

Abstract
Background
The Abdominal Inflammatory Myofibroblastic Tumor (AIMT) is a rare tumor with unknown etiology which usually occurs in children and adolescents. It is composed of myofibroblastic spindle cells intermixed with inflammatory cells. We present four cases of AIMT.
Cases Presentation
We herein present four cases of AIMT in different ages (range: 3.5 to 13 years) and in different organs (stomach, periduodenal, mesenteric, and colon). There were two females and two males. The main symptoms were abdominal pain/mass/obstruction, vomiting, and weight loss. In all four patients, diagnosis was made by laparatomy and pathologic examination of excised mass lesion. Three patients underwent complete excision and no residual disease was present, one patient received chemotherapy due to tumor recurrences. The patients were followed up in average for four years.
Conclusion
As the imaging and laboratory tests are non-specific, the diagnosis of AIMT is rarely made before surgery. AIMT should, therefore, be considered when a mass arises in an unusual location in the pediatric age group. Complete surgical resection should be performed whenever possible and the child should be kept on long-term follow-up.
Keywords: Inflammatory myofibroblastic tumor, Malignancy, Abdomen, Stomach, Duodenum, Mesenteric, Colon
Introduction
Inflammatory myofibroblastic tumor (IMT) is a rare neoplasm usually seen in children and adolescents [1], mostly occurring between 2–16 years[2]. Females are affected slightly more commonly than males[3]. It is also known as cellular inflammatory pseudo tumor, plasma cell granuloma, mixed hamartoma, and inflammatory fibrosarcoma and is a spindle cell proliferation with a characteristic fibro- inflammatory appearance; IMT has a wide variation in histological appearance, with three major subtypes: fibromyxoid and vascular pattern, proliferating pattern, and sclerosing pattern[3]. Lung is the most common site of involvement, and the abdomen is the most extra-pulmonary region. The most prevalent symptoms are fever, weight loss, and systemic symptoms[4, 5]. the usual laboratory findings are hypochromic microcytic anemia, thrombo-cytosis, leukocytosis, hypergammaglobulinemia, and a high sedimentation rate,[1]. These symptoms and laboratory findings often regress following excision[4, 5]. Microscopically, it is composed of spindle cell proliferation with heavy infiltration of plasma cells which may mimic plasmacytoma [2]. Debate exists as to neoplastic or inflammatory nature of inflammatory myofibroblastic Tumor [4]. Hereby we report on four cases of abdominal inflammatory myofibroblastic tumor in children and review the literature to delineate the natural history of this entity in children.
Case Presentation
Case 1: A 13 year old boy admitted because of weight loss and abdominal pain. An epigastric mass was detected in examination as well as splenomegaly and generalized abdominal tenderness. Hypochromic microcytic anemia was reported in complete blood count. Barium studies showed remarkable stricture and several filling defects as well as mild mucosal thickness in proximal part of duodenum (Fig. 1A).
Fig. 1A.
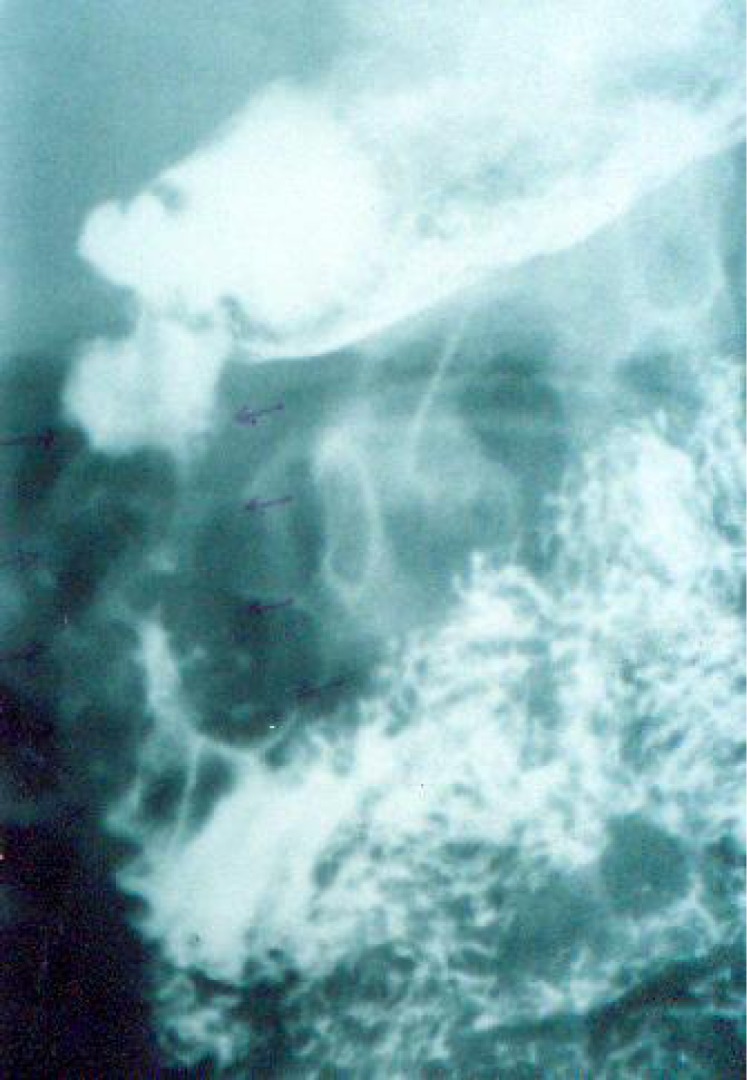
Barium study showing stricture and mucosal thickening of duodenum
Gastroscopy showed post bulbar stricture followed by biopsy of duodenum in which nonspecific chronic inflammation was reported.
Following surgical resection of the tumor, duodeno-duodenostomy was carried out. Clinical manifestation resolved and inflammatory myofibroblastic tumor was reported after pathologic examination (Fig. 1B).
Fig. 1B.

The infiltrate is mainly composed of lymphoplasma cells, eosinophils and the spindle cell proliferation
Case 2: An 8-year-old girl presented with the acute onset of abdominal pain which had developed since two days ago. Physical examination revealed mild abdominal tenderness in the hypogastrium, but no palpable mass. Laboratory findings revealed normocytic, normochromic anemia (Hg: 9gr/dl and Hct 28%) and elevated sedimentation rate (54mm in the first hour), tumor markers were unremarkable (Ca 125, Cea, Ca19-9 were within normal limits). Abdominal CT and MRI demonstrated a mass of approximately 5 cm in diameter in the gastro-colic ligament or gastric wall (Fig 2A). Endoscopic findings were unremarkable. The patient underwent surgical exploration and on laparatomy, there was hemopritoneum (approximately one liter blood) and an exophytic gastric mass measuring 6 cm in greatest diameter in the anterior wall of body along the greater curvature. Gastric wedge resection, including the tumor and greater omentum was performed. The final pathologic diagnosis was consistent with IMT, which originated from the gastric wall (Fig 2B). The patient had an uneventful postoperative course and has been followed up for any recurrences including performing of positron emission tomography.
Fig. 2A.

Abdominal CT showing large solid mass at gastrocolic ligament of the gastric wall, which showed heterogeneous density on a non-enhanced image
Fig. 2B.

Loosely arranged spindle cells in a prominent myxoid stroma admixed with inflammatory cell infiltrate.
Abdominal CT revealed a large solid mass at the gastrocolic ligament of the gastric wall, which showed heterogeneous density on a non-enhanced image, and microscopic examination showed loosely arranged spindle cells in a prominent myxoid stroma admixed with inflammatory cell infiltrate.
Case 3: A 3.5 year old girl, referred with anorexia and abdominal distention since one month prior to admission.
Physical examination revealed a tense, mobile abdominal mass with round border in left upper quadrant measuring 5×5 cm in diameter.
Laboratory findings revealed normocytic, normochromic anemia (Hb 8g/l and Hct 26%), high sedimentation rate (60 mm in the first hour) and normal tumor markers. Chest Xray was normal and MRI revealed a round mass, approximately 5×5cm in diameter of the mesenteric site (Fig 3A). She underwent surgical exploration and mesenteric mass was totally excised. Pathologic examination revealed IMT (Fig 3B).
Fig. 3A.

MRI showing a round mass, approximately 5×5cm in diameter of the mesenteric site
Fig. 3B.

Mesenteric mass presented during laparatomy
The child is well with no evidence of recurrence one year after surgery and is under follow-up.
Case 4: A 12 year old boy presented with abdominal pain and vomiting.
Physical examination revealed an abdominal mass in the left upper quadrant. Abdominal CT demonstrated a mass measuring 6×7cm in the colon next to splenic flexure. Colonoscopy showed presence of mass effect. Laparatomy was carried out and mass was totally resected.
Pathologic examination revealed IMT. The was under observation as after a year and half he developed a midline abdominal wall mass, which was resected (Fig 4A, B) and pathology diagnosis was recurrent IMT. As he had recurrences after complete excision, patient received chemotherapy. After a 5 year follow-up, he was in complete remission, and is still under follow-up.
Fig. 4A.

Recurred mass in the abdominal wall
Fig. 4B.

Abdominal wall after mass resection
The diagnosis of IMT on all cases was made only based on histologic findings and IHC studies were not performed in either case.
Discussion
Inflammatory myofibroblastic tumor mostly occurs between 2–16 years; however, it could be seen in adults[1, 2], the age of our four patients was 3.5 to 13 years. Females are affected slightly more commonly than males[3], while in our study the ratio was equal. It usually involves the lung and the most prevalent extrapulmonary site is abdominal cavity[4]. Other sites include pelvis, head and neck, trunk, extremities and skin[6]. When tumor is located in the abdomen, mesentery is usually involved. Gastric, colonic, appendiceal and esophageal involvement are also reported[6, 9]. All our four patients had intra-abdominal IMT. The most prevalent symptoms are fever, weight loss, and constitutional symptoms which regress after excision[4, 5], but the most common symptoms in our study were, weight loss and abdominal pain. Abdominal pain is a common complaint in those with abdominal mass, as it was seen in our cases. The incidence of other signs and symptoms depends on site of involvement such as dysphagia in esophageal involvement or jaundice in liver involvement[6, 7, 8]. The most common data includes microcytic hypochromic anemia, thrombocytosis, polyclonal hypergamma-globulinemia, and elevated sedimentation rate[2, 6, 7, 9], as we had these findings in our patients, but hypochromic anemia and thrombocytosis. There is no specific radiological finding; however, it may show local extension that indicates malignancy.
Primary intraluminal intestinal lesion is rare, although mesenteric lesion could penetrate intestinal wall and present as polypoid intraluminal mass and show macroscopic features of botyroid sarcoma[10].
In inflammatory myofibroblastic tumor the predominant cell is myofibroblast as is confirmed by immunohistochemical and ultrastructural studies[11, 12]. The other lesion that should be differentiated from IMT in spite of immunohistochemical and ultrastructural similarities is inflammatory fibroid polyp[4]. However, some authors believe that intestinal inflammatory fibroid polyp which is seen mostly in stomach is a variant of IMT, similar to our case with gastric tumor, and could be seen in small and large bowel. There are three histologic variants for IMT according to Coffin et al, including spindle cell, mixed, and desmoids variant [6].
There are many conflicting opinions regarding the inflammatory or neoplastic nature of this lesion [4]. Su et al showed some monoclonal chromosomal anomaly in three cases that support the neoplastic nature of this lesion[13, 14].
Mesenteric inflammatory myofibroblastic tumor can be solitary or multicentric. Our patient with mesenteric tumor had solitary type mass. The solitary lesions can be large, circumscribed and lobular with extensive adhesion to adjacent structures that need radical excision [2]. Surgical excision is the only treatment for IMT [4, 15]; we performed surgical excision of the mass in all of our patients. Although several cases of recurrence have been reported after primary excision, most of them cured after resection of recurred mass. This means that the tumor is often curable with surgical excision, close follow up and re-excision in even multiple recurrent cases [6–9, 16], but in one of our cases it was not curable, and we had to perform chemotherapy after consultation with pediatric oncologist. Coffin et al reported results of follow up in 53 patients, 44 of which were alive with no evidence of disease, four were alive with IMT, and five were dead [6]. We had no mortality in our series.
Although recurrences are seen after complete surgical resection, ITM is considered a benign lesion [6, 9, 11, 12]. However Mies et al have reported cases with distant metastasis and death because of tumoral involvement, they termed these tumors as inflammatory fibrosarcoma. We had no metastasis in our study. There is also a report of IMT with bone marrow metastasis [17]. Coffin et al believe that IMT and inflammatory fibrosarcoma are related histogenetically considering several similar clinical presentations and pathologic features, and that they are the extremes of a spectrum according to the degree of differentiation [18–23]. More studies are needed to confirm this hypothesis.
The presented cases in this report have rather the same clinical and paraclinical findings as in other reports except for fever and thrombocytosis. Malignant lymphoma was laid in differential diagnosis according to the site of involvement and the mucosal thickening seen in imaging studies, but it was ruled out in microscopic examination. However, it is important to know that IMT could be associated with other malignancies such as mature B cell lymphoma [9].
Conclusion
As the imaging and laboratory tests are non-specific, the diagnosis of AIMT is rarely made before surgery. AIMT should, therefore, be considered when a mass arises in an unusual location in the pediatric age group. Complete surgical resection should be performed whenever possible and the child should be kept on long term follow up
Acknowledgment
The author would like to thank Mrs. M Saeedi for her kind help to prepare this manuscript
References
- 1.Mills SE, Catter D, Greenson JK, et al. Sternberg's Diagnostic Surgical Pathology. 4th ed. Lippincott: Williams & Wilkins, Philadelphia; 2004. [Google Scholar]
- 2.Wick MR, Humpherg PA, Rich JH. Pathology of pseudoneoplastic lesions. Lippincott: Williams & Wilkins, Philadelphia; 1997. pp. 134–6. [Google Scholar]
- 3.Yimyaem P, Saranrittichai S, Sinawat P, et al. Inflammatory myofibroblastic tumor of the small intestine: A case report of a 2 month-old infant. Med Assoc Thai. 2009;1:114–9. [PubMed] [Google Scholar]
- 4.Dial DH. Plasma cell granuloma-histiocytoma complex. In: Daile DH, Hamma SP, editors. Pulmonary Pathology. New York: Springer Verlag; 1988. pp. 889–93. [Google Scholar]
- 5.Tanf TT, Segura AD, Oechier HW, et al. Inflammatory myofibrohistiocytic proliferation simulating sarcoma in children. Cancer. 1990;65(7):1626–34. doi: 10.1002/1097-0142(19900401)65:7<1626::aid-cncr2820650729>3.0.co;2-v. [DOI] [PubMed] [Google Scholar]
- 6.Coffin CM, Watterson J, Priest JR, et al. Extrapulmonary inflammatory myofibroblastic tumor (inflammatory pseudotumor). A clinical, pathologic and immunohistochemical study of 84 cases. Am J Surge Pathol. 1995;19(8):859–72. doi: 10.1097/00000478-199508000-00001. [DOI] [PubMed] [Google Scholar]
- 7.Gupta CR, Mohta A, Khurana N, et al. Inflammatory pseudotumor of the omentum: An uncommon pediatric tumor. I J Patho Microbiol. 2009;52(2):219–21. doi: 10.4103/0377-4929.48924. [DOI] [PubMed] [Google Scholar]
- 8.Sanders BM, West KW, Gingalewski C, et al. Inflammatory pseudotumor of the alimentary tract: clinical and surgical experience. J Pediatr Surg. 2001;36(1):169–73. doi: 10.1053/jpsu.2001.20045. [DOI] [PubMed] [Google Scholar]
- 9.Souid AK, Ziemba MC, Dubansky AS, et al. Inflammatory myofibroblastic tumor in children. Cancer. 1993;72(6):2042–8. doi: 10.1002/1097-0142(19930915)72:6<2042::aid-cncr2820720641>3.0.co;2-i. [DOI] [PubMed] [Google Scholar]
- 10.Demirkan NC, Akalin T, Yilmaz F, et al. Inflammatory myofibroblastic tumor of small bowel wall in childhood: Report of a case and a review of the literature. Pathol Int. 2001;51(1):47–9. doi: 10.1046/j.1440-1827.2001.01158.x. [DOI] [PubMed] [Google Scholar]
- 11.Meis JM, Enzinger FM. Inflammatory fibrosarcoma of mesentery and retroperitoneum: A tumor closely simulating inflammatory pseudotumor. Am J Surg Pathol. 1991;15(12):1146–56. doi: 10.1097/00000478-199112000-00005. [DOI] [PubMed] [Google Scholar]
- 12.Pettinato G, Manivel JC, de Rosa N, Dehen LP. Inflammatory myofibroblastic tumor (plasma cell granuloma). Clinicopathological study of 20 cases with immunohistochemical and ultra structural observation. Am J Clin Pathol. 1990;94(5):588–96. doi: 10.1093/ajcp/94.5.538. [DOI] [PubMed] [Google Scholar]
- 13.Su LD, Atayde-Perez A, Sheldon S, et al. Inflammatory myofibroblastic tumor: cytogenetic evidence supporting clonal origin. Mod Pathol. 1998;11(4):364–8. [PubMed] [Google Scholar]
- 14.Bezrodnik L, Giovanni D, Matteo E, et al. Thymus inflammatory myofibroblastic tumor in child with an interleukin-12-receptor beta 1 deficiency (IL12rB1) J Pediatr Infec Dis. 2010;5(1):107–10. [Google Scholar]
- 15.Saleem MI, Mahfud AB, Barret AM, et al. Lower abdominal inflammatory myofibroblastic tumor - an unusual presentation - a case report and brief literature review. Eur J Pediatr. 2007;166(7):679–83. doi: 10.1007/s00431-006-0305-y. [DOI] [PubMed] [Google Scholar]
- 16.Sawant S, Kasturi L, Amin A. Inflammatory myofibroblastic tumor. Indian J Peditr. 2002;69(11):1001–2. doi: 10.1007/BF02726027. [DOI] [PubMed] [Google Scholar]
- 17.Hagenstad CT, Kilpatrick SE, Pettenati MJ, et al. Inflammatory myofibroblastic tumor with bone marrow involvement. A case report and review of the literature. Arch Pathol Lab Med. 2003;127(7):865–7. doi: 10.5858/2003-127-865-IMTWBM. [DOI] [PubMed] [Google Scholar]
- 18.Coffin CM, Dehner LP, Meis-Kindblom JM. Inflammatory myofibroblastic tumor, inflammatory fibrosarcoma, and related lesions: an historical review with differential diagnostic considerations. Semin Diagn Pathol. 1998;15(2):102–10. [PubMed] [Google Scholar]
- 19.Coffin CM, Watterson J, Priest JR, Dehner LP. Inflammatory myofibroblastic tumor (inflammatory pseudotumor). A clinicopatho-logic and immunohistochemical study of 84 cases. Mod Pathol. 1994;7:5A. doi: 10.1097/00000478-199508000-00001. [DOI] [PubMed] [Google Scholar]
- 20.Gonzalez-Crussi F, de Mello DE, Sotelo-Avila C. Omental-mesenteric myxoid hamartomas. Infantile lesions simulating malignant tumors. Am J Surg Pathol. 1983;7(3):567–78. doi: 10.1097/00000478-198309000-00007. [DOI] [PubMed] [Google Scholar]
- 21.Bonnet JP, Basset T, Dijoux D. Abdominal inflammatory myofibroblastic tumors in children: report of an appendiceal case and review of the literature. J Pediatr Surg. 1996;31(9):1311–4. doi: 10.1016/s0022-3468(96)90262-6. [DOI] [PubMed] [Google Scholar]
- 22.Kojimahara K, Mukai M, Yamazaki K, et al. Inflammatory pseudotumor of the stomach: report of a highly infiltrative case with electron microscopic and immunohistochemical studies. Acta Pathol Jpn. 1993;43(1–2):65–70. doi: 10.1111/j.1440-1827.1993.tb02916.x. [DOI] [PubMed] [Google Scholar]
- 23.Walsh SV, Evangelista F, Khettry U. Inflammatory myofibroblastic tumor of the pancreaticobiliary region: morphologic and immunocytochemical study of three cases. Am J Surg Pathol. 1998;22(4):412–8. doi: 10.1097/00000478-199804000-00004. [DOI] [PubMed] [Google Scholar]