

Well-ordered mesoporous silica has been successfully prepared using cationic quaternary ammonium surfactants (M41S family)[1], nonionic surfactants (SBA family) [2], and recently, anionic surfactants [3]. As a result of their attractive properties, i.e. large surface areas and uniform pore sizes, numerous versatile applications of mesoporous silica have been reported in catalysis, bioabsorption, drug delivery and nanoreactors [4]. It is generally accepted that the formation of well-ordered mesoporous silica involves both charge matching and cooperative assembly of surfactant micelles and silicate into 3D lattice structures such as hexagonal, cubic, or lamellar[5].
Biological particles are also good templates for the formation of silica.[6] Moreover, nature has designed filamentous viruses such as M13 and fd bacteriophage that are morphologically similar to rod-like micelles yet possess greater monodispersity than surfactant micelles. In addition, they are facile to form a lyotropic liquid crystalline (LC) phase through self-assembly. As an example, semiflexible rod-like bacteriophages (fd or M13) used in this study are bacteria-specific viruses (~880 nm long and ~7 nm wide) that can be pictured as an ordered assembly of coat proteins along circular ssDNA (Fig. S1). They have high aspect ratios and are capable of self-assembling into LC structures [7]. Such a high degree of monodispersity and anisotropy of bacteriophage, however, has never been successfully employed in synthesizing mesoporous silica with ordered pore lattices.
Here, we report the preparation of well-ordered mesoporous silica fibers with hexagonally arranged pores (termed OU-880) via utilization of filamentous bacteriophages as templates. In addition, we discuss successful utilization of the resultant mesoporous silica as a means for fabricating 3D arrays of PbS nanoparticles. Nature creates incredibly sophisticated mineral structures through the self-assembly of specific proteins that direct biomineralization at ambient conditions[8]. As one of the most prominent examples, diatoms form fine micro-/nano-structured silica walls under the direction of assembled siliffins[9]. Therefore, we also explore the ability of bacteriophages to control the nanostructures of silica by controlling their self-assembly behavior.
Since foreign peptides can be genetically fused to the coat proteins of bacteriophage to produce highly decorated viruses[10], and the assembly behavior of bacteriophage is strongly affected by its surface charge properties, bacteriophage presents itself as an excellent candidate template for controlling resulting silica nanostructures[11]. By displaying peptides with different charges on the side wall of filamentous bacteriophage, we demonstrate the successful controlling of silica nanostructures via control of the bacteriophage surface charge density and the concomitant bacteriophage assembly behaviors.
Both wild-type and genetically engineered bacteriophage were used as templates for fabricating silica. Wild-type bacteriophage fd is a polyelectrolyte with a molecular weight of MW = 1.67 × 107 g/mol, and a charge density of 10e/nm in water at pH = 8.1[12]. M13 bacteriophage is almost identical to fd with the exception that the 12th negatively charged residue (Asp) of each major coat protein (pVIII, Fig. S1) in fd is replaced by a polar one (Asn) in M13[13]. Each virus consists of a ssDNA molecule packed in a sheath of ~3000 identical coat proteins. As a result, M13 has an approximate one quarter reduction in surface charge density. Four minor coat proteins (pIII, pVI, pVII and pIX) additionally cap the two ends of the filamentous phage (Fig. S1)[14]. A foreign peptide can be genetically fused to the N-terminal of pVIII, enabling genetic controlling of the side wall surface chemistry of bacteriophage [15].
The preparation of mesoporous silica was achieved by employing 3-aminopropyltriethoxysilane (APTES) as a co-structure-directing agent and prehydrolyzed tetraethoxysilane (TEOS), which was prepared by stirring TEOS in deionized (DI) water overnight, as a growth agent. In a typical experiment, APTES was first added to an fd virus suspension (20 mg/ml) in DI water. The resulting mixture was incubated at room temperature for about 3 min followed by the addition of excessive prehydrolyzed TEOS. Vortexing was applied for about 3-5 min after addition of TEOS to the virus suspension to ensure a good dispersion of TEOS within the mixture. Although white flocculates appeared within 15 min, the reaction mixture was allowed to stand at room temperature for 8 h. OU-880 was then collected by centrifugation and washed with ethanol and water several times before being dried at 70 °C. Some samples were further calcined at 550 °C for 4 h for characterization and applications in templating the growth of nanoparticles.
Both SEM and TEM image showed the formation of silica fibers with a length ranging from several micrometers to tens of micrometers (Fig. 1). High resolution SEM images (Fig. 1a, inset) revealed that these fibers were indeed bundles of narrower silica fibers with uniform diameters ranging from 70-80 nm. High resolution TEM images of as-synthesized small silica fibers showed uniform and parallel aligned lattice fringes (Fig. 1b), indicating a LC array of bacteriophages in the resulting OU-880.
Figure 1.

Characterization of bacteriophage-governed mesoporous silica fibers (OU-880). a, SEM images of OU-880 with a range of the length from several to ten micrometers. Inset shows the high resolution SEM image of OU-880, indicating that the large fibers were indeed bundles of small uniform nanofibers with diameter of 70-80 nm. b and c, High resolution TEM image of as-synthesized (b) and calcined (c) OU-880, respectively. d, SAXS pattern of the as-synthesized and calcined mesoporous silica fibers (OU-880). e and f, Cross-sectional TEM images of the as-synthesized (e) and calcined, TEM images of the formed bacteriophage bundles (f) OU-880. g induced by APTES interactions. Inset shows the corresponding high magnification TEM image.
Removal of bacteriophages by calcination at 550 °C for 4 h in air gave parallel aligned and uniform sized channels in OU-880 which ran through the entire silica fiber (Fig. 1c). To gain insight into the bacteriophage alignment pattern and lattice spacing within OU-880, the silica fibers were cut by microtome to form thin cross sections which were viewed by TEM. As shown in Fig. 1d and 1e for the cross-sectional TEM images of the as-synthesized and calcined OU-880, respectively, a hexagonal array of bacteriophages or channels in the obtained silica fibers was observed. The calculated average lattice spacing (channel center-to-center distance) for the as-synthesized and calcined OU-880 was determined as 7.8 and 7.6 nm, respectively. Since the thickness of the silica side wall (between two neighboring bacteriophages) was found to be 2 ± 0.5 nm, the diameter of bacteriophages calculated from lattice spacing (~5.8 ± 0.5 nm) was slightly less than that of native fd bacteriophage (6.6 nm)[12], which was possibly due to compression of biomolecules during silicification[16].
Hexagonal lattices of the bacteriophage templated OU-880 were then investigated by small angle X-ray scattering (SAXS). SAXS patterns of both the as-synthesized and calcined OU-880 showed a Bragg peak of low index planes that is typical for a hexagonal lattice, suggesting a long range order of bulky materials (Fig. 1d). The center-to-center pore spacing was calculated to be 7.8 nm and 7.6 nm for as-synthesized and calcined OU-880, respectively, which match the result from TEM observation. The decrease of d-spacing in the calcined OU-880 was due to further condensation of silica during the calcinations. The surface area of OU-880 obtained from the BET method is 341.57 m2/g which is close to the reported results of silica nanotubes (328 m2/g [17] or 332±21 m2/g [18]) with similar structure as OU-880. And the calculated pore diameter (4.871 nm) from BJH method matches the pore (channel) diameter measured from TEM image (~5.5 nm) (See Fig. S2 and supporting information for details).
The hexagonal pore structures of the resulting silica arose from synergistic interactions between bacteriophages, APTES (functioning as co-structure-directing agent) and silicic acid (from prehydrolyzed TEOS)[19]. In our experiment, we noted that addition of APTES to the fd virus suspension induced the aggregation of viruses, forming a bundle structure (Figs. 1g). APTES binds to individual fd bacteriophages by electrostatic interactions and hydrogen bonding between its amino group and surface proteins of virus (See supporting information). Nucleophilic residues at the N-terminal of pVIII such as Lys 8 and Ser 13 may catalyze and initiate the hydrolysis of APTES, forming oligomers or even nanoparticles[5b, 20].
Due to the propyl amine arm in APTES, the formed oligomeric silicic acids contained both positively charged amino groups and negatively charged polysilicic acid (Fig. S3). The formed APTES intermediates functioned as multidentate counterions, attracting neighboring viruses together to form an ordered assembly through electrostatic and hydrogen bonding interactions (Figs. 2, S1 & S3 and supporting information). The APTES-induced virus assembly was also confirmed by dynamitic light scattering (DLS) and zeta potentials (ξ) analysis. DLS results showed a kinetic formation of larger aggregates once APTES was added to the bacteriophage solution (Fig. S4). Zeta potential analysis of bacteriophage before and after the addition of APTES showed a remarkable decrease of mobility and effective surface-charge (Fig. S5) which was attributed to the virus assembly and phase segregation as observed in the nanofabrication of silica in diatom biosilica[21].
Figure 2.
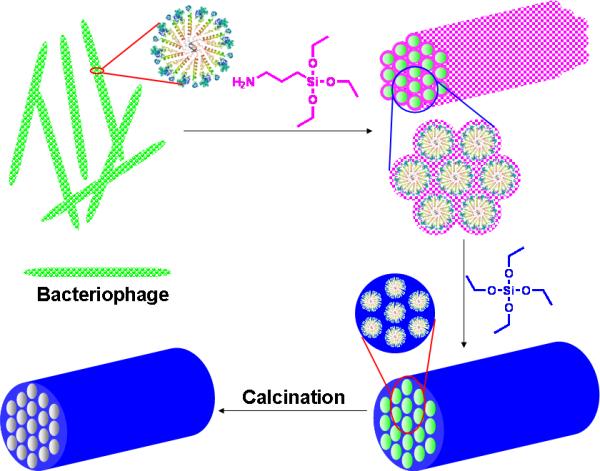
Proposed mechanism for the bacteriophage directed formation of hexagonal mesoporous silica. The insets show the cross-section view of bacteriophages assembly. The magenta spots stand for mono- and oligo-hydrolysized APTES that are anchored to bacteriophage surface and function as nuclei for subsequent TEOS polycondensation. The blue color illustrates the formed silica net work through TEOS polycondensation on the nuclei.
When prehydrolyzed TEOS was added to the mixture of APTES and bacteriophage, the formed APTES intermediates (oligomeric silicic acids) functioned as seeds initiating polycondensation of TEOS on the surface of bacteriophages (Fig. 2). As TEOS polycondensation proceeded, the charge density of the inorganic layer changed with the increase in thickness of the silica wall. The matching of charge density between the bacteriophage surface and silica species may finally govern the 3D assembly with the lowest interface energy[5a, 22]. Furthermore, contributions to the hexagonal organization of bacteriophages in the resulting mesoporous silica include the condensation of bacteriophage from solution through APTES interactions, the semiflexible rod-like structure of bacteriophage, the high degree of monodispersity and the anisotropic polarity of bacteriophages (Fig. 2).
Due to the uniform cylindrical pores and unidirectional array of the pores, mesoporous silica has been successfully exploited as templates for the preparation of metal and semiconductor nanowires[23]. However, a unidirectional ordered array of nanoparticles in mesoporous silica has never been successfully attempted. Here, we show the preparation of an ordered 3D array of nanoparticles with preferred nanocrystal orientation along the fiber by taking advantage of the hexagonal arrangement of uniform pores within OU-880.
To prepare a 3D PbS nanoparticle array, OU-880 was first calcined at 550 °C for 4 h to remove any organic composition, followed by immersion in an aqueous solution of saturated lead acetate (PbAc). OU-880 absorbed with Pb2+ solution was filtered and washed with DI water twice, and placed into a chamber filled with H2S gas. TEM imaging (Fig. S6a) confirms that an ordered 3D array of PbS nanocrystals was formed within the channels of OU-880. The uniform diameter of PbS nanoparticles was determined to be 5.5 ± 0.2 nm, which is consistent with the diameter of an individual channel (ca. 5.5 nm). Corresponding selected area electron diffraction (SAED) analysis indicated that the PbS nanoparticles in the individual channels were oriented with a [100] axis preferentially parallel to the channel (Fig. S6b). Energy-dispersive spectroscopy (EDS) analysis (Fig. S6c) gave a Pb to S molar ratio of ~1:1, consistent with the stoichiometry of a PbS crystal. Our proposed model for the formation of a 3D array of PbS nanoparticles in OU-880 is illustrated in Fig. S6d.
Since the formation of the hexagonal 3D assembly of bacteriophage in the resulting silica was governed by charge density matching between the bacteriophage surface and silica species, it was anticipated that fine controlling of the resulting silica structures could be achieved by control of the bacteriophage surface charge properties, which can be fulfilled by displaying differently charged peptides on the side walls. In our experiment, control of the bacteriophage surface charge density was achieved via genetic engineering of the ~3000 copies of pVIII on the side wall of the M13 bacteriophage. Fusion of a foreign peptide to the N-terminal of pVIII on the bacteriophage was acquired by inserting a single DNA fragment that encoded the foreign peptide sequence into the phage DNA. In this manner, we displayed 8 negatively charged glutamate residues (E8-M13) or 4 positively charged arginine residues (R4-M13) on the side walls of bacteriophage. Thus, three bacteriophages were prepared with different negative charge densities in the following order: E8-M13 > M13 (wild type) > R4-M13. This order was also confirmed with zeta potential measurements. The purified engineered bacteriophage (E8-M13 and R4-M13) and wild type M13 were used to prepare silica under the similar conditions as in preparation of OU-880.
Electron microscopy images exhibited remarkable morphological evolution and obvious change of the bacteriophage assembly pattern in the resulting silica nanofibers (See supporting information for explanation of the mechanism). For example, silica fibers with a length of 2-3 μm, 10-15 μm, and ~ 1 μm were obtained when E8-M13, M13, and R4-M13 were employed as templates, respectively (Fig. 3a,c,e). TEM micrographs revealed that the surface charge properties of bacteriophages influenced virus assembly in the presence of APTES, thus controlling pore structures of the resulting silica nanotubes. Parallel aligned viruses were observed in the E8-M13 templated silica nanofibers (Fig. 3b), while wild type M13 bacteriophage were assembled into bundle-like structures (Fig. 3d). As expected, APTES did not induce aggregation of bacteriophage R4-M13. When R4-M13 bacteriophages were used as templates, uniform silica nanofibers with a length of 1 μm containing a single virus within each individual silica nanofiber was observed (Fig. 3f).
Figure 3.
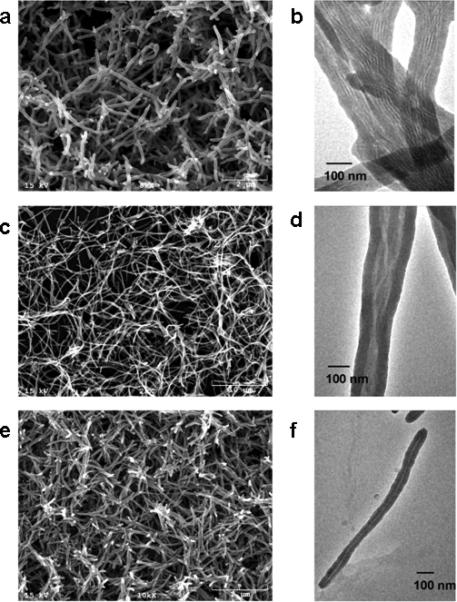
Electron microscopy of silica fibers synthesized by employing E8-M13 (a, SEM, b, TEM), wild type M13 (c, SEM; d, TEM), and R4-M13 bacteriophages (e, SEM; f, TEM) as templates. E8-M13 and R4-M13 are bacteriophages that display anionic and cationic peptide on the side walls, respectively.
To further understand the virus alignment patterns in the resulting silica structures, bacteriophages (E8-M13 and M13) were conjugated with Alexa Fluor® 488-labeled anti-pIII antibody that reccognizes pIII (the tip of phage). The dye-labeled bacteriophages were employed as templates to prepare silica fibers under the same conditions as described above. The fluorescence imaging of the obtained silica was consistent with TEM imaging (Fig. 3) and confirms the proposed assembly of bacteriophages in silica (Fig. S7). For the silica fibers with bundle aligned viruses (Fig. 3d), the whole fiber is fluorescent as shown in Fig. S7B. In this kind of structure, viruses were supposed to align in a nemetic structure, with fluorescent dyes evenly distributed along the fiber. However, for the silica fibers with defined layers (Fig. S7C), there is a smectic alignment of viruses as can be seen from the periodic alignment of the dyes along the fiber, with an interval distance of 800-1000 nm, which is consistent with the length of a single phage (Fig. S7D). However, the silica fibers prepared from the antibody conjugated phages are shorter than the fibers from phages without conjugation, generally in the range of 1~2 μm. This may be due to the disturbance of the antibody.
To the best of our knowledge, this is the first successful synthesis of nano-structured mesoporous silica with hexagonally aligned pore structures using biotemplates via a synergistic co-assembly of bacteriophage and silica precursor species. This particular method can provide new families of mesoporous materials for potential applications in photonic and electronic nanodevices, drug release, and bioreactors. Furthermore, we demonstrated the versatility of bacteriophage in controlling the nanostructures of silica by controlling the assembly behavior through surface charge. Thus, great promise is seen in the future for the preparation of inorganic materials which includes a broad range of structural diversity.
Experimental Section
The general procedure for synthesizing mesoporous silica
The display of peptides on the side wall of phage was reported in our earlier publication.[24] APTES was added to an aqueous solution of virus (20 mg/ml, determined using spectrophotometry with an absorption coefficient of 3.84 cm2/mg at 270 nm) at room temperature. The solution was then mixed by vortexing for about 3 min and kept in an ice-water bath for about 10 min. Next, prehyolyzed TEOS with a final concentration of 0.1 M was added and mixed by vortexing for another 3 min. The resulting mixture was allowed to stand at room temperature for 8 h. Generally, the solution became an opalescent gel after 15 min due to the formation of long silica fibers. Shaking broke the gel to form suspended flocculates which were then taken out by centrifugation and washed with water and alcohol.
Supplementary Material
Footnotes
We thank National Science Foundation (DMR-0847758, CBET-0854414, CBET-0854465), National Institutes of Health (5R01HL092526-02, 5R21EB009909-0 2 and 4R03AR056848-03), Department of Defense Congressionally Directed Medical Research ProgramOklahoma Center for Adult Stem Cell Research Center, and Oklahoma Center for the Advancement of Science and Technology (HR11-006) for financial support. We also thank Drs. B. Grady, A. Madden, W. T. Yip, H. Lu and P. Larson for their kind help during the study.
Supporting information for this article is available on the WWW under http://www.angewandte.org or from the author.
References
- 1.Kresge CT, Leonowicz ME, Roth WJ, Vartuli JC, Beck JS. Nature. 1992;359:710–712. [Google Scholar]
- 2.Zhao DY, Feng JL, Huo QS, Melosh N, Fredrickson GH, Chmelka BF, Stucky GD. Science. 1998;279:548–552. doi: 10.1126/science.279.5350.548. [DOI] [PubMed] [Google Scholar]
- 3.Che S, Garcia-Bennett AE, Yokoi T, Sakamoto K, Kunieda H, Terasaki O, Tatsumi T. Nat. Mater. 2003;2:801–805. doi: 10.1038/nmat1022. [DOI] [PubMed] [Google Scholar]
- 4.a Hartmann M. Chem. Mater. 2005;17:4577–4593. [Google Scholar]; b Trewyn BG, Slowing II, Giri S, Chen HT, Lin VSY. Acc. Chem. Res. 2007;40:846–853. doi: 10.1021/ar600032u. [DOI] [PubMed] [Google Scholar]; c Ying JY, Mehnert CP, Wong MS. Angew. Chem. Int. Ed. 1999;38:56–77. [Google Scholar]
- 5.a Wan Y, Zhao DY. Chem. Rev. 2007;107:2821–2860. doi: 10.1021/cr068020s. [DOI] [PubMed] [Google Scholar]; b Monnier A, Schuth F, Huo Q, Kumar D, Margolese D, Maxwell RS, Stucky GD, Krishnamurty M, Petroff P, Firouzi A, Janicke M, Chmelka BF. Science. 1993;261:1299–1303. doi: 10.1126/science.261.5126.1299. [DOI] [PubMed] [Google Scholar]
- 6.a Wang F, Li D, Newton S, Klebba P, Mao CB. Advanced Functional Materials. 2008;18:4007–4013. [Google Scholar]; b Wang F, Mao CB. Chemical Communications. 2009:1222–1224. doi: 10.1039/b818652a. [DOI] [PubMed] [Google Scholar]; c Niu Z, Kabisatpathy S, He J, Lee LA, Rong J, Yang L, Sikha G, Popov BN, Emrick TS, Russell TP, Wang Q. Nano Res. 2009;2:474–483. [Google Scholar]
- 7.Tang JX, Fraden S. Liq. Cryst. 1995;19:459–467. [Google Scholar]
- 8.Sarikaya M, Tamerler C, Jen AKY, Schulten K, Baneyx F. Nat. Mater. 2003;2:577–585. doi: 10.1038/nmat964. [DOI] [PubMed] [Google Scholar]
- 9.a Kroger N, Deutzmann R, Sumper M. Science. 1999;286:1129–1132. doi: 10.1126/science.286.5442.1129. [DOI] [PubMed] [Google Scholar]; b Round FEC, R. M., Mann DG. The Diatom: Biology & Morphology of the Genera. Cambridge University Press; Cambridge: 1990. [Google Scholar]
- 10.Flynn CE, Lee SW, Peelle BR, Belcher AM. Acta Mater. 2003;51:5867–5880. [Google Scholar]
- 11.Dogic Z, Purdy KR, Grelet E, Adams M, Fraden S. Phys. Rev. E. 2004;69:051702. doi: 10.1103/PhysRevE.69.051702. [DOI] [PubMed] [Google Scholar]
- 12.a Dogic Z, Fraden S. Curr. Opin. Colloid In. 2006;11:47–55. [Google Scholar]; b Dogic Z, Fraden S. Phys. Rev. Lett. 1997;78:2417–2420. [Google Scholar]
- 13.Day LA, Marzec CJ, Reisberg SA, Casadevall A. Ann. Rev. Biophys. Biophys. Chem. 1988;17:509–539. doi: 10.1146/annurev.bb.17.060188.002453. [DOI] [PubMed] [Google Scholar]
- 14.Webster RE. Display of Peptides and proteins. Academic Press; London, U.K.: 1996. [Google Scholar]
- 15.a Smith GP, Petrenko VA. Chem. Rev. 1997;97:391–410. doi: 10.1021/cr960065d. [DOI] [PubMed] [Google Scholar]; b Merzlyak A, Lee SW. Curr. Opin. Chem. Biol. 2006;10:246–252. doi: 10.1016/j.cbpa.2006.04.008. [DOI] [PubMed] [Google Scholar]
- 16.Fowler CE, Shenton W, Stubbs G, Mann S. Adv. Mater. 2001;13:1266–1269. [Google Scholar]
- 17.Lei S, Zhang J, Wang JR, Huang JB. Langmuir. 2010;26:4288–4295. doi: 10.1021/la9033707. [DOI] [PubMed] [Google Scholar]
- 18.Qiao Y, Chen HF, Lin YY, Yang ZY, Cheng XH, Huang JB. J Phys Chem C. 2011;115:7323–7330. [Google Scholar]
- 19.a Dickerson MB, Sandhage KH, Naik RR. Chem. Rev. 2008;108:4935–4978. doi: 10.1021/cr8002328. [DOI] [PubMed] [Google Scholar]; b Tomczak MM, Glawe DD, Drummy LF, Lawrence CG, Stone MO, Perry CC, Pochan DJ, Deming TJ, Naik RR. J. Am. Chem. Soc. 2005;127:12577–12582. doi: 10.1021/ja0524503. [DOI] [PubMed] [Google Scholar]
- 20.a Cha JN, Stucky GD, Morse DE, Deming TJ. Nature. 2000;403:289–292. doi: 10.1038/35002038. [DOI] [PubMed] [Google Scholar]; b Caruso RA, Antonietti M. Chem. Mater. 2001;13:3272–3282. [Google Scholar]; c Morse DE. Trends Biotechnol. 1999;17:230–232. [Google Scholar]
- 21.a Sumper M, Kroger N. J. Mater. Chem. 2004;14:2059–2065. [Google Scholar]; b Sumper M. Science. 2002;295:2430–2433. doi: 10.1126/science.1070026. [DOI] [PubMed] [Google Scholar]
- 22.Huo QS, Margolese DI, Ciesla U, Feng PY, Gier TE, Sieger P, Leon R, Petroff PM, Schuth F, Stucky GD. Nature. 1994;368:317–321. [Google Scholar]
- 23.a Han YJ, Kim JM, Stucky GD. Chem. Mater. 2000;12:2068–2069. [Google Scholar]; b Coleman NRB, O'Sullivan N, Ryan KM, Crowley TA, Morris MA, Spalding TR, Steytler DC, Holmes JD. J. Am. Chem. Soc. 2001;123:7010–7016. doi: 10.1021/ja015833j. [DOI] [PubMed] [Google Scholar]
- 24.Liu A, Abbineni G, Mao CB. Adv. Mater. 2009;21:1001–1005. [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.