

Abstract
Gamma oscillations appear to be dependent on inhibitory neurotransmission from parvalbumin (PV)-containing gamma-amino butyric acid neurons. Thus, the abnormalities in PV neurons found in schizophrenia may underlie the deficits of gamma-band synchrony in the illness. Because gamma-band synchrony is thought to be crucial for cognition, cognitive deficits in schizophrenia may reflect PV neuron dysfunction in cortical neural circuits. Interestingly, it has been suggested that PV alterations in schizophrenia are the consequence of a hypofunction of signaling through N-methyl-D-aspartate (NMDA) receptors (NMDARs). Here, we review recent findings that address the question of how NMDAR hypofunction might produce deficits of PV neuron–mediated inhibition in schizophrenia. We conclude that while dysregulation of NMDARs may play an important role in the pathophysiology of schizophrenia, additional research is required to determine the particular cell type(s) that mediate dysfunctional NMDAR signaling in the illness.
Keywords: glutamate, GABA, inhibition
Introduction
Cognitive deficits, the major determinant of long-term functional outcome for schizophrenia patients,1 reflect dysfunction of neural circuits in different cortical regions.2 Because the neural substrate for cognitive function is thought to involve synchronization of neural activity at gamma-band frequency, abnormalities in cortical gamma oscillations may contribute to the cognitive deficits in schizophrenia.1
Gamma oscillations in cortical circuits appear to be dependent on inhibitory neurotransmission from the parvalbumin (PV)-containing subclass of gamma-amino butyric acid (GABA) neurons.3,4 Thus, the abnormalities in molecular markers of PV neurons in schizophrenia5 suggest that alterations of PV neuron–mediated inhibition may play a critical role in the deficits of gamma-band synchrony in the illness.
The dysfunction of PV neurons in schizophrenia has been suggested to be a consequence of a hypofunction of signaling through N-methyl-d-aspartate (NMDA) receptors (NMDARs) (for a recent review, see Nakazawa et al6). Although schizophrenia involves alterations in other interneuron subtypes and in oscillations of other frequencies,7 this review is focused on how NMDAR hypofunction may produce deficits of PV neuron–mediated inhibition that could lead to the impairments of gamma oscillations that are correlated with cognitive dysfunction in schizophrenia.1
Specifically, we review: (1) studies in human subjects supporting the idea of NMDAR hypofunction in schizophrenia; (2) recent evidence supporting the role of PV neurons in gamma rhythm production; (3) circuit models of inhibition-based gamma oscillations; (4) the distinctive properties of alpha-amino-3-hydroxy-5-methyl-4-isoxazole propionic acid (AMPA) receptor and NMDAR-mediated synaptic responses and their contributions to excitation of PV neurons; and (5) studies testing the effects of NMDAR antagonism on gamma oscillations. We conclude with a discussion of alternative scenarios regarding the role of NMDAR hypofunction and gamma oscillations in schizophrenia.
NMDAR Hypofunction Hypothesis of Schizophrenia
The idea that the pathophysiology of schizophrenia might be attributable to NMDAR hypofunction was originally based on clinical observations that NMDAR antagonists, such as phencyclidine and ketamine, transiently reproduced key clinical features of schizophrenia.8 Attempts to demonstrate that this hypofunction was due to lower levels of NMDAR in the brains of deceased individuals with schizophrenia have produced mixed results, with variable alterations in transcript and protein expression depending upon the NMDAR subunit and cortical area examined,9 findings that contrast with the highly consistent evidence for alterations of cortical GABA synthesis in schizophrenia.5
In contrast, convergent lines of evidence suggest that NMDAR hypofunction in schizophrenia might reflect dysregulation of the receptor rather than a deficit in the number of NMDARs.10 A variety of postmortem findings, albeit not yet widely replicated, are consistent with this idea. For example, schizophrenia has been associated with (1) alterations in the levels of NMDAR-associated postsynaptic proteins11; (2) less NMDAR-mediated signaling following neuregulin 1 activation of ErbB4 receptors12; (3) reduced levels of glutathione which modulates the redox-sensitive site of NMDARs13; and (4) downregulation of kynurenine 3-monooxygenase which could lead to elevated levels of kynurenic acid, an NMDAR antagonist.14
The idea of an NMDAR dysregulation in schizophrenia is also indirectly supported by the findings that the protein products of a number of putative risk genes for schizophrenia can affect NMDAR function.15 For example, allelic variants in both the neuregulin 1 and ErbB4 genes have been associated with an increased risk for schizophrenia,15 their gene products have been reported to be increased in the illness,16 and a gain of function in neuregulin 1-ErbB4 signaling has been shown to suppress NMDAR upregulation.17 In addition, the gene encoding GluN2B subunits has been implicated as a risk gene for schizophrenia in a recent meta-analysis.18 Finally, both preclinical studies and some clinical trials suggest that NMDAR-enhancing agents might reduce the severity of certain clinical symptoms of schizophrenia.19
Role of PV-Positive Basket Neurons in Production of Gamma Oscillations
As reviewed elsewhere,7,20,21 GABA-mediated inhibition is a very efficient way of synchronizing large numbers of cortical pyramidal neurons, and particular subclasses of GABA neurons are preferentially involved in generating inhibition-based rhythms. For example, GABA neurons targeting the perisomatic pyramidal cell membrane (proximal dendrites, soma and axon initial segment) may be strongly inhibitory because they synapse near the proximal axon, where action potentials are usually triggered. Three subtypes of perisomatic-targeting GABA neurons are prominent in cortical circuits (figure 1): PV-positive and cholecystokinin-positive basket cells (PVBCs and CCKBCs) and PV-positive chandelier cells (PVChCs). Among these, interestingly, PVChCs may not participate in gamma rhythm mechanisms because their GABAergic inputs onto the axon initial segment appear to be excitatory.22 CCKBCs are inhibitory but their synapses release GABA in a desynchronized manner23 that is imprecise for gamma-band synchronization. Consistent with their lack of contribution to gamma oscillations, PVChC and CCKBC synapses onto pyramidal neurons are insensitive to pharmacological manipulations that abolish gamma oscillations.24 Furthermore, PVChC and CCKBC activity is weakly coupled to the gamma oscillation rhythm.25 In contrast, synapses from PVBCs are inhibitory22 and are highly sensitive to pharmacological compounds that abolish gamma oscillations,24 as expected for inputs involved in gamma rhythm production. Optogenetic experiments also suggest a crucial role of PV neurons in gamma oscillations,3,4 although whether this approach preferentially targeted PVBCs could not be determined.
Fig. 1.
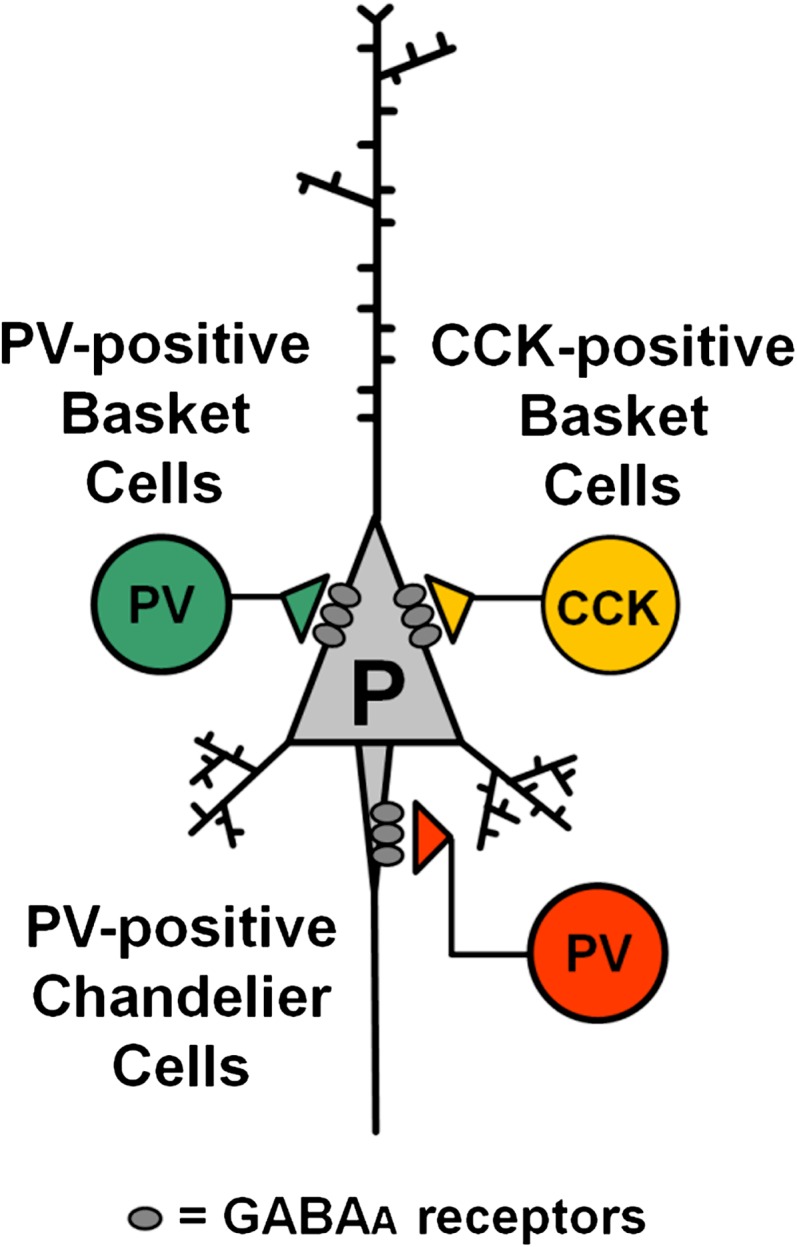
Major subtypes of perisomatic-targeting gamma-amino butyric acid (GABA) neurons in cortical circuits. Both parvalbumin (PV)-positive and cholecystokinin (CCK)-positive basket cells (green and yellow, respectively) target the soma and proximal dendrites of the pyramidal cell membrane. PV-positive chandelier cells (red) target the axon initial segment. These 3 GABA neuron subtypes signal via GABAA receptors, although different GABAA receptor subtypes may mediate the response to GABA at each type of synapse (not shown in the scheme). As described in the main text, current data suggest that gamma oscillation production depends mostly on pyramidal neuron inhibition by PV-positive basket cells.
Role of Glutamate-Mediated Excitation in Circuit Models of Gamma Oscillations
Two main circuit models have been proposed for the mechanisms producing rhythmic PVBC activity during gamma oscillations (figure 2).20 In the Interneuron Network Gamma (ING) model, oscillations depend on mutual inhibition between reciprocally connected GABA neurons; that is, interneurons are driven by continuous (tonic) excitatory currents and are synchronized rhythmically by their mutual inhibitory inputs.20 In ING, pyramidal cells are synchronized rhythmically by the interneuron network but are not directly involved in rhythm production (figure 2). In the second model, termed Pyramidal Interneuron Network Gamma (PING), oscillations depend on the interplay between pyramidal cells and GABA neurons via recurrent synaptic connections.20 In PING models, interneurons are driven by the phasic (synaptic) excitatory glutamate–mediated currents arriving from the pyramidal cells, which are synchronized rhythmically by feedback inhibition (figure 2). Monosynaptic excitation from pyramidal cells is the main source of interneuron activation in PING.
Fig. 2.

Two different circuit-based models for gamma oscillation production. In the Interneuron Network Gamma (ING) model (left panel), the oscillation depends on reciprocal inhibitory interactions between parvalbumin (PV)-positive basket cells. Some form of continuous (tonic) excitatory current is the main source of interneuron activation in ING. Important for the ING model are the electrical gap-junction connections (zig-zag wires) between PV-positive basket cells. In ING, pyramidal cells are synchronized rhythmically by the interneuron network but are not directly involved in rhythm production. In the Pyramidal Interneuron Network Gamma (PING) model (right panel), oscillations depend on the interplay between pyramidal cells and gamma-amino butyric acid neurons via recurrent synaptic connections. In PING models, interneurons are driven by the phasic (synaptic) excitatory glutamate–mediated currents arriving from the pyramidal cells, which are synchronized rhythmically by feedback inhibition. Monosynaptic excitation from pyramidal cells is the main source of interneuron activation in PING. Therefore, in PING, pyramidal cells are directly involved in the gamma rhythm mechanisms. As reviewed in the main text, some current data favor the PING over the ING model.
Computational simulations in networks of artificial neurons may generate gamma oscillations via either ING or PING mechanisms. Interestingly, whereas the mechanisms producing gamma synchrony in cortical networks are still poorly understood, findings from both in vivo and in vitro experimental studies favor the PING model.
Since in the PING model, phasic excitatory input from pyramidal neurons is the main source of interneuron excitation, during the gamma cycle, the pyramidal cell population should fire first, followed by interneurons firing with a delay consistent with monosynaptic excitation. Recordings of the timing of neuronal firing during the gamma oscillation cycle in fact demonstrated such a pattern of firing for pyramidal cells and interneurons in vivo26,27 and as well as in in vitro studies where the interneurons were identified as PVBCs.28
Additional support for the PING model was obtained from studies of genetically modified mice. For example, in mice with a deletion of GABAA receptors selectively from PV-positive cells, synaptic inhibition of PVBCs, which is required in the ING model, is completely abolished.29 Interestingly, in a manner inconsistent with ING, these mice display deficits in theta oscillations but have intact gamma oscillations.29 On the other hand, in mice with a deletion of AMPARs selectively in PV-positive cells,30 synaptic excitation of PVBCs is strongly reduced, and gamma oscillation power is severely depressed.30 Such an effect of AMPAR deletion is expected if PING mechanisms mediate gamma rhythm production because AMPARs are highly efficient at mediating the phasic excitation that drives PV neuron activity in PING. In contrast, AMPARs cannot mediate the tonic excitation of PV neurons in ING because they are rapidly and strongly desensitized when tonically exposed to glutamate. Therefore, several lines of evidence support the idea that phasic activation of synaptic glutamate receptors in PVBCs is crucial for gamma rhythm production, as in PING models.
Roles of NMDAR- and AMPA-Mediated Glutamate Synaptic Transmission in Gamma Oscillations
AMPA and NMDA are the main receptor subtypes at cortical and subcortical glutamate synapses, where they coexist in the postsynaptic density and are activated nearly simultaneously by synaptically released glutamate. The AMPAR/NMDAR ratio varies during development and with synaptic plasticity and may characterize subtypes of glutamate synapses. What distinctive functional properties are conferred by NMDARs vs AMPARs?
NMDAR and AMPAR currents differ in at least 3 fundamental properties (figure 3). First, at the resting potential or depolarized potentials below firing threshold, NMDAR channels are largely blocked by magnesium ions, whereas AMPAR channels are not. Their voltage-dependent magnesium block makes NMDARs unlikely to initiate neuronal excitation on their own, unless the synaptic membrane is depolarized via AMPARs or voltage-dependent channels. Second, NMDAR channels typically are highly permeable to calcium, unlike most AMPAR channels subtypes. The high calcium permeability of NMDARs is crucial for induction of synaptic plasticity because NMDAR-mediated calcium influx activates calcium-calmodulin kinase II-alpha, which mediates NMDAR-dependent changes in synaptic strength. Third, in response to synaptic glutamate, NMDARs generate significantly longer currents (∼100–400 ms) than AMPARs (∼2–10 ms). The long-lasting NMDAR–mediated currents generate a prolonged time window of increased excitability, leading to delayed action potential firing.31,32
Fig. 3.

Properties of N-methyl-d-aspartate receptor (NMDAR)- and AMPAR-mediated excitatory synaptic currents (EPSCs). Various properties distinguishing NMDAR-EPSCs and AMPAR-EPSCs are described in the main text. Especially, important for models of gamma oscillations is the much shorter duration of the AMPAR-EPSC relative to the NMDAR-EPSC. Whereas short-lasting AMPAR-EPSCs are sufficient to support robust gamma oscillations,31 long-lasting NMDAR-EPSCs produce asynchronous and delayed postsynaptic interneuron firing32 that is not locked to the incoming inputs from pyramidal cells31 and in a PING model of gamma oscillations reduces gamma oscillation power.31 Such an inverse relation between gamma power and NMDAR contribution31 suggests that the reduction of NMDAR contribution at synapses onto parvalbumin (PV)-positive basket cells that occurs during normal postnatal development33,34 helps optimize gamma oscillation production during cortical circuit development. Thus, during gamma oscillations in adult cortex, PV-positive basket cells appear to be mostly driven by fast AMPAR-mediated excitation. See main text for additional details.
Since the PING model is favored by recent findings, a key issue is the relative role of NMDARs and AMPARs in the synaptic activation of PVBCs in general and particularly during gamma oscillations. Studies of neuronal activity in the prefrontal cortex of awake rats showed that NMDARs may be crucial to drive inhibitory interneuron activity.35 In such studies, systemic NMDAR antagonist administration enhanced pyramidal cell firing and decreased the activity of putative GABA neurons,35 suggesting that NMDAR blockade leads to pyramidal cell disinhibition. However, in such recordings in awake animals, identification of neurons is challenging and relies on measuring extracellular action potential duration, which is typically shorter for interneurons than pyramidal cells, as determined for the NMDAR antagonist-sensitive interneurons.35 Because GABA neurons are extremely diverse and most of the subclasses have shorter action potentials than pyramidal cells, it is not clear if the NMDAR antagonist-sensitive interneurons recorded in awake animals include the PVBCs, whose activity is crucial for gamma oscillation production.
Using in vitro recordings, PVBCs can be easily distinguished from pyramidal cells and from other interneuron subtypes based on their electrophysiological and morphological properties. Interestingly, results from in vitro studies suggest that, compared with pyramidal cells, NMDAR contribution to synaptic activation of PV-positive neurons is relatively modest. For example, in PVBCs from various cortical regions, excitatory postsynaptic potentials (EPSPs) display minimal NMDAR contribution at subthreshold membrane potentials.31,36 Similarly, NMDAR contribution to excitatory postsynaptic currents (EPSCs) in PVBCs is weaker than in pyramidal cells.31,33,36 Driving PVBCs to fire action potentials from rest is significantly more dependent on AMPARs than on NMDARs, compared with pyramidal neurons.31 Consistent with these physiological findings, electron microscopy studies have shown low levels of NMDAR subunits in glutamate synapses onto PV-positive cells.37 Finally, the AMPAR/NMDAR ratio in synapses onto PV-positive neurons suggests a ∼3 times stronger AMPAR than NMDAR contribution,27,31,33,36,38 a significantly higher ratio than in pyramidal cells.
As mentioned, NMDAR-mediated currents have the following 3 distinctive properties: voltage-dependent magnesium block, high calcium permeability, and long-lasting EPSCs. Studying these properties in synaptic responses in PVBCs revealed a number of features consistent with relatively weak NMDAR contribution. First, NMDAR magnesium block is stronger in PVBCs than in excitatory neurons,36 weakening NMDAR contribution to EPSP-spike coupling in PVBCs.36 Second, glutamate synapses generate intracellular calcium transients that are less sensitive to NMDAR antagonists in PV-positive neurons than in pyramidal cells or other interneuron subtypes.39 Third, synaptically evoked calcium transients in PVBCs are short lasting and sensitive to blockers of the GluA2 subunit-lacking AMPARs,39 which are, atypically for AMPARs, calcium permeable. Whereas the data reviewed above suggest that the NMDAR contribution at glutamate synapses onto PV cells is relatively weak, it is possible that the calcium permeability conferred by NMDARs plays some role independent of directly mediating PV cell excitation. For instance, it is well established that calcium influx through the NMDAR channel participates in the mechanisms of long-term synaptic plasticity. Interestingly, consistent with weak calcium influx through NMDARs, long-term potentiation at glutamate synapses onto PVBCs is NMDAR independent,40,41 even though the same stimulation protocols produce NMDAR-dependent long-term potentiation in pyramidal cells.40 Moreover, long-term potentiation at glutamate synapses onto PVBCs is enhanced by postsynaptic hyperpolarization,41 which increases NMDAR channel magnesium block but enhances calcium influx via GluA2-lacking AMPARs.41 Therefore, additional research is necessary to determine the role of calcium influx through NMDAR channels at synapses onto PV neurons.
Compared with pyramidal neurons and other interneuron subtypes, EPSCs in PVBCs are short lasting.31,33 Interestingly, the purely AMPAR-mediated EPSCs (measured after NMDARs are blocked) have shorter duration in PVBCs compared with pyramidal neurons.31 Among AMPARs, the GluA2-lacking subtype determines the shortest AMPAR-EPSC duration, and, interestingly, GluA2-lacking AMPARs were shown to mediate short-lasting AMPAR-EPSCs in PVBCs.33,34 Among NMDARs, the GluN2A subunit–containing subtype determines the shortest NMDAR-EPSC duration, and, interestingly, PV-positive neurons express higher GluN2A/GluN2B subunit ratio than pyramidal cells.42 Thus, the AMPAR/NMDAR ratio and subunit composition of AMPARs and NMDARs in PVBCs suggest that their glutamatergic synaptic inputs are optimized for very fast activation. In fact, synaptic activation of PVBCs is extremely fast,43 apparently due to specific biophysical properties of their synapses and dendrites.43
Interestingly, the NMDAR antagonist MK801 applied acutely to brain slices from the posterior cingulate or retrospenial cortices, decreases the frequency of IPSCs in pyramidal cells.44 Furthermore, prolonged exposure to ketamine in vivo decreases inhibitory postsynaptic current (ISPC) frequency in pyramidal cells recorded in prefrontal cortex slices.45 NMDAR antagonists suppress disynaptic IPSPs evoked by stimulation of excitatory inputs onto hippocampal pyramidal cells,46,47 but similar disynaptic IPSPs evoked in neocortical pyramidal neurons are insensitive to NMDAR antagonists.31,46 Whereas the results reviewed above support the idea that NMDAR antagonists decrease inhibition onto pyramidal cells in some cortical regions, the interneuronal source of the IPSCs sensitive to NMDAR antagonism was not identified in such previous studies.44,45,47 Because several studies reviewed here suggest that NMDAR contribution to synaptic activation of PV neurons is weak, an interesting possibility is that the NMDAR antagonist-induced disinhibition is mediated by interneurons of some class different from the PV cells. For instance, in vivo exposure to ketamine increases IPSCs in pyramidal cells in the prefrontal cortex of 3-month-old but not of 1-month-old rats.45 Such findings are inconsistent with reports showing that the NMDAR contribution in synapses onto PV neurons is strong in the immature prefrontal cortex but is weak in mature prefrontal cortex.31,33,34
Effects of NMDAR Antagonism on Gamma Oscillations
In the favored PING model for circuit mechanisms of gamma oscillations (see ‘Role of Glutamate-Mediated Excitation in Circuit Models of Gamma Oscillations’ section), weakening the activation of PVBCs by pyramidal neurons would significantly reduce PVBC recruitment and thus gamma oscillation power. Consistent with this prediction, genetically engineered deletion of AMPARs selectively from PVBCs markedly impairs gamma oscillations.30 However, since the NMDAR contribution to PVBC activation is relatively modest, does NMDAR antagonism or hypofunction affect gamma oscillation production? To address this issue, we built a computational model network of artificial pyramidal cells and interneurons generating gamma oscillations via PING-like mechanisms.31 In this network, fast AMPAR–mediated excitation of PVBCs produced a robust gamma rhythm.31 In contrast, we observed an inverse relation between NMDAR contribution and gamma power such that increasing NMDAR currents decreased gamma-band power.31 In the model, consistent with experimental observations,32 the introduction of long-lasting NMDAR currents in synapses onto PVBCs produced firing of delayed action potentials that were not locked to the excitation arriving from the pyramidal cells,31 and such additional asynchronous firing of some PVBCs decreased gamma power.31 Our simulations therefore predict that, if the NMDAR contribution at inputs onto PVBCs is normally low, then further reducing NMDAR contribution would not decrease gamma oscillation power. In fact, NMDAR antagonists acting on PVBCs would be predicted to somewhat enhance gamma-band activity.
Consistent with these computational modeling findings, gamma oscillations in hippocampal or neocortical brain slices were reduced or abolished by AMPAR antagonists but were unaffected by NMDAR antagonists in most studies (see references in Rotaru et al31). In entorhinal cortex slices, the NMDAR antagonist ketamine did not alter gamma power in layers 4–5 but reduced gamma power in layer 2.48 In primary auditory cortex, ketamine increased gamma power.48 The results of most in vitro studies therefore suggest that the NMDAR contribution to PVBC activation during the gamma cycle is relatively modest. However, ketamine, in addition to blocking NMDARs, has several other pharmacological effects that limit its utility for testing the role of NMDARs in PVBCs.
Several studies of gamma oscillations recorded in vivo in animal models found that NMDAR antagonists increase basal gamma oscillation power.49,50 Ketamine also enhanced gamma oscillation power in healthy human subjects.51 Although in vivo studies clearly have advantages over in vitro studies, the systemic administration of NMDAR antagonists simultaneously affects NMDARs in multiple brain regions and various cell types within a region, limiting the interpretation of findings in terms of the role of NMDARs in PVBCs.
To model NMDAR hypofunction in vivo, an alternative approach to systemic administration of antagonists is cell-type specific, genetically engineered reduction of NMDAR expression. Recently, mice were generated with an ablation of the NMDAR subunit GluN1 restricted to PV-positive neurons (PV-GluN1−/− mice).27,38 In the hippocampus of awake freely moving PV-GluN1−/− mice, theta oscillation power and theta modulation of gamma oscillation amplitude were decreased, but gamma power was increased.27 Similarly, gamma oscillations were enhanced in somatosensory cortex of anesthetized or awake PV-GluN1−/− mice.38
Overall, the in vitro and in vivo pharmacological studies and experiments in PV-GluN1−/− mice do not support the idea that NMDARs mediate PVBC recruitment during gamma oscillations. In fact, the findings that NMDAR hypofunction increases gamma power suggest the opposite.
Conclusions
In concert, the findings reviewed above raise an important question regarding the mechanisms by which NMDAR hypofunction could contribute to the neural substrate for cortical circuit dysfunction in schizophrenia. Specifically, given that NMDARs appear to make a small contribution to activation of PVBCs, how could impaired NMDAR regulation contribute to the alterations in markers of PVBC function (eg, lower levels of PV and glutamic acid decarboxylase 67 mRNAs) observed in schizophrenia? The idea that the effects are not directly mediated by NMDARs on mature PVBCs is supported by findings that neither mice deficient in serine racemase, which produces the endogenous NMDAR coagonist D-serine, nor the chronic administration of ketamine or phencyclidine to adult mice or rats, resulted in altered phenotype of PV neurons.52 Interestingly, glutamate inputs onto immature PV neurons display a significant NMDAR contribution, which declines during development until reaching low levels in adulthood.33,34 Moreover, early postnatal ablation of NMDAR subunits from PV neurons produces behavioral abnormalities that are not observed if the ablation is produced postadolescence.53 Therefore, NMDAR antagonists may affect the phenotype of PV neurons if delivered early in development when these neurons have high levels of NMDAR. This view would suggest that the NMDAR hypofunction must arise early in development in individuals who do not manifest the diagnostic psychotic features of schizophrenia until years or decades later. This interpretation might provide the neural substrate for the findings that certain cognitive abnormalities reflective of cortical dysfunction are present during childhood in persons later diagnosed with schizophrenia.54
Alternatively, cortical NMDAR hypofunction in schizophrenia may be mediated by NMDAR on pyramidal neurons, other classes of GABA neurons or neurons that furnish projections to the cortex. This interpretation is supported by some experimental models. For example, genetic manipulations that lower the expression levels of the schizophrenia risk gene dysbindin, which exhibits lower mRNA and protein levels in schizophrenia, leads to lower NMDA currents in cortical pyramidal cells, and impaired cognition.55
Thus, although substantial evidence supports a role for dysregulation of NMDARs in the pathophysiology of schizophrenia, additional research is required to determine the particular cell type(s) that mediate dysfunctional NMDAR signaling in the illness.
Funding
National Institute of Health (MH084053, MH043784)
Acknowledgments
The content is solely the responsibility of the authors and does not necessarily represent the official views of the National Institute of Mental Health or the National Institutes of Health. D.A.L. currently receives investigator-initiated research support from the BMS Foundation, Bristol-Myers Squibb, Curridium Ltd, and Pfizer and in 2009–2011, served as a consultant in the areas of target identification and validation and new compound development to BioLine RX, Bristol-Myers Squibb, Merck, and SK Life Science. Guillermo Gonzalez-Burgos has no conflicts of interest.
References
- 1.Lesh TA, Niendam TA, Minzenberg MJ, Carter CS. Cognitive control deficits in schizophrenia: mechanisms and meaning. Neuropsychopharmacology. 2011;36:316–338. doi: 10.1038/npp.2010.156. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2.Lewis DA, Sweet RA. Schizophrenia from a neural circuitry perspective: advancing toward rational pharmacological therapies. J Clin Invest. 2009;119:706–716. doi: 10.1172/JCI37335. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.Sohal VS, Zhang F, Yizhar O, Deisseroth K. Parvalbumin neurons and gamma rhythms enhance cortical circuit performance. Nature. 2009;459:698–702. doi: 10.1038/nature07991. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Cardin JA, Carlen M, Meletis K, et al. Driving fast-spiking cells induces gamma rhythm and controls sensory responses. Nature. 2009;459:663–667. doi: 10.1038/nature08002. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Gonzalez-Burgos G, Hashimoto T, Lewis DA. Alterations of cortical GABA neurons and network oscillations in schizophrenia. Curr Psychiatry Rep. 2010;12:335–344. doi: 10.1007/s11920-010-0124-8. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Nakazawa K, Zsiros V, Jiang Z, et al. GABAergic interneuron origin of schizophrenia pathophysiology. Neuropharmacology. 2011;62:1574–1583. doi: 10.1016/j.neuropharm.2011.01.022. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Gonzalez-Burgos G, Lewis DA. GABA neurons and the mechanisms of network oscillations: implications for understanding cortical dysfunction in schizophrenia. Schizophr Bull. 2008;34:944–961. doi: 10.1093/schbul/sbn070. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Javitt DC, Zukin SR. Recent advances in the phencyclidine model of schizophrenia. Am J Psychiatry. 1991;148:1301–1308. doi: 10.1176/ajp.148.10.1301. [DOI] [PubMed] [Google Scholar]
- 9.Kristiansen LV, Huerta I, Beneyto M, Meador-Woodruff JH. NMDA receptors and schizophrenia. Curr Opin Pharmacol. 2007;7:48–55. doi: 10.1016/j.coph.2006.08.013. [DOI] [PubMed] [Google Scholar]
- 10.Kantrowitz JT, Javitt DC. N-methyl-d-aspartate (NMDA) receptor dysfunction or dysregulation: the final common pathway on the road to schizophrenia? Brain Res Bull. 2010;83:108–121. doi: 10.1016/j.brainresbull.2010.04.006. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Beneyto M, Meador-Woodruff JH. Lamina-specific abnormalities of NMDA receptor-associated postsynaptic protein transcripts in the prefrontal cortex in schizophrenia and bipolar disorder. Neuropsychopharmacology. 2008;33:2175–2186. doi: 10.1038/sj.npp.1301604. [DOI] [PubMed] [Google Scholar]
- 12.Hahn CG, Wang HY, Cho DS, et al. Altered neuregulin 1-erbB4 signaling contributes to NMDA receptor hypofunction in schizophrenia. Nat Med. 2006;12:824–828. doi: 10.1038/nm1418. [DOI] [PubMed] [Google Scholar]
- 13.Steullet P, Neijt HC, Cuenod M, Do KQ. Synaptic plasticity impairment and hypofunction of NMDA receptors induced by glutathione deficit: relevance to schizophrenia. Neuroscience. 2006;137:807–819. doi: 10.1016/j.neuroscience.2005.10.014. [DOI] [PubMed] [Google Scholar]
- 14.Wonodi I, Stine OC, Sathyasaikumar KV, et al. Downregulated kynurenine 3-monooxygenase gene expression and enzyme activity in schizophrenia and genetic association with schizophrenia endophenotypes. Arch Gen Psychiatry. 2011;68:665–674. doi: 10.1001/archgenpsychiatry.2011.71. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Harrison PJ, Weinberger DR. Schizophrenia genes, gene expression, and neuropathology: on the matter of their convergence. Mol Psychiatry. 2005;10:40–68. doi: 10.1038/sj.mp.4001558. [DOI] [PubMed] [Google Scholar]
- 16.Law AJ, Lipska BK, Weickert CS, et al. Neuregulin 1 transcripts are differentially expressed in schizophrenia and regulated by 5' SNPs associated with the disease. Proc Natl Acad Sci U S A. 2006;103:6747–6752. doi: 10.1073/pnas.0602002103. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Pitcher GM, Kalia LV, Ng D, et al. Schizophrenia susceptibility pathway neuregulin 1-ErbB4 suppresses Src upregulation of NMDA receptors. Nat Med. 2011;17:470–478. doi: 10.1038/nm.2315. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Allen NC, Bagade S, McQueen MB, et al. Systematic meta-analyses and field synopsis of genetic association studies in schizophrenia: the SzGene database. Nat Genet. 2008;40:827–834. doi: 10.1038/ng.171. [DOI] [PubMed] [Google Scholar]
- 19.Lin CH, Lane HY, Tsai GE. Glutamate signaling in the pathophysiology and therapy of schizophrenia. Pharmacol Biochem Behav. 2012;100:665–677. doi: 10.1016/j.pbb.2011.03.023. [DOI] [PubMed] [Google Scholar]
- 20.Whittington MA, Traub RD, Kopell N, Ermentrout B, Buhl EH. Inhibition-based rhythms: experimental and mathematical observations on network dynamics. Int J Psychophysiol. 2000;38:315–336. doi: 10.1016/s0167-8760(00)00173-2. [DOI] [PubMed] [Google Scholar]
- 21.Wang XJ. Neurophysiological and computational principles of cortical rhythms in cognition. Physiol Rev. 2010;90:1195–1268. doi: 10.1152/physrev.00035.2008. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Szabadics J, Varga C, Molnar G, Olah S, Barzo P, Tamas G. Excitatory effect of GABAergic axo-axonic cells in cortical microcircuits. Science. 2006;311:233–235. doi: 10.1126/science.1121325. [DOI] [PubMed] [Google Scholar]
- 23.Jonas P, Hefft S. GABA release at terminals of CCK-interneurons: synchrony, asynchrony and modulation by cannabinoid receptors (commentary on Ali & Todorova) Eur J Neurosci. 2010;31:1194–1195. doi: 10.1111/j.1460-9568.2010.07189.x. [DOI] [PubMed] [Google Scholar]
- 24.Gulyas AI, Szabo GG, Ulbert I, et al. Parvalbumin-containing fast-spiking basket cells generate the field potential oscillations induced by cholinergic receptor activation in the hippocampus. J Neurosci. 2010;30:15134–15145. doi: 10.1523/JNEUROSCI.4104-10.2010. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Klausberger T, Somogyi P. Neuronal diversity and temporal dynamics: the unity of hippocampal circuit operations. Science. 2008;321:53–57. doi: 10.1126/science.1149381. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Senior TJ, Huxter JR, Allen K, O'Neill J, Csicsvari J. Gamma oscillatory firing reveals distinct populations of pyramidal cells in the CA1 region of the hippocampus. J Neurosci. 2008;28:2274–2286. doi: 10.1523/JNEUROSCI.4669-07.2008. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Korotkova T, Fuchs EC, Ponomarenko A, von EJ, Monyer H. NMDA receptor ablation on parvalbumin-positive interneurons impairs hippocampal synchrony, spatial representations, and working memory. Neuron. 2010;68:557–569. doi: 10.1016/j.neuron.2010.09.017. [DOI] [PubMed] [Google Scholar]
- 28.Hajos N, Paulsen O. Network mechanisms of gamma oscillations in the CA3 region of the hippocampus. Neural Netw. 2009;22:1113–1119. doi: 10.1016/j.neunet.2009.07.024. [DOI] [PubMed] [Google Scholar]
- 29.Wulff P, Ponomarenko AA, Bartos M, et al. Hippocampal theta rhythm and its coupling with gamma oscillations require fast inhibition onto parvalbumin-positive interneurons. Proc Natl Acad Sci U S A. 2009;106:3561–3566. doi: 10.1073/pnas.0813176106. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Fuchs EC, Zivkovic AR, Cunningham MO, et al. Recruitment of parvalbumin-positive interneurons determines hippocampal function and associated behavio. Neuron. 2007;53:591–604. doi: 10.1016/j.neuron.2007.01.031. [DOI] [PubMed] [Google Scholar]
- 31.Rotaru DC, Yoshino H, Lewis DA, Ermentrout GB, Gonzalez-Burgos G. Glutamate receptor subtypes mediating synaptic activation of prefrontal cortex neurons: relevance for schizophrenia. J Neurosci. 2011;31:142–156. doi: 10.1523/JNEUROSCI.1970-10.2011. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Maccaferri G, Dingledine R. Control of feedforward dendritic inhibition by NMDA receptor-dependent spike timing in hippocampal interneurons. J Neurosci. 2002;22:5462–5472. doi: 10.1523/JNEUROSCI.22-13-05462.2002. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.Wang HX, Gao WJ. Cell type-specific development of NMDA receptors in the interneurons of rat prefrontal cortex. Neuropsychopharmacology. 2009;34:2028–2040. doi: 10.1038/npp.2009.20. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.Wang HX, Gao WJ. Development of calcium-permeable AMPA receptors and their correlation with NMDA receptors in fast-spiking interneurons of rat prefrontal cortex. J Physiol. 2010;588:2823–2838. doi: 10.1113/jphysiol.2010.187591. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35.Homayoun H, Moghaddam B. NMDA receptor hypofunction produces opposite effects on prefrontal cortex interneurons and pyramidal neurons. J Neurosci. 2007;27:11496–11500. doi: 10.1523/JNEUROSCI.2213-07.2007. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36.Hull C, Isaacson JS, Scanziani M. Postsynaptic mechanisms govern the differential excitation of cortical neurons by thalamic inputs. J Neurosci. 2009;29:9127–9136. doi: 10.1523/JNEUROSCI.5971-08.2009. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37.Nyiri G, Stephenson BM, Freund TF, Somogyi P. Large variability in synaptic N-methyl-D-aspartate receptor density on interneurons and a comparison with pyramidal-cell spines in the rat hippocampus. Neuroscience. 2003;119:347–363. doi: 10.1016/s0306-4522(03)00157-x. [DOI] [PubMed] [Google Scholar]
- 38.Carlen M, Meletis K, Siegle JH, et al. A critical role for NMDA receptors in parvalbumin interneurons for gamma rhythm induction and behavior. doi: 10.1038/mp.2011.31. [published online ahead of print April 5, 2011]. Mol Psychiatry. doi:10.1038/mp.2011.31. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39.Goldberg JH, Yuste R, Tamas G. Ca2+ imaging of mouse neocortical interneurone dendrites: contribution of Ca2+-permeable AMPA and NMDA receptors to subthreshold Ca2+dynamics. J Physiol. 2003;551(pt 1):67–78. doi: 10.1113/jphysiol.2003.042598. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40.Sarihi A, Jiang B, Komaki A, Sohya K, Yanagawa Y, Tsumoto T. Metabotropic glutamate receptor type 5-dependent long-term potentiation of excitatory synapses on fast-spiking GABAergic neurons in mouse visual cortex. J Neurosci. 2008;28:1224–1235. doi: 10.1523/JNEUROSCI.4928-07.2008. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41.Kullmann DM, Lamsa KP. LTP and LTD in cortical GABAergic interneurons: emerging rules and roles. Neuropharmacology. 2011;60:712–719. doi: 10.1016/j.neuropharm.2010.12.020. [DOI] [PubMed] [Google Scholar]
- 42.Kinney JW, Davis CN, Tabarean I, Conti B, Bartfai T, Behrens MM. A specific role for NR2A-containing NMDA receptors in the maintenance of parvalbumin and GAD67 immunoreactivity in cultured interneurons. J Neurosci. 2006;26:1604–1615. doi: 10.1523/JNEUROSCI.4722-05.2006. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43.Hu H, Martina M, Jonas P. Dendritic mechanisms underlying rapid synaptic activation of fast-spiking hippocampal interneurons. Science. 2010;327:52–58. doi: 10.1126/science.1177876. [DOI] [PubMed] [Google Scholar]
- 44.Li Q, Clark S, Lewis DV, Wilson WA. NMDA receptor antagonists disinhibit rat posterior cingulate and retrosplenial cortices: a potential mechanism of neurotoxicity. J Neurosci. 2002;22:3070–3080. doi: 10.1523/JNEUROSCI.22-08-03070.2002. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 45.Zhang Y, Behrens MM, Lisman JE. Prolonged exposure to NMDAR antagonist suppresses inhibitory synaptic transmission in prefrontal cortex. J Neurophysiol. 2008;100:959–965. doi: 10.1152/jn.00079.2008. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 46.Ling DS, Benardo LS. Recruitment of GABAA inhibition in rat neocortex is limited and not NMDA dependent. J Neurophysiol. 1995;74:2329–2335. doi: 10.1152/jn.1995.74.6.2329. [DOI] [PubMed] [Google Scholar]
- 47.Grunze HC, Rainnie DG, Hasselmo ME, et al. NMDA-dependent modulation of CA1 local circuit inhibition. J Neurosci. 1996;16:2034–2043. doi: 10.1523/JNEUROSCI.16-06-02034.1996. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 48.Roopun AK, Cunningham MO, Racca C, Alter K, Traub RD, Whittington MA. Region-specific changes in gamma and beta2 rhythms in NMDA receptor dysfunction models of schizophrenia. Schizophr Bull. 2008;34:962–973. doi: 10.1093/schbul/sbn059. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 49.Pinault D. N-methyl d-aspartate receptor antagonists ketamine and MK-801 induce wake-related aberrant gamma oscillations in the rat neocortex. Biol Psychiatry. 2008;63:730–735. doi: 10.1016/j.biopsych.2007.10.006. [DOI] [PubMed] [Google Scholar]
- 50.Hakami T, Jones NC, Tolmacheva EA, et al. NMDA receptor hypofunction leads to generalized and persistent aberrant gamma oscillations independent of hyperlocomotion and the state of consciousness. PLoS One. 2009;4:e6755. doi: 10.1371/journal.pone.0006755. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 51.Hong LE, Summerfelt A, Buchanan RW, et al. Gamma and delta neural oscillations and association with clinical symptoms under subanesthetic ketamine. Neuropsychopharmacology. 2010;35:632–640. doi: 10.1038/npp.2009.168. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 52.Benneyworth MA, Roseman AS, Basu AC, Coyle JT. Failure of NMDA receptor hypofunction to induce a pathological reduction in PV-positive GABAergic cell markers. Neurosci Lett. 2011;488:267–271. doi: 10.1016/j.neulet.2010.11.043. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 53.Belforte JE, Zsiros V, Sklar ER, et al. Postnatal NMDA receptor ablation in corticolimbic interneurons confers schizophrenia-like phenotypes. Nat Neurosci. 2010;13:76–83. doi: 10.1038/nn.2447. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 54.Reichenberg A, Caspi A, Harrington H, et al. Static and dynamic cognitive deficits in childhood preceding adult schizophrenia: a 30-year study. Am J Psychiatry. 2010;167:160–169. doi: 10.1176/appi.ajp.2009.09040574. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 55.Karlsgodt KH, Robleto K, Trantham-Davidson H, et al. Reduced dysbindin expression mediates N-methyl-D-aspartate receptor hypofunction and impaired working memory performance. Biol Psychiatry. 2011;69:28–34. doi: 10.1016/j.biopsych.2010.09.012. [DOI] [PMC free article] [PubMed] [Google Scholar]