

Abstract
Efficient editing of Trypanosoma brucei mitochondrial RNAs involves the actions of multiple accessory factors. T. brucei RGG2 (TbRGG2) is an essential protein crucial for initiation and 3′-to-5′ progression of editing. TbRGG2 comprises an N-terminal G-rich region containing GWG and RG repeats and a C-terminal RNA recognition motif (RRM)-containing domain. Here, we perform in vitro and in vivo separation-of-function studies to interrogate the mechanism of TbRGG2 action in RNA editing. TbRGG2 preferentially binds preedited mRNA in vitro with high affinity attributable to its G-rich region. RNA-annealing and -melting activities are separable, carried out primarily by the G-rich and RRM domains, respectively. In vivo, the G-rich domain partially complements TbRGG2 knockdown, but the RRM domain is also required. Notably, TbRGG2's RNA-melting activity is dispensable for RNA editing in vivo. Interactions between TbRGG2 and MRB1 complex proteins are mediated by both G-rich and RRM-containing domains, depending on the binding partner. Overall, our results are consistent with a model in which the high-affinity RNA binding and RNA-annealing activities of the G-rich domain are essential for RNA editing in vivo. The RRM domain may have key functions involving interactions with the MRB1 complex and/or regulation of the activities of the G-rich domain.
INTRODUCTION
Trypanosome RNA editing entails the precise addition and removal of uridine nucleotides in mitochondrial RNAs. In Trypanosoma brucei, 12 of the 18 mitochondrially encoded mRNAs require editing for maturation prior to their translation. Essential players in this process are mitochondrially encoded 50- to 60-nucleotide (nt)-long guide RNAs (gRNAs), which direct the positions of uridine insertion and deletion through base-pairing interactions. The editing cycle is initiated upon association of a cognate gRNA with preedited mRNA by formation of a short anchor duplex. Editing catalysis is mediated by multiprotein complexes called editosomes or RNA editing core complexes (RECCs), and editing efficiency is achieved through the actions of transiently associating accessory factors (8, 16, 45, 55, 56, 62, 63). Following annealing of gRNA/preedited mRNA, a gRNA-directed endonuclease cleaves the premRNA at the site of gRNA/mRNA mismatch, and U insertion or deletion is catalyzed by terminal uridylyl transferase or U-specific exoribonuclease activities, respectively. The mRNA is then resealed by RNA ligase in preparation for a subsequent editing cycle. The editing cycles continue until gRNA/mRNA base pairing is extended along the entire length of the gRNA. gRNAs are then presumably exchanged, and the process continues, proceeding in a general 3′-to-5′ direction along the mRNA. While “minimally edited mRNAs” are edited only in small regions, the majority of mRNAs are edited throughout their lengths and thus are termed panedited. Complete editing of panedited mRNAs requires sequential utilization of dozens of gRNAs.
RNA-editing accessory factors are thought to coordinate recruitment of RNAs to the editosome, to direct correct gRNA/mRNA annealing, and to regulate editing progression by modulating RNA-RNA and RNA-protein interactions. Accessory factors studied to date include RBP16, MRP1/2, and TbRGG2, all of which bind and anneal RNAs, T. brucei RGG1 (TbRGG1), and the RNA helicase REH1 (2, 28, 33, 40, 44, 50, 57, 65). TbRGG2 is a component of a multiprotein complex, Mitochondrial RNA Binding Complex 1 (MRB1, also known as GRBC), which contains numerous proteins that affect RNA editing and stability and exhibits a dynamic composition maintained by a network of protein-protein and protein-RNA contacts (1, 3, 4, 32, 34, 53, 66). Here, we focus on the mechanism of action of TbRGG2, which is required for editing of all panedited RNAs and is thus essential for growth of both the insect procyclic form (PF) and the bloodstream form (BF) of T. brucei (1, 28). Previously, we showed that repression of TbRGG2 impacts both initiation of RNA editing at mRNA 3′ ends and the 3′-to-5′ progression of editing, while gRNA levels remain unaffected. In vitro, TbRGG2 binds mRNA and gRNA and possesses robust gRNA/mRNA-annealing activity (5, 28). It also exhibits RNA-melting activity in an Escherichia coli model system. The ability of TbRGG2 to modulate RNA-RNA interactions was suggested as a key function in RNA editing, likely impacting gRNA utilization (5). TbRGG2 forms multiple mitochondrial complexes that are partially RNA independent (28), and yeast two-hybrid analysis identified several direct TbRGG2 binding partners in the MRB1 complex (3). TbRGG2 is organized into two distinct domains: an N-terminal glycine-rich (G-rich) region and a C-terminal RNA recognition motif (RRM)-containing domain (Fig. 1). Within the G-rich domain are two slightly overlapping regions of eight glycine-tryptophan-glycine (GWG) repeats and eight arginine-glycine (RG) repeats. The RRM contains two conserved sequence motifs, RNP1 and RNP2. How the distinct TbRGG2 domains contribute to its multiple biochemical functions and its essential role in RNA editing remains obscure.
Fig 1.

Recombinant TbRGG2 proteins. (A) Domain structure of TbRGG2 variants used in the study. The numbers refer to amino acid positions. RGG2 FV-AA contains two point mutations in the RRM domain (shown by ×). (B) GST-tagged TbRGG2 proteins and GST were purified from E. coli cells and analyzed by 15% SDS-PAGE and Coomassie blue staining.
In other proteins, G-rich and RRM domains bind RNA and/or contribute to protein-protein interactions (30, 31, 35, 46). For example, GWG repeats in TNRC6 family proteins bind Argonaute (Ago), and this interaction is essential for microRNA (miRNA)-mediated repression (23, 25, 42). Interactions between the RGG boxes of Sm proteins and SMN are critical in formation of spliceosomal snRNPs (26). Many RG- and RGG-containing sequences also bind RNA directly (20, 22), and it has been proposed that some RGG-containing proteins regulate RNA processing and enhance the proper assembly of mature RNPs through interactions with sequence-specific RNA binding proteins (31). The RRM is a commonly occurring motif in eukaryotic proteins that function in numerous aspects of RNA processing, translation, decay, and transport (11, 47). Structural analyses of RRM domains show that RRMs are extremely diverse in terms of structure and function. Classical RRMs contain 80 to 90 amino acids (aa), comprising a four-stranded antiparallel β-sheet enclosed by two α-helices (7). RRMs serve as RNA binding elements, often utilizing two conserved sequence motifs, RNP1 and RNP2, located on the central two β-strands to mediate RNA contacts (47). Many structural studies of RRM domains in complex with different RNAs show that this small domain is a central component of RNA recognition but not the only determinant. N- and C-terminal extensions, simultaneous actions of multiple RRM domains, or protein cofactors can play an important role in RNA binding specificity (14, 49, 58). RNA binding affinities of RRM domains range from very high to low (10, 11, 38). Additionally, a number of proteins have been described in which RRM domains exclusively mediate protein-protein interactions (13, 29, 36, 39, 61).
Here, we report in vitro and in vivo separation-of-function studies that provide insights into the mechanisms by which TbRGG2 contributes to RNA editing. We show that full-length TbRGG2 preferentially binds preedited mRNA rather than gRNA or edited mRNA. The N-terminal G-rich domain is the primary mediator of high-affinity RNA binding and RNA annealing, and both GWG and RG repeats contribute to these activities. In vivo, the G-rich domain partially complements the knockdown of endogenous TbRGG2, but RRM-mediated functions are also essential. The C-terminal RRM-containing domain mediates RNA melting and negatively regulates the activities of the G-rich domain, although additional mutations demonstrate that the RNA-melting activity of TbRGG2 is dispensable for growth and RNA editing in vivo. Interactions between TbRGG2 and MRB1 complex components are mediated by both G-rich and RRM-containing domains, depending on the binding partner. Overall, our results are consistent with a model in which the high-affinity RNA binding and RNA-annealing activities of the G-rich domain are essential for RNA editing in vivo. The RRM domain may have key functions involving interactions with the MRB1 complex and/or regulation of the activities of the G-rich domain.
MATERIALS AND METHODS
Cloning and expression of recombinant proteins.
The entire TbRGG2 open reading frame (ORF) was PCR amplified from oligo(dT)-primed cDNA, which was synthesized from PF T. brucei total RNA using the primers RGG2-5 (5′-GCGAATTCATGAAGCGCACACCTGTTAG-3′) and RGG2-3 (5′-GGAAGCTTTTCCTTCTGACTGGCATC-3′). The product was cloned into the pJET1.2/blunt cloning vector (Clone JET PCR Cloning Kit; Fermentas) to yield pJET1.2-TbRGG2. The TbRGG2 fragment was excised from pJET1.2-TbRGG2 and ligated into the EcoRI/HindIII sites of pET42a (Novagen) to yield pET42-TbRGG2. This plasmid was transformed into E. coli strain Rosetta cells (Novagen) for expression of N-terminally glutathione S-transferase (GST)-tagged TbRGG2. The transformed cells were grown to an optical density (OD) of 0.6, and protein expression was induced with 1 mM IPTG (isopropyl-β-d-thiogalactopyranoside) for 3 h at 37°C. Recombinant TbRGG2 protein was purified according to standard protocols using glutathione-agarose beads (Invitrogen). The pJET1.2-TbRGG2 plasmid was used as a template for making all TbRGG2 variants. The primer sets used for amplification were as follows: G-rich domain (aa 1 to 160), RGG2-5 and G-rich-3 (5′-GGAAGCTTGTTTGGTTGACCCCAGAC-3′); GWG region (aa 1 to 107), RGG2-5 and GWG-3 (5′-GGAAGCTTGAGCCCCAGCCGCCGTTG-3′); RRM domain (aa 161 to 330), RRM-5 (5′-GGGAATTCGTGGTCGACGAGGAGGCA-3′) and RGG2-3; and FL-GWG region (aa 108 to 330), FL-GWG-5 (5′-GGAATTCGGCTGGGGCTCTGGTCGG-3′) and RGG2-3. To make the RGG2VF-AA mutant, we used the QuikChange kit (Stratgene) with primers RGG2VF-AA-fwd (5′-TCAGGGGGAAGAGCTGTGGCGGAAGCTGTCACCCCGGAAGACGCT-3′) and RGG2-VF-AA-rev (5′-AGCGTCTTCCGGGGTGACAGCTTCCGCCACAGCTCTTCCCCCTGA-3′).
Filter binding assay.
Filter binding assays were performed by incubating increasing concentrations (0.25 to 1,000 nM) of GST-tagged purified proteins with 0.5 fmol of internally [α-32P]UTP-labeled RNAs (∼30,000 cpm) at 27°C for 30 min. Here, we tested three different in vitro-transcribed RNAs, 221-nt preedited RPS12 (PE-RPS12), 325-nt fully edited RPS12 (FE-RPS12) (37), and 79-nt gA6[14] (see Fig. S1 in the supplemental material) (43, 59). The reaction was performed in a total volume of 15 μl containing 1× binding buffer (phosphate-buffered saline [PBS] [pH 7.6], 2.1 mM MgCl2, 0.5 mM dithiothreitol [DTT], 0.1 mM EDTA, 6% glycerol, 1.5 mM ATP, 5 mM phosphocreatine, 10 μg/ml torula yeast RNA, and 50 μg/ml bovine serum albumin). A microfiltration apparatus (Bio-Rad) was used to filter the RNA binding reaction mixture on two membranes, an upper nitrocellulose and a lower Nytran-SPC (Whatman), as described previously (67). The membranes were presoaked in 1× filter buffer (phosphate-buffered saline [pH 7.6], 2.1 mM MgCl2, 0.5 mM DTT, 0.1 mM EDTA, and 6% glycerol). Following filtration, the membranes were washed twice with 1 ml of 1× binding buffer and left to dry for 15 min at room temperature. We used a Bio-Rad phosphorimager scanner and Quantity One software to measure the radiolabeled signals of both bound and free RNA. Sigma Plot 11.2 software (Systat Software, Inc.) was used to analyze the data and calculate the apparent dissociation constant (Kd) by the best fit to a nonlinear regression curve.
In vitro RNA annealing.
RNA-annealing reactions were performed as previously described (5). Briefly, we used increasing concentrations of GST-tagged recombinant proteins to promote the annealing of equivalent concentrations (∼10 nM) of 5′-radiolabeled A6U5 41-nt pre-mRNA with gA6[14]NX gRNA. The annealed product was analyzed via native PAGE and quantified using a Bio-Rad phosphorimager scanner and Quantity One software.
E. coli RNA-melting assay.
RNA-melting assays were performed as previously described (5). The sequences of each primer set used to introduce TbRGG2 variants into the pINIII plasmid are as follows. Full-length TbRGG2 was amplified by TbRGG2-5′NdeI (5′-GCGCATATGAAGCGCACACCTGTTAG-3′) and TbRGGm-3-21 (5′-GGAAGCTTTTACACCTTCTGACTGGC-3′). The G-rich domain was amplified with TbRGG2-5′NdeI and G-rich-3 (see above). The RRM domain was amplified with RRM-3 (5′-GCGCATGTGGTCGACGAGGAGGCA-3′) and TbRGGm-3-21. The RGG2VF-AA mutant was amplified using the pET42-RGG2VF-AA plasmid as a template. DNA fragments were excised and cloned into the NdeI/HindIII sites of the pINIII vector. To test the transcriptional antitermination activity, the various pINIII plasmids were transformed into RL211 cells provided by Robert Landick (University of Wisconsin—Madison) (9). The transformed cells were grown overnight in Luria broth (LB) with 100 μg/ml ampicillin. The cell cultures were then diluted into fresh medium and grown to an optical density at 600 nm (OD600) of 1. Expression of TbRGG2 variant proteins was induced with 1 mM IPTG for 1 h. About 5 μl of each cell culture was spotted on LB plates containing 100 μg/ml ampicillin and 1 mM IPTG in the absence or presence of 15 μg/ml chloramphenicol. The plates were grown at 37°C for up to 4 days.
Cell cultures and cell lines.
PF strain 29-13 (kindly provided by George Cross, Rockefeller University), expressing the T7 polymerase under the control of a tetracycline-inducible promoter, was grown as indicated previously (57). Generation of the 3′ untranslated region (UTR)-based TbRGG2 RNA interference (RNAi) line and the corresponding add-back line complemented with wild-type myc-tagged TbRGG2 was previously described (3). All the other add-back cell lines were created using the same strategy and the following primer sets: G-rich domain, RGG2-5pLEW and G-rich-3pLEW (5′-GGTCTAGAGTTTGGTTGACCCCAGAC-3′); RRM domain, RRM-5pLEW (5′-GGAAGCTTGTGGTCGACGAGGAGGCA-3′) and RGG2-3pLEW. To construct the RGG2VF-AA mutant plasmid, RGG2-5pLEW and RGG2-3pLEW primers were used to amplify the mutant from the pET42-RGG2VF-AA template.
qRT-PCR.
Total RNA was extracted from uninduced and induced cells cultured for 3 days using TRIzol reagent (Invitrogen). RNA was treated with a DNA-free-DNase kit (Ambion) to remove any residual DNA. cDNA was synthesized using the Taq-Man reverse transcription kit (Applied Biosciences) and used as a template in quantitative reverse transcription-PCRs (qRT-PCRs) with primers specific for never-edited, preedited, and fully edited mRNAs of the T. brucei mitochondrion (15, 16). The results were analyzed with iQ5 software (Bio-Rad), and all data are normalized to β-tubulin mRNA.
Immunofluorescence microscopy.
Fluorescence microscopy was performed as described previously (24, 69). Cells were grown for 1 day in the presence of 2.5 μg/ml tetracycline. To visualize mitochondria, we treated the growing cells with 250 nM MitoTracker Red CMXRos for 15 min at 27°C. MitoTracker Red-stained cells were fixed for 30 min with 4% paraformaldehyde at 4°C. The cells were then permeabilized by gentle suspension in 0.5 ml of 0.1 M Na2HPO4-0.1 M glycine for 10 min, followed by suspension in 0.5 ml of PBS containing 0.2% Triton X-100 for 5 min. The cells were then washed and incubated for 1 h with 1:50-diluted mouse monoclonal c-Myc antibody (Santa Cruz Biotechnology). Following another wash, the cells were incubated with DAPI (4′,6-diamidino-2-phenylindole) and a 1:200 dilution of Cy5-conjugated anti-mouse IgG antibody (Chemicon) and then mounted. A Zeiss Axioimager Z1 fluorescence microscope and AxioVision software were used to visualize trypanosomes.
Yeast two-hybrid analysis.
We analyzed the interaction of TbRGG2 variants with six different mitochondrial proteins that are components of the MRB1 complex: MRB3010 (Tb927.5.3010), MRB4160 (Tb927.4.4160), MRB8170 (Tb927.8.8170), MRB8620 (Tb11.01.8620), MRB8180 (Tb927.8.8180), and MRB10130 (Tb927.10.10130). The entire ORFs were PCR amplified from either T. brucei procyclic form 39 strain 29-13 genomic DNA or cDNA. pET42a constructs were used to shuttle out the DNA fragments of TbRGG2 variants using the EcoRI/XhoI cut sites. The products were ligated into EcoRI/XhoI cut sites of the activation domain pGADT7 vector. The PCR products of the MRB1-interacting partners were cloned into pGBKT7 binding domain vectors (3). Saccharomyces cerevisiae strain PJ69-4A was cotransformed with 1 μg of each plasmid by the lithium acetate method. The cotransformed cells were grown on plates contain synthetically defined (SD) medium lacking leucine and tryptophan for 3 days at 30°C. Postincubation, 5 to 10 colonies of cotransformed yeast were inoculated onto both SD media to select for the two cotransformed plasmids and SD medium lacking histidine to screen for protein-protein interaction. The SD plates lacking histidine contained 1, 2, 3.5, or 5 mM 3-amino-1,2,4-triazole (3-AT) to inhibit strain PJ69-4A yeast growth resulting from the leaky expression of the HIS gene and to allow for increasingly stringent conditions. We incubated the inoculated plates for 3 days at 30°C.
IP.
Mitochondrial vesicles were enriched from approximately 2 × 1010 cells of each add-back line grown for 2 days in the presence of 2.5 μg/ml tetracycline (33). Mitochondrial vesicles corresponding to 1 × 1010 cells were lysed in 1 ml lysis buffer (25 mM Tris [pH 8.0], 15 mM Mg acetate, 50 mM KCl, 0.2% NP-40, 1 μg/ml pepstatin, 2 μg/ml leupeptin, 1 mM phenylmethylsulfonyl fluoride [PMSF]). Each 1 × 1010-cell equivalent was treated with DNase 1 (0.002 U/μl). RNase treatment was performed for 15 min at room temperature on 1 × 1010 cells using a nuclease cocktail that included RNase A (0.1 U/μl), RNase T1 (0.1 U/μl), RNase H (0.01 U/μl), RNase 1 (0.1 U/μl), RNase V1 (0.002 U/μl), and micrococcal nuclease (0.25 U/μl) (Fermentas). RNase-treated or untreated lysates were incubated with anti-myc polyclonal antibody (ICL Laboratories) cross-linked to protein A-Sepharose beads (GE Healthcare) overnight at 4°C. The flowthrough was collected, and the beads were washed several times with phosphate-buffered saline containing 0.1% Tween. The bound proteins were eluted with 100 mM glycine (pH 2.5) and neutralized using 1 M Tris buffer (pH 8.5). Non-RNase-treated mitochondrial lysate of 29-13 parental cells was incubated with anti-myc antibody cross-linked to protein A-Sepharose beads and used as a negative control for the immunoprecipitation (IP) experiments.
Western blotting and antibodies.
Protein samples were separated on 10 or 15% SDS-polyacrylamide gels, as indicated in the figure legends for each experiment, and then transferred to a nitrocellulose membrane. Immunoblotting was performed using polyclonal antibodies directed against TbRGG2 (28), GAP1 (32), MRB3010 (4), and MRB8170 (3). Rabbit anti-MRB10130 polyclonal antibody was produced by Bethyl Laboratories using the oligopeptide CNSMPKEVSVPEEDAISPES as an antigen. myc-tagged TbRGG2 variants were detected with rabbit polyclonal anti-myc antibodies (ICL, Inc.).
RESULTS
Recombinant TbRGG2 variants.
To analyze the RNA binding and annealing activities of TbRGG2 domains in vitro, we generated several recombinant GST-tagged TbRGG2 variants, including full-length TbRGG2, multiple truncated versions, and a double point mutant (Fig. 1). Truncation constructs included the ORFs of the N-terminal G-rich region (G-rich; amino acids 1 to 160) and the C-terminal RRM-containing domain (RRM; amino acids 161 to 330). To further dissect the functions of the G-rich domain, we created a construct expressing only the GWG region (GWG; amino acids 1 to 107) and a construct expressing full-length TbRGG2 lacking the GWG repeats but retaining RG repeats (FL-GWG; amino acids 108 to 330). We also created the double point mutant RGG2VF-AA by mutating 2 residues in the highly conserved RRM signature motif, RNP1. Homologous RNP1 residues are critical for both in vitro and in vivo RNA binding in other RRM-containing proteins (21, 68). The observed sizes of all purified recombinant proteins were consistent with their expected molecular weights plus that of the GST tag with linker (Fig. 1B).
The G-rich domain exhibits high-affinity binding to preedited RNA in vitro.
We showed previously using UV cross-linking that TbRGG2 can directly interact with preedited mRNA, fully edited mRNA, and gRNA (28). Because both G-rich and RRM domains in other proteins have been reported to bind RNA (30, 31, 35), it was important to define the roles of TbRGG2 domains in binding different RNA classes so that we could correlate the different RNA-based activities of TbRGG2 with the RNA binding properties of each domain. We tested three different RNAs, PE-RPS12, FE-RPS12, and gA6[14] (see Fig. S1 in the supplemental material), and calculated the Kds (Table 1). Initially, we asked whether wild-type TbRGG2 exhibits markedly different affinities for the three different RNAs. Indeed, TbRGG2 exhibited a significantly higher affinity for the preedited RNA than for the fully edited RNA or gRNA, with a Kd for PE-RPS12 in the low nM range (4.3 ± 1.3). The Kds of wild-type TbRGG2 for FE-RPS12 and gA6[14] were 9 and 18 times higher than for PE-RPS12, respectively. These data suggest that preedited RNA is a preferred target of TbRGG2 in vivo.
Table 1.
RNA binding activity of TbRGG2 variants
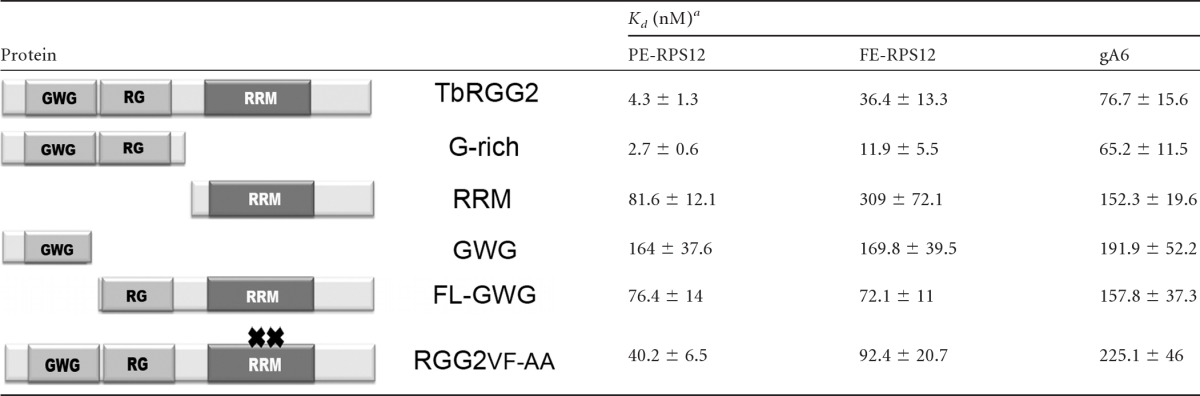
The Kd was determined by the best fit to a nonlinear regression curve as described in Materials and Methods. Shown are the means and standard deviations of three measurements.
We next determined the binding affinities of TbRGG2 variants. Interestingly, the G-rich region exhibited RNA binding affinities equal to or better than those of the wild-type protein for all three RNAs tested (Table 1), suggesting that the domain largely contributes to the RNA binding activity of the full-length protein. The RRM domain displayed low affinity compared to either wild-type TbRGG2 or the G-rich region, with Kds approximately 10 and 20 times higher for fully edited and preedited RNA, respectively. Like wild-type TbRGG2, both G-rich and RRM domains bound PE-RPS12 with higher affinity than FE-RPS12 or gA6[14]. However, the RRM domain was unique in that it appeared to display a slight preference for gRNA over fully edited RNA.
To further define the contributions of the G-rich and RRM-containing domains to RNA binding, we assayed additional TbRGG2 variants. We began with proteins harboring truncated G-rich regions, the GWG and FL-GWG variants (Fig. 1A). Both truncations severely impacted the RNA binding of the G-rich domain, as well as its specificity for PE-RPS12 (Table 1). Hence, the high-affinity RNA binding activity of TbRGG2 requires an intact G-rich domain. To further investigate the contribution of RRM to RNA binding, we mutated the RRM signature motif, RNP1, in a manner predicted to abrogate its RNA binding (Fig. 1, RGG2VF-AA) (21, 68). Mutating the RNP1 motif decreased the RNA binding affinity of TbRGG2 by 3- to 10-fold, depending on the RNA tested. This was somewhat surprising, given that RGG2VF-AA retains an intact G-rich domain. The decreased RNA binding affinity of the RGG2FV-AA protein suggests that the mutated RRM domain has an indirect effect on the RNA binding capacity of the G-rich domain, possibly resulting from a conformational change of TbRGG2. In addition, we cannot rule out a limited direct contribution of the RRM domain to the high-affinity RNA binding of TbRGG2 that was disrupted by mutating RNP1. Collectively, the RNA binding data indicate that TbRGG2 preferentially binds preedited RNA through a high-affinity interaction that requires the entire G-rich domain. The RRM domain may contribute modestly to in vitro TbRGG2-RNA interactions.
RNA-annealing activity is mediated by the N-terminal G-rich region.
TbRGG2 exhibits robust in vitro RNA-annealing activity (5). To define the roles of TbRGG2 domains in facilitating gRNA/mRNA annealing, we incubated increasing concentrations of each TbRGG2 variant with equivalent molar concentrations of 5′-radiolabeled A6U5 mRNA and cognate gA6[14] RNA and resolved single and double-stranded RNAs on a native gel following protease treatment. The maximal annealing activity of TbRGG2 was approximately 55% after subtracting the 10% activity observed in control reaction mixtures containing GST (Fig. 2A). We reproducibly observed a modest decrease in annealed RNA at the highest concentrations of TbRGG2. In comparison with TbRGG2, the G-rich region displayed even more robust RNA-annealing activity (87% at maximum) (Fig. 2B), and unlike the wild-type protein, the activity did not decrease at high protein concentrations. Both TbRGG2 and G-rich proteins facilitated gRNA/mRNA annealing at low concentrations, with about 30% annealing observed at 10 nM protein (Fig. 2A and B). In contrast, the RRM domain displayed very weak activity. Annealing activity reached a maximum of 45%, but this required 500 nM protein, and no activity was observed below 100 nM RRM (Fig. 2C). We next determined whether the robust annealing activity of the G-rich protein was attributable to either of its constituent GWG and RG regions. The annealing activity of the GWG protein (Fig. 2D) was higher than that of FL-GWG (Fig. 2E) in terms of both maximal activity (76% versus 48%) and the minimal concentration at which activity was detected (5 nM versus 50 nM). However, neither protein with a truncated G-rich region exhibited activity equivalent to that of the intact G-rich domain (compare Fig. 2D and E with B). Thus, we conclude that the G-rich domain mediates the annealing activity of TbRGG2 and that an intact G-rich domain is required to achieve maximal annealing. Finally, we tested the gRNA/mRNA-annealing activity of RGG2VF-AA. The activity of this mutant was lower than that of the G-rich protein, reaching 53% maximal annealing, despite the fact that RGG2VF-AA contains an intact G-rich domain (compare Fig. 2F and B). Nevertheless, RGG2VF-AA still retains moderate annealing activity, especially compared to RRM, exhibiting annealing activity between 1.8- and 49-fold higher than that of RRM, depending on the protein concentration (Table 2). These data suggest that the RNP1 mutations have an indirect negative effect on the annealing activity of the G-rich domain. Overall, the RNA-annealing results are very consistent with the RNA binding data and indicate that the high-affinity RNA binding activity of the N-terminal G-rich domain is utilized to accelerate annealing of complementary RNAs.
Fig 2.

The G-rich domain mediates RNA-annealing activity in vitro. RNA-annealing assays were performed with equivalent concentrations (∼10 nM) of 5′-radiolabeled A6U5 41-nt pre-mRNA and unlabeled gA6[14]NX gRNA at the indicated protein concentrations. Negative-control reactions were performed in the absence of gA6[14]NX (Probe) or with both RNAs in the presence of 250 nM GST. Reaction mixtures were incubated for 20 min, treated with proteinase K, and analyzed on 12% native PAGE. The blots represent RNA-annealing assays at the indicated concentrations of TbRGG2 (A), G-rich protein (B), RRM (C), GWG (D), FL-GWG (E), and RGG2 FV-AA (F). The graphs represent means and standard deviations of the percentages annealed in three independent experiments.
Table 2.
Annealing activities of RGG2VF-AA and RRM
| Variant | Annealing activity (%) at protein concn (nM)a: |
||||
|---|---|---|---|---|---|
| 25 | 50 | 100 | 250 | 500 | |
| RRM | 0.2 | 4.4 | 8.5 | 22.1 | 43.3 |
| RGG2VF-AA | 9.8 | 20.7 | 29.5 | 40.6 | 52.6 |
| Ratio of RGG2VF-AA/RRM | 49 | 4.7 | 3.5 | 1.8 | 1.2 |
Shown are the percentages of annealing activities, after subtracting self-annealing, at the indicated increasing concentrations of the RGG2VF-AA mutant or RRM. The RGG2VF-AA/RRM ratios show the fold differences in RNA-annealing activities between the two variants at the different protein concentrations.
RNA-melting activity is performed by the C-terminal RRM domain.
In addition to RNA annealing, TbRGG2 possesses RNA-melting activity, as measured by an E. coli transcription antitermination assay (5, 41). To evaluate the contributions of TbRGG2 domains to RNA melting, we used E. coli cells containing a chloramphenicol resistance gene preceded by the trpL terminator, which forms an RNA hairpin upon transcription (Fig. 3A). These cells can grow in the presence of the antibiotic only if they overexpress a protein able to resolve the terminator structure and enable expression of the resistance gene. We transformed the E. coli cells with TbRGG2, RGG2VF-AA, G-rich, or RRM proteins, which were expressed at similar levels (Fig. 3B). Like wild-type TbRGG2 and the positive-control CSPE, the RRM domain enabled the cells to grow in the presence of chloramphenicol (Fig. 3C). In contrast, neither the G-rich nor the RGG2VF-AA protein could support growth on chloramphenicol. These results indicate that RNA melting is attributable solely to the RRM domain. The inability of RGG2VF-AA to facilitate melting indicates that the RNP1 motif is crucial for this activity.
Fig 3.

The RRM-containing domain mediates RNA melting and interferes with RNA annealing. (A) E. coli strain RL211 contains the trpL terminator that forms a hairpin loop upstream of a chloramphenicol resistance gene (CAT). Expression of proteins capable of unwinding the RNA results in chloramphenicol resistance. (B) (Top) Anti-TbRGG2 Western blot of RL211 cells expressing TbRGG2, RGG2 FV-AA, RRM, or G-rich protein. (Bottom) A portion of the corresponding Coomassie-stained gel showing equal loading. (C) RL211 cells were transformed with empty pINIII vector, CspE (positive control), TbRGG2, RGG2FV-AA, RRM, or G-rich constructs and grown in the absence (− chl) or presence (+ chl) of chloramphenicol. (D) Annealing assays performed as in Fig. 2 using a constant amount of G-rich protein and increasing concentrations (nM) of RRM protein.
Notably, our results to this point indicate that the G-rich and RGG2VF-AA proteins promote RNA annealing but cannot facilitate RNA melting. Melting activity is exerted only by an intact RRM domain, which lacks substantial annealing activity. Thus, there is a separation of these two opposing activities within TbRGG2. Moreover, the RNA-annealing activity of the G-rich domain exceeds that of full-length TbRGG2, suggesting possible negative effects of RRM (Fig. 2A and B). These data led us to ask whether the RRM domain can affect the annealing activity of the G-rich domain. To address the interplay between the two domains, we performed annealing assays with a constant concentration (100 nM) of G-rich protein and increasing concentrations of RRM. We observed an almost complete suppression of G-rich-domain-mediated RNA annealing even in the presence of equimolar amounts of RRM (Fig. 3D). The small amount of annealing activity observed at the highest concentrations of RRM presumably reflects the weak annealing activity of the RRM itself (see Fig. 2C). These data suggest that the RNA-melting activity mediated by the RRM domain can interfere with RNA annealing mediated by the G-rich domain or that the RRM domain directly masks the RNA binding surface of the G-rich domain or affects its structure. The ability of the RRM domain to impact the activity of the G-rich domain suggests a potential mechanism for regulation of TbRGG2.
Both G-rich and RRM domains are essential for cell growth.
Having established structure-function relationships for TbRGG2 RNA binding, -annealing, and -melting activities in vitro, we next wanted to define the contributions of these activities to trypanosome growth and RNA editing in vivo. Our previous studies utilizing RNAi targeted against the TbRGG2 ORF demonstrated that the protein is essential for PF and BF growth and for editing of panedited RNAs (5, 28). To analyze the ability of TbRGG2 mutants to complement growth and editing, we established a system in which endogenous TbRGG2 is repressed in PF T. brucei using RNAi targeted against its 3′ UTR, and myc-tagged TbRGG2 variants are expressed with a heterologous 3′ UTR that is refractory to RNAi. Repression of endogenous TbRGG2 and expression of TbRGG2 variants are simultaneously induced by tetracycline treatment. Cell lines stably transfected with constructs expressing wild-type TbRGG2, RGG2VF-AA, RRM, or G-rich proteins expressed equivalent levels of myc-tagged TbRGG2 variants within the TbRGG2 knockdown background (Fig. 4A). Indirect immunofluorescence confirmed that all exogenous proteins were mitochondrially localized (Fig. 4B). Upon tetracycline induction, growth of TbRGG2 3′ UTR-RNAi cells ceased, with a time course very similar to that of the previously described ORF knockdown (28) (Fig. 5A), accompanied by an approximately 50% reduction of TbRGG2 protein at day 2 postinduction (Fig. 4A) and 60% at day 3 (data not shown). Complementation with wild-type myc-TbRGG2 completely restored cell growth in the 3′ UTR-RNAi background, indicating that exogenous myc-tagged TbRGG2 is functional (Fig. 5B). Thus, we have established a genetic system that will allow us to analyze the functions of TbRGG2 domains and their associated biochemical activities in vivo.
Fig 4.

T. brucei RNAi cell lines complemented with TbRGG2 variants. (A) Lysates of 1 × 107 PF T. brucei cell lines grown in the absence (−) or presence (+) of tetracycline (Tet) for 2 days were immunoblotted with anti-TbRGG2 antibodies (top) to reveal the expression levels of the endogenous TbRGG2 before (−Tet) and after (+Tet) induction of the 3′ UTR-RNAi vector. RNAi indicates the uncomplemented 3′ UTR RNAi cell line. The anti-myc (αmyc) immunoblots reveal the expression levels of exogenously expressed myc-tagged TbRGG2 variants, which are also Tet induced. P22, loading control. (B) The subcellular localization of myc-tagged TbRGG2 proteins was determined by indirect immunofluorescence (Cy5) (green). Mitochondria were detected using MitoTracker Red CMXRos (Mito) (red). Nuclei and kinetoplasts were stained blue with DAPI. The signals are shown merged on the right. DIC, differential interference contrast.
Fig 5.

Growth of RNAi knockdown and complemented cell lines. Cells were either left untreated (−Tet) or treated with 2.5 μg/ml tetracycline (+Tet) to induce expression of both the 3′ UTR-RNAi vector and myc-tagged proteins. Growth was measured for 10 days. Cumulative cell numbers are shown as the means and standard deviations of triplicate cell cultures.
Having established this system, we examined the growth of cells predominantly expressing mutant versions of TbRGG2. Neither the G-rich nor the RRM domain was able to restore normal growth to cells depleted of endogenous TbRGG2 (Fig. 5C and D), demonstrating that both domains provide the cell with essential functions. However, the growth defect of the G-rich cell line is less severe than that of either the 3′ UTR-RNAi or RRM add-back cell line. Growth of the G-rich line essentially plateaued beyond day 4, while by day 10 postinduction, growth of the 3′ UTR-RNAi line diminished nearly 10-fold and the RRM line contained no live cells. Thus, the G-rich region can partially restore cell growth, suggesting that the G-rich domain performs a critical function and that the RRM domain is required to facilitate or complement the G-rich region's function. The dramatic negative effect of RRM expression on cell growth suggests that the RRM-containing domain, in the absence of the G-rich domain, has a dominant-negative effect on residual endogenous TbRGG2. We next tested the ability of the myc-tagged RGG2VF-AA mutant protein to restore cell growth in the 3′ UTR-RNAi background (Fig. 5E). Remarkably, the RGG2FV-AA line grew at the same rate as uninduced cells or those expressing myc-tagged wild-type TbRGG2 (Fig. 5A and E), even under low serum stress (data not shown). Thus, the RGG2FV-AA protein possesses the activities that mediate the essential functions of TbRGG2. This mutant protein does not possess RNA-melting activity (Fig. 3C), implying that RNA melting is dispensable for cell growth under the tested conditions. The RGG2VF-AA mutant displayed in vitro RNA binding and -annealing activities with moderate reduction compared to the wild-type protein. Given that RGG2FV-AA protein is completely competent for growth restoration, either its reduced RNA binding and/or annealing activities are sufficient to maintain essential functions or the cellular exogenous mutant protein is more effective than the recombinant protein. Collectively, these results demonstrate that both G-rich and RRM domains contribute to the essential functions mediated by TbRGG2 and that the protein's RNA-melting activity is dispensable.
Both G-rich and RRM domains are required for RNA editing.
We next asked whether defects in RNA editing parallel growth defects in cells expressing TbRGG2 variants. We performed qRT-PCR analysis of RNA collected at day 3 postinduction, using primer sets designed to analyze representatives of different mitochondrial RNA classes, including never-edited COI, minimally edited CYb, and panedited A6 and RPS12 RNAs (15, 16). Similar to the ORF RNAi cell line, the induced 3′ UTR-RNAi line displayed 93% and 82% reductions in edited A6 and RPS12 RNAs, respectively, compared to uninduced cells, without corresponding accumulation of preedited A6 or RPS12 RNA (Fig. 6A). Also in keeping with previous results, we did not observe significant changes in the levels of COI, CYb preedited, or CYb edited RNA. Thus, the 3′ UTR-based RNAi line behaves similarly to the ORF-based RNAi line in terms of RNA-editing defects.
Fig 6.

Mitochondrial RNA levels in RNAi knockdown and complemented cell lines. Shown is qRT-PCR analysis of RNA from cell lines cultured for 3 days and either untreated or treated with 2.5 μg/ml tetracycline. COI is never edited, Cyb is minimally edited, and A6 and RPS12 are panedited. RNA levels were normalized to β-tubulin mRNA and represent the means and standard errors (SE) of 12 determinations. P, preedited; E, edited.
We then used qRT-PCR to determine RNA levels in the complemented cells. The 3′ UTR-RNAi line complemented with wild-type TbRGG2 displayed substantially restored levels of both edited A6 and RPS12 RNAs (Fig. 6B). The levels of the other RNAs examined were essentially unchanged, ruling out nonspecific effects. Thus, exogenous wild-type TbRGG2 compensated for the downregulation of endogenous TbRGG2 and restored RNA editing. To determine whether restoration of RNA editing can account for growth rescue, we measured the RNA levels in the other cell lines and found that edited RNA levels were very consistent with the growth curves. We observed substantially reduced edited A6 and RPS12 RNA levels in cells complemented with either G-rich or RRM domains and restored levels in RGG2VF-AA-complemented cells. The correlation between RNA editing and growth was particularly evident when examining edited A6 RNA levels. Edited A6 RNA was not as dramatically decreased in cells expressing the G-rich domain as in 3′ UTR-RNAi cells, consistent with the more robust growth of G-rich-domain-expressing cells (Fig. 5C). Likewise, cells expressing RRM exhibited a somewhat more dramatic decrease in edited A6 RNA than the 3′ UTR-RNAi line, possibly accounting for the more severe growth defects in the RRM-expressing cell line (Fig. 5). The restoration of nearly normal edited RNA levels in cells expressing the RGG2VF-AA mutant protein indicates that the mutant restored RNA editing without a requirement for RNA melting and that it did not cause nonspecific effects on preedited or never-edited RNAs (Fig. 6E). Together, the results of mitochondrial RNA analysis indicate that both G-rich and RRM domains are crucial for RNA-editing-related functions mediated by TbRGG2. The growth defects observed in cells expressing truncated versions of TbRGG2 reflect RNA-editing defects in these cells.
G-rich and RRM domains mediate distinct protein-protein interactions.
TbRGG2 is a component of the MRB1 complex, a dynamic macromolecular complex involved in RNA editing and stability and mediated by numerous protein-protein, protein-RNA, and RNA-enhanced interactions (1, 3, 5, 32, 34, 53, 66). Previous yeast two-hybrid analyses showed that TbRGG2 interacts strongly with six other MRB1 components, including the MRB1 core proteins MRB3010 and MRB8620, and MRB10130, which may act as an MRB1 organizer. The three other yeast two-hybrid interacting proteins are those with which TbRGG2 appears to associate in mutually exclusive subcomplexes: the paralogous MRB8170 and MRB4160 proteins and MRB8180 (3). Using yeast two-hybrid analysis, we determined the roles of TbRGG2 domains in mediating these interactions to better understand the function of TbRGG2 in the context of MRB1 (Fig. 7A). Core components MRB3010 and MRB8620 and the ARM protein MRB10130 interact with TbRGG2 primarily through its G-rich domain. MRB10130 interacts as strongly with the G-rich protein as with TbRGG2 and thus appears to utilize exclusively G-rich contacts. The attenuated interactions of MRB3010 and MRB8620 with the G-rich domain compared to TbRGG2 suggest that the RRM-containing domain may assist in stabilizing these interactions. In contrast, MRB8170 and MRB8180 interact with TbRGG2 primarily through its RRM domain, with a potential modest contribution of the G-rich region. We did not detect interaction of MRB4160 with either the G-rich or RRM domain. We also tested interactions between the RGG2VF-AA mutant and MRB1 proteins (Fig. 7A). While MRB8170 interacted as strongly with RGG2VF-AA as with wild-type TbRGG2, the RGG2VF-AA mutant displayed reduced interaction with the other proteins. The attenuated interactions of MRB10130 and MRB3010 with RGG2VF-AA compared to their interactions with the G-rich domain are consistent with our previous results showing that the mutated RRM domain appears to negatively impact some functions of the G-rich region (Fig. 2 and Table 1). Together, these results demonstrate that the G-rich and RRM domains of TbRGG2 mediate distinct protein-protein interactions within the MRB1 complex.
Fig 7.

Protein interaction profiles of TbRGG2 variants. (A) Summary of yeast two-hybrid assays monitoring interactions between TbRGG2 variants and six MRB1 components. Genes encoding TbRGG2 variants and MRB1 components were ligated into pGADT7 and pGBKT7, respectively. Empty pGBKT7 vector was the negative control. Cotransformed S. cerevisiae was grown on SD media lacking leucine and tryptophan (SD−Leu/−Trp) to confirm transformation and on SD−Leu/−Trp/−His with increasing concentrations of 3-AT to screen for protein interaction. Growth was scored according to the highest concentration on which growth of cotransformed cells was observed: 5 mM 3-AT (strong; +++), 3.5 mM 3-AT (moderate; ++), 2 mM 3-AT (weak; +), or no growth (negative; −). (B) Coimmunoprecipitation assays with myc-tagged TbRGG2 variants. Mitochondrial lysate was either RNase-treated (+) or left untreated (−). Tagged proteins were precipitated with anti-myc polyclonal antibody cross-linked to protein A-Sepharose. Immunoprecipitated proteins were equally loaded according to the anti-myc signal. 29-13, whole-cell lysate of approximately 1 × 107 29-13 parental cells. Negative, untreated mitochondrial lysate of 29-13 parental cells was incubated with anti-myc antibody cross-linked to protein A-Sepharose. Each row represents a single blot and was probed with antibodies recognizing the proteins indicated on the left.
Having defined the roles of TbRGG2 domains in binary protein-protein interactions, we next wanted to examine the interactions of TbRGG2 variants in vivo. This allowed us to establish the interaction capacity of each domain in an environment containing both RNA and the complex protein milieu of the mitochondrion. We carried out coimmunoprecipitations using mitochondrial extracts from cells expressing different TbRGG2 variants and investigated the role of RNA by comparing untreated and nuclease-treated extracts. The myc-tagged TbRGG2 variants were immunoprecipitated with anti-myc antibody and detected by anti-myc Western blotting (Fig. 7B, top). Associated proteins were detected by Western blotting with specific antibodies. In general, MRB1 components for which specific antibodies are available displayed interactions with TbRGG2 domains in vivo that were consistent with yeast two-hybrid results. For example, MRB3010 and MRB10130 both interact strongly with the G-rich domain in vivo and display weak or no interaction with RRM. Indeed, MRB3010 interacts much more strongly with the G-rich domain than with full-length TbRGG2, again pointing to an inhibitory or regulatory effect of the RRM domain within the context of TbRGG2. Interactions with the G-rich domain mimic those with TbRGG2 regarding the contribution of RNA, which is modest in the case of MRB3010 and substantial with MRB10130. The apparent role of RNA in the MRB10130/G-rich domain co-IP is at odds with the strong yeast two-hybrid signal between the two proteins, which suggests a direct protein-protein interaction. It is likely this discrepancy reflects the previously described RNA-enhanced nature of the MRB10130-TbRGG2 interaction (3), which is evident in the co-IP with RGG2VF-AA and which may be obscured here due to the relatively low signal levels with the full-length and G-rich proteins. In keeping with the two-hybrid results, coimmunoprecipitation of MRB8170 with RRM was equivalent to that with TbRGG2, and both were enhanced by the presence of RNA, whereas MRB8170 interaction with the G-rich domain was at background levels. All three proteins interacted strongly and in an RNA-enhanced manner with the RGG2VF-AA mutant, indicating that the RNP1 mutations did not compromise TbRGG2 protein-protein interactions in vivo. We also examined the interactions of the GAP1 gRNA binding component of the MRB1 complex with TbRGG2 variants. By the yeast two-hybrid assay, GAP1 interacts weakly with TbRGG2 itself but strongly with TbRGG2 binding partners MRB3010, MRB8620, and MRB8170 (3). GAP1 precipitated with full-length TbRGG2 and all TbRGG2 variants. These interactions were RNA independent (G-rich), RNA enhanced (full-length TbRGG2 and RGG2VF-AA), or RNA dependent (RRM). In summary, combined yeast two-hybrid and in vivo coimmunoprecipitation experiments demonstrate that protein-mediated interactions between TbRGG2 and the MRB1 core (which includes MRB8620, MRB3010, and GAP1), as well as the ARM protein MRB10130, are primarily mediated by the G-rich domain. In contrast, interactions with MRB8170 and MRB8180 are mediated primarily by the RRM-containing domain. Mutations of the RNP1 domain did not impact TbRGG2 protein-protein interactions.
DISCUSSION
Repression of TbRGG2 causes a dramatic reduction in panedited RNAs and a severe growth defect in both BF and PF T. brucei (1, 28). TbRGG2 is crucial for initiation and 3′-to-5′ progression of RNA editing, suggesting a key role in modulating gRNA-mRNA interactions. TbRGG2 exhibits multiple RNA-based activities, including RNA binding, RNA annealing, RNA melting, and RNA-enhanced protein interactions (3, 5, 28). The TbRGG2 protein comprises G-rich and RRM domains, which in other proteins function in diverse RNA modification processes and are implicated in both protein-RNA and protein-protein interactions (31, 47). Due to the reported functional similarities of G-rich and RRM domains in other proteins, we performed in vitro and in vivo separation-of-function analyses of TbRGG2 to gain insight into the activities of its constituent domains and thereby into the mechanism of TbRGG2 action. We show that cells depleted of endogenous TbRGG2 but expressing one or the other domain are dramatically impaired for RNA editing and growth. Together with functional and protein-protein interaction studies, these data reveal distinct roles for the G-rich and RRM-containing domains of TbRGG2, which can be utilized in RNA editing.
Our results strongly suggest that the essential functions of the G-rich domain are RNA binding and annealing. We show that TbRGG2 exhibits high affinity for preedited RNA that is 10 to 20 times its affinity for edited RNA or gRNA. TbRGG2 depletion has modest, if any, effects on preedited RNA levels, precluding any major impact on RNA stability (references 1 and 28 and this study). Hence, our data suggest that the high affinity of TbRGG2 for preedited RNA is directly correlated with its role in RNA editing. The G-rich domain of TbRGG2 is the primary mediator of preedited RNA binding. Indeed, we observed that for all RNAs tested, binding by the G-rich domain was comparable to, or slightly better than, that of the full-length protein. Further truncation of the domain demonstrated that both GWG and RG repeats are essential for high-affinity RNA binding and for specificity for preedited RNA. Thus, the structure of the TbRGG2 N-terminal domain may contribute to its RNA binding capacity, which is surprising, as G-rich domains often lack significant structure. GW/WG repeats have been studied primarily in TNRC6 family proteins, where they are critical for interaction with Ago and also serve as effector motifs in miRNA-mediated silencing, apart from Ago binding (19). To our knowledge, ours is the first report of GW/WG repeats being critical for a protein's RNA binding activity. While our data implicate binding of preedited RNA by the G-rich region of TbRGG2 as a primary function, it is also possible that lower-affinity binding of edited RNA and gRNA, potentially with modest assistance from the RRM, contributes to TbRGG2 function (see below). Future studies aimed at assessing TbRGG2-RNA interactions in vivo will further illuminate the RNA binding properties of the protein.
The gRNA/mRNA-annealing activity of TbRGG2 closely parallels its RNA binding activity, in that the G-rich domain is the primary effector of annealing. Proteins truncated in or lacking the G-rich domain display relatively limited gRNA/mRNA-annealing capacities. This is especially true of the RRM domain. While involvement of high-affinity preedited RNA binding by the G-rich domain is likely important for annealing, our data suggest that gRNA binding by the G-rich domain also contributes to this activity. Comparison of the intact G-rich domain with the GWG protein shows a correlation between their affinities for gRNA and their RNA-annealing capacities. The GWG protein exhibits 2- to 3-fold-reduced annealing at all concentrations compared to the G-rich domain (Fig. 2). Similarly, its gRNA binding affinity is approximately 3-fold reduced compared to the G-rich domain, whereas its preedited RNA binding affinity is 60-fold reduced compared to the G-rich region (Table 1). The MRP1/2 and RBP16 proteins also exhibit gRNA/mRNA-annealing activity (6, 52). Moreover, MRP1/2 has been sporadically reported to be associated with editosomes, cocrystallized with gRNAs, and rigorously shown to promote gRNA/mRNA matchmaking activity (51, 60). However, in vivo depletion of MRP1/2 and RBP16, either singly or together, affects only editing of a small subset of RNAs, suggesting that these are not primary RNA-annealing factors during RNA editing (27, 57, 65). In contrast, TbRGG2 depletion leads to dramatic repression in editing of all panedited RNAs (1, 28). TbRGG2 has been identified in immunoprecipitated editosomes (28, 54) and is a component of the MRB1 complex (1, 3, 4, 32, 34, 53). Thus, the annealing function of TbRGG2 is presumably utilized during RNA editing of panedited RNAs, whose complete editing requires dozens of gRNAs and myriad RNA-annealing events. We show here that complementation with the G-rich domain reduces the severe growth defect caused by repression of endogenous TbRGG2, although it does not restore growth to wild-type levels. Collectively, these data provide strong evidence that the RNA binding and -annealing activities of the G-rich domain constitute its essential functions in vivo.
If the RNA binding and -annealing activities of the G-rich domain do not completely restore editing, what then is the essential role of the RRM-containing domain? Its RNA-melting activity cannot constitute its essential function because RGG2VF-AA, which lacks RNA-melting activity, efficiently rescues RNA editing and growth. Nevertheless, RNA annealing and melting may both contribute to TbRGG2 function in vivo, even if the latter is apparently not essential. These two opposing activities reportedly occur in some RNA helicases and in the Prp24 splicing factor, where they may work together to create functional RNA secondary structures and/or for regulation (18, 48, 64). The RNA-melting activity of TbRGG2 may simply be redundant due to the presence of other proteins with similar activities. For example, the REH1 helicase can unwind double-stranded RNA and is essential for editing of a subset of RNAs (44), and the editosome itself was recently reported to exhibit a chaperone-type RNA-unwinding activity (12). Regarding the essential function of RRM, one potential role is as a regulator of G-rich-domain-mediated activities. Both RNA binding and -annealing activities of G-rich protein exceed those of TbRGG2. In addition, the annealing activity of TbRGG2 reproducibly decreases at high protein concentrations, while that of the G-rich protein does not, again implying a negative regulatory effect of the RRM domain. We directly demonstrated such an effect, showing that equimolar amounts of RRM protein completely abrogate the strong annealing activity of the G-rich protein. These results suggest a model in which, in vivo, expression of unregulated G-rich domain alone may actually arrest RNA editing and growth due to excessive RNA binding and/or -annealing activities. Within full-length TbRGG2, the RRM domain may control the activity of the G-rich region by a direct intramolecular interaction that either masks the RNA binding surface or distorts the domain's structure. Intermolecular interactions may also contribute to RRM-mediated repression, as endogenous TbRGG2 is present in myc-TbRGG2 pulldowns (3). This hypothesis is supported by our data showing that the RRM add back not only failed to restore RNA editing or cell growth, but exacerbated these defects, possibly by inhibiting the function of residual endogenous protein. The capacity of the RRM domain to dramatically affect the RNA-based activities of the G-rich domain presents an opportunity for regulation of TbRGG2. Additional protein-protein interactions and/or posttranslational modifications of either domain could modulate their cross talk and thereby regulate TbRGG2 RNA binding and -annealing activities.
TbRGG2 is transiently associated with the editosome and is a component of the MRB1 complex (1, 3, 4, 28, 32, 34, 53, 54, 66). Several MRB1 components are essential for RNA editing, and TbRGG2 binding partners within MRB1 are candidate regulatory factors. Association of TbRGG2 with the MRB1 core (3) appears to be mediated by interaction of the G-rich domain with core proteins, MRB3010 and MRB8620. The G-rich domain also mediates interaction with the putative MRB1 organizer, MRB10130, likely in an RNA-enhanced manner. On the other hand, the RRM-containing domain mediates interactions with MRB8170 and MRB8180. It is tempting to speculate that MRB8170, MRB8180, and/or MRB4160 (an MRB8170 paralog) interact with the RRM domain to influence its effects on the RNA binding and -annealing properties of the G-rich domain. Importantly, like RRM, the RGG2FV-AA mutant that restores growth in TbRGG2-depleted cells also interacts with MRB8170, suggesting that this interaction could represent an essential RRM function. We previously identified weak interactions between TbRGG2 and four other MRB1 components (3). The binding of these and other as yet unidentified binding partners may impact TbRGG2 functions and underlie essential RRM domain functions.
In summary, we show that the essential RNA-based activities of TbRGG2 are carried out by its N-terminal G-rich domain. The C-terminal RRM domain may play a regulatory role whereby it modulates G-rich-domain-mediated activities. The RRM also participates in some protein interactions of TbRGG2, which may affect the RRM's regulatory functions and facilitate editosome association. Collectively, our data suggest a model for TbRGG2 function during RNA editing (Fig. 8). Initially, TbRGG2 binds preedited RNA through its G-rich domain (Fig. 8A). At this stage, TbRGG2 adopts a conformation in which the RRM domain represses the annealing activity of the G-rich region, and thus GAP1/2-bound gRNA does not anneal with mRNA bound to TbRGG2. These and subsequent events may take place within the context of the dynamic MRB1 complex (Fig. 8A and B, not shown; C, lavender). In Fig. 8B, the RRM domain associates with a protein partner (possibly, but not necessarily, MRB8170), which leads to TbRGG2 rearrangement so that RRM-mediated repression of annealing is released. Rearrangement then permits the annealing of GAP1/2-bound gRNA with TbRGG2-bound mRNA (Fig. 8C). Annealing may occur in association with MRB1 and the editosome, although the precise order of events is unknown. Here, we also illustrate the possibility that unidentified RRM domain binding proteins help recruit the essential RNA binding and -annealing functions of TbRGG2 to the editosome. Numerous cycles of the events in Fig. 8 must occur to complete the editing of a panedited RNA, as one gRNA is released and a subsequent gRNA is annealed. Involvement of TbRGG2 annealing activity in each gRNA exchange event would explain our previous results showing that both initiation and 3′-to-5′ progression of editing are compromised in TbRGG2-depleted cells (5).
Fig 8.

Proposed model for the interplay between TbRGG2 domains, mRNA, gRNA, MRB1 components, and the editosome that takes place during RNA editing. See the text for details.
Overall, the studies presented here provide significant insight into the mechanism of TbRGG2 action during trypanosome RNA editing. Important questions regarding the precise order of events and the identities of TbRGG2 binding partners that function at key steps of the editing process remain, providing fertile ground for future research.
Supplementary Material
ACKNOWLEDGMENTS
This work was supported by NIH grant number RO1 AI061580 to L.K.R. B.M.F. was supported in part by a fellowship from the Egyptian Ministry of Higher Education, and J.C.F. was supported in part by NIH postdoctoral fellowship number F32 AI07718501.
We thank Julius Lukes for anti-GAP1 antibodies and Michelle Ammerman for critical reading of the manuscript. We thank Robert Landick (University of Wisconsin) and Sangita Phadtare (Robert Wood Johnson Medical School) for providing us with E. coli strain RL211 and the PINIII vector, respectively.
Footnotes
Published ahead of print 13 July 2012
Supplemental material for this article may be found at http://ec.asm.org/.
REFERENCES
- 1. Acestor N, Panigrahi AK, Carnes J, Zikova A, Stuart KD. 2009. The MRB1 complex functions in kinetoplastid RNA processing. RNA 15:277–286 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2. Allen TE, et al. 1998. Association of guide RNA binding protein gBP21 with active RNA editing complexes in Trypanosoma brucei. Mol. Cell. Biol. 18:6014–6022 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3. Ammerman ML, et al. 2012. Architecture of the trypanosome RNA editing accessory complex, MRB1. Nucleic Acids Res. 40:5637–5650 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4. Ammerman ML, et al. 2011. MRB3010 is a core component of the MRB1 complex that facilitates an early step of the kinetoplastid RNA editing process. RNA 17:865–877 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5. Ammerman ML, Presnyak V, Fisk JC, Foda BM, Read LK. 2010. TbRGG2 facilitates kinetoplastid RNA editing initiation and progression past intrinsic pause sites. RNA 16:2239–2251 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6. Ammerman ML, Fisk JC, Read LK. 2008. gRNA/pre-mRNA annealing and RNA chaperone activities of RBP16. RNA 14:1069–1080 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7. Antson AA. 2000. Single-stranded-RNA binding proteins. Curr. Opin. Struct. Biol. 10:87–94 [DOI] [PubMed] [Google Scholar]
- 8. Aphasizhev R, Aphasizheva I. 2011. Uridine insertion/deletion editing in trypanosomes: a playground for RNA-guided information transfer. Wiley Interdiscip. Rev. RNA 2:669–685 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9. Bae W, Xia B, Inouye M, Severinov K. 2000. Escherichia coli CspA-family RNA chaperones are transcription antiterminators. Proc. Natl. Acad. Sci. U. S. A. 97:7784–7789 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10. Bandziulis RJ, Swanson MS, Dreyfuss G. 1989. RNA-binding proteins as developmental regulators. Genes Dev. 3:431–437 [DOI] [PubMed] [Google Scholar]
- 11. Birney E, Kumar S, Krainer AR. 1993. Analysis of the RNA-recognition motif and RS and RGG domains: conservation in metazoan pre-mRNA splicing factors. Nucleic Acids Res. 21:5803–5816 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12. Bohm C, Katari VS, Brecht M, Goringer HU. 2012. Trypanosoma brucei 20S editosomes have one RNA substrate-binding site and execute RNA unwinding activity. J. Biol. Chem. 287:26268–26277 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13. Bono F, et al. 2004. Molecular insights into the interaction of PYM with the Mago-Y14 core of the exon junction complex. EMBO Rep. 5:304–310 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14. Calero G, et al. 2002. Structural basis of m7GpppG binding to the nuclear cap-binding protein complex. Nat. Struct. Biol. 9:912–917 [DOI] [PubMed] [Google Scholar]
- 15. Carnes J, Trotter JR, Ernst NL, Steinberg A, Stuart K. 2005. An essential RNase III insertion editing endonuclease in Trypanosoma brucei. Proc. Natl. Acad. Sci. U. S. A. 102:16614–16619 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16. Carnes J, Trotter JR, Peltan A, Fleck M, Stuart K. 2008. RNA editing in Trypanosoma brucei requires three different editosomes. Mol. Cell Biol. 28:122–130 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17. Reference deleted.
- 18. Chamot D, Colvin KR, Kujat-Choy SL, Owttrim GW. 2005. RNA structural rearrangement via unwinding and annealing by the cyanobacterial RNA helicase, CrhR. J. Biol. Chem. 280:2036–2044 [DOI] [PubMed] [Google Scholar]
- 19. Chekulaeva M, Parker R, Filipowicz W. 2010. The GW/WG repeats of Drosophila GW182 function as effector motifs for miRNA-mediated repression. Nucleic Acids Res. 38:6673–6683 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20. Chen T, Côté J, Carvajal HV, Richard S. 2001. Identification of Sam68 arginine glycine-rich sequences capable of conferring nonspecific RNA binding to the GSG domain. J. Biol. Chem. 276:30803–30811 [DOI] [PubMed] [Google Scholar]
- 21. Cho S, et al. 2011. Interaction between the RNA binding domains of Ser-Arg splicing factor 1 and U1-70K snRNP protein determines early spliceosome assembly. Proc. Natl. Acad. Sci. U. S. A. 108:8233–8238 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22. Corbin-Lickfett KA, Souki SK, Cocco MJ, Sandri-Goldin RM. 2010. Three arginine residues within the RGG box are crucial for ICP27 binding to herpes simplex virus 1 GC-rich sequences and for efficient viral RNA export. J. Virol. 84:6367–6376 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23. El-Shami M, et al. 2007. Reiterated WG/GW motifs form functionally and evolutionarily conserved ARGONAUTE-binding platforms in RNAi-related components. Genes Dev. 21:2539–2544 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24. Engstler M, Boshart M. 2004. Cold shock and regulation of surface protein trafficking convey sensitization to inducers of stage differentiation in Trypanosoma brucei. Genes Dev. 18:2798–2811 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25. Eulalio A, Helms S, Fritzsch C, Fauser M, Izaurralde E. 2009. A C-terminal silencing domain in GW182 is essential for miRNA function. RNA 15:1067–1077 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26. Fischer U, Liu Q, Dreyfuss G. 1997. The SMN-SIP1 complex has an essential role in spliceosomal snRNP biogenesis. Cell 90:1023–1029 [DOI] [PubMed] [Google Scholar]
- 27. Fisk JC, Presnyak V, Ammerman ML, Read LK. 2009. Distinct and overlapping functions of MRP1/2 and RBP16 in mitochondrial RNA metabolism. Mol. Cell. Biol. 29:5214–5225 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28. Fisk JC, Ammerman ML, Presnyak V, Read LK. 2008. TbRGG2, an essential RNA editing accessory factor in two Trypanosoma brucei life cycle stages. J. Biol. Chem. 283:23016–23025 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29. Fribourg S, Gatfield D, Izaurralde E, Conti E. 2003. A novel mode of RBD-protein recognition in the Y14-Mago complex. Nat. Struct. Biol. 10:433–439 [DOI] [PubMed] [Google Scholar]
- 30. Gilbert W, Guthrie C. 2004. The Glc7p nuclear phosphatase promotes mRNA export by facilitating association of Mex67p with mRNA. Mol. Cell 13:201–212 [DOI] [PubMed] [Google Scholar]
- 31. Godin KS, Varani G. 2007. How arginine-rich domains coordinate mRNA maturation events. RNA Biol. 4:69–75 [DOI] [PubMed] [Google Scholar]
- 32. Hashimi H, Cicová Z, Novotná L, Wen YZ, Lukes J. 2009. Kinetoplastid guide RNA biogenesis is dependent on subunits of the mitochondrial RNA binding complex 1 and mitochondrial RNA polymerase. RNA 15:588–599 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33. Hashimi H, ZíkovÁ A, Panigrahi AK, Stuart KD, Lukes J. 2008. TbRGG1, an essential protein involved in kinetoplastid RNA metabolism that is associated with a novel multiprotein complex. RNA 14:970–980 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34. Hernandez A, et al. 2010. REH2 RNA helicase in kinetoplastid mitochondria: ribonucleoprotein complexes and essential motifs for unwinding and guide RNA (gRNA) binding. J. Biol. Chem. 285:1220–1228 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35. Hom RA, et al. 2010. Molecular mechanism of MLL PHD3 and RNA recognition by the Cyp33 RRM domain. J. Mol. Biol. 400:145–154 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36. Kadlec J, Izaurralde E, Cusack S. 2004. The structural basis for the interaction between nonsense mediated mRNA decay factors UPF2 and UPF3. Nat. Struct. Mol. Biol. 11:330–337 [DOI] [PubMed] [Google Scholar]
- 37. Kao CY, Read LK. 2005. Opposing effects of polyadenylation on the stability of edited and unedited mitochondrial RNAs in Trypanosoma brucei. Mol. Cell. Biol. 25:1634–1644 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38. Kenan DJ, Query CC, Keene JD. 1991. RNA recognition: towards identifying determinants of specificity. Trends Biochem. Sci. 16:214–220 [DOI] [PubMed] [Google Scholar]
- 39. Kielkopf CL, Lücke S, Green MR. 2004. U2AF homology motifs: protein recognition in the RRM world. Genes Dev. 18:1513–1526 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40. Lambert L, Müller UF, Souza AE, Göringer HU. 1999. The involvement of gRNA-binding protein gBP21 in RNA editing—an in vitro and in vivo analysis. Nucleic Acids Res. 27:1429–1436 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41. Landick R, Stewart J, Lee DN. 1990. Amino acid changes in conserved regions of the beta-subunit of Escherichia coli RNA polymerase alter transcription pausing and termination. Genes Dev. 4:1623–1636 [DOI] [PubMed] [Google Scholar]
- 42. Lazzaretti D, Tournier I, Izaurralde E. 2009. The C-terminal domains of human TNRC6A, TNRC6B, and TNRC6C silence bound transcripts independently of Argonaute proteins. RNA 15:1059–1066 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43. Leung SS, Koslowsky DJ. 1999. Mapping contacts between gRNA and mRNA in trypanosome RNA editing. Nucleic Acids Res. 27:778–787 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44. Li F, Herrera J, Zhou S, Maslov DA, Simpson L. 2011. Trypanosome REH1 is an RNA helicase involved with the 3′-5′ polarity of multiple gRNA-guided uridine insertion/deletion RNA editing. Proc. Natl. Acad. Sci. U. S. A. 108:3542–3547 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 45. Lukes J, Hashimi H, ZíkovÁ A. 2005. Unexplained complexity of the mitochondrial genome and transcriptome in kinetoplastid flagellates. Curr. Genet. 48:277–299 [DOI] [PubMed] [Google Scholar]
- 46. Ma AS, et al. 2002. Heterogeneous nuclear ribonucleoprotein A3, a novel RNA trafficking response element-binding protein. J. Biol. Chem. 277:18010–18020 [DOI] [PubMed] [Google Scholar]
- 47. Maris C, Dominguez C, Allain FH. 2005. The RNA recognition motif, a plastic RNA-binding platform to regulate post-transcriptional gene expression. FEBS J. 272:2118–2131 [DOI] [PubMed] [Google Scholar]
- 48. Martin-Tumasz S, Richie AC, Clos LJ, II, Brow DA, Butcher SE. 2011. A novel occluded RNA recognition motif in Prp24 unwinds the U6 RNA internal stem loop. Nucleic Acids Res. 39:7837–7847 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 49. Mazza C, Segref A, Mattaj IW, Cusack S. 2002. Co-crystallization of the human nuclear cap-binding complex with a m7GpppG cap analogue using protein engineering. Acta Crystallogr. D Biol. Crystallogr. 58:2194–2197 [DOI] [PubMed] [Google Scholar]
- 50. Miller MM, Halbig K, Cruz-Reyes J, Read LK. 2006. RBP16 stimulates trypanosome RNA editing in vitro at an early step in the editing reaction. RNA 12:1292–1303 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 51. Muller UF, Goringer HU. 2002. Mechanism of the gBP21-mediated RNA/RNA annealing reaction: matchmaking and charge reduction. Nucleic Acids Res. 30:447–455 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 52. Muller UF, Lambert L, Goringer HU. 2001. Annealing of RNA editing substrates facilitated by guide RNA-binding protein gBP21. EMBO J. 20:1394–1404 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 53. Panigrahi AK, et al. 2008. Mitochondrial complexes in Trypanosoma brucei: a novel complex and a unique oxidoreductase complex. Mol. Cell. Proteomics 7:534–545 [DOI] [PubMed] [Google Scholar]
- 54. Panigrahi AK, Allen TE, Stuart K, Haynes PA, Gygi SP. 2003. Mass spectrometric analysis of the editosome and other multiprotein complexes in Trypanosoma brucei. J. Am. Soc. Mass Spectrom. 14:728–735 [DOI] [PubMed] [Google Scholar]
- 55. Panigrahi AK, et al. 2006. Compositionally and functionally distinct editosomes in Trypanosoma brucei. RNA 12:1038–1049 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 56. Panigrahi AK, Schnaufer A, Stuart KD. 2007. Isolation and compositional analysis of trypanosomatid editosomes. Methods Enzymol. 424:3–24 [DOI] [PubMed] [Google Scholar]
- 57. Pelletier M, Read LK. 2003. RBP16 is a multifunctional gene regulatory protein involved in editing and stabilization of specific mitochondrial mRNAs in Trypanosoma brucei. RNA 9:457–468 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 58. Price SR, Evans PR, Nagai K. 1998. Crystal structure of the spliceosomal U2B′-U2A′ protein complex bound to a fragment of U2 small nuclear RNA. Nature 394:645–650 [DOI] [PubMed] [Google Scholar]
- 59. Read LK, Goringer HU, Stuart K. 1994. Assembly of mitochondrial ribonucleoprotein complexes involves specific guide RNA (gRNA)-binding proteins and gRNA domains but does not require preedited mRNA. Mol. Cell. Biol. 14:2629–2639 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 60. Schumacher MA, Karamooz E, Zikova A, Trantirek L, Lukes J. 2006. Crystal structures of T. brucei MRP1/MRP2 guide-RNA binding complex reveal RNA matchmaking mechanism. Cell 126:701–711 [DOI] [PubMed] [Google Scholar]
- 61. Selenko P, et al. 2003. Structural basis for the molecular recognition between human splicing factors U2AF65 and SF1/mBBP. Mol. Cell 11:965–976 [DOI] [PubMed] [Google Scholar]
- 62. Simpson L, Aphasizhev R, Gao G, Kang X. 2004. Mitochondrial proteins and complexes in Leishmania and Trypanosoma involved in U-insertion/deletion RNA editing. RNA 10:159–170 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 63. Stuart KD, Schnaufer A, Ernst NL, Panigrahi AK. 2005. Complex management: RNA editing in trypanosomes. Trends Biochem. Sci. 30:97–105 [DOI] [PubMed] [Google Scholar]
- 64. Valdez BC, Henning D, Perumal K, Busch H. 1997. RNA-unwinding and RNA-folding activities of RNA helicase II/Gu—two activities in separate domains of the same protein. Eur. J. Biochem. 250:800–807 [DOI] [PubMed] [Google Scholar]
- 65. Vondruskova E, et al. 2005. RNA interference analyses suggest a transcript-specific regulatory role for mitochondrial RNA-binding proteins MRP1 and MRP2 in RNA editing and other RNA processing in Trypanosoma brucei. J. Biol. Chem. 280:2429–2438 [DOI] [PubMed] [Google Scholar]
- 66. Weng J, et al. 2008. Guide RNA-binding complex from mitochondria of trypanosomatids. Mol. Cell 32:198–209 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 67. Wong I, Lohman TM. 1993. A double-filter method for nitrocellulose-filter binding: application to protein-nucleic acid interactions. Proc. Natl. Acad. Sci. U. S. A. 90:5428–5432 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 68. Zhou J, Hidaka K, Futcher B. 2000. The Est1 subunit of yeast telomerase binds the Tlc1 telomerase RNA. Mol. Cell. Biol. 20:1947–1955 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 69. Zimmer SL, McEvoy SM, Li J, Qu J, Read LK. 2011. A novel member of the RNase D exoribonuclease family functions in mitochondrial guide RNA metabolism in Trypanosoma brucei. J. Biol. Chem. 286:10329–10340 [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.