

Abstract
The fibrin(ogen) receptor, integrin αIIbβ3, has a well-established role in platelet spreading, aggregation and clot retraction. How αIIbβ3 contributes to platelet-dependent coagulation is less well resolved. Here, we demonstrate that the potent suppressing effect of clinically used αIIbβ3 blockers on tissue factor-induced thrombin generation is linked to diminished platelet Ca2+ responses and phosphatidylserine (PS) exposure. The same blockers suppress these responses in platelets stimulated with collagen and thrombin receptor agonists, whereas added fibrinogen potentiates these responses. In platelets spreading on fibrinogen, outside-in αIIbβ3 signaling similarly enhances thrombin-induced Ca2+ rises and PS exposure. These responses are reduced in αIIbβ3-deficient platelets from patients with Glanzmann’s thrombasthenia. Furthermore, the contribution of αIIbβ3 to tissue factor-induced platelet Ca2+ rises, PS exposure and thrombin generation in plasma are fully dependent on Syk kinase activity. Tyrosine phosphorylation analysis confirms a key role of Syk activation, which is largely but not exclusively dependent on αIIbβ3 activation. It is concluded that the majority of tissue factor-induced procoagulant activity of platelets relies on Syk activation and ensuing Ca2+ signal generation, and furthermore that a considerable part of Syk activation relies on αIIbβ3 signaling. These results hence point to a novel role of Syk in integrin-dependent thrombin generation.
Electronic supplementary material
The online version of this article (doi:10.1007/s00018-012-1033-2) contains supplementary material, which is available to authorized users.
Keywords: Platelets, Integrin αIIbβ3, Thrombin generation, Syk kinase, PS exposure
Introduction
Integrin αIIbβ3 (glycoprotein IIb/IIIa) is among the most abundantly expressed glycoproteins at the platelet surface, which strongly regulates the adhesion and aggregation of platelets. Platelet agonists induce a conformational change in the extracellular part of αIIbβ3 via so-called inside-out signaling events, accumulating in Rap1b activation and talin complex formation, which increases the adhesiveness of the integrin [1–3]. Conversely, binding of αIIbβ3 to its ligands, such as fibrinogen, fibrin and von Willebrand factor, stimulates outside-in signal transduction by a pathway involving activation of Src and Syk protein tyrosine kinases [4–6]. Platelet responses known to rely on αIIbβ3 outside-in signaling include the shedding of microparticles, the formation of lamellipods of fibrinogen-adhered platelets, and the contraction of platelets within a fibrin clot [7–9].
It has been known for a long time that platelets stimulated with strong agonists promote the processes of thrombin generation and blood coagulation [10, 11]. This procoagulant function is triggered by agonist combinations like collagen/thrombin, which evoke prolonged and high rises in cytosolic [Ca2+]i, and also by stimulating platelets in plasma with tissue factor [12, 13]. The procoagulant activity is caused by exposure of the negatively charged phosphatidylserine (PS), at the membrane surface via a transmembrane protein encoded by TMEM16F [14], which promotes the local assembly of vitamin K-dependent coagulation factors and hence generation of thrombin [15]. Early findings have pointed to a significant role of αIIbβ3 in the development of platelet procoagulant activity, in that integrin antagonists were found to suppress tissue factor-induced thrombin generation [12, 16, 17]. Yet, the mechanism by which αIIbβ3 blockage interferes with platelet-dependent coagulation has not been resolved. Suggestions are that the integrin is (1) involved in the formation of procoagulant microparticles [17–19], (2) directly binds prothrombin [20], or (3) provides binding sites for factor Va and other coagulation factors [21, 22]. Studies so far are complicated by data that show that distinct αIIbβ3 antagonists may differ in their effects on platelet activation and procoagulant activity [19, 23]. Another complicating factor is that part of the role of αIIbβ3 may be secondary to that of autocrine ADP, which enhances PS exposure via P2Y12 receptor stimulation [24–26].
In this paper, we hypothesize that αIIbβ3 interaction with its principal ligand, fibrinogen, enhances platelet-dependent thrombin generation via an outside-in signaling mechanism. We demonstrate that signaling via Syk kinase is responsible for the majority of tissue factor-induced thrombin generation of platelets in plasma by stimulating PS exposure.
Materials and methods
Materials
Human α-thrombin was obtained from Enzyme Research Laboratories, recombinant human tissue factor came from Dade Behring, abciximab (reopro) from Centocor; tirofiban (aggrastat) from Merck Sharp & Dohme, and eptifibatide (integrilin) from GlaxoSmithKline. Dimethyl BAPTA (DM-BAPTA), Fura-2 and Fura-Red acetoxymethyl esters were from Molecular Probes, while Syk inhibitor II and IV were from Merck Biosciences. Apyrase, bovine serum albumin (BSA), human and bovine fibrinogen (fraction 1, type III), and non-radioactive protein tyrosine kinase assay kit were all from Sigma. Cangrelor (AR-C69931MX) was kindly provided by The Medicine Company. Ancrod came from NIBSC; fluorescein isothiocyanate (FITC)-labeled annexin A5 from PharmaTarget, and FITC-labeled monoclonal antibody (mAb) against platelet-bound human fibrinogen from WAK Chemie Medical. Rabbit anti-phospho-Syk (Tyr525/526) mAb and HRP-linked anti-rabbit IgG were from Cell Signaling Technology, mouse anti-Syk mAb was from Santa Cruz Biotechnology, rabbit anti-α-tubulin Ab from Abcam, and HRP-linked anti-mouse IgG from GE Healthcare. Pefabloc (Gly-Pro-Arg-Pro-amide, GPRP) was obtained from Kordia Life Sciences. Microbeads coated with human anti-CD31 mAb and MS columns were a kind gift from Miltenyi Biotec. Procoagulant phospholipid vesicles (PS:phosphatidyl choline:phosphatidyl ethanolamine 1:3:1; mol/mol) were prepared as described [27]. Convulxin was purified to homogeneity from the venom of Crotalus durissus terrificus [28]. Other materials including fibrinogen were from sources indicated before [27].
Platelet and plasma preparation
Blood was taken from healthy volunteers and from two patients with Glanzmann’s thrombasthenia, with established deficiencies in integrin αIIbβ3 [29], after informed consent and in accordance with the Declaration of Helsinki. Approval was received from the local medical ethical committee. Blood was collected into 1/10 volume of 129 mM trisodium citrate. PRP was obtained by centrifuging at 240g for 15 min and platelet-free plasma (PFP) by centrifuging twice at 2,630g for 10 min. Platelet count in PRP was determined with a thrombocounter (Coulter Electronics) and normalized with autologous PFP. Citrate-anticoagulated PFP was partly defibrinated by a 10-min treatment with low ancrod protease (1.3 U/mL). After centrifuging the fibrin clots that were formed, non-turbid plasma was isolated. The remaining fibrinogen content was determined at ~0.5 mg/mL according to the conventional Claus method based on turbidimetric measurements [30]. When supplemented with platelets, the ancrod-treated plasma showed normal collagen-induced platelet aggregation responses.
For the preparation of washed platelets, blood was collected into 1/6 volume of acid-citrate glucose solution (ACD, 80 mM trisodium citrate, 52 mM citric acid and 180 mM glucose). Platelets were obtained by centrifugation, washed in the presence of apyrase (0.1 U/mL ADPase), and resuspended in Hepes buffer pH 7.45 (10 mM Hepes, 136 mM NaCl, 2.7 mM KCl, 2 mM MgCl2, 0.1 % glucose and 0.1 % BSA) at a count of 1.0 × 108/mL [27].
For experiments with reconstituted PRP, partly defibrinated plasma was supplemented with washed platelets (1.0 × 108/mL). Apyrase was not added, because of the presence of autologous exonucleotidase activity in plasma.
Flow cytometry
Washed, unstirred platelets in Hepes buffer were activated with PAR1 agonist SFLLRN (15 μM) or thrombin (10 nM) in combination with convulxin (50 ng/mL). Alternatively, the washed platelets were resuspended in ancrod-treated citrate plasma at 1.0 × 108/mL. The reconstituted PRP was activated with tissue factor (2 pM) and CaCl2 (16.6 mM) at 37 °C. After 15 min of activation, PS exposure and integrin activation were determined with FITC-labeled annexin A5 or FITC-labeled mAb against platelet-bound human fibrinogen, respectively, using flow cytometry [13]. For cytosolic Ca2+ measurements, platelets were loaded with Fura-Red (22 μM) and pluronic (400 μg/mL) in the presence of apyrase (0.1 U/mL ADPase). After a washing step, the loaded platelets were resuspended in ancrod-treated citrate plasma, which was triggered by tissue factor (2 pM) and CaCl2 (16.6 mM) at 37 °C. Increases in cytosolic Ca2+, apparent as decreases in fluorescence, were recorded in time by flow cytometry [26].
Thrombin generation
Thrombin generation was determined in normalized PRP (1.5 × 108 platelets/mL) or, as a control, in PFP supplemented with phospholipid vesicles (10 μM). The normalized PRP from control subjects or a Glanzmann patient was activated with tissue factor/CaCl2, and fluorescence accumulation was measured according to the thrombogram method under non-stirred conditions in a Fluoroskan Ascent well-plate reader at 37 °C [12]. Nanomolar thrombin concentrations were obtained by comparison with a human thrombin standard using Thrombinoscope software.
Spectrofluorometry
Platelets were loaded with Fura-2 when rises in cytosolic Ca2+ concentration were determined in the absence of plasma [31]. Fura-2-loaded platelets were activated in the presence of 2 mM CaCl2 at slow stirring (100 rpm, 37 °C); inhibitors were given before (10 min) or after agonist addition, as indicated. Nanomolar changes in Ca2+ level were obtained by calibration procedures, described in detail elsewhere [32].
Fluorescence microscopy
Glass coverslips were coated with 25 μL of fibrinogen solution (1 mg/mL), rinsed twice with saline, and incubated with washed (Fura-2-loaded) platelets in Hepes buffer pH 7.45 (1.0 × 108/mL) [32]. Where indicated, the coverslips were coated with a low fibrinogen solution (10 μg/mL). After 30 min, non-adherent platelets were removed, and the adhered spreading platelets were stimulated with thrombin (10 nM) in the presence of 2 mM CaCl2. Microscopic phase-contrast and fluorescence images of PS exposure (FITC-labeled annexin A5) were taken using a dual camera imaging system, controlled by Visitech software [33]. Fluorescence ratio images of Fura-2 fluorescence were taken to obtain rises in [Ca2+]i. For calibration, fluorescence values were obtained from Ca2+-saturated and Ca2+-free lysed platelets containing the fluorescent probe, using the microscopic and camera settings as described [34].
Platelet isolation from coagulating plasma
Washed platelets (5 × 108 platelets/mL) were reconstituted in ancrod-defibrinated plasma in the presence of GPRP (1 mg/mL) and cangrelor (20 μM). Samples of reconstituted PRP were preincubated with vehicle, Syk inhibitor II (5 μM), and tirofiban (10 μg/mL), as indicated, and activated with tissue factor (2 pM) and CaCl2 (16.6 mM). Initial attempts were made to isolate platelets from the activated PRP by centrifugation or gel filtration, but these were unsuccessful. Hence, a novel method was developed, in which platelets were captured from activated PRP by addition of anti-CD31 mAb-coated magnetic microbeads. After 15 min of activation, these platelets were isolated by passage of the PRP through a separation column, and an immediate rinse to remove all plasma proteins. Isolated platelets in the separation column were immediately lysed by flowing with lysis buffer (600 mM NaCl, 40 mM Tris, 4 mM EGTA, 4 mM EDTA, 4 % nonidet-P40, 10 mM Na3VO4, 4 mM PMSF, 20 μg/mL leupeptin, 20 μg/mL aprotinin, 5 μg/mL pepstatin A). Lysates were frozen at −80 °C until use.
Protein separation and western blotting
Platelet lysates were separated by polyacrylamide gel electrophoresis and subjected to standard western blotting. Blots were stained for phosphorylated Syk with anti-Syk Tyr525/526 mAb (1:1,000) and secondary HRP-conjugated secondary Ab (1:500). Total Syk was determined by reprobing with anti-Syk mAb (1:1,000) and HRP-conjugated secondary Ab (1:1,000). To control for total platelet proteins, parallel blots were probed for α-tubulin (1:1,000). Antibody staining was quantified by densitometric analysis [35].
Statistics
Data are given as mean ± SEM. Significance of differences was determined with the Mann–Whitney U test or the independent samples t test, as appropriate, using the statistical package for social sciences (SPSS 15.0).
Results
Roles of integrin αIIbβ3 in tissue factor-stimulated thrombin generation and platelet activation
Early reports suggest that various integrin αIIbβ3 antagonists differently affect thrombin generation in platelet-rich plasma (PRP) [19, 23]. To verify this, we prepared human PRP and determined the effects on tissue factor-induced thrombin generation of three αIIbβ3 blockers, all in clinical use: the human/mouse chimeric monoclonal antibody fragment abciximab (blocking integrins αIIbβ3 and αvβ3), the peptide mimetic eptifibatide, and the non-peptide sulfonamido compound, tirofiban. When added to recalcified PRP, tirofiban dose-dependently suppressed and delayed thrombin generation induced by tissue factor (Fig. 1a, b). A similar reduction in thrombin generation was seen with the other αIIbβ3 blockers (Online Resource Fig. 1a, b). Tirofiban, abciximab and eptifibatide reduced the thrombin peak with 60 % at IC50 values of 0.1, 2 and 1 μg/mL, respectively. In spite of this reduction, a thrombin peak of 20 nM was still reached, which is sufficiently high for maximal thrombin-induced platelet activation. Control experiments indicated that the integrin blockers were similarly effective in suppressing platelet aggregation of PPACK-anticoagulated PRP, stimulated with the PAR1 agonist SFLLRN (not shown). In contrast, none of the αIIbβ3 blockers affected thrombin generation in plasma containing phospholipids instead of platelets (Fig. 1b and Online Resource Fig. 1b), thus demonstrating that the blocker effects required the presence of platelets.
Fig. 1.
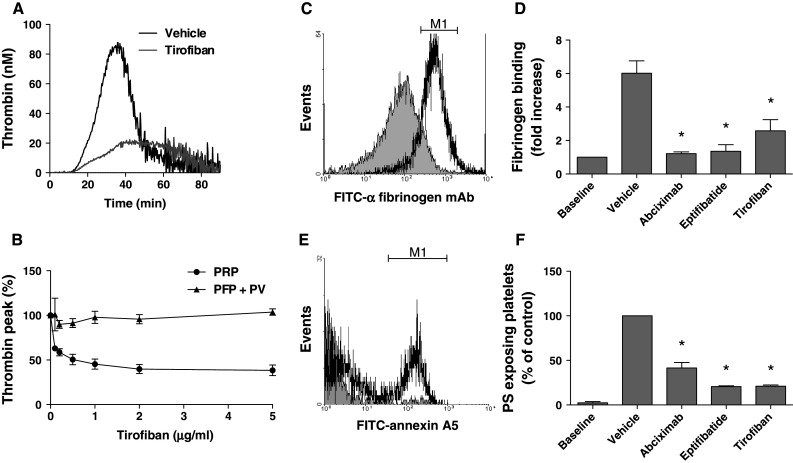
Blocking of integrin αIIbβ3 suppresses PS exposure and thrombin generation in tissue factor-stimulated PRP. a, b PRP (1.0 × 108 platelets/mL) or PFP supplemented with phospholipid vesicles (PV, 10 μM) was preincubated with vehicle or tirofiban (0.1–5 μg/mL) for 20 min. Thrombin generation was stimulated with tissue factor (1 pM) and CaCl2. a Representative thrombin generation curves with PRP, b dose-dependent inhibition of thrombin generation only in the presence of platelets. Mean ± SEM (n = 3–7). c–f Ancrod-treated PRP was preincubated with vehicle, abciximab (10 μg/mL), eptifibatide (10 μg/mL) or tirofiban (1 μg/mL), and thrombin generation was stimulated as above. After 15 min, platelet activation was evaluated by flow cytometry. c, d Platelet fibrin(ogen) binding measured with FITC-labeled anti-fibrinogen mAb. Data are fold increase in fluorescence relative to baseline (prior to activation). e, f Platelet PS exposure measured with FITC-annexin A5. Data are fractions of PS-exposing platelets (compared to vehicle control). M1 indicates platelet populations with increased fluorescence. Mean ± SEM (n = 3–6); *p < 0.05 versus vehicle
In order to study tissue factor-induced platelet activation in plasma, formation of disturbing fibrin clots needed to be prevented. Therefore, human plasma was partly defibrinated with a low concentration (1.3 U/mL) of the snake venom ancrod, which induces fibrin clotting without thrombin generation [36]. After removal of the ancrod clots by centrifugation, the remaining plasma contained a residual concentration of ~0.5 mg/mL fibrinogen, which does not form large clots. In this plasma reconstituted with platelets, αIIbβ3 blockers suppressed tissue factor-induced thrombin generation with 40–50 % (data not shown). Flow cytometry demonstrated that, after tissue factor stimulation, fibrin(ogen) binding to platelets was antagonized by any of the three αIIbβ3 blockers (Fig. 1c, d). Furthermore, tissue factor induced PS exposure in 35 % of the platelets, as determined by staining with FITC-annexin A5 (Fig. 1e). In the absence of tissue factor, fibrin(ogen) binding and PS exposure were quite low. Strikingly, blockage of αIIbβ3 greatly reduced the tissue factor-induced PS exposure with 60 % (abciximab) or 80 % (eptifibatide, tirofiban) at maximally effective concentrations (Fig. 1f). Flow cytometry furthermore indicated that the integrin blockers suppressed formation of PS-exposing microparticles by >50 %, as reported before [17]. Similar experiments were performed in the presence of apyrase (0.1 U/mL), in which case again abciximab, eptifibatide, and tirofiban reduced the number of PS-exposing platelets by 75–85 %.
A frequently used way of provoking PS exposure is by stimulating washed platelets with thrombin in combination with collagen receptor agonist, convulxin [37]. Considering that these platelets secrete fibrinogen which binds to αIIbβ3 in an autocrine way, we investigated whether in this condition αIIbβ3 blocking may also influence PS exposure. Flow cytometry indicated that the co-stimulation of isolated platelets with thrombin (10 nM) and convulxin (50 ng/mL) resulted in large fractions of platelets binding fibrin(ogen) and exposing PS (Fig. 2a). Dose–response experiments indicated that this concentration of thrombin (10 nM) was maximally effective (not shown), similarly as described before [38]. All integrin blockers caused a substantial decrease in PS exposure of 35 % (abciximab) or 50 % (eptifibatide, tirofiban) of the control condition (Fig. 2b). This suggested a supportive role of αIIbβ3 in thrombin and collagen receptor-induced PS exposure via interaction with (secreted) fibrinogen.
Fig. 2.

Blocking of integrin αIIbβ3 suppresses PS exposure of convulxin/thrombin-stimulated platelets. Washed platelets containing apyrase were activated with thrombin (10 nM) or SFLLRN (15 μM) plus convulxin (50 ng/mL), as indicated, in the presence of CaCl2 (2 mM) for 15 min. Pretreatment with abciximab (10 μg/mL), eptifibatide (10 μg/mL) or tirofiban (1 μg/mL). a Histograms of fibrin(ogen) binding (FITC-anti-fibrinogen mAb) and PS exposure (FITC-annexin A5). b Quantitative effect of αIIbβ3 blockage on platelet PS exposure. c Effect of added human fibrinogen (0.1–2.0 mg/mL) on PS exposure. Mean ± SEM (n = 3); *p < 0.05 versus vehicle
To further study this under conditions where fibrin clot formation was prevented, the platelets were activated with convulxin plus PAR1 agonist SFLLRN. Addition of exogenous human fibrinogen resulted in a dose-dependent stimulatory effect on PS exposure, increasing the fraction of PS-exposing platelets from 35 up to 70% (Fig. 2c). In contrast, addition of fibrinogen alone, without other agonists, did not stimulate PS exposure. Pretreatment with tirofiban (Fig. 2c) or other integrin blockers (not shown) completely reversed the stimulating effect of fibrinogen. Comparable results were obtained with bovine and human fibrinogen (not shown). Together, these results point to a role of integrin αIIbβ3, likely via interaction with fibrin(ogen) on platelet PS exposure both in tissue factor-stimulated PRP (resulting in increased thrombin generation), and in washed platelets stimulated with thrombin and collagen receptor agonists.
Signaling role of integrin αIIbβ3 in Ca2+ and procoagulant platelet responses
Others have suggested that inhibitory effects of αIIbβ3 blockers on platelet PS exposure may occur independently of modulating Ca2+ responses [22, 39]. Also, in stored platelets, PS exposure can occur independently of elevated Ca2+ [40]. We therefore re-examined a role of αIIbβ3 in Ca2+-signaling by stimulating washed suspensions of Fura-2-loaded platelets with thrombin and convulxin. While measuring rises in Ca2+, samples were taken for flow cytometric determination of PS exposure. The agonists caused a potent increase in Ca2+ peak, which was followed by a sustained elevated level (450 nM), persisting during 15 min (Fig. 3a). Pretreatment with eptifibatide or tirofiban did not influence the initial Ca2+ peak, but it markedly reduced the sustained high Ca2+ level to 56 ± 10 or 60 ± 8 % of the control value, respectively (Fig. 3a, b). The reduction in sustained Ca2+ response was accompanied by a proportional decrease in PS exposure from 36 ± 3 to 16 ± 3 or 14 ± 3 %, respectively (Fig. 3c). Reasoning that persistent integrin signaling may prolong these Ca2+ responses and then contribute to PS exposure, we added the integrin blockers at various time points after thrombin/convulxin. Addition of eptifibatide or tirofiban at 1 min after activation still caused a substantial reduction in the fraction of PS-exposing platelets, whereas addition after 5–10 min resulted in progressively less inhibition (Fig. 3d).
Fig. 3.

Blocking of αIIbβ3 suppresses long-term platelet Ca2+ responses and PS exposure. Fura-2-loaded platelets containing apyrase were preincubated with vehicle (control), eptifibatide (10 μg/mL) or tirofiban (1 μg/mL). Cells were then activated with thrombin (10 nM) plus convulxin (50 ng/mL) and 1 mM CaCl2. a Representative platelet Ca2+ responses. b Effect of integrin blockers on Ca2+ peak levels (light gray) and 15-min end levels (dark gray). c Effect of blockers on fractions of PS-exposing platelets after 15 min, analyzed by flow cytometry. d Effect of addition of eptifibatide or tirofiban at different time points before (t = −10 min) or after (t = 1–10 min) activation. Fractions of PS-exposing platelets after 15 min. Data are relative to control condition without integrin blocker. Mean ± SEM (n = 3–5); *p < 0.05 versus vehicle
To further confirm the contribution of integrin signaling in PS exposure, platelets were obtained from two Glanzmann patients with complete deficiency in αIIbβ3 expression. Loaded with Fura-2, the platelets showed high peak rises in Ca2+ in response to thrombin/convulxin, but at later time points Ca2+ levels declined to ~150 nM (Fig. 4a, b). This corresponded to a low amount of 10–12 % PS-exposing platelets (Fig. 4c). Addition of tirofiban altered neither the late Ca2+ response nor the low exposure of PS. Collectively, these data point to a role of αIIbβ3-mediated signaling in long-term Ca2+ rises induced by thrombin and collagen receptor agonists and thereby in development of platelet procoagulant activity.
Fig. 4.

Impaired long-term Ca2+ responses and PS exposure in activated Glanzmann platelets. Suspensions of Fura-2-loaded platelets containing apyrase from two Glanzmann patients (1, 2) were pretreated with vehicle or tirofiban (1 μg/mL). Platelets were then activated with thrombin (10 nM) plus convulxin (50 ng/mL) in the presence of 1 mM CaCl2. a Representative traces of Ca2+ responses. b Averaged Ca2+ peak levels (light gray) and 15-min end levels (dark gray). c Fractions of PS-exposing platelets after 15 min, showing no effect of tirofiban. Mean ± SEM (n = 3 experiments)
Signaling role of integrin αIIbβ3 in Ca2+ and procoagulant responses of platelets spreading on fibrinogen
Outside-in signaling by integrin αIIbβ3 mediates the spreading of platelets on fibrinogen surfaces [4]. In Fura-2-loaded platelets, we found that spreading was accompanied by irregular, low-amplitude Ca2+ spikes (Fig. 5a). In most of these platelets, thrombin induced a persistently high Ca2+ level and stimulated the spreading process (Fig. 5a, b). To investigate a role of integrin signaling, the platelets were preincubated with tirofiban prior to thrombin stimulation. This retarded the spreading process, and suppressed the Ca2+ response, in a way that the persistent Ca2+ rise changed into a pattern of repetitive Ca2+ spiking. Furthermore, tirofiban reduced the fractions of PS-exposing platelets with thrombin from ~15 to only 2.5 % (Fig. 5c). The inhibiting effects of tirofiban were preserved on a surface coated with a low fibrinogen solution of 10 μg/mL (not shown). Thrombin stimulation of adhered, non-spread platelets (5 min fibrinogen adhesion) similarly resulted in a low fraction of PS-exposing platelets of 2.5 ± 0.4 %. Control experiments were carried out with DM-BAPTA-loaded platelets, where basal levels of Ca2+ amounted ~20 nM, and thrombin addition resulted in neither Ca2+ rises nor PS exposure (data not shown).
Fig. 5.

Blocking of integrin αIIbβ3 reduces Ca2+ responses and PS exposure of spreading platelets. Fura-2-loaded platelets were adhered to coated fibrinogen in the absence (control) or presence of tirofiban (1 μg/mL) for 30 min. Rises in Ca2+ in single, adhered platelets in response to thrombin (10 nM) measured by fluorescence ratio imaging. a Traces of Ca2+ rises of two representative platelets; also brightfield contrast images after 10 min (bars 10 μm). b Fractions of platelets with elevated Ca2+ (>1.2-fold signal) after 10 min. c Fractions of platelets binding FITC-annexin A5 after 10 min. Mean ± SEM (n = 4–6); *p < 0.05 versus control
To further verify a role of αIIbβ3 in thrombin-induced responses of fibrinogen-adhered platelets, similar experiments were performed with the platelets from two Glanzmann patients. While these platelets hardly spread on fibrinogen, they also remained low in PS exposure with only ~2 % annexin A5 binding after thrombin stimulation (Online Resource Fig. 2). These data thus suggest that integrin outside-in signaling during platelet spreading stimulates thrombin-induced procoagulant activity.
Contribution of integrin αIIbβ3 to platelet procoagulant response via Syk kinase activation
Integrin αIIbβ3-mediated outside-in signaling triggers inactivation of RhoA and activation of the protein tyrosine kinase Syk, resulting in phospholipase Cγ2 activation [6, 41]. Employing several approaches, we investigated a role of Syk in αIIbβ3-dependent Ca2+ rises and procoagulant activity. First, we used the pharmacologic blockers, Syk inhibitor II and IV, which abolished collagen-induced aggregation of platelets in plasma at maximally effective concentrations of 10 μM. In tissue factor-stimulated PRP, both inhibitors suppressed platelet PS exposure at a similar degree as tirofiban (Fig. 6a). Next, platelets were loaded with the Ca2+ probe Fura-Red, which allows the monitoring of Ca2+ rises in the presence of plasma by flow cytometry [42]. Stimulation with tissue factor resulted in a prolonged rise in Ca2+ in the majority of platelets in plasma (observed as a decrease in Fura-Red fluorescence). This Ca2+ rise was reduced in the presence of tirofiban and even more so with Syk inhibitor II or IV (Fig. 6b).
Fig. 6.
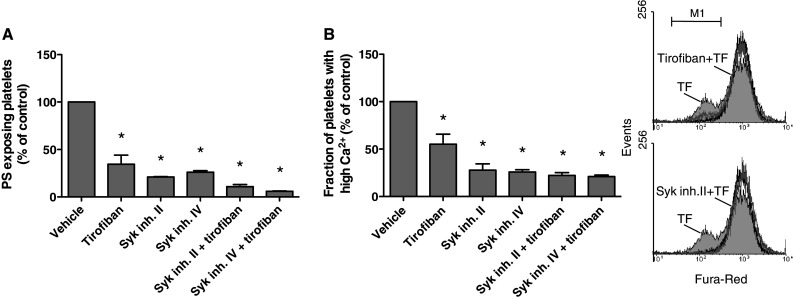
Contribution of Syk kinase to platelet activation in tissue factor-stimulated plasma. Fura-Red loaded platelets in ancrod-treated plasma (1.0 × 108/mL) were preincubated with vehicle, Syk inhibitor II or IV (10 μM), and/or tirofiban (1 μg/mL), as described for Fig. 1. Cangrelor (AR–C, 10 μM) was present to eliminate P2Y12-dependent signaling events. PRP was then stimulated with 1 pM tissue factor and CaCl2 for 15 min. a Fractions of PS-exposing platelets determined by FITC-annexin A5 binding. b Fractions of platelets with high Ca2+ as determined by flow cytometry (M1). Flow cytometric histograms of Fura-Red fluorescence. Note decreased Fura-Red fluorescence points to high Ca2+ (M1). Histograms of unstimulated platelets (black), and tissue factor-stimulated platelets with vehicle (light gray), tirofiban or Syk inhibitor II (dark gray). Mean ± SEM (n = 4); *p < 0.05 versus vehicle
Third, we directly examined the activation of Syk in tissue factor-stimulated PRP. It appeared not to be possible to collect these platelets from plasma by centrifugation or gel filtration. Hence, we developed a method to isolate platelets after tissue factor stimulation by using magnetic beads coupled to anti-CD31 mAb. Lysates of the isolated platelets were subjected to gel electrophoresis and western blotting, and probed for phosphorylation of Syk at Tyr525/526, which is an essential step in Syk activation [43]. While no Syk phosphorylation was detected in the absence of tissue factor, platelet stimulation with tissue factor stimulation markedly increased the phosphorylation, which event was completely prevented by Syk inhibitor II (Fig. 7a, b). Pretreatment with tirofiban substantially but not completely antagonized Tyr525/526 phosphorylation, suggesting a prominent role of αIIbβ3-dependent signaling to Syk in the tissue factor-activated PRP.
Fig. 7.
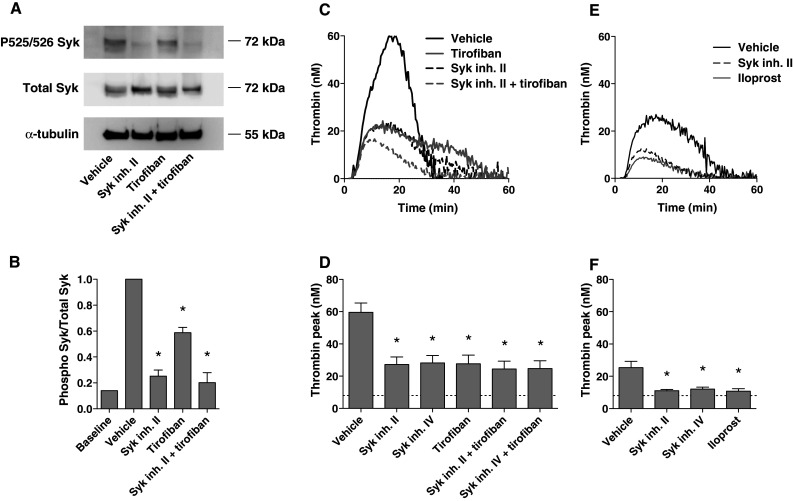
Contribution of integrin αIIbβ3 signaling and Syk kinase to thrombin generation in tissue factor-stimulated plasma. Reconstituted PRP was preincubated with vehicle, Syk inhibitor II (10 μM), iloprost (1 μM) and/or tirofiban (1 μg/mL), as indicated, and then stimulated with tissue factor (1 pM) and CaCl2. Cangrelor (AR–C, 10 μM) was present to eliminate P2Y12-dependent signaling events. Control condition (baseline) was without tissue factor. a, b Effects of tirofiban and Syk inhibitor on Tyr525/526 phosphorylation of Syk. Platelets were isolated from activated plasma (15 min) using anti-CD31-coupled magnetic beads and examined for protein phosphorylation. Representative western blots after probing for phospho-Syk (Tyr525/526), and reprobing for total Syk; parallel blots were stained for α-tubulin. Shown is the ratio of phospho-Syk/total Syk assessed by densitometric analysis (n = 6). c–f Effects of tirofiban and Syk inhibitor on tissue factor-induced thrombin generation in PRP. Blood was collected on citrate (n = 8) (c, d); or on citrate plus tirofiban (1 μg/mL) (n = 3) (e, f). Representative thrombin generation curves and thrombin peak heights. Dotted lines in bar graphs indicate residual thrombin formed in plasma not containing platelets. Mean ± SEM; *p < 0.05 versus vehicle
Fourth, we directly investigated the effects of Syk inhibitors II and IV on tissue factor-induced thrombin generation. Using citrate-anticoagulated PRP, it appeared that both compounds were similarly effective as tirofiban in the suppression of thrombin generation, while there was no additional effect of a combination with tirofiban (Fig. 7c, d). As platelet-dependent thrombin generation was still incompletely blocked, we considered the possibility that the platelets exhibited residual αIIbβ3-dependent signaling during blood collection and PRP preparation. To investigate this, blood was collected on citrate anticoagulant plus tirofiban. Indeed, with tirofiban initially present, tissue factor-induced thrombin generation was reduced, while either Syk inhibitor fully reduced the thrombin peak to the level obtained with the strong platelet inhibitor, iloprost (Fig. 7e, f). In fact, this residual, low thrombin generation was also present in plasma devoid of platelets, and could be ascribed to the presence of microparticles. Similar results were obtained with blood collected on Syk inhibitor II (not shown).
Finally, thrombin generation experiments were performed with PRP from a Glanzmann patient. In the patient PRP, tissue factor stimulation evoked limited thrombin generation, which, however, was not influenced by the presence of tirofiban (Online Resource Fig. 3). Similar to control PRP with tirofiban, addition of Syk inhibitor caused an additional decrease in thrombin generation. Taken together, these various sets of data show a substantial role of αIIbβ3 and Syk kinase in tissue factor-induced PS exposure and thrombin generation. Furthermore, they point to the existence of a pathway of thrombin generation that is dependent on Syk, but not on integrin activation.
Discussion
This paper reveals a new role of integrin αIIbβ3-dependent signaling via Syk kinase in tissue factor-induced platelet procoagulant activity and thrombin generation in plasma. The results point to a pathway where initial traces of thrombin triggered by tissue factor activate platelets to expose procoagulant PS, resulting in a cycle of thrombin generation and platelet activation that is greatly enforced and prolonged by integrin-dependent signaling to Syk activation and Ca2+ rises. The data furthermore identify a Syk-dependent, but integrin-independent pathway of thrombin generation.
In tissue factor-stimulated PRP, we found that the blockage of αIIbβ3 with different antagonists suppresses platelet Ca2+ responses, PS exposure and thrombin generation in a dose-dependent way. However, in washed platelets stimulated via thrombin and collagen receptors, integrin blockage also suppressed long-term Ca2+ rises along with PS exposure, while added fibrinogen enhanced these responses. Furthermore, during platelet spreading on fibrinogen, a process known to rely on αIIbβ3 outside-in signaling, blocking of the integrin resulted in Ca2+ signaling and PS exposure in response to maximally effective concentrations of thrombin. Confirmative evidence for a signaling role of αIIbβ3 came from the observation that long-term Ca2+ responses and PS exposure were reduced in platelets from two patients with Glanzmann’s thrombasthenia, lacking αIIbβ3. Together, these data indicate that, in platelets stimulated with Ca2+ mobilizing agonists, αIIbβ3 outside-in signaling prolongs the Ca2+ signal, increases procoagulant activity, and hence supports tissue factor-stimulated thrombin generation on the platelet surface.
Anticoagulant effects of platelet αIIbβ3 antagonists have been reported by several authors [12, 16, 17], but the mechanism was not disclosed. Several groups reported that integrin blockers were unable to change platelet Ca2+ responses to collagen and/or thrombin [22, 23, 39]. However, the measurements mostly concerned initial Ca2+ rises, while in our hands only late Ca2+ signals appear to be affected. Interestingly, one study does describe long-term inhibition of collagen/thrombin-induced Ca2+ responses with abciximab but not with other integrin blockers specifically under conditions of stirring [23]. This contrasts with the present findings where appropriate concentrations of different integrin blockers all had similar effects. Another published finding that αIIbβ3 blockage reduces shear-dependent Ca2+ responses and microparticle release [19, 44] can be explained by increased fibrinogen secretion of platelets subjected to a high shear rate.
The (patho)physiological relevance of this work comes from recent data that β3-mutated mice with a deficiency in platelet outside-in signaling and tyrosine phosphorylation are protected from arterial thrombus formation after carotid artery injury with FeCl3 [45], i.e. a mouse thrombosis model where thrombus formation depends on tissue factor activity and on procoagulant, PS-exposing platelets [13, 46–48].
Platelets from mice deficient in phospholipase Cγ2 have shown reduced Ca2+ signals and spreading on immobilized fibrinogen [44, 49]. Based on these and other data a scheme has been proposed of αIIbβ3-induced signaling via Src and Syk kinases to activation of phospholipase Cγ2 [6]. This signaling scheme was confirmed by recent proteomic analyses demonstrating the presence of many tyrosine phosphorylated proteins in human platelets spread on fibrinogen, among which multiple tyrosine kinases [5]. The present finding points to a particular role of Syk phosphorylation and subsequent phospholipase Cγ2 activation in αIIbβ3-dependent procoagulant activity and thrombin generation upon triggering with tissue factor. Evidence for this came from the reduction in Syk tyrosine phosphorylation by integrin blockage and by two Syk inhibitors. These experiments were carried out under conditions eliminating a contribution of P2Y12 signaling [24, 50].
Interestingly, in addition to a novel αIIbβ3-dependent role of Syk kinase in platelet procoagulant activity, our data also point to an αIIbβ3-independent role of Syk in this process. We have not yet unraveled the mechanism of αIIbβ3-independent Syk activation, but according to the literature this activation pathway can include FcγRIIa [51], glycoprotein Ib–IX–V complex [52, 53], or CLEC2 [54], all of which have been shown to activate Syk. Altogether, our results point to a signaling scheme where fibrin(ogen)-induced integrin activation supports a Syk/phospholipase Cγ2 pathway, resulting in prolonged Ca2+ and PS exposure and thrombin generation in plasma.
Electronic supplementary material
Below is the link to the electronic supplementary material.
Acknowledgments
We thank Dr. S. Cauwenberghs for contribution in initial experiments. We acknowledge support from the Cardiovascular Centre, MUMC+, Maastricht, and the Landsteiner Foundation for Blood Transfusion Research, LSBR1006. Supported by the Thrombosis Expertise Center Maastricht, the Landsteiner Foundation for Transfusion Research and the Marie-Curie Early Stage Researcher Program 2005-020706-3.
Open Access
This article is distributed under the terms of the Creative Commons Attribution License which permits any use, distribution, and reproduction in any medium, provided the original author(s) and the source are credited.
Footnotes
M. A. H. Feijge and F. Swieringa contributed equally to this work.
References
- 1.Cosemans JM, Iserbyt BF, Deckmyn H, Heemskerk JW. Multiple ways to switch platelet integrins on and off. J Thromb Haemost. 2008;6:1253–1261. doi: 10.1111/j.1538-7836.2008.03041.x. [DOI] [PubMed] [Google Scholar]
- 2.Ma YQ, Qin J, Plow EF. Platelet integrin αIIbβ3: activation mechanisms. J Thromb Haemost. 2007;5:1345–1352. doi: 10.1111/j.1538-7836.2007.02537.x. [DOI] [PubMed] [Google Scholar]
- 3.Nieswandt B, Moser M, Pleines I, Varga-Szabo D, Monkley S, Critchley D, Fassler R. Loss of talin1 in platelets abrogates integrin activation, platelet aggregation, and thrombus formation in vitro and in vivo. J Exp Med. 2007;204:3113–3118. doi: 10.1084/jem.20071827. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Shattil SJ, Newman PJ. Integrins: dynamic scaffolds for adhesion and signaling in platelets. Blood. 2004;104:1606–1615. doi: 10.1182/blood-2004-04-1257. [DOI] [PubMed] [Google Scholar]
- 5.Senis YA, Antrobus R, Severin S, Parguina AF, Rosa I, Zitzmann N, Watson SP, Garcia A. Proteomics analysis of integrin αIIbβ3 outside-in signaling reveals Src-kinase-independent phosphorylation of Dok-1 and Dok-3 leading to SHIP-1 interactions. J Thromb Haemost. 2009;7:1718–1726. doi: 10.1111/j.1538-7836.2009.03565.x. [DOI] [PubMed] [Google Scholar]
- 6.Watson SP, Auger JM, McCarty OJ, Pearce AC. GPVI and integrin αIIbβ3 signaling in platelets. J Thromb Haemost. 2005;3:1752–1762. doi: 10.1111/j.1538-7836.2005.01429.x. [DOI] [PubMed] [Google Scholar]
- 7.Cauwenberghs S, Feijge MAH, Harper AG, Sage SO, Curvers J, Heemskerk JWM. Shedding of procoagulant microparticles from unstimulated platelets by integrin-mediated destabilization of actin cytoskeleton. FEBS Lett. 2006;580:5313–5320. doi: 10.1016/j.febslet.2006.08.082. [DOI] [PubMed] [Google Scholar]
- 8.Schoenwaelder SM, Yuan Y, Cooray P, Salem HH, Jackson SP. Calpain cleavage of focal adhesion proteins regulates the cytoskeletal attachment of integrin αIIbβ3 (platelet glycoprotein IIb/IIIa) and the cellular retraction of fibrin clots. J Biol Chem. 1997;272:1694–1702. doi: 10.1074/jbc.272.40.24876. [DOI] [PubMed] [Google Scholar]
- 9.Shattil SJ, Kashiwagi H, Pampori N. Integrin signaling: the platelet paradigm. Blood. 1998;91:2645–2657. [PubMed] [Google Scholar]
- 10.Heemskerk JWM, Kuijpers MJE, Munnix ICA, Siljander PRM. Platelet collagen receptors and coagulation. A characteristic platelet response as possible target for antithrombotic treatment. Trends Cardiovasc Med. 2005;15:86–92. doi: 10.1016/j.tcm.2005.03.003. [DOI] [PubMed] [Google Scholar]
- 11.Monroe DM, Hoffman M, Roberts HR. Platelets and thrombin generation. Arterioscler Thromb Vasc Biol. 2002;22:1381–1389. doi: 10.1161/01.ATV.0000031340.68494.34. [DOI] [PubMed] [Google Scholar]
- 12.Vanschoonbeek K, Feijge MAH, van Kampen RJ, Kenis H, Hemker HC, Giesen PLA, Heemskerk JWM. Initiating and potentiating role of platelets in tissue factor-induced thrombin generation in the presence of plasma: subject-dependent variation in thrombogram characteristics. J Thromb Haemost. 2004;2:476–484. doi: 10.1111/j.1538-7933.2004.00618.x. [DOI] [PubMed] [Google Scholar]
- 13.Munnix ICA, Kuijpers MJE, Auger J, Thomassen CM, Panizzi P, van Zandvoort MA, Rosing J, Bock PE, Watson SP, Heemskerk JWM. Segregation of platelet aggregatory and procoagulant microdomains in thrombus formation: regulation by transient integrin activation. Arterioscler Thromb Vasc Biol. 2007;27:2484–2490. doi: 10.1161/ATVBAHA.107.151100. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Castoldi E, Collins PW, Williamson PL, Bevers EM. Compound heterozygosity for 2 novel TMEM16F mutations in a patient with Scott syndrome. Blood. 2011;117:4399–4400. doi: 10.1182/blood-2011-01-332502. [DOI] [PubMed] [Google Scholar]
- 15.Zwaal RF, Schroit AJ. Pathophysiologic implications of membrane phospholipid asymmetry in blood cells. Blood. 1997;89:1121–1132. [PubMed] [Google Scholar]
- 16.Ilveskero S, Lassila R. Abciximab inhibits procoagulant activity but not the release reaction upon collagen- or clot-adherent platelets. J Thromb Haemost. 2003;1:805–813. doi: 10.1046/j.1538-7836.2003.00136.x. [DOI] [PubMed] [Google Scholar]
- 17.Reverter JC, Béguin S, Kessels H, Kumar R, Hemker HC, Coller BS. Inhibition of platelet-mediated, tissue-factor-induced thrombin generation by the mouse/human chimeric 7E3 antibody. J Clin Invest. 1996;98:863–874. doi: 10.1172/JCI118859. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Gemmell CH, Sefton MV, Yeo EL. Platelet-derived microparticle formation involves glycoprotein IIb/IIIa. Inhibition by RGDS and a Glanzmann’s thrombasthenia defect. J Biol Chem. 1993;268:14586–14589. [PubMed] [Google Scholar]
- 19.Goto S, Tamura N, Li M, Handa M, Ikeda Y, Handa S, Ruggeri ZM. Different effects of various anti-GPIIb/IIIa agents on shear-induced platelet activation and expression of procoagulant activity. J Thromb Haemost. 2003;1:2022–2030. doi: 10.1046/j.1538-7836.2003.00349.x. [DOI] [PubMed] [Google Scholar]
- 20.Byzova TV, Plow EF. Networking in the hemostatic system. Integrin αIIbβ3 binds prothrombin and influences its activation. J Biol Chem. 1997;272:27183–27188. doi: 10.1074/jbc.272.43.27183. [DOI] [PubMed] [Google Scholar]
- 21.Furman MI, Krueger LA, Frelinger AL, Barnard MR, Mascelli MA, Nakada MT, Michelson AD. GPIIb/IIIa antagonist-induced reduction in platelet surface factor V/Va binding and phosphatidylserine expression in whole blood. Thromb Haemost. 2000;84:492–498. [PubMed] [Google Scholar]
- 22.Pedicord DL, Thomas BE, Mousa SA, Dicker IB. Glycoprotein IIb/IIIa receptor antagonists inhibit the development of platelet procoagulant activity. Thromb Res. 1998;90:247–258. doi: 10.1016/S0049-3848(98)00029-2. [DOI] [PubMed] [Google Scholar]
- 23.Lages B, Weiss HJ. Greater inhibition of platelet procoagulant activity by antibody-derived glycoprotein IIb/IIIa inhibitors than by peptide and peptidomimetic inhibitors. Br J Haematol. 2001;113:65–71. doi: 10.1046/j.1365-2141.2001.02721.x. [DOI] [PubMed] [Google Scholar]
- 24.Léon C, Ravanat C, Freund M, Cazenave JP, Gachet C. Differential involvement of the P2Y1 and P2Y12 receptors in platelet procoagulant activity. Arterioscler Thromb Vasc Biol. 2003;23:1941–1947. doi: 10.1161/01.ATV.0000092127.16125.E6. [DOI] [PubMed] [Google Scholar]
- 25.Storey RF, Sanderson HM, White AE, May JA, Cameron KE, Heptinstall S. The central role of the P2T receptor in amplification of human platelet activation, secretion and procoagulant activity. Br J Haematol. 2000;110:925–934. doi: 10.1046/j.1365-2141.2000.02208.x. [DOI] [PubMed] [Google Scholar]
- 26.Van der Meijden PE, Feijge MAH, Giesen PL, Huijberts M, van Raak LP, Heemskerk JWM. Platelet P2Y12 receptors enhance signalling towards procoagulant activity and thrombin generation. A study with healthy subjects and patients at thrombotic risk. Thromb Haemost. 2005;93:1128–1136. doi: 10.1160/TH04-09-0597. [DOI] [PubMed] [Google Scholar]
- 27.Van der Meijden PEJ, Munnix ICA, Auger JM, Govers-Riemslag JW, Cosemans JMEM, Kuijpers MJE, Spronk HM, Watson SP, Renné T, Heemskerk JWM. Dual role of collagen in factor XII-dependent thrombus and clot formation. Blood. 2009;114:881–890. doi: 10.1182/blood-2008-07-171066. [DOI] [PubMed] [Google Scholar]
- 28.Siljander P, Farndale RW, Feijge MAH, Comfurius P, Kos S, Bevers EM, Heemskerk JWM. Platelet adhesion enhances the glycoprotein VI-dependent procoagulant response: involvement of p38 MAP kinase and calpain. Arterioscler Thromb Vasc Biol. 2001;21:618–627. doi: 10.1161/01.ATV.21.4.618. [DOI] [PubMed] [Google Scholar]
- 29.Rosado JA, Meijer EM, Hamulyak K, Novakova I, Heemskerk JWM, Sage SO. Fibrinogen binding to the integrin αIIbβ3 modulates store-mediated calcium entry in human platelets. Blood. 2001;97:2648–2656. doi: 10.1182/blood.V97.9.2648. [DOI] [PubMed] [Google Scholar]
- 30.Clauss A. Rapid physiological coagulation method in determination of fibrinogen. Acta Haematol. 1957;17:237–246. doi: 10.1159/000205234. [DOI] [PubMed] [Google Scholar]
- 31.Feijge MAH, van Pampus ECM, Lacabaratz-Porret C, Hamulyak K, Lévy-Toledano S, Enouf J, Heemskerk JWM. Inter-individual variability in Ca2+ signalling in platelets from healthy volunteers: effects of aspirin and relationship with expression of endomembrane Ca2+-ATPases. Br J Haematol. 1998;102:850–859. doi: 10.1046/j.1365-2141.1998.00844.x. [DOI] [PubMed] [Google Scholar]
- 32.Heemskerk JWM, Vuist WMJ, Feijge MAH, Reutelingsperger CPM, Lindhout T. Collagen but not fibrinogen surfaces induce bleb formation, exposure of phosphatidylserine, and procoagulant activity of adherent platelets: evidence for regulation by protein tyrosine kinase-dependent Ca2+ responses. Blood. 1997;90:2615–2625. [PubMed] [Google Scholar]
- 33.Siljander PRM, Munnix ICA, Smethurst PA, Deckmyn H, Lindhout T, Ouwehand WH, Farndale RW, Heemskerk JWM. Platelet receptor interplay regulates collagen-induced thrombus formation in flowing human blood. Blood. 2004;103:1333–1341. doi: 10.1182/blood-2003-03-0889. [DOI] [PubMed] [Google Scholar]
- 34.van Gorp RM, Feijge MA, Vuist WM, Rook MB, Heemskerk JW. Irregular spiking in free calcium concentration in single, human platelets. Regulation by modulation of the inositol trisphosphate receptors. Eur J Biochem. 2002;269:1543–1552. doi: 10.1046/j.1432-1033.2002.02806.x. [DOI] [PubMed] [Google Scholar]
- 35.Nergiz-Unal R, Lamers MM, Van Kruchten R, Luiken JJ, Cosemans JM, Glatz JF, Kuijpers MJ, Heemskerk JW. Signaling role of CD36 in platelet activation and thrombus formation on immobilized thrombospondin or oxidized low-density lipoprotein. J Thromb Haemost. 2011;JTH 9:1835–1846. doi: 10.1111/j.1538-7836.2011.04416.x. [DOI] [PubMed] [Google Scholar]
- 36.Dempfle CE, Argiriou S, Kucher K, Muller-Peltzer H, Rubsamen K, Heene DL. Analysis of fibrin formation and proteolysis during intravenous administration of ancrod. Blood. 2000;96:2793–2802. [PubMed] [Google Scholar]
- 37.Heemskerk JWM, Bevers EM, Lindhout T. Platelet activation and blood coagulation. Thromb Haemost. 2002;88:186–193. [PubMed] [Google Scholar]
- 38.Heemskerk JW, Feijge MA, Henneman L, Rosing J, Hemker HC. The Ca2+-mobilizing potency of alpha-thrombin and thrombin-receptor-activating peptide on human platelets: concentration and time effects of thrombin-induced Ca2+ signaling. Eur J Biochem. 1997;249:547–555. doi: 10.1111/j.1432-1033.1997.00547.x. [DOI] [PubMed] [Google Scholar]
- 39.Razmara M, Hu H, Masquelier M, Li N. Glycoprotein IIb/IIIa blockade inhibits platelet aminophospholipid exposure by potentiating translocase and attenuating scramblase activity. Cell Mol Life Sci. 2007;64:999–1008. doi: 10.1007/s00018-007-6546-8. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40.Schoenwaelder SM, Yuan Y, Josefsson EC, White MJ, Tao Y, Mason KD, O’Reilly LA, Henley KJ, Ono A, Hsiao S, Willcox A, Roberts AW, Huang DCS, Salem HH, Kile BT, Jackson SP. Two distinct pathways regulate platelet phosphatidylserine exposure and procoagulant function. Blood. 2009;114:663–666. doi: 10.1182/blood-2009-01-200345. [DOI] [PubMed] [Google Scholar]
- 41.Li Z, Delaney MK, O’Brien KA, Du X. Signaling during platelet adhesion and activation. Arterioscler Thromb Vasc Biol. 2010;30:2341–2349. doi: 10.1161/ATVBAHA.110.207522. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42.Dachary-Prigent J, Pasquet JM, Nurden AT. Simultaneous detection of changes in cytoplasmic Ca2+, aminophospholipid exposure and micro-vesiculation in activated platelets. Platelets. 1997;8:405–412. doi: 10.1080/09537109777096. [DOI] [PubMed] [Google Scholar]
- 43.Zhang J, Billingsley ML, Kincaid RL, Siraganian RP. Phosphorylation of Syk activation loop tyrosines is essential for Syk function. An in vivo study using a specific anti-Syk activation loop phosphotyrosine antibody. J Biol Chem. 2000;275:35442–35447. doi: 10.1074/jbc.M004549200. [DOI] [PubMed] [Google Scholar]
- 44.Goncalves I, Hughan SC, Schoenwaelder SM, Yap CL, Yuan Y, Jackson SP. Integrin αIIbβ3-dependent calcium signals regulate platelet–fibrinogen interactions under flow. Involvement of phospholipase Cγ2. J Biol Chem. 2003;278:34812–34822. doi: 10.1074/jbc.M306504200. [DOI] [PubMed] [Google Scholar]
- 45.Ablooglu AJ, Kang J, Petrich BG, Ginsberg MH, Shattil SJ. Antithrombotic effects of targeting alphaIIbbeta3 signaling in platelets. Blood. 2009;113:3585–3592. doi: 10.1182/blood-2008-09-180687. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 46.Kuijpers MJ, Munnix IC, Cosemans JM, Vlijmen BV, Reutelingsperger CP, Egbrink MO, Heemskerk JW. Key role of platelet procoagulant activity in tissue factor- and collagen-dependent thrombus formation in arterioles and venules in vivo differential sensitivity to thrombin inhibition. Microcirculation. 2008;15:269–282. doi: 10.1080/10739680701653517. [DOI] [PubMed] [Google Scholar]
- 47.Falati S, Gross P, Merrill-Skoloff G, Furie BC, Furie B. Real-time in vivo imaging of platelets, tissue factor and fibrin during arterial thrombus formation in the mouse. Nat Med. 2002;8:1175–1181. doi: 10.1038/nm782. [DOI] [PubMed] [Google Scholar]
- 48.Hechler B, Nonne C, Eckly A, Magnenat S, Rinckel JY, Denis CV, Freund M, Cazenave JP, Lanza F, Gachet C. Arterial thrombosis: relevance of a model with two levels of severity assessed by histologic, ultrastructural and functional characterization. J Thromb Haemost. 2010;8:173–184. doi: 10.1111/j.1538-7836.2009.03666.x. [DOI] [PubMed] [Google Scholar]
- 49.Wonerow P, Pearce AC, Vaux DJ, Watson SP. A critical role for phospholipase Cgamma2 in alphaIIbbeta3-mediated platelet spreading. J Biol Chem. 2003;278:37520–37529. doi: 10.1074/jbc.M305077200. [DOI] [PubMed] [Google Scholar]
- 50.Van der Meijden PEJ, Schoenwaelder SM, Feijge MAH, Cosemans JMEM, Munnix ICA, Wetzker R, Heller R, Jackson SP, Heemskerk JWM. Dual P2Y12 receptor signaling in thrombin-stimulated platelets. Involvement of phosphoinositide 3-kinase β but not γ isoform in Ca2+ mobilization and procoagulant activity. FEBS J. 2008;275:371–385. doi: 10.1111/j.1742-4658.2007.06207.x. [DOI] [PubMed] [Google Scholar]
- 51.Boylan B, Gao C, Rathore V, Gill JC, Newman DK, Newman PJ. Identification of FcgammaRIIa as the ITAM-bearing receptor mediating alphaIIbbeta3 outside-in integrin signaling in human platelets. Blood. 2008;112:2780–2786. doi: 10.1182/blood-2008-02-142125. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 52.Suzuki-Inoue K, Wilde JI, Andrews RK, Auger JM, Siraganian RP, Sekiya F, Rhee SG, Watson SP. Glycoproteins VI and Ib–IX–V stimulate tyrosine phosphorylation of tyrosine kinase Syk and phospholipase Cgamma2 at distinct sites. Biochem J. 2004;378:1023–1029. doi: 10.1042/BJ20031430. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 53.Cosemans JM, Schols SE, Stefanini L, de Witt S, Feijge MA, Hamulyak K, Deckmyn H, Bergmeier W, Heemskerk JW. Key role of glycoprotein Ib/V/IX and von Willebrand factor in platelet activation-dependent fibrin formation at low shear flow. Blood. 2011;117:651–660. doi: 10.1182/blood-2010-01-262683. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 54.Hughes CE, Pollitt AY, Mori J, Eble JA, Tomlinson MG, Hartwig JH, O’Callaghan CA, Futterer K, Watson SP. CLEC-2 activates Syk through dimerization. Blood. 2010;115:2947–2955. doi: 10.1182/blood-2009-08-237834. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.