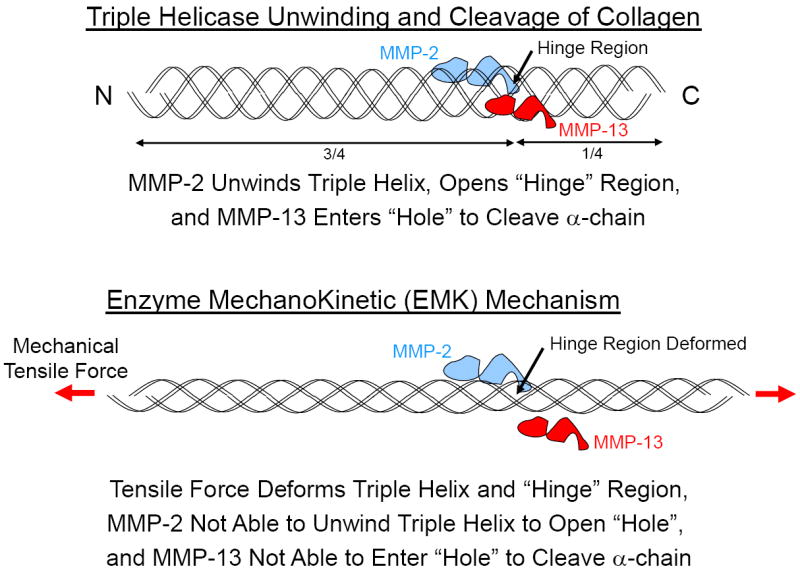
Triple helicase unwinding and cleavage of collagen (top). Collagen cleavage by MMPs occurs at the 3/4-1/4 site (hinge region). MMPs enter the triple helix (hole) to cleave the α-chains. MMP-2 (gelatinase A) has been shown to aid in the cleavage process by unwinding the α-chains at the hinge region (triple helicase activity) for easier MMP access [42, 43]. Inhibition of cleavage by enzyme mechanokinetic (EMK) mechanism (bottom). As collagen is stretched it becomes resistant to enzyme cleavage from changes in the collagen’s molecular conformation induced by the mechanical tensile force [44].
We have previously demonstrated that when collagen is stretched in tension it becomes resistant to enzyme cleavage [44]. The mechanism responsible for this was hypothesized to arise from changes in the collagen’s molecular conformation induced by the mechanical tensile force, such that the enzyme no longer had access to the cleavage site on the collagen molecule. We term this phenomena an Enzyme MechanoKinetic or EMK effect (Fig. 9). There is ample scientific evidence that in the microenvironment the collagen in the stroma is stretched by the expanding tumor [4, 16, 18, 20, 21]. Thus, as the tumor expands, the collagen in the stroma will realign and stretch perpendicular to the expanding tumor to resist the tumor expansion and enzymatic degradation. The tumor cells must now overcome the increased collagen alignment, density and tension before the cells can invade the stroma. An important question arises as to whether the cells are capable of exerting sufficiently high traction forces to reorient the tensed collagen to allow cell invasion and migration through the stroma? While it has already been shown that the cells are capable of significantly increasing their traction force in response to the increasing ECM stiffness (1 kPa to 100 kPa) as they transition to a differentiated myofibroblastic phenotype [5, 22, 32], it is doubtful that this is enough in itself, without ECM degradation, to allow cell invasion and migration through the ECM. Thus, the cells must also degrade the ECM through synthesis of MMPs specific to collagen, PG and fibronectin [36, 37, 41]. However MMP synthesis alone may not be sufficient in itself to overcome the EMK effect as the tumor expands. Thus it appears that a combination of cell traction forces and ECM degradation is required. An important question, however, and one not previously discussed, is what role the cell’s traction forces may have in assisting the MMPs in their ability to open, unwind and cleave the collagen?
Steered Molecular Dynamics Suggests Cell-Assisted Collagen Unwinding
Collagen is one of the most abundant matrix proteins in the body and at the cellular level (nano and microscales) collagen and fibronectin are the primary ECM components that physically connect the cell to the surrounding matrix through attachments such as integrins. Therefore these matrix proteins serve as key structural elements through which intracellular systems physically attach to and connect with the extracellular environment.
Based on the ECM structure, basic mechanics principles imply that as a tumor expands the collagen immediately adjacent to the tumor experiences increasing tension. Experimental data previously suggested that tension dramatically inhibits the rate (kinetics) of enzymatic degradation of the collagen proteins in the matrix [44-48]. Of interest, this phenomenon likely has a natural containment effect as tumor cell proliferation and expansion is likely simultaneously increasing matrix resistance to MMPs and preserving the very ECM encapsulating the tumor. At some point however, the MMPs begin to degrade the collagen and the tumor cells are now able to pass through the surrounding stroma.
In order to simulate the effects of mechanical forces on protein conformation a number of groups, including our own, have utilized steered molecular dynamics (SMD) to model collagen mechanical loading at the molecular level (nanoscale) [49-52]. Molecular dynamics simulations integrate many physical variables such as thermal and bond energies over femtosecond (1×10-15 second) time steps to predict atomic movements of the protein as a function of time. By adding an external steering force vector to the molecular dynamics simulation, SMD can predict the system’s conformation in response to external force [53].
Our laboratory has recently described how mechanical forces are transmitted from the microscale down to the nanoscale and atomic regimes, based on the multi-scale structure commonly observed within the collagen triple helix. We developed novel SMD analytical approaches to characterize and quantify changes in the collagen’s protein conformation from forces acting parallel to (tensile, Fig. 10) and perpendicular to (Fig. 11) the long axis of the triple helix [49]. Previous SMD simulations indicated that during tensile loading collagen unwinds and changes its conformation [50, 52]. However many of these conformational changes in the collagen are likely undetectable using traditional force spectroscopy methods, and require atomistic resolution techniques to be distinguished. An example of this is provided in Figure 10, where a tensile force applied to a single α-chain results in both α-chain slippage (~400 pN) and triple helix chain unwinding (~800 pN), with nearly undetectable landmarks in the force – displacement curves.
Figure 10.
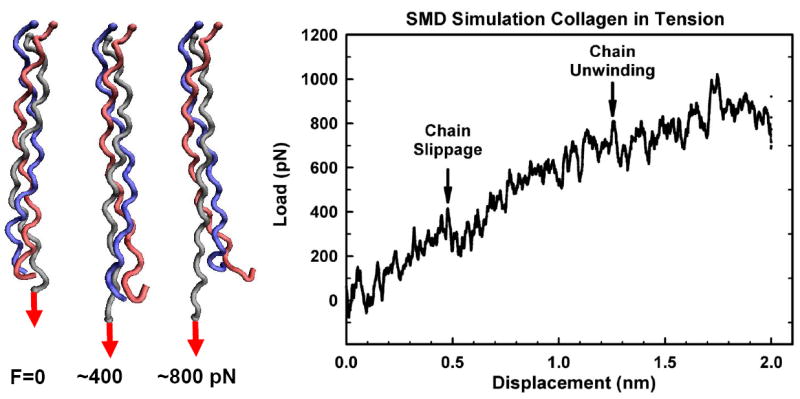
Force-induced α-chain slippage and helical microunwinding of the collagen molecule. Steered molecular dynamic (SMD) simulation of helical deformation from a tensile (axial) mechanical force applied to the end of a single α-chain (gray). The α-chain is pulled out of the triple helix at low force (slippage ~400 pN) while at higher forces (~800 pN) the triple helix begins to unwind. In this simulation, all three chains (gray, red, blue) are fixed at the opposite end of the triple helix.
Figure 11.
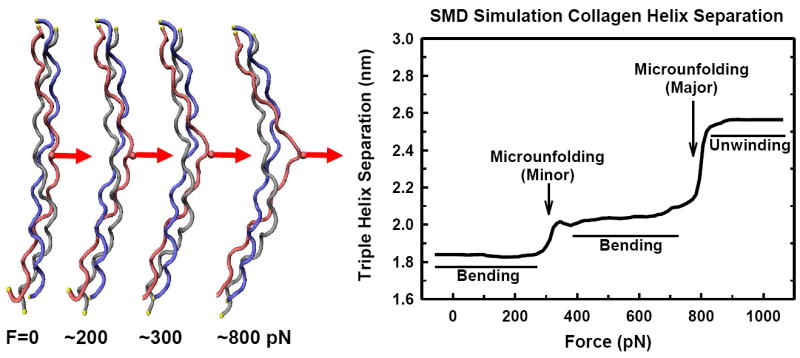
Force-induced microunfolding of the collagen triple helix [49]. Steered molecular dynamic (SMD) simulation of triple helical peptide deformation from a perpendicularly (transversely) applied mechanical force to a single α-chain (amino acid side chain, red). The triple helix first bends with local triple helix microunfolding (~300 pN). Bending continues at higher forces (~400-800 pN) without significant separation of the individual chains. Fundamental mechanics principles show that tumor growth and expansion would cause increasing tension in the surrounding collagen (see Fig. 14), suggesting that this tension likely assists in trapping and containment at early stages of cancer cell proliferation by preventing matrix degradation. At some point however, the enzymes begin to degrade the collagen and the tumor cells are now able to pass through the surrounding stroma. One potential contributing mechanism may be due to increasing cellular mechanical forces being triggered through process such as TGFβ signaling and upregulation of α-smooth muscle actin. As cellular forces on the matrix increase, SMD data shows that local helix disruption would be induced thus facilitating MMP proteolytic activity against the otherwise previously protected collagen. At forces greater then ~900 pN the α-chains separate with loss of the triple helix conformation (α-chain destabilization). In this simulation, all three chains (gray, red, blue) are fixed at both ends of the triple helix.
Our SMD simulations of the non-axial loading directions are of particular interest as they represent some of the first attempts to simulate force transmission between adjacent collagen molecules (across cross-links), collagen and other ECM components (e.g., decorin), and forces applied to collagen through cellular attachment sites (integrins). Previously, disruption of the collagen triple helices by an applied force was predicted to occur in the range from 4,000 to 11,500 pN [54, 55 [56]. A major finding from our collagen model was that local helix disruption, referred to as microunfolding, was predicted to occur below 1,000 pN of applied force (Fig. 11) [49]. This force is well below previously described mechanical failure mechanisms, and suggests that mechanical perturbations such as cellular traction forces may actually cause local microunfolding of the collagen, possibly sensitizing the collagen triple helix to MMP degradation. Of great interest, this molecular model provides a framework for the EMT process whereby the cellular traction forces exerted on the ECM during invasion and migration are a molecular-based mechanism which might enable increased enzymatic degradation of the collagen. That is, we hypothesize that the cell can exert sufficiently large traction forces to the collagen to assist in the unwinding of the collagen to enhance the enzymes (MMPs) ability to cleave the collagen.
A New Paradigm Based on Cellular Assisted Enzyme MechanoKinetics
Our SMD results clearly showed that the collagen triple helix could easily be unwound at a force well below what a cell can exert, thus suggesting that cells can physically assist in the enzymatic cleavage of collagen. Based on our SMD and EMK findings, we can now postulate a new paradigm for tumor cell invasion and migration through the collagen-dense stroma, shown schematically in Fig. 12. We term this new cell-matrix interaction a Cellular Assisted Enzyme MechanoKinetic (CAEMK) effect or mechanism. In this model the expanding tumor generates an increased mechanical tensile force in the collagen which inhibits an MMP’s ability to cleave the collagen molecule. As the tumor and/or stroma cell(s) attempt to reorient the collagen fibrils the cells can “sense” the increased tension (stiffness) in the collagen and respond by increasing the magnitude of the cell-generated traction force, thus unwinding the collagen molecule (single α-chain) and opening the “hole” for MMP entrance and cleavage. Still to be determined is whether the mechanical force deforms the hinge region, thus closing the “hole” through which the MMPs enter to cleave the collagen molecule, changes the MMP’s attachment site on the collagen to inhibit MMP attachment, or possibly changing the distance between the MMP’s attachment and cleavage sites, thus modifying the MMP’s ability to reach the hinge region.
Figure 12.
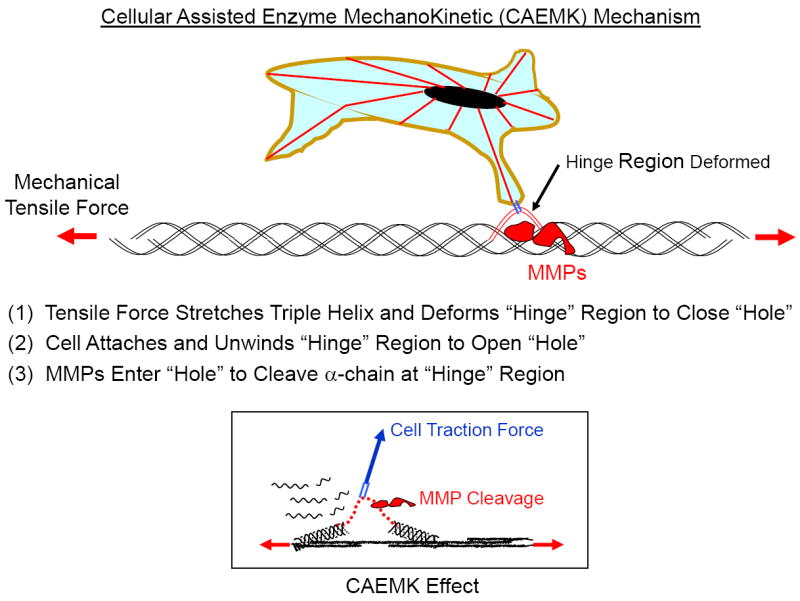
Schematic representation of proposed Cellular Assisted Enzyme MechanoKinetic (CAEMK) mechanism. Experimental data indicates that tension dramatically inhibits the enzymatic degradation of matrix collagen (Enzyme MechanoKinetic or EMK response, Fig. 9) [44]. However since cells can apply significant tensile traction forces to the α-chains, our SMD model (Fig. 11) indicates that a cell can separate the α-chain from the other α-chains (microunfolding) to deform (open) the hinge region for MMP access and cleavage.
Our SMD simulation of collagen deformation showed that the unwinding of the collagen monomer occurs at two distinct but different length scales (Fig. 11). We can now apply this unwinding mechanism to describe how a cell can deform the collagen to assist the enzyme degradation of the ECM. When the cell traction force is directed perpendicular to the long axis of the triple helix, as shown in Fig. 13, the collagen monomer will first bend (cellular micro-deformation) at low forces (~100-200 pN), with small changes in triple helix spacing (minor microunfolding, ~300 pN), followed by further bending of the monomer at higher forces (~400-800 pN) which will cause an increase in triple helix spacing but no significant separation of the individual α-chains (fibril destabilization). However when the cell traction force reaches ~900 pN, the α-chains separate (cellular nano-deformation or major microunfolding) causing loss of the triple helix conformation (α-chain destabilization). At cell traction forces >900 pN the triple helix unwinds, allowing MMPs to cleave the α-chain and further unravel the triple helix for further enzyme cleavage. Based on this new mechanism (CAEMK), in which the cell has to increase its traction force on the collagen to overcome the increased stiffness in the collagen as the tumor expands, we hypothesize that there exists a critical magnitude of the cell-generated traction force that will act as a biomechanical trigger for the initiation of the metastatic invasive process. This cell-generated traction force is transmitted from the ECM to the cell through the cell’s integrins and focal adhesion attachments, specifically to the collagen. What magnitude of force causes the biomechanical trigger is not known. However we believe there exists a cell-specific magnitude of traction force that acts as a mechanotransduction signal to the cell to aggressively remodel the ECM in order to decrease the internal stresses (tension and compression) caused by the expanding tumor on the stroma (Fig. 14). In addition to a force-dependent biomechanical trigger, the cells may also sense the change in the conformation of the stretched collagen, for instance through recognition of changes in the cell’s integrin binding sites on the deformed collagen. Either or both of these mechanisms may act as a biomechanical trigger for the initiation of tumor invasion.
Figure 13.
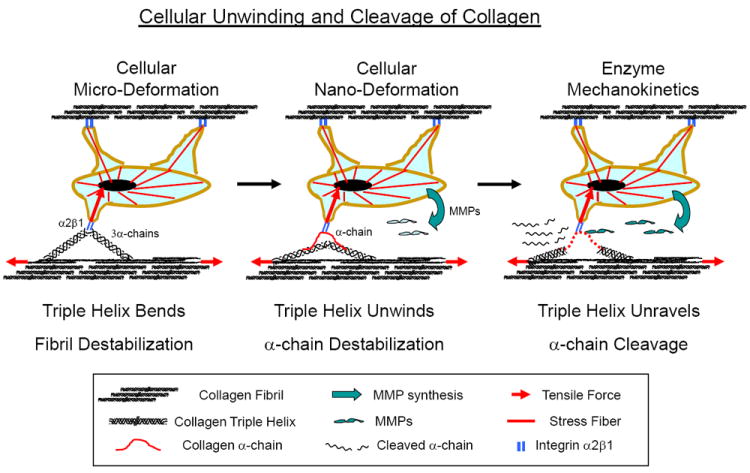
Schematic representation of cellular unwinding and cleavage of collagen by the proposed Cellular Assisted Enzyme MechanoKinetic (CAEMK) mechanism. The cell’s traction force on the collagen monomer will first bend the monomer (cellular micro-deformation, left figure) to destabilize the collagen fibril. Then the α-chain will separate (cellular nano-deformation, middle figure) to destabilize and unwind the triple helix, allowing MMPs to cleave the α-chain (EMK, right figure). Based on the CAEMK mechanism, there may be a critical cell traction force magnitude that will acts as a biomechanical trigger to initiate the metastatic invasive process.
Figure 14.
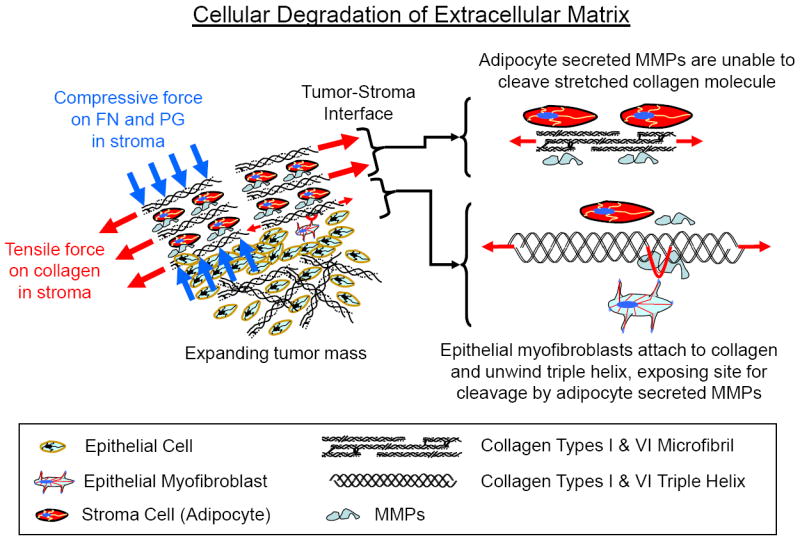
Proposed model for cellular degradation of the extracellular matrix (ECM) in response to tensile and compressive forces generated during tumor growth. Fundamental mechanics principles show that tumor growth and expansion would cause increasing tension and compression in the surrounding stroma. This suggests that the tension in the collagen within the stroma likely assists in trapping and containment at early stages of cancer cell proliferation by preventing matrix degradation (EMK effect). MMPs secreted by adipocytes within the stroma are unable to cleave the collagen. In response to the increased forces within the ECM, epithelial myofibroblasts then attach to the ECM to reorient the collagen, sense the increased tension (stiffness) in the collagen and increase their tensile traction force on the collagen, which results in unwinding of the triple helix, allowing MMP entrance and cleavage of the α-chains.
Based on these new mechanisms we propose the following new paradigm for EMT (Figs. 15a-c). As the tumor grows the stroma will also expand, with a concomitant increase in collagen density and mechanical tension in the collagen within the stroma. Both the increased collagen density and tension will act to constrain the tumor’s expansion [4, 6, 20, 32]. Initially the increased tensile forces in the collagen will be protective and inhibit MMP cleavage of the collagen due to mechano-conformational changes in the collagen from the EMK effect. These mechanobiological mechanisms will act to encapsulate the expanding tumor and inhibit tumor invasion of the stroma (Fig. 15a, right). However as the tumor continues to expand there will be increased stroma stretching and compression, causing increased mechanical forces (tensile and compressive) on the cells within both the tumor and stroma microenvironment (Fig. 14). The increase in the mechanical forces will initially cause both the tumor and stroma cells to transition from a quiescent state (Fig. 15a, left) to a proto-fibroblast state, with the cells having little ability to exert traction forces on the ECM (Fig. 15a, right).
Figures 15.
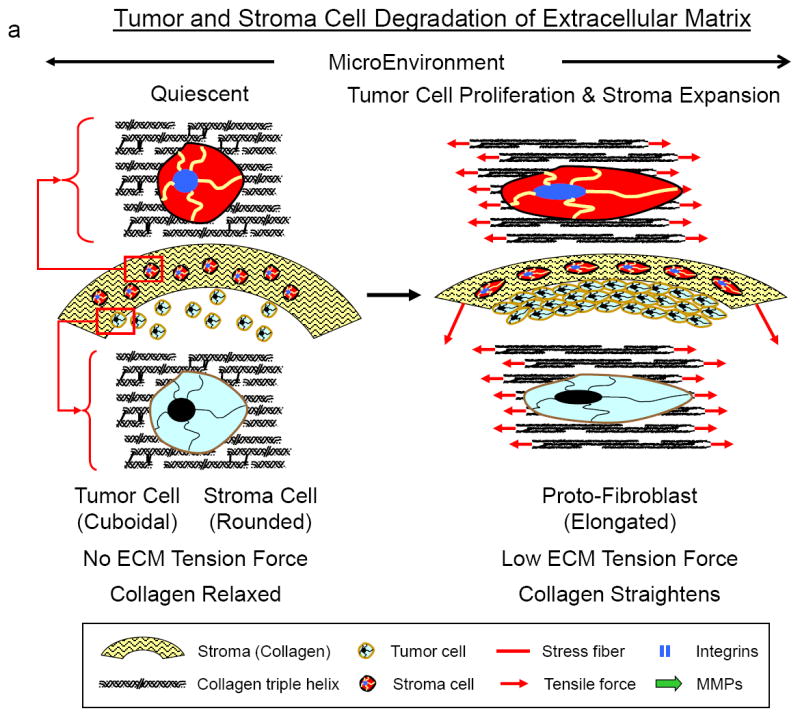
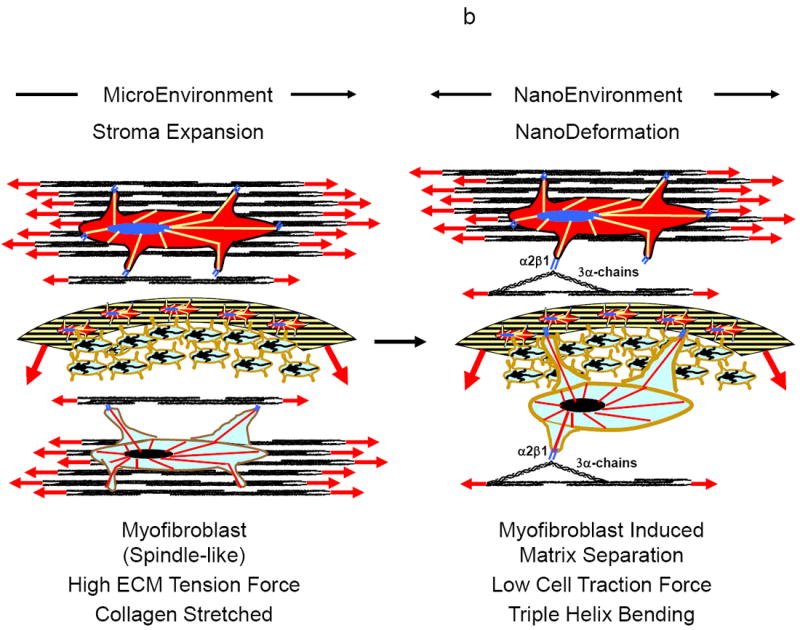
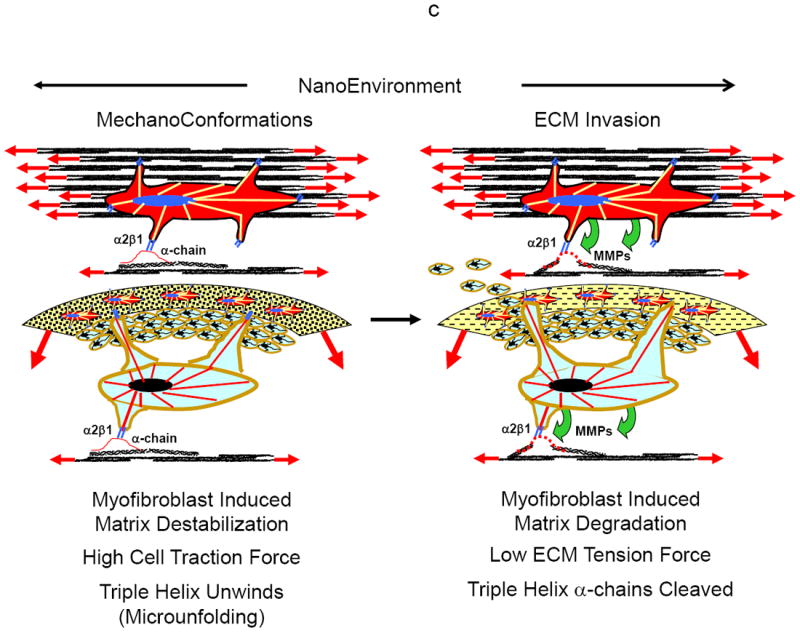
a-c Shown in Figures 15a-c is a hypothesized paradigm for how tumor and stroma cells respond to tumor growth and expansion to degrade the extracellular matrix for tumor cell invasion and migration through the stroma.
a Transition of quiescent tumor cells (left) to proliferating cells expanding the stroma extracellular matrix (right). As the tumor expands the surrounding increased mechanical forces (tensile and compressive) will be generated in the cells within the tumor and stroma microenvironment. The increase in the mechanical forces will cause tumor and stroma cells to transition from a quiescent state (left) to a proto/mesenchymal-fibroblast state (right). In this state the cells will have little ability to exert large traction forces on the ECM.
b As the stroma continues to expand with tumor growth, there will be increased mechanical forces (tension and compression) on the tumor and stroma cells within the extracellular matrix (left). When a critical magnitude of intrinsic force is reached (biomechanical trigger), a signal will be sent to the tumor and stroma cells to remodel the extracellular matrix in order to lower the mechanical forces. The cells will then transition to a more myofibroblastic phenotype with the ability to exert larger traction forces on the extracellular matrix, eventually bending the collagen monomer (right).
c Traction forces exerted by the tumor and stroma cells on the extracellular matrix will destabilize the collagen monomer and unwind the collagen triple helix (left). Once the collagen triple helix is destabilized it will become accessible to MMPs for cleavage of the collagen’s α-chains, allowing the tumor cells to invade and migrate through the degraded stroma (right).
However as the mechanical forces on the cells increase with further tumor expansion, rather than being protective for ECM degradation (EMK effect), the force will become a key biomechanical trigger signaling both the tumor and stroma cells to remodel the ECM (reorient and cleave the collagen) to reduce the stresses on the cell (e.g., hydrostatic pressure), and thus initiating tumor invasion of the stroma (EMT effect). The cells will now gain the ability to exert significant traction forces on the ECM as they transition from an epithelial state to a more mesenhymal/myofibroblastic phenotype (Fig. 15b, left). Eventually the cells will exert sufficiently high enough traction forces to separate and bend the collagen monomer (Fig. 15b, right), and then unwind and destabilize the collagen triple helix (Fig. 15c, left) for MMP cleavage of the collagen’s α-chains (CAEMK effect) (Fig. 15c, right).
Both these mechanobiological mechanisms (EMK and CAEMK effects) can mediate dual roles in cancer by acting as a tumor suppressor or protective shield in early tumor development, and paradoxically, by promoting tumor cell invasion in later stages. This is similar to how certain growth factors act, such as TGFβ, which can both inhibit and promote cancer by acting at different stages in tumor progression [5, 19]. We can not specify or even hypothesize which mechanism, cytokine, chemokine or MMP is more important in tumor metastasis and invasion (EMT), or if they even act independently. Rather we can assume that they do not act independently and are equally important.
Summary
So what is the influence of biomechanical forces on the invasive capacity of tumor cells in the metastatic process? In this paper we have proposed a new and exciting paradigm about fundamental biomechanical (force) and biophysical (molecular conformation) mechanisms involved in cancer growth and metastasis, and how these mechanisms are interrelated to be both protective (EMK effect) and destructive (CAEMK effect). These mechanobiological mechanisms directly challenge current paradigms that are focused chiefly at the cellular level on the biological and biochemical mechanisms associated with tumor metastasis, namely the intracellular pathways of signal transduction from the ECM to the nucleus (outside-in signaling), and the cell’s metabolic response for synthesizing proteinases to degrade the ECM (inside-out signaling). Our new mechanobiological mechanisms are focused not only on how tumor expansion generates mechanical forces (tensile and compressive) within the stroma to resist tumor expansion at the tissue level, but more important, how these same forces can inhibit or enhance tumor invasion of the stroma at the molecular level by, respectively, inhibiting MMP degradation of the tensed interstitial collagen (EMK effect) or triggering the invading cell to unwind the collagen for cleavage (CAEMK effect). This approach is also different from the concept of cells sensing and responding to changes (increases) in the ECM stiffness. We are proposing that the biomechanical trigger is not so much the ECM stiffness (i.e., force-deformation response) but rather the magnitude of traction force the cell must generate (i.e., peak cell-traction force) to overcome the intrinsic ECM structural resistance to deform and unwind the collagen triple helix for α-chain cleavage. These two new conceptual mechanobiological mechanisms have not been previously applied to the study of tumor metastasis, and we believe they may have the potential to open new areas of research in our understanding of the tumor invasive process.
Acknowledgments
Support for this study was made possible by Grant Numbers AR46574 (PAT), AR45748 (PAT), AR059203 (PAT) and AR051636 (PAT) from the National Institute of Arthritis and Musculoskeletal and Skin Diseases of the National Institutes of Health (NIH), Grant Numbers TL1RR024998 (JWB) and UL1RR024996 (PAT) from the Weill Cornell Medical College’s National Center for Research Resources (NIH) Clinical and Translational Science Center, and the Weill Cornell Graduate School of Medical Sciences (JWB). CTV was supported by the Swedish Research Council, Georg and Eva Klein Visiting Junior Scientist Award, Sigurd och Elsa Goljes minne, American Scandinavia Foundation, VINNMER–Marie Curie international qualification. Its contents are solely the responsibility of the authors and do not necessarily represent the official views of the NIH-NIAMS and other supporting agencies. Investigations cited in this manuscript were conducted in a facility constructed with support from Research Facilities Improvement Program Grant Number C06-RR12538-01 from the National Center for Research Resources, National Institutes of Health.
Footnotes
Conflict of Interest
The authors declare that there are no conflicts of interest.
Publisher's Disclaimer: This is a PDF file of an unedited manuscript that has been accepted for publication. As a service to our customers we are providing this early version of the manuscript. The manuscript will undergo copyediting, typesetting, and review of the resulting proof before it is published in its final citable form. Please note that during the production process errors may be discovered which could affect the content, and all legal disclaimers that apply to the journal pertain.
References
- 1.Jemal A, Siegel R, Ward E, Hao Y, Xu J, Thun MJ. Cancer statistics, 2009. CA Cancer J Clin. 2009;59(4):225–49. doi: 10.3322/caac.20006. [DOI] [PubMed] [Google Scholar]
- 2.Liedtke C, Mazouni C, Hess KR, Andre F, Tordai A, Mejia JA, et al. Response to neoadjuvant therapy and long-term survival in patients with triple-negative breast cancer. J Clin Oncol. 2008;26(8):1275–81. doi: 10.1200/JCO.2007.14.4147. [DOI] [PubMed] [Google Scholar]
- 3.Lee JM, Dedhar S, Kalluri R, Thompson EW. The epithelial-mesenchymal transition: new insights in signaling, development, and disease. J Cell Biol. 2006;172(7):973–81. doi: 10.1083/jcb.200601018. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Provenzano PP, Eliceiri KW, Campbell JM, Inman DR, White JG, Keely PJ. Collagen reorganization at the tumor-stromal interface facilitates local invasion. BMC Med. 2006;4(1):38–54. doi: 10.1186/1741-7015-4-38. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Hinz B. The myofibroblast: paradigm for a mechanically active cell. J Biomech. 2011;43(1):146–55. doi: 10.1016/j.jbiomech.2009.09.020. [DOI] [PubMed] [Google Scholar]
- 6.Provenzano PP, Alejandro-Osorio AL, Valhmu WB, Jensen KT, Vanderby R., Jr Intrinsic fibroblast-mediated remodeling of damaged collagenous matrices in vivo. Matrix Biol. 2005;23(8):543–55. doi: 10.1016/j.matbio.2004.09.008. [DOI] [PubMed] [Google Scholar]
- 7.Wolf K, Wu YI, Liu Y, Geiger J, Tam E, Overall C, et al. Multi-step pericellular proteolysis controls the transition from individual to collective cancer cell invasion. Nat Cell Biol. 2007;9(8):893–904. doi: 10.1038/ncb1616. [DOI] [PubMed] [Google Scholar]
- 8.Rouyer N, Wolf C, Chenard MP, Rio MC, Chambon P, Bellocq JP, et al. Stromelysin-3 gene expression in human cancer: an overview. Invasion Metastasis. 1994;14(1-6):269–75. [PubMed] [Google Scholar]
- 9.Motrescu ER, Blaise S, Etique N, Messaddeq N, Chenard MP, Stoll I, et al. Matrix metalloproteinase-11/stromelysin-3 exhibits collagenolytic function against collagen VI under normal and malignant conditions. Oncogene. 2008;27(49):6347–55. doi: 10.1038/onc.2008.218. [DOI] [PubMed] [Google Scholar]
- 10.Frantz C, Stewart KM, Weaver VM. The extracellular matrix at a glance. J Cell Sci. 2011;123(Pt 24):4195–200. doi: 10.1242/jcs.023820. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Vincent T, Neve EP, Johnson JR, Kukalev A, Rojo F, Albanell J, et al. A SNAIL1-SMAD3/4 transcriptional repressor complex promotes TGF-beta mediated epithelial-mesenchymal transition. Nat Cell Biol. 2009;11(8):943–50. doi: 10.1038/ncb1905. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Tan JL, Tien J, Pirone DM, Gray DS, Bhadriraju K, Chen CS. Cells lying on a bed of microneedles: an approach to isolate mechanical force. Proc Natl Acad Sci U S A. 2003;100(4):1484–9. doi: 10.1073/pnas.0235407100. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Kumar S, Weaver VM. Mechanics, malignancy, and metastasis: the force journey of a tumor cell. Cancer Metastasis Rev. 2009;28(1-2):113–27. doi: 10.1007/s10555-008-9173-4. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Hoffman BD, Grashoff C, Schwartz MA. Dynamic molecular processes mediate cellular mechanotransduction. Nature. 2011;475(7356):316–23. doi: 10.1038/nature10316. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Mierke CT, Frey B, Fellner M, Herrmann M, Fabry B. Integrin alpha5beta1 facilitates cancer cell invasion through enhanced contractile forces. J Cell Sci. 2011;124(Pt 3):369–83. doi: 10.1242/jcs.071985. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Paszek MJ, Zahir N, Johnson KR, Lakins JN, Rozenberg GI, Gefen A, et al. Tensional homeostasis and the malignant phenotype. Cancer Cell. 2005;8(3):241–54. doi: 10.1016/j.ccr.2005.08.010. [DOI] [PubMed] [Google Scholar]
- 17.Levental I, Georges PC, Janmey PA. Soft biological materials and their impact on cell function. Soft Matter. 2006;2:1–9. doi: 10.1039/b610522j. [DOI] [PubMed] [Google Scholar]
- 18.Levental KR, Yu H, Kass L, Lakins JN, Egeblad M, Erler JT, et al. Matrix crosslinking forces tumor progression by enhancing integrin signaling. Cell. 2009;139(5):891–906. doi: 10.1016/j.cell.2009.10.027. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Gjorevski N, Boghaert E, Nelson CM. Regulation of Epithelial-Mesenchymal Transition by Transmission of Mechanical Stress through Epithelial Tissues. Cancer Microenviron. 2011 doi: 10.1007/s12307-011-0076-5. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Provenzano PP, Inman DR, Eliceiri KW, Knittel JG, Yan L, Rueden CT, et al. Collagen density promotes mammary tumor initiation and progression. BMC Med. 2008;6:1–15. doi: 10.1186/741-7015-6-11. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Shieh AC. Biomechanical forces shape the tumor microenvironment. Ann Biomed Eng. 2011;39(5):1379–89. doi: 10.1007/s10439-011-0252-2. [DOI] [PubMed] [Google Scholar]
- 22.Solon J, Levental I, Sengupta K, Georges PC, Janmey PA. Fibroblast adaptation and stiffness matching to soft elastic substrates. Biophys J. 2007;93(12):4453–61. doi: 10.1529/biophysj.106.101386. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Wipff PJ, Hinz B. Myofibroblasts work best under stress. J Bodyw Mov Ther. 2009;13(2):121–7. doi: 10.1016/j.jbmt.2008.04.031. [DOI] [PubMed] [Google Scholar]
- 24.Chandler EM, Berglund CM, Lee JS, Polacheck WJ, Gleghorn JP, Kirby BJ, et al. Stiffness of photocrosslinked RGD-alginate gels regulates adipose progenitor cell behavior. Biotechnol Bioeng. 2011;108(7):1683–92. doi: 10.1002/bit.23079. [DOI] [PubMed] [Google Scholar]
- 25.Keely PJ. Mechanisms by which the extracellular matrix and integrin signaling act to regulate the switch between tumor suppression and tumor promotion. J Mammary Gland Biol Neoplasia. 2011;16(3):205–19. doi: 10.1007/s10911-011-9226-0. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Provenzano PP, Keely PJ. Mechanical signaling through the cytoskeleton regulates cell proliferation by coordinated focal adhesion and Rho GTPase signaling. J Cell Sci. 2011;124(Pt 8):1195–205. doi: 10.1242/jcs.067009. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Holton SE, Walsh MJ, Bhargava R. Subcellular localization of early biochemical transformations in cancer-activated fibroblasts using infrared spectroscopic imaging. Analyst. 2011;136(14):2953–8. doi: 10.1039/c1an15112f. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Gullberg D, Reed RK. Tumor-Stroma Interactions: Focus on Fibroblasts. In: Brakebusch C, Pihlajaniemi T, editors. Mouse as a Model Organism. Springer Netherlands; 2011. pp. 117–30. [Google Scholar]
- 29.Brown TD. Techniques for mechanical stimulation of cells in vitro: a review. J Biomech. 2000;33(1):3–14. doi: 10.1016/s0021-9290(99)00177-3. [DOI] [PubMed] [Google Scholar]
- 30.Humphrey JD. Stress, strain, and mechanotransduction in cells. J Biomech Eng. 2001;123(6):638–41. doi: 10.1115/1.1406131. [DOI] [PubMed] [Google Scholar]
- 31.Janmey PA, Georges PC, Hvidt S. Basic rheology for biologists. Methods Cell Biol. 2007;83:3–27. doi: 10.1016/S0091-679X(07)83001-9. [DOI] [PubMed] [Google Scholar]
- 32.Tang X, Kuhlenschmidt TB, Zhou J, Bell P, Wang F, Kuhlenschmidt MS, et al. Mechanical force affects expression of an in vitro metastasis-like phenotype in HCT-8 cells. Biophys J. 2010;99(8):2460–9. doi: 10.1016/j.bpj.2010.08.034. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.Samani A, Bishop J, Luginbuhl C, Plewes DB. Measuring the elastic modulus of ex vivo small tissue samples. Phys Med Biol. 2003;48(14):2183–98. doi: 10.1088/0031-9155/48/14/310. [DOI] [PubMed] [Google Scholar]
- 34.Nathanson SD, Nelson L. Interstitial fluid pressure in breast cancer, benign breast conditions, and breast parenchyma. Ann Surg Oncol. 1994;1(4):333–8. doi: 10.1007/BF03187139. [DOI] [PubMed] [Google Scholar]
- 35.Nathan SS, Huvos AG, Casas-Ganem JE, Yang R, Linkov I, Sowers R, et al. Tumor interstitial fluid pressure may regulate angiogenic factors in osteosarcoma. J Orthop Res. 2008;26(11):1520–5. doi: 10.1002/jor.20633. [DOI] [PubMed] [Google Scholar]
- 36.Gonzalez LO, Gonzalez-Reyes S, Junquera S, Marin L, Gonzalez L, Del Casar JM, et al. Expression of metalloproteases and their inhibitors by tumor and stromal cells in ductal carcinoma in situ of the breast and their relationship with microinvasive events. J Cancer Res Clin Oncol. 2010;136(9):1313–21. doi: 10.1007/s00432-010-0782-2. [DOI] [PubMed] [Google Scholar]
- 37.Gonzalez LO, Junquera S, del Casar JM, Gonzalez L, Marin L, Gonzalez-Reyes S, et al. Immunohistochemical study of matrix metalloproteinases and their inhibitors in pure and mixed invasive and in situ ductal carcinomas of the breast. Hum Pathol. 2010;41(7):980–9. doi: 10.1016/j.humpath.2009.08.027. [DOI] [PubMed] [Google Scholar]
- 38.Del Casar JM, Gonzalez-Reyes S, Gonzalez LO, Gonzalez JM, Junquera S, Bongera M, et al. Expression of metalloproteases and their inhibitors in different histological types of breast cancer. J Cancer Res Clin Oncol. 2010;136(6):811–9. doi: 10.1007/s00432-009-0721-2. [DOI] [PubMed] [Google Scholar]
- 39.del Casar JM, Carreno G, Gonzalez LO, Junquera S, Gonzalez-Reyes S, Gonzalez JM, et al. Expression of metalloproteases and their inhibitors in primary tumors and in local recurrences after mastectomy for breast cancer. J Cancer Res Clin Oncol. 2011;136(7):1049–58. doi: 10.1007/s00432-009-0750-x. [DOI] [PubMed] [Google Scholar]
- 40.Folgueras AR, Pendas AM, Sanchez LM, Lopez-Otin C. Matrix metalloproteinases in cancer: from new functions to improved inhibition strategies. Int J Dev Biol. 2004;48(5-6):411–24. doi: 10.1387/ijdb.041811af. [DOI] [PubMed] [Google Scholar]
- 41.Xu X, Wang Y, Chen Z, Sternlicht MD, Hidalgo M, Steffensen B. Matrix metalloproteinase-2 contributes to cancer cell migration on collagen. Cancer Res. 2005;65(1):130–6. [PubMed] [Google Scholar]
- 42.Overall CM. Molecular determinants of metalloproteinase substrate specificity: matrix metalloproteinase substrate binding domains, modules, and exosites. Mol Biotechnol. 2002;22(1):51–86. doi: 10.1385/MB:22:1:051. [DOI] [PubMed] [Google Scholar]
- 43.Tam EM, Moore TR, Butler GS, Overall CM. Characterization of the distinct collagen binding, helicase and cleavage mechanisms of matrix metalloproteinase 2 and 14 (gelatinase A and MT1-MMP): the differential roles of the MMP hemopexin c domains and the MMP-2 fibronectin type II modules in collagen triple helicase activities. J Biol Chem. 2004;279(41):43336–44. doi: 10.1074/jbc.M407186200. [DOI] [PubMed] [Google Scholar]
- 44.Wyatt KE, Bourne JW, Torzilli PA. Deformation-dependent enzyme mechanokinetic cleavage of type I collagen. J Biomech Eng. 2009;131(5):051004. doi: 10.1115/1.3078177. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 45.Huang C, Yannas IV. Mechanochemical studies of enzymatic degradation of insoluble collagen fibers. J Biomed Mater Res. 1977;11(1):137–54. doi: 10.1002/jbm.820110113. [DOI] [PubMed] [Google Scholar]
- 46.Bhole AP, Flynn BP, Liles M, Saeidi N, Dimarzio CA, Ruberti JW. Mechanical strain enhances survivability of collagen micronetworks in the presence of collagenase: implications for load-bearing matrix growth and stability. Philos Transact A Math Phys Eng Sci. 2009;367(1902):3339–62. doi: 10.1098/rsta.2009.0093. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 47.Camp RJ, Liles M, Beale J, Saeidi N, Flynn BP, Moore E, et al. Molecular Mechanochemistry: Low Force Switch Slows Enzymatic Cleavage of Human Type I Collagen Monomer. J Am Chem Soc. 2011 doi: 10.1021/ja110098b. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 48.Nabeshima Y, Grood ES, Sakurai A, Herman JH. Uniaxial tension inhibits tendon collagen degradation by collagenase in vitro. J Orthop Res. 1996;14(1):123–30. doi: 10.1002/jor.1100140120. [DOI] [PubMed] [Google Scholar]
- 49.Bourne JW, Torzilli PA. Molecular simulations predict novel collagen conformations during cross-link loading. Matrix Biol. 2011;30(5-6):356–60. doi: 10.1016/j.matbio.2011.03.010. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 50.in ’t Veld PJ, Stevens MJ. Simulation of the mechanical strength of a single collagen molecule. Biophys J. 2008;95(1):33–9. doi: 10.1529/biophysj.107.120659. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 51.Gautieri A, Vesentini S, Montevecchi FM, Redaelli A. Mechanical properties of physiological and pathological models of collagen peptides investigated via steered molecular dynamics simulations. J Biomech. 2008;41(14):3073–7. doi: 10.1016/j.jbiomech.2008.06.028. [DOI] [PubMed] [Google Scholar]
- 52.Gautieri A, Vesentini S, Redaelli A, Buehler MJ. Single molecule effects of osteogenesis imperfecta mutations in tropocollagen protein domains. Protein Sci. 2009;18(1):161–8. doi: 10.1002/pro.21. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 53.Phillips JC, Braun R, Wang W, Gumbart J, Tajkhorshid E, Villa E, et al. Scalable molecular dynamics with NAMD. J Comput Chem. 2005;26(16):1781–802. doi: 10.1002/jcc.20289. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 54.Uzel SG, Buehler MJ. Molecular structure, mechanical behavior and failure mechanism of the C-terminal cross-link domain in type I collagen. J Mech Behav Biomed Mater. 2011;4(2):153–61. doi: 10.1016/j.jmbbm.2010.07.003. [DOI] [PubMed] [Google Scholar]
- 55.Buehler M. Atomistic and continuum modeling of mechanical properties of Collagen: Elasticity, fracture and self-assembly. Journal of Materials Research. 2006;21(8):1947–61. [Google Scholar]
- 56.Grandbois M, Beyer M, Rief M, Clausen-Schaumann H, Gaub HE. How strong is a covalent bond? Science. 1999;283(5408):1727–30. doi: 10.1126/science.283.5408.1727. [DOI] [PubMed] [Google Scholar]
- 57.Eilaghi A, Flanagan JG, Tertinegg I, Simmons CA, Wayne Brodland G, Ross Ethier C. Biaxial mechanical testing of human sclera. J Biomech. 2010;43(9):1696–701. doi: 10.1016/j.jbiomech.2010.02.031. [DOI] [PubMed] [Google Scholar]
- 58.Pai S, Ledoux WR. The compressive mechanical properties of diabetic and non-diabetic plantar soft tissue. J Biomech. 2010;43(9):1754–60. doi: 10.1016/j.jbiomech.2010.02.021. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 59.Sasaki N, Odajima S. Stress-strain curve and Young’s modulus of a collagen molecule as determined by the X-ray diffraction technique. J Biomech. 1996;29(5):655–8. doi: 10.1016/0021-9290(95)00110-7. [DOI] [PubMed] [Google Scholar]