

Abstract
The peroxisome proliferator-activated receptors (PPARs) are a set of three nuclear hormone receptors that together play a key role in regulating metabolism, particularly the switch between the fed and fasted state and the metabolic pathways involving fatty-acid oxidation and lipid metabolism. In addition, they have a number of important developmental and regulatory roles outside metabolism. The PPARs are also potent targets for treating type II diabetes, dyslipidemia and obesity, although a number of individual agonists have also been linked to unwanted side effects, and there is a complex relationship between the PPARs and the development of cancer. This review examines the part that metabolomics, including lipidomics, has played in elucidating the roles PPARs have in regulating systemic metabolism, as well as their role in aspects of drug-induced cancer and xenobiotic metabolism. These studies have defined the role PPARδ plays in regulating fatty-acid oxidation in adipose tissue and the interaction between aging and PPARα in the liver. The potential translational benefits of these approaches include widening the role of PPAR agonists and improved monitoring of drug efficacy.
Keywords: metabolic syndrome, obesity, peroxisome proliferator-activated receptors, type II diabetes, functional genomics, the metabolic syndrome
An introduction to the peroxisome proliferator-activated receptors as key nutritional sensors
The peroxisome proliferator-activated receptors (PPARs) are transcription factors that translate nutritional signals into specific gene-expression patterns that control cellular bioenergetics. The receptors act as nutritional sensors, regulating metabolism across organs to modify systemic metabolism. There are three members of the PPAR family: PPARα, γ and δ (also known as PPARβ), which have tissue-specific distributions correlating with their pro-oxidative or storage functions [1]. Each PPAR initially binds a ligand and then heterodimerizes with the retinoid × receptor (RXR), before the complex binds to DNA sequences referred to as peroxisome proliferator hormone-response elements (PPREs), which are generally found in the promoter region of PPAR-targeted genes [2]. This action of heterodimerization and binding to PPREs is further modulated by the presence of co-activator and co-repressor proteins. The ligands for the PPARs are a diverse range of metabolites, including certain free fatty acids, eicosanoids and xenobiotics. PPARα and PPARγ have been extensively studied, as they are important pharmacological targets for the treatment of obesity, atherosclerosis and insulin insensitivity. PPARδ has been less well studied but is increasingly of interest to researchers. Understanding how the PPAR receptors regulate normal systemic metabolism and energy homeostasis in the whole organism is particularly important given the global increase in obesity. From the UK's Department of Health Statistics, almost one in four adults and one in ten children in England are obese. This translates to estimated costs of £4.2 billion for the National Health Service (NHS) and £16 billion for the wider economy in England [3], with these costs set to double by 2050. In addition, obesity can ultimately lead to a range of diseases, including hypertension, cardiovascular disease and type II diabetes.
Numerous drugs have been developed that are agonists for PPARα and PPARγ, with the global market for this class of drugs being $5.8 billion in 2008 compared with the total market for anti-diabetes drugs of $24 billion for that year. However, agonists that target the PPARs have raised a number of safety concerns, with PPARα agonists being linked to hepatic cancer, and all three receptors thought to play a role in protecting or predisposing to a variety of other cancers (Table 1). This is an example of the complications of targeting transcription factors that have such a wide variety of roles in the body. More recently, thiazolidinedione drugs, compounds derived from a ring structure containing a sulfur and two ketone groups that target PPARγ, have been linked to cardiovascular disease, and specifically myocardial ischemia [4], prompting the United States (US) Food and Drug Administration (FDA) to issue a safety alert on one of the most widely prescribed classes of anti-diabetes drugs. However, since the publication of this alert, the American Heart Association and American College of Cardiology Foundation have published an advisory note stating that health professionals should weigh up the benefits from improved insulin sensitivity and glycemic control afforded by thiazolidinedione drugs such as rosiglitazone and pioglitazone against the inconclusive role in ischemic heart disease [5].
Table 1.
PPAR agonists and their treatment of type II diabetes and obesity
| Drug class | Target | Mode of action | Examples | Concerns |
|---|---|---|---|---|
| Fibrates | PPARα | Target cholesterol metabolism Increase fatty acid oxidation |
Clofibrate, gemfibrozil, ciprofibrate, bezafibrate, fenofibrate | Increased risk of cancer (especially liver) |
| Thiazolidinediones | PPARγ | Insulin resistance is decreased, adipocyte differentiation is increased, leptin decreased and adiponectin increased | Rosiglitazone (Avandia®) pioglitazone (Actos®) troglitazone (Rezulin®) |
Rosiglitazone - concern over cardiovascular events; pioglitazone - concerns over bladder cancer; troglitazone withdrawn due to increased drug-induced hepatitis |
| PPAR-delta agonists | PPARδ | Stimulate fatty acid oxidation in skeletal muscle and adipose tissue, decrease insulin resistance, stimulate glucose metabolism | None currently on the market | They have been linked with increased and decreased risks of cancer; drug-induced myopathy |
| Dual agonists and PPARpana agonists | Two or all three receptors | A combination of the effects of the pure agonists | Aleglitazar, muraglitazar and tesaglitazar | Increased risk of certain cancers; increased risk of myocardial infarction |
This table shows some of the different types of drugs that are used to treat type II diabetes and obesity by targeting at least one of the PPAR receptors. Given the wide range of drugs that fall into this class of compounds, the list is not comprehensive but used to illustrate the diverse range of drugs available. aA PPARpan drug targets all three PPAR receptors.
Metabolomics, also referred to by some as metabonomics, is the comprehensive analysis of the metabolic complement of a cell, tissue, biofluid or organism. The approach makes use of techniques of analytical chemistry to profile as wide a range of metabolites as possible, in a quantitative or semi-quantitative manner, to follow a variety of physiological and pathophysiological stimuli such as disease progression, gene function or a toxic insult. Metabolomics has been used as part of personalized medicine to identify populations that will respond to a particular drug intervention [6], identify predictive biomarkers of disease presence and future prognosis [7-10], conduct functional genomics [11-13] and identify genes involved in diseases such as diabetes [14,15].
In this review we assess the use of metabolomics to elucidate the part PPARs play in regulating systemic metabolism. Although physiology-based approaches have been highly successful at defining the broad actions of these receptors, much of the detail in terms of how regulation is exerted has still to be defined. Metabolomics is viewed in its broadest sense to also include lipidomic approaches, which focus on the analysis of intact lipids, as well as fluxomic approaches, which assess a range of pathway activities using stable isotope techniques. We begin by briefly reviewing the important role animal models have had in the understanding of PPAR activities, before describing the advantages of a metabolomics approach and the insights gained. The focus of much of the research into PPARs has been on their role in treating type II diabetes, obesity and dyslipidemia, but it has also been known since the original discovery of these receptors that agonists for all three receptors alter the relative risks for a variety of cancers. In addition we are now beginning to understand how the PPAR signaling systems interact with other molecular signaling pathways to modulate cellular metabolism, proliferation and inflammation, indicating their profound and diverse roles in regulation within the cell (Figure 1).
Figure 1.
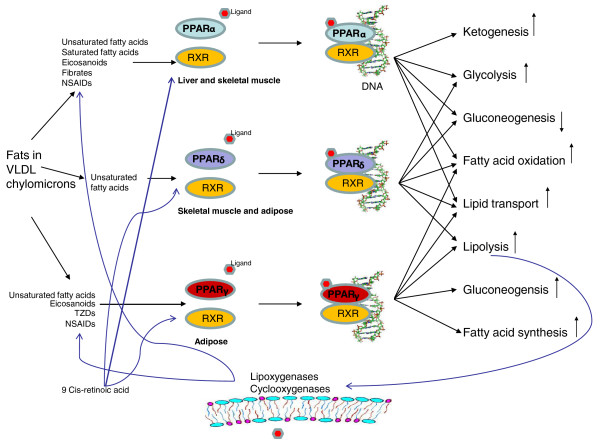
A schematic diagram illustrating the cross-talk of the three PPAR receptors and the metabolic pathways they interact with. For each PPAR, the initial step involves the receptor binding a ligand to activate it. Then, the retinoid × receptor (RXR), a nuclear receptor activated by 9-cis retinoic acid, heterodimerizes with a PPAR prior to the subsequent binding of the complex with DNA. Key: NSAID, non-steroidal anti-inflammatory drug; TZD, thiazolidinedione, a class of drugs that bind to PPARγ and have insulin-sensitizing properties; VLDL, very low density lipoprotein (used to transport lipids in the blood).
Understanding PPARs through animal models: what classic physiology tells us about the receptors
Of the three PPARs, the first discovered was PPARα. It was found in rodents following the observation that it was the target of drugs that caused peroxisomes (organelles that primarily break down lipids) to proliferate in the liver [16]. Remarkably, despite the important regulatory roles that PPARα plays in regulating metabolism in key organs, PPARα-knockout mice have been generated [17]. In addition to investigating the role PPARα plays in peroxisome proliferation, the mouse also became a useful research tool for following its role in regulating metabolism. Kersten and co-workers [18] either fed the null mice a high-fat diet or fasted them for 24 hours. Both challenges induced an accumulation of lipid in the liver, but fasting demonstrated that the PPARα-null mouse had reduced capacity for gluconeogenesis and ketogenesis, experienced hypothermia and had increased levels of circulating free fatty acids. Similarly, Leone and colleagues [19] used the knockout mouse to investigate how fat metabolism is regulated by PPARα during the fasting response. They found that fasting induced steatosis (retention of fatty acids) in the liver and heart, with a failure to upregulate the ketogenic and glucogenic pathways in the liver. These and numerous other studies indicate that PPARα regulates a number of catabolic pathways; by contrast, the activity of PPARγ appears to be mainly one of lipid storage and synthesis, as well as playing an important role in adipogenesis [6,7]. PPARγ is also the target of a number of potent insulin-sensitizing agents, including the PPARγ agonists the thiazolidinediones. The expression of PPARγ is also regulated by diet, with marked reductions in its expression detected in adipose tissue during fasting, but induction by high-fat feeding, demonstrating its role in fat storage [20].
There are three major isoforms of PPARγ, with PPARγ1 found in nearly all tissue apart from muscle, PPARγ2 found mostly in adipose tissue [21] and PPARγ3 found in macrophages, the large intestine and adipose tissue. Failure to express PPARγ causes embryonic lethality in mice, but heterozygous mice and humans with a Pro12Ala polymorphism in PPARγ2 appear protected from the development of type II diabetes, suggesting that PPARγ is part of the thrifty genotype thought to predispose the individual to insulin resistance [22]. Although PPARγ is required for life, PPARγ2-null mice are viable and have been used to investigate its role in insulin sensitivity, where it appears to be an important factor in insulin sensitivity in adipose tissue [23].
PPARδ has been much less studied than the other PPARs. Despite the important role the receptor is thought to play in controlling systemic metabolism, selective agonists have only recently been developed for PPARδ. This receptor has a role in adipocyte differentiation, lipoprotein metabolism, atherosclerosis and β-oxidation in adipocytes, skeletal muscle and the heart [24]. It also, in part, regulates fat metabolism in skeletal muscle, and over-expression of PPARδ in the mouse almost doubles this animal's endurance exercise capacity [25]. PPARδ also regulates glucose homeostasis, with PPARδ-null mice having reduced capabilities for glucose handling; PPARδ agonists also reduce insulin resistance and weight in obese obob mice [26], which, as they lack the leptin receptor, have such a high appetite that they develop obesity and type II diabetes as a result of over-nutrition.
Of course the role of PPARs in regulating metabolism in humans has been the subject of great interest, particularly within the pharmaceutical industry. It would be beyond the scope of this review to do full justice to the pharmacology of PPAR agonists in humans, and instead it seems more profitable to consider the rare human mutations of these receptors. Dominant negative mutations have been reported in PPARγ in humans [27]. Savage and colleagues [27] reported a dominant negative mutation of PPARγ arising from a proline-467-leucine mutation, which resulted in severe peripheral and hepatic insulin resistance, partial lipodystrophy (the loss/reduction of certain fat depots) and hepatic steatosis. Although relatively rare, individuals with this mutation have proved to be invaluable in investigating the role of PPARγ in humans, and a useful contrast with their rodent counterparts.
Advantages of the metabolomic approach to understanding the roles of the PPARs
Before examining in detail the information gained from metabolomic studies, it seems prudent to make a few points concerning the relevance of the approaches to studying functional genomics, and in particular to investigating systems that have a strong metabolic component. Firstly, any functional genomic study focusing on a system with a strong metabolic component is well suited to metabolomics, as many of the key outputs and regulatory stages will involve a significant number of metabolites. Secondly, the technologies used in metabolomics have largely been developed as high-throughput analytical chemistry tools, capable of analyzing relatively large numbers (50 to 200) of samples per day, with relatively low consumable costs, relying on relatively cheap solvents and derivatization agents for mass spectrometry and off-the-peg chromatography. Being cheap on a per-sample basis allows the approach to be used to analyze the range of tissues and biofluids necessary to study the function of transcription factors across the whole organism as part of a systems biology approach.
This system-wide approach is important for the PPARs because these nuclear hormone receptors are expressed across a range of tissues, and each plays a key role in regulating systemic metabolism in underfed or fasted conditions. In addition, metabolomics allows the analysis of large numbers of replicates of the same sample type. Although most animal models of type II diabetes and obesity have centered on rare monogenic causes, the disease is generally considered to be polygenic in the vast majority of humans; hence, to understand the role of the PPARs in predisposing individuals to metabolic syndromes, or in the treatment of this disease, it is necessary to either investigate a range of monogenic models or examine crosses between strains that are relatively predisposed to and relatively protected from developing a particular disorder. Both approaches necessitate a large number of samples, which makes metabolomics, an approach that is cheap on a per-sample basis, ideal for performing 'functional genomic triage'. As part of a polyomic approach, it may be too expensive to perform transcriptomics on multiple tissues or too time consuming to perform proteomics on such a large scale. However, high-throughput metabolomics can be used to identify the tissues that are most interesting to follow up as part of a triage process.
Metabolomic studies of PPARα: targeting gluconeogenesis to treat diabetes and examining the interaction with aging
The nuclear receptor PPARα plays a central role in regulating the oxidation of fatty acids in the liver and skeletal muscle and gluconeogenesis in the liver, particularly during the fasted state or following high-fat feeding. As already mentioned, one of the surprises of the PPARα-null mouse was that this mouse was viable, and indeed phenotypes were only apparent under relatively large physiological stresses. The role of PPARα in terms of regulating gluconeogenesis was elegantly demonstrated by Xu and co-workers [28], using gas chromatographic mass spectrometry (GC-MS) analysis of labeled isotopic isomers (called isotopomers) following subcutaneous infusion, via Alza miniosmotic pumps, of either [2-13C]glycerol, [U-13C3]lactate or [U-13C6]glucose. Following a 17-hour fast, while hepatic glucose production was found to be increased in the PPARα-null mouse, the stable isotope approach demonstrated reduced capacity for converting lactate into glucose, but increased capacity for the conversion of glycerol into glucose via gluconeogenesis. This identified the perturbation in gluconeogenesis as being at the start of the pathway, with the expression of pyruvate kinase found to be reduced 16-fold in the fasted PPARα-null mouse compared with the fasted wild-type control. In a follow-up study, Xu and colleagues [29] used the same stable isotope approach to demonstrate that a failure to express PPARα reduces futile cycling (the cyclic conversion between two substrates, which consumes adenosine triphosphate (ATP) during the process) in the liver but, to compensate for this, peripheral uptake of glucose is increased.
To examine the role of PPARα across the whole animal, Atherton and co-workers [30] used a combination of high-resolution 1H nuclear magnetic resonance (NMR) spectroscopy, GC-MS and liquid chromatograph-mass spectrometry (LC-MS) to profile liver, skeletal muscle, smooth muscle, cardiac tissue and white adipose tissue in one-month-old PPARα-null mice and compared them to wild-type controls. Despite conducting the study in mice fed on standard chow, the metabolomic approach readily distinguished the null mouse from the wild-type control in all tissues, with the most marked effects detected in the liver associated with alterations in glycolysis, the citric acid cycle, amino acid metabolism and fatty acid oxidation. One of the biggest surprises was that white adipose tissue demonstrated metabolic alterations, despite very low expression of PPARα in control adipose tissue. In support of this, Knauf and colleagues [31] report that glucose clearance by adipose tissue is increased in the PPARα-null mouse, even when expression is restored in the liver, and may even be mediated by the brain.
Expanding their metabolomic approach to investigate the interaction of PPARα expression with age, Atherton and co-workers [32] modeled the metabolic changes between 3 and 13 months of age in the PPARα-null mouse, using multivariate statistics (Figure 2). Hepatic glycogen and glucose decreased with age for both the mutant and wild-type mice, but the tissue from the null mouse had lower concentrations, with evidence of age-onset hepatic steatosis on a normal chow diet. This approach demonstrated that any phenotype should be placed within the context of the normal aging process, to understand the role of the gene across the lifetime of the organism.
Figure 2.
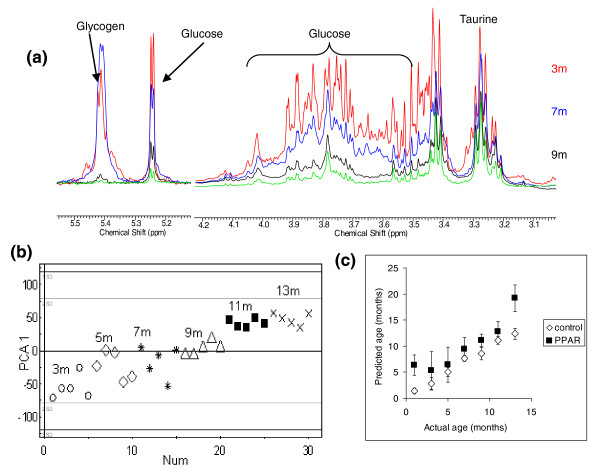
Metabolic changes in the PPARα-null mouse. (a) 1H-NMR spectra showing the difference in glucose and glycogen concentration between 3 and 13 months for liver tissue extracts from PPARα-null mice. Each spectrum is the average of the five spectra obtained from all animals at that age. Key: red, 3 months; blue, 5 months; black, 11 months; green, 13 months. (b) Principal components analysis (PCA) plot showing the clustering of 3-month (open circles), 5-month (open diamonds), 7-month (stars), 9-month (open triangles), 11-month (black squares) and 13-month (crosses) liver tissue across principal component 1. (c) Partial least squares plot regressing age of animal (y-axis) against the metabolic profile of the liver tissue (x-axis) in control mice as measured by 1H NMR spectroscopy. PPARα-null mice were then mapped on to the same model. Error bars indicate standard error. Reproduced from [20] with permission.
An important area in drug development is the identification of potential markers of drug efficacy, to gauge how the individual responds to a pharmacological stimulus. To address this, Zhen and co-workers [33] used LC-MS to profile urinary metabolites in control and PPARα-null mice and examined the action of Wy-14,643 ([4-chloro-6-(2,3-xylidino)-2-pyrimidinylthio] acetic acid), a potent PPARα agonist. Using principal components analysis (PCA) to interrogate the data, the PPARα-null groups of mice co-clustered (occupied similar positions on the PCA plot, indicating similar metabolic changes across the group compared with the control group), regardless of drug treatment, while the control groups formed separate clusters. This agonist increased urinary concentrations of 11beta-hydroxy-3,20-dioxopregn-4-en-21-oic acid, 11beta,20-dihydroxy-3-oxopregn-4-en-21-oic acid, nicotinamide, nicotinamide 1-oxide, 1-methylnicotinamide, hippuric acid, and 2,8-dihydroxyquinoline-beta-d-glucuronide, and decreased concentrations of xanthurenic acid, hexanoylglycine, phenylpropionylglycine and cinnamoylglycine. This highlighted the action of PPARα in regulating tryptophan, corticosterone and fatty-acid metabolism and on glucuronidation. This approach was extended by Patterson and colleagues [34], who examined the use of fenofibrate (a fibrate drug that targets PPARα; Table 1) treatment in humans, identifying pantothenic acid and short-chain acyl carnitines as markers of increased β-oxidation, and validating their results using the PPARα-null mouse. Following up the production of 11beta-hydroxy-3,20-dioxopregn-4-en-21-oic acid and altered steroid production by PPARα, Wang and co-workers [35] demonstrated cross-talk between the receptor, neuropeptide Y and agouti-related protein interacting with the hypothalamic-pituitary-adrenal axis.
In addition to its role in regulating the fasted response, PPARα also plays an important role in toxicology, and the original drive for the production of the PPARα-null mouse was to investigate the role PPARα plays in drug-induced hepatocarcinogenicity. Ohta and colleagues [36] investigated the early stages of fenofibrate toxicity in the Fischer rat. High doses of fenofibrate are known to be carcinogenic in the liver, via a non-genotoxic mechanism. In this study, Ohta and colleagues used a combination of LC-MS and GC-MS to highlight that part of the toxicity was caused by the generation of reactive oxygen species in the liver. Manna and co-workers [37] used metabolomics and the PPARα-null mouse to investigate alcohol-induced hepatotoxicity. The PPARα-null mouse is particularly predisposed to alcohol-induced hepatotoxicity, as a failure to express PPARα reduces the conversion of tryptophan to NAD+ (nicotine adenine dinucleotide+), which exacerbates the action of alcohol and its reducing potential on the liver. The combination of these events produced a characteristic increase in indole-3-lactic acid, which could be followed in the urine of these animals [37]. This suggests that PPARα agonists may be a viable treatment for alcohol-induced hepatic steatosis, and urinary indole-3-lactic acid would provide a marker of drug efficacy.
Metabolomic studies of PPARγ
Given its central role in regulating lipid storage and its importance as a potent target for restoring insulin sensitivity, PPARγ has been the subject of a number of influential lipidomic studies. One of the first studies, and an elegant demonstration that one does not necessarily need expensive equipment to perform lipidomics, was a study by Watkins and co-workers [38] to investigate the action of rosiglitazone in obese mice. Chronic treatment with rosiglitazone restored insulin sensitivity but increased hepatic steatosis. To assess these lipid changes, a combination of preparative thin-layer chromatography and gas-chromatograph flame-ionization detector (GC-FID) analysis was used to separate different lipid classes and analyze their fatty acid composition in plasma and key tissues. This provided enough detail to follow the increased synthesis of fatty acids within the liver and the remodeling of cardiolipin in the heart. Many of these changes were reflected in the plasma, suggesting that drug efficacy can be monitored effectively using metabolomics.
LC-MS-based lipidomics has been instrumental in determining the role of PPARγ in regulating adipose tissue morphology and expandability. There are three isoforms of PPARγ, with PPARγ2 being the most potent isoform for adipogenesis in vitro. Medina-Gomez and co-workers [39] used LC-MS, magnetic resonance imaging (MRI), transcriptomics and comprehensive physiology to investigate the role of PPARγ2 in regulating lipid storage in adipose tissue. Intriguingly, while ablation of PPARγ2 induced increased insulin resistance in chow-fed mice, this effect was not apparent in animals fed a high-fat diet. The increased basal insulin resistance was not associated with lipodystrophy, although adipocytes were larger in the transgenic mice following high-fat feeding. However, there is a high degree of compensation following the ablation of PPARγ2. To address this, Medina-Gomez and colleagues [39] crossed the PPARγ2-null mouse with the obese Lep(ob)/Lep(ob) mouse to generate a PPARγ2 (-/-) Lep(ob)/Lep(ob) mouse, referred to as the POKO mouse. This animal has severe insulin resistance, β-cell failure, reduced fat mass and dyslipidemia. This demonstrates the importance of the PPARγ2 isoforms in regulating normal adipose tissue expandability. The LC-MS analysis of tissues and blood plasma also demonstrated that the isoform plays an important role in reducing potentially lipotoxic intermediates. Medina-Gomez and colleagues [40] followed up this initial study with a further characterization of the β-cell failure in the pancreas of the POKO mouse. They followed changes in the pancreas between 4 and 16 weeks of age, highlighting that the lipidome changes of the pancreas were relatively mild in comparison to the large changes detected in the plasma, liver, muscle and adipose tissue of the mice. Lipotoxic species were only detected at the later time point, suggesting the POKO mouse may be a useful tool to follow β-cell dysfunction secondary to peripheral insulin resistance or lipid infiltration.
Metabolomic studies of PPARδ/PPARβ
Metabolomic and lipidomic studies of PPARδ have been relatively rare, as much less work has focused on PPARδ, in part because of difficulties in developing selective ligands that do not target the other PPARs as well, and also because of the importance of PPARδ in normal development, complicating its genetic manipulation in rodent models. However, Roberts and co-workers [41] have used a comprehensive metabolomic and lipidomic approach to study the role of PPARδ in regulating systemic metabolism, by focusing on the obese and diabetic obob mouse and a highly selective PPARδ agonist. Given the high expression of PPARδ in both liver and skeletal muscle, both tissues were examined, alongside plasma, to understand how tissue changes influence the composition of plasma. This was contrasted with the metabolic changes induced in these tissues by a highly selective PPARγ ligand. While both ligands restored insulin sensitivity, only the agonist for PPARδ reduced the body weight of the obese obob mice. Both ligands induced an increase in glycolysis in both tissues and a decrease in gluconeogensis in the liver, partly explaining how both compounds reduce blood glucose levels. However, PPARδ induced increased citric acid cycle activity, and mobilized free fatty acids, ketone bodies and triglycerides and activity in the linoleic and α-linolenic acid essential-fatty-acid pathways. Thus, PPARδ has a profound role in hepatic and skeletal muscle fatty-acid oxidation.
One problem with metabolomic approaches that rely on taking tissue samples at set time points is that it is difficult to talk about fluxes from the total concentration changes that are measured using this approach. To address this, Roberts and colleagues [42] used 3T3-L1 adipocytes in conjunction with stable isotope labeling, as well as an in vivo study in the obob mouse, to investigate the role of PPARδ in adipocytes (Figure 3). Stimulation of the receptor using a highly selective agonist induced increased oxygen consumption in 3T3-L1 adipocytes, and in both adipose tissue and cultured cells there were increases in fatty acid β-oxidation, tricarboxylic acid cycle rate and oxidation of extracellular branch chain amino acids. This highlights the potential of PPARδ agonists to act as both anti-diabetes and anti-obesity treatments, addressing both these consequences of dyslipidemia.
Figure 3.
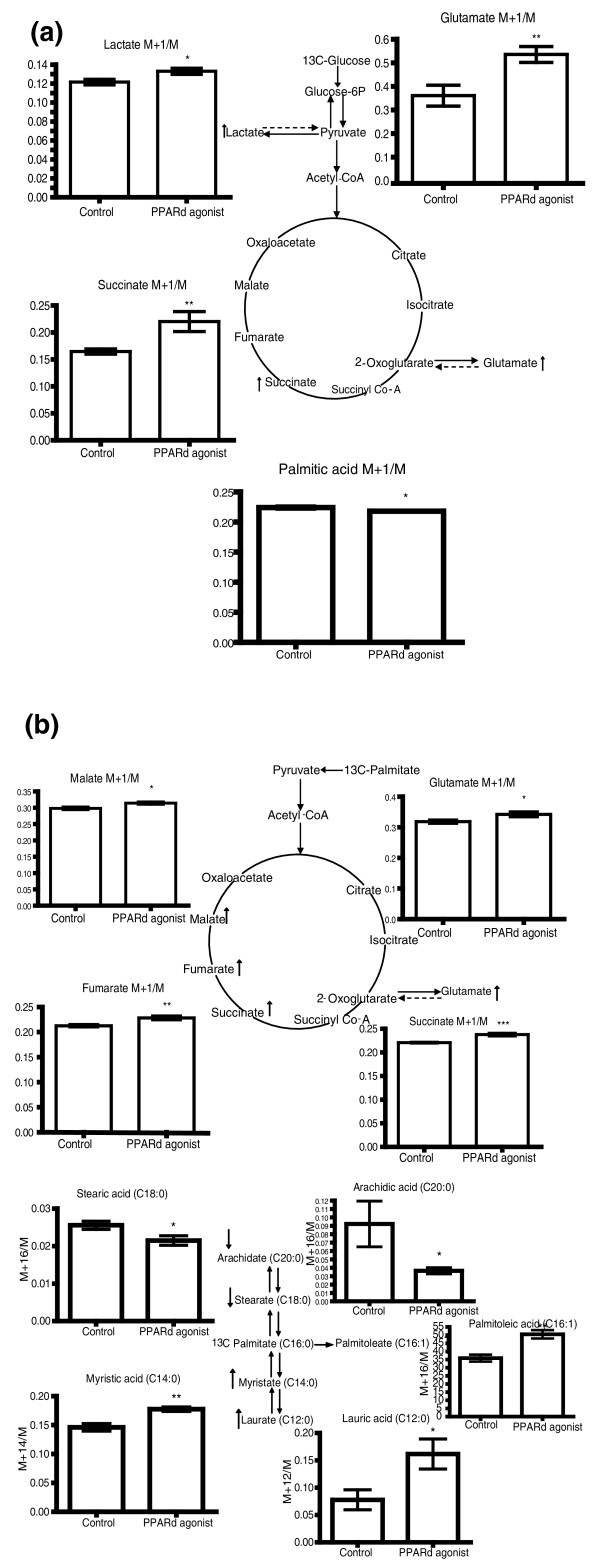
Stable Isotope flux analysis of PPARδ-agonist-treated 3T3-L1 adipocytes. (a) Graphs showing the M+1/M isotope ratio 13C enrichment of lactate, glutamate and succinate analyzed by GC-MS of the aqueous fraction and M+1/M isotope ratio 13C enrichment of palmitic acid analyzed by GC-MS of the organic fraction from control (n = 6) and PPARδ-agonist-dosed (n = 6) 3T3-L1 cells incubated with 1-13C glucose. *P < 0.05, **P < 0.01. The metabolites have been mapped to the glycolysis and tricarboxylic acid cycle metabolic pathways. Up arrow indicates a metabolite increased, and down arrow indicates a metabolite decreased in 13C enrichment by PPARδ activation. (b) Graphs showing the M+1/M isotope ratio 13C enrichment of malate, glutamate, fumarate and succinate analyzed by GC-MS of the aqueous fraction and enrichment of arachidic acid, stearic acid, palmitoleic acid, myristic acid and lauric acid analyzed by GC-MS of the organic fraction from control (n = 6) and PPARδ-agonist-dosed (n = 6) 3T3-L1 cells incubated with U-13C palmitate. *P < 0.05, **P < 0.01,***P < 0.005. Up arrow indicates a metabolite increased, and down arrow indicates a metabolite decreased in 13C enrichment by PPARδ activation. Parent ions were used to calculate the ion ratio. Reproduced from [30] with permission.
PPARδ agonists have also been studied in moderately overweight humans, using a combination of stable isotope techniques to monitor changes in flux and classic physiology measures [43]. Treatment with GW501516, a selective PPARδ agonist, resulted in a 30% reduction in plasma triglycerides, 20% reduction in liver fat and 30% reduction in pro-inflammatory isoprostanes in the urine. The stable isotope measurements were able to show an increase in fatty-acid oxidation, thus demonstrating that PPARδ was exerting its beneficial effects by upregulating β-oxidation, with skeletal muscle being a major target for the drug action.
Metabolomic studies of the systems that interact with the PPAR signaling pathways
PPARγ co-activator (PGC)1α and PGC1β are transcriptional co-activators that interact with PPARγ and, in turn, allow interaction with the multiple proteins involved in the regulation of cellular metabolism, including cAMP-response-element-binding protein (CREB) and nuclear respiratory factors (NRFs). PGC1α has been implicated in mitochondrial biogenesis, muscle fiber-type determination and adaptation to endurance exercise. While the PGC1 co-activators have been studied in depth in terms of physiology, they have been the focus of much less attention in terms of metabolomics and lipidomics. However, a recent study by Gurung and colleagues [44] provided intriguing evidence that PGC1β is involved in cardiac arrhythmia. PGC1β(-/-) mice had increased concentrations of pro-arrhythmic and pro-inflammatory lipids, including lysophosphatidylcholine. The pro-arrhythmogenic phenotype was confirmed by Langendorff heart perfusion, with the hearts showing ventricular tachycardia. In turn this was related to altered calcium handling in the heart tissue of PGC1β(-/-) mice.
The PPARs play a critical physiological role as lipid sensors and regulators of lipid metabolism. Polyunsaturated fatty acids (PUFAs) such as arachidonic acid and docosahexaenoic acid (DHA) are natural ligands with medium affinity for the PPARs and subsequently induce the heterodimerization of the PPAR receptors with the RXR [45,46], linking eicosanoid and related lipid mediator signaling to the PPAR systems. Once the PPAR receptor binds to its ligands, it heterodimerizes and activates the subsequent complex to induce genes for various functions such as adipocyte proliferation [47], lipid homeostasis in cardiomyocytes and skeletal muscle, stem cell proliferation [48] and the regulation of inflammation [49]. There are a number of proposed potential molecular mechanisms for the anti-inflammatory effects of PPARs. For example, PPARα activation regulates inflammation by inhibiting nuclear factor-κB (NF-κB) activation and consequently the inhibition of cyclooxygenase (COX), which modulates the production of inflammatory lipid mediators. The RXR/PPAR complex can also inhibit expression of other inflammatory mediators, including inducible nitric oxide synthase (iNOS), tumor necrosis factor-α (TNF-α), interferon-γ (IFN-γ) and interleukin-12 (IL-12) [50].
Although the identities of high-affinity natural ligands for the PPARs are currently unknown, there is evidence that fatty acids and their oxidized metabolites can serve as activating ligands for PPARs. In particular, COX-related metabolites have demonstrated their physiological function via binding to PPARs. For instance, prostaglandin D2 (PGD2) metabolites, such as 15-deoxy Δ12,14prostaglandin J2 (PGJ2), promote adipocyte differentiation via activation of PPARγ [51], whereas prostacyclin (PGI2) activates PPARδ in pregnant mouse uterus during natural implantation of the placenta [52]. In addition, a number of studies have indicated that PPAR signaling is activated by lipoxygenase (LOX)-derived metabolites. For instance, 8(S)-hydroxyeicosatetraenoic acid (8(S)-HETE) has been reported to be an effective activator of PPARα [53]. Linoleic-acid-oxidized metabolites, 9-HODE and 13-HODE, have been reported to activate PPARγ-dependent gene expression in monocytes. In another study, PPARγ and 15-LOX were coordinately induced by IL-4 in macrophages, and it has been suggested that the 15-LOX-derived metabolites act as endogenous ligands of PPARγ to mediate transcriptional induction of the CD36 gene in these cells [54,55].
Lipidomic approaches are beginning to piece together how these different signaling pathways interact with one another. For example, Bazan and co-workers [56] have recently reported DHA-derived neuroprotectin (NPD1) as a potential PPARγ ligand and demonstrated that NPD1 anti-amyloidogenic bioactivity is mediated via activation of the PPARγ. Kim and co-workers [57] have described a novel metabolomic approach aimed at profiling metabolite-protein interactions on columns, to look at lipids that bind to PPARγ and PPARα. Following a pull-down assay approach, they ran crude lipid extracts down the columns containing immobilized PPARγ and PPARα, identifying arachidonic acid (C20:4), oleate (C18:1), linoleic acid (C18:2), palmitoleic acid (C16:2) and C18:1 lysophosphatidic acid binding to PPARγ and arachidonic acid, linoleic acid and oleic acid binding to PPARα. Although naturally occurring lipid mediators that activate PPARs have been identified in vitro, establishing the physiological relevance of many of these substances as regulators of PPARs in vivo is challenging, due to their low concentrations and relatively low affinities to PPARs. However, advancements in analytic techniques and profiling of these metabolites could overcome the difficulties of detecting these ligands in vivo and could advance our understanding of their mechanisms of action. Consequently, due to its specificity and sensitivity, lipidomics analysis using LC-MS/MS is currently the most powerful tool for the analysis of lipid mediators (ms/ms: tandem mass spectrometry where mass spectrometry is performed on the fragments of the parent ion) [58].
PPARδ signaling is also intricately linked with lipid mediator signaling, and this has been investigated in the pancreas using high-resolution LC-MS. Cohen and colleagues [59] observed that high glucose levels in the media of INS-1E β-cells activated cPLA(2), releasing arachidonic and linoleic acid from phospholipids in the cell membrane. The high glucose and PPARδ activation both stimulated the generation of 4-hyroxy-2E-nonenal, an endogenous ligand for PPARδ, which in turn amplifies insulin secretion in β-pancreatic cells.
Metabolomic studies of PPAR-induced cancer
The PPARs have a long and complex history in terms of cancer biology. Agonists for all three receptors have been linked to increased relative risks of certain cancers in carcinogenicity studies in rodents. However, for many of these agonists, the increased risks for certain types of cancer are offset by a reduction in the relative risk for other types of cancers. However, one area that has received much attention from those using metabolomics is the role peroxisome proliferation plays in PPARα-induced cancer.
Peroxisome proliferators (PPs), many of them substrates of PPARα, are non-DNA reactive, non-genotoxic (epigenetic) carcinogens. They cause peroxisome proliferation, hyperplasia and ultimately hepatocarcinoma in rats and mice. As described earlier, the property of PPs to cause hepatocellular carcinoma eventually led to the discovery of PPARα [16]. The mechanisms underlying the carcinogenesis are not fully understood, although the hepatocarcinogenicity of PPs provides one of the best-studied [60-64] and perhaps the most informative system for understanding non-genotoxic mechanisms, as well as key aspects of general metabolism.
Many efforts in the pharmaceutical industry have focused on the early detection of carcinogenic potential, in an attempt to replace the costly, long-term rodent bioassays and histopathology for the detection of PP-induced non-genotoxic carcinogenicity. Non-invasive techniques, such as urinary metabolite profiling, are particularly attractive in terms of low costs for both time and animals. Studies by Delaney and colleagues describe urinary measurements of N-methylnicotinamide (NMN) and N-methyl-4-pyridone-3-carboxamide (4PY) (end-products of the tryptophan-NAD+ pathway) as being correlated with PP as measured by electron microscopy [65]. Urinary metabolites were determined by high-resolution 1H NMR spectroscopy and NMN, 2PY and 4PY were determined using high-pressure liquid chromatography (HPLC)-MS/MS for accurate quantification. A follow-up report by Connor and co-workers [66] further characterized the NMR spectra obtained in the same animal study.
The discovery that peroxisome proliferation leads to epigenetic events gave grounds for the investigation of S-adenosylmethionine (SAMe) and S-adenosylhomocysteine (SAH) [67], as well as other metabolites in the methionine cycle, and even phospholipid metabolism following peroxisomal proliferation [68]. The increased production of H2O2 following peroxisome proliferation evidently links oxidative stress to non-genotoxic carcinogenesis and to metabolites of oxidative stress, including glutathione (GSH)/oxidized glutathione (GSSG) [69], epoxides, uric acid [70], peroxisomal and mitochondrial ω-oxidation (carnitine and acyl-carnitine species) and other products of lipid oxidation [71]. It has been well documented that PPs mediate liver P450s, which carry out ω-hydroxylation of lauric acid (and other lipids) [72]. Metabolism of lauric acid has since been of interest as a useful tool for the detection of possible toxicity associated ω-hydroxylation [73,74].
Metabolomics has also been used to investigate the role PPARδ plays in breast cancer [70]. In this study, to induce tumors 3-phosphoinositide-dependent kinase-1 (PDK1) was expressed in mammary epithelium. The expression of the gene induced increased expression of PPARδ. In addition, treatment of both the transgenic mouse and its wild-type control with GW501516 (a selective PPARδ agonist) increased rates of tumor formation, particularly in the transgenic animal. LC-MS-based metabolomics of the methanol extract of breast tumors demonstrated that the agonist induced a distinctive metabolomic profile in breast tumors [70]. Intriguingly, this was associated with increased phospholipid and fatty acid accumulation, in contrast with the action of PPARδ agonists in healthy liver, skeletal muscle and adipose tissue, where increased β-oxidation appears central.
Conclusions
Because of the diverse roles of the PPAR transcription factors, and in particular their key part in regulating systemic metabolism, this area of biology has been a useful 'sandbox' for researchers to develop metabolomic techniques. In the opinion of the authors, there is adequate evidence that metabolomics has an integral part to play alongside more established physiology and transcriptomic techniques. This has included monitoring the role individual PPARs play in regulating systemic metabolism and how the normal aging process interacts with these receptors. The use of metabolomics has also opened up novel areas of research, such as the role PPARα plays in alcohol-induced steatohepatitis or the induction of liver cancer by non-genotoxic carcinogenicity. However, the interest in PPARs as a direct target for drugs has somewhat diminished recently, due to concerns of unwanted side effects. The capability of metabolomics to monitor some of the changes in lipid mediators may provide alternative targets either to bypass the PPAR receptor itself, or to provide a more controlled manner of stimulation for these key regulators of cellular metabolism. Metabolomics-derived biomarkers of drug efficacy may also allow the deployment of drugs as part of a personalized medicine approach and ensure that some of the side effects are avoided in their future use.
Abbreviations
ATP: adenosine triphosphate; COX: cyclooxygenase; cPLA(2): cytoplasmic phospholipase 2; CREB: cAMP-response-element-binding protein; DHA: docosahexaenoic acid; FDA: Food and Drug Administration; GC-FID: gas-chromatograph flame-ionization detector; GC-MS: gas chromatographic mass spectrometry; 8(S)-HETE, 8(S)-hydroxyeicosatetraenoic acid; GSH: glutathione; GSSH: oxidized glutathione; HPLC: high-pressure liquid chromatography; IFN-γ: interferon-γ; IL-12: interleukin-12; iNOS: inducible nitric oxide synthase; LC-MS: liquid chromatograph-mass spectrometry; LOX: lipoxygenase; NAD: nicotine adenine dinucleotide; NHS: National Health Service; NMN: N-methylnicotinamide; NMR: nuclear magnetic resonance; NF-κB: nuclear factor-κB; NPD1: DHA-derived neuroprotectin; NRF: nuclear respiratory factor; PCA: principal components analysis; PDK1: 3-phosphoinositide-dependent kinase-1; PGC: PPARγ co-activator; PGI2: prostacyclin; PP: peroxisome proliferator; PPAR: peroxisome proliferator-activated receptor; PPRE: peroxisome proliferator hormone-response element; PUFA: polyunsaturated fatty acid; 4PY: N-methyl-4-pyridone-3-carboxamide; RXR: retinoid × receptor; SAH: S-adenosylhomocysteine; SAMe: S-adenosylmethionine; TNF-α: tumor necrosis factor-α; US: United States; WY-14,643: 4-chloro-6-(2,3-xylidino)-2-pyrimidinylthio] acetic acid.
Competing interests
The authors declare that they have no competing interests.
Contributor Information
Zsuzsanna Ament, Email: amentzs@gmail.com.
Mojgan Masoodi, Email: mojgan.masoodi@mrc-hnr.cam.ac.uk.
Julian L Griffin, Email: jules.griffin@mrc-hnr.cam.ac.uk.
Acknowledgements
JLG and MM are funded by the Medical Research Council (UD99999906). Work in the JLG lab is funded in addition by the BBSRC (BB/H013539/1), the Wellcome Trust (093148/Z/10/Z) and the EU (INHERITANCE). ZA is a recipient of an MRC ITTP training PhD studentship.
References
- Evans RM, Barish GD, Wang Y-X. PPARs and the complex journey to obesity. Nat Med. 2004;10:355–361. doi: 10.1038/nm1025. [DOI] [PubMed] [Google Scholar]
- Peters JM, Shah YM, Gonzalez FJ. The role of peroxisome proliferator-activated receptors in carcinogenesis and chemoprevention. Nat Rev Cancer. 2012;12:181–195. doi: 10.1038/nrc3214. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Delaney J, Hodson MP, Thakkar H, Connor SC, Sweatman BC, Kenny SP, McGill PJ, Holder JC, Hutton KA, Haselden JN, Waterfield CJ. Tryptophan?NAD+ pathway metabolites as putative biomarkers and predictors of peroxisome proliferation. Arch Toxicol. 2004;79:208–223. doi: 10.1007/s00204-004-0625-5. [DOI] [PubMed] [Google Scholar]
- Nissen SE, Wolski K. Effect of rosiglitazone on the risk of myocardial infarction and death from cardiovascular causes. N Engl J Med. 2007;356:2457–2471. doi: 10.1056/NEJMoa072761. [DOI] [PubMed] [Google Scholar]
- Kaul S, Bolger AF, Herrington D, Giugliano RP, Eckel RH. Thiazolidinedione drugs and cardiovascular risks: a science advisory from the American Heart Association and American College of Cardiology Foundation. Circulation. 2010;121:1868–1877. doi: 10.1161/CIR.0b013e3181d34114. [DOI] [PubMed] [Google Scholar]
- Clayton TA, Lindon JC, Cloarec O, Antti H, Charuel C, Hanton G, Provost J-P, Le Net J-L, Baker D, Walley RJ, Everett JR, Nicholson JK. Pharmaco-metabonomic phenotyping and personalized drug treatment. Nature. 2006;440:1073–1077. doi: 10.1038/nature04648. [DOI] [PubMed] [Google Scholar]
- Kirschenlohr HL, Griffin JL, Clarke SC, Rhydwen R, Grace AA, Schofield PM, Brindle KM, Metcalfe JC. Proton NMR analysis of plasma is a weak predictor of coronary artery disease. Nat Med. 2006;12:705–710. doi: 10.1038/nm1432. [DOI] [PubMed] [Google Scholar]
- Salek RM, Maguire ML, Bentley E, Rubtsov DV, Hough T, Cheeseman M, Nunez D, Sweatman BC, Haselden JN, Cox RD, Connor SC, Griffin JL. A metabolomic comparison of urinary changes in type 2 diabetes in mouse, rat, and human. Physiol Genomics. 2007;29:99–108. doi: 10.1152/physiolgenomics.00194.2006. [DOI] [PubMed] [Google Scholar]
- Wang TJ, Larson MG, Vasan RS, Cheng S, Rhee EP, McCabe E, Lewis GD, Fox CS, Jacques PF, Fernandez C, O'Donnell CJ, Carr SA, Mootha VK, Florez JC, Souza A, Melander O, Clish CB, Gerszten RE. Metabolite profiles and the risk of developing diabetes. Nat Med. 2011;17:448–453. doi: 10.1038/nm.2307. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kettunen J, Tukiainen T, Sarin A-P, Ortega-Alonso A, Tikkanen E, Lyytikäinen L-P, Kangas AJ, Soininen P, Würtz P, Silander K, Dick DM, Rose RJ, Savolainen MJ, Viikari J, Kähönen M, Lehtimäki T, Pietiläinen KH, Inouye M, McCarthy MI, Jula A, Eriksson J, Raitakari OT, Salomaa V, Kaprio J, Järvelin MR, Peltonen L, Perola M, Freimer NB, Ala-Korpela M, Palotie A. et al. Genome-wide association study identifies multiple loci influencing human serum metabolite levels. Nat Genet. 2012;44:269–276. doi: 10.1038/ng.1073. [DOI] [PMC free article] [PubMed] [Google Scholar]
- King RD, Whelan KE, Jones FM, Reiser PGK, Bryant CH, Muggleton SH, Kell DB, Oliver SG. Functional genomic hypothesis generation and experimentation by a robot scientist. Nature. 2004;427:247–252. doi: 10.1038/nature02236. [DOI] [PubMed] [Google Scholar]
- Griffin JL, Bonney SA, Mann C, Hebbachi AM, Gibbons GF, Nicholson JK, Shoulders CC, Scott J. An integrated reverse functional genomic and metabolic approach to understanding orotic acid-induced fatty liver. Physiol Genomics. 2004;17:140–149. doi: 10.1152/physiolgenomics.00158.2003. [DOI] [PubMed] [Google Scholar]
- Griffin JL, Williams HJ, Sang E, Clarke K, Rae C, Nicholson JK. Metabolic profiling of genetic disorders: a multitissue (1)H nuclear magnetic resonance spectroscopic and pattern recognition study into dystrophic tissue. Anal Biochem. 2001;293:16–21. doi: 10.1006/abio.2001.5096. [DOI] [PubMed] [Google Scholar]
- Dumas M-E, Wilder SP, Bihoreau M-T, Barton RH, Fearnside JF, Argoud K, D'Amato L, Wallis RH, Blancher C, Keun HC, Baunsgaard D, Scott J, Sidelmann UG, Nicholson JK, Gauguier D. Direct quantitative trait locus mapping of mammalian metabolic phenotypes in diabetic and normoglycemic rat models. Nat Genet. 2007;39:666–672. doi: 10.1038/ng2026. [DOI] [PubMed] [Google Scholar]
- Illig T, Gieger C, Zhai G, Römisch-Margl W, Wang-Sattler R, Prehn C, Altmaier E, Kastenmüller G, Kato BS, Mewes H-W, Meitinger T, de Angelis MH, Kronenberg F, Soranzo N, Wichmann HE, Spector TD, Adamski J, Suhre K. A genome-wide perspective of genetic variation in human metabolism. Nat Genet. 2009;42:137–141. doi: 10.1038/ng.507. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Issemann I, Green S. Activation of a member of the steroid hormone receptor superfamily by peroxisome proliferators. Nature. 1990;347:645–650. doi: 10.1038/347645a0. [DOI] [PubMed] [Google Scholar]
- Lee SS, Pineau T, Drago J, Lee EJ, Owens JW, Kroetz DL, Fernandez-Salguero PM, Westphal H, Gonzalez FJ. Targeted disruption of the alpha isoform of the peroxisome proliferator-activated receptor gene in mice results in abolishment of the pleiotropic effects of peroxisome proliferators. Mol Cell Biol. 1995;15:3012–3022. doi: 10.1128/mcb.15.6.3012. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kersten S, Seydoux J, Peters JM, Gonzalez FJ, Desvergne B, Wahli W. Peroxisome proliferator-activated receptor α mediates the adaptive response to fasting. J Clin Invest. 1999;103:1489–1498. doi: 10.1172/JCI6223. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Leone TC, Weinheimer CJ, Kelly DP. A critical role for the peroxisome proliferator-activated receptor alpha (PPARalpha) in the cellular fasting response: the PPARalpha-null mouse as a model of fatty acid oxidation disorders. Proc Natl Acad Sci USA. 1999;96:7473–7478. doi: 10.1073/pnas.96.13.7473. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Vidal-Puig A, Jimenez-Liñan M, Lowell BB, Hamann A, Hu E, Spiegelman B, Flier JS, Moller DE. Regulation of PPAR gamma gene expression by nutrition and obesity in rodents. J Clin Invest. 1996;97:2553–2561. doi: 10.1172/JCI118703. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Fajas L, Auboeuf D, Raspé E, Schoonjans K, Lefebvre AM, Saladin R, Najib J, Laville M, Fruchart JC, Deeb S, Vidal-Puig A, Flier J, Briggs MR, Staels B, Vidal H, Auwerx J. The organization, promoter analysis, and expression of the human PPARgamma gene. J Biol Chem. 1997;272:18779–18789. doi: 10.1074/jbc.272.30.18779. [DOI] [PubMed] [Google Scholar]
- Kadowaki T, Hara K, Kubota N, Tobe K, Terauchi Y, Yamauchi T, Eto K, Kadowaki H, Noda M, Hagura R, Akanuma Y. The role of PPARgamma in high-fat diet-induced obesity and insulin resistance. J Diabetes Complications. 2002;16:41–45. doi: 10.1016/S1056-8727(01)00206-9. [DOI] [PubMed] [Google Scholar]
- Medina-Gomez G, Virtue S, Lelliott C, Boiani R, Campbell M, Christodoulides C, Perrin C, Jimenez-Linan M, Blount M, Dixon J, Zahn D, Thresher RR, Aparicio S, Carlton M, Colledge WH, Kettunen MI, Seppänen-Laakso T, Sethi JK, O'Rahilly S, Brindle K, Cinti S, Oresic M, Burcelin R, Vidal-Puig A. The link between nutritional status and insulin sensitivity is dependent on the adipocyte-specific peroxisome proliferator-activated receptor-gamma2 isoform. Diabetes. 2005;54:1706–1716. doi: 10.2337/diabetes.54.6.1706. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Takahashi S, Tanaka T, Kodama T, Sakai J. Peroxisome proliferator-activated receptor delta (PPARdelta), a novel target site for drug discovery in metabolic syndrome. Pharmacol Res. 2006;53:501–507. doi: 10.1016/j.phrs.2006.03.019. [DOI] [PubMed] [Google Scholar]
- Wang Y-X, Zhang C-L, Yu RT, Cho HK, Nelson MC, Bayuga-Ocampo CR, Ham J, Kang H, Evans RM. Regulation of muscle fiber type and running endurance by PPARdelta. PLoS Biol. 2004;2:e294–e294. doi: 10.1371/journal.pbio.0020294. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lee C-H, Olson P, Hevener A, Mehl I, Chong L-W, Olefsky JM, Gonzalez FJ, Ham J, Kang H, Peters JM, Evans RM. PPARdelta regulates glucose metabolism and insulin sensitivity. Proc Natl Acad Sci USA. 2006;103:3444–3449. doi: 10.1073/pnas.0511253103. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Savage DB, Tan GD, Acerini CL, Jebb SA, Agostini M, Gurnell M, Williams RL, Umpleby AM, Thomas EL, Bell JD, Dixon AK, Dunne F, Boiani R, Cinti S, Vidal-Puig A, Karpe F, Chatterjee VK, O'Rahilly S. Human metabolic syndrome resulting from dominant-negative mutations in the nuclear receptor peroxisome proliferator-activated receptor-gamma. Diabetes. 2003;52:910–917. doi: 10.2337/diabetes.52.4.910. [DOI] [PubMed] [Google Scholar]
- Xu J, Xiao G, Trujillo C, Chang V, Blanco L, Joseph SB, Bassilian S, Saad MF, Tontonoz P, Lee WNP, Kurland IJ. Peroxisome proliferator-activated receptor alpha (PPARalpha) influences substrate utilization for hepatic glucose production. J Biol Chem. 2002;277:50237–50244. doi: 10.1074/jbc.M201208200. [DOI] [PubMed] [Google Scholar]
- Xu J, Chang V, Joseph SB, Trujillo C, Bassilian S, Saad MF, Lee WNP, Kurland IJ. Peroxisomal proliferator-activated receptor alpha deficiency diminishes insulin-responsiveness of gluconeogenic/glycolytic/pentose gene expression and substrate cycle flux. Endocrinology. 2004;145:1087–1095. doi: 10.1210/en.2003-1173. [DOI] [PubMed] [Google Scholar]
- Atherton HJ, Bailey NJ, Zhang W, Taylor J, Major H, Shockcor J, Clarke K, Griffin JL. A combined 1H-NMR spectroscopy- and mass spectrometry-based metabolomic study of the PPAR-{alpha} null mutant mouse defines profound systemic changes in metabolism linked to the metabolic syndrome. Physiol Genomics. 2006;27:178–186. doi: 10.1152/physiolgenomics.00060.2006. [DOI] [PubMed] [Google Scholar]
- Knauf C, Rieusset J, Foretz M, Cani PD, Uldry M, Hosokawa M, Martinez E, Bringart M, Waget A, Kersten S, Desvergne B, Gremlich S, Wahli W, Seydoux J, Delzenne NM, Thorens B, Burcelin R. Peroxisome proliferator-activated receptor-alpha-null mice have increased white adipose tissue glucose utilization, GLUT4, and fat mass: role in liver and brain. Endocrinology. 2006;147:4067–4078. doi: 10.1210/en.2005-1536. [DOI] [PubMed] [Google Scholar]
- Atherton HJ, Gulston MK, Bailey NJ, Cheng K-K, Zhang W, Clarke K, Griffin JL. Metabolomics of the interaction between PPAR-alpha and age in the PPAR-alpha-null mouse. Mol Syst Biol. 2009;5:259–259. doi: 10.1038/msb.2009.18. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zhen Y, Krausz KW, Chen C, Idle JR, Gonzalez FJ. Metabolomic and genetic analysis of biomarkers for peroxisome proliferator-activated receptor alpha expression and activation. Mol Endocrinol. 2007;21:2136–2151. doi: 10.1210/me.2007-0150. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Patterson AD, Slanar O, Krausz KW, Li F, Höfer CC, Perlík F, Gonzalez FJ, Idle JR. Human urinary metabolomic profile of PPARalpha induced fatty acid beta-oxidation. J Proteome Res. 2009;8:4293–4300. doi: 10.1021/pr9004103. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wang T, Shah YM, Matsubara T, Zhen Y, Tanabe T, Nagano T, Fotso S, Krausz KW, Zabriskie TM, Idle JR, Gonzalez FJ. Control of steroid 21-oic acid synthesis by peroxisome proliferator-activated receptor alpha and role of the hypothalamic-pituitary-adrenal axis. J Biol Chem. 2010;285:7670–7685. doi: 10.1074/jbc.M109.090175. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ohta T, Masutomi N, Tsutsui N, Sakairi T, Mitchell M, Milburn MV, Ryals JA, Beebe KD, Guo L. Untargeted metabolomic profiling as an evaluative tool of fenofibrate-induced toxicology in Fischer 344 male rats. Toxicol Pathol. 2009;37:521–535. doi: 10.1177/0192623309336152. [DOI] [PubMed] [Google Scholar]
- Manna SK, Patterson AD, Yang Q, Krausz KW, Li H, Idle JR, Fornace AJ, Gonzalez FJ. Identification of noninvasive biomarkers for alcohol-induced liver disease using urinary metabolomics and the Ppara-null mouse. J Proteome Res. 2010;9:4176–4188. doi: 10.1021/pr100452b. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Watkins SM, Reifsnyder PR, Pan H-j, German JB, Leiter EH. Lipid metabolome-wide effects of the PPARgamma agonist rosiglitazone. J Lipid Res. 2002;43:1809–1817. doi: 10.1194/jlr.M200169-JLR200. [DOI] [PubMed] [Google Scholar]
- Medina-Gomez G, Gray SL, Yetukuri L, Shimomura K, Virtue S, Campbell M, Curtis RK, Jimenez-Linan M, Blount M, Yeo GS, Lopez M, Seppänen-Laakso T, Ashcroft FM, Oresic M, Vidal-Puig A. PPAR gamma 2 prevents lipotoxicity by controlling adipose tissue expandability and peripheral lipid metabolism. PLoS Genet. 2007;3:e64–e64. doi: 10.1371/journal.pgen.0030064. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Medina-Gomez G, Yetukuri L, Velagapudi V, Campbell M, Blount M, Jimenez-Linan M, Ros M, Oresic M, Vidal-Puig A. Adaptation and failure of pancreatic beta cells in murine models with different degrees of metabolic syndrome. Dis Model Mech. 2009;2:582–592. doi: 10.1242/dmm.003251. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Roberts LD, Hassall DG, Winegar DA, Haselden JN, Nicholls AW, Griffin JL. Increased hepatic oxidative metabolism distinguishes the action of peroxisome proliferator-activated receptor delta from Peroxisome proliferator-activated receptor gamma in the ob/ob mouse. Genome Med. 2009;1:115–115. doi: 10.1186/gm115. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Roberts LD, Murray AJ, Menassa D, Ashmore T, Nicholls AW, Griffin JL. The contrasting roles of PPARδ and PPARγ in regulating the metabolic switch between oxidation and storage of fats in white adipose tissue. Genome Biol. 2011;12:R75–R75. doi: 10.1186/gb-2011-12-8-r75. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Risérus U, Sprecher D, Johnson T, Olson E, Hirschberg S, Liu A, Fang Z, Hegde P, Richards D, Sarov-Blat L, Strum JC, Basu S, Cheeseman J, Fielding BA, Humphreys SM, Danoff T, Moore NR, Murgatroyd P, O'Rahilly S, Sutton P, Willson T, Hassall D, Frayn KN, Karpe F. Activation of peroxisome proliferator-activated receptor (PPAR)delta promotes reversal of multiple metabolic abnormalities, reduces oxidative stress, and increases fatty acid oxidation in moderately obese men. Diabetes. 2008;57:332–339. doi: 10.2337/db07-1318. [DOI] [PubMed] [Google Scholar]
- Gurung IS, Medina-Gomez G, Kis A, Baker M, Velagapudi V, Neogi SG, Campbell M, Rodriguez-Cuenca S, Lelliott C, McFarlane I, Oresic M, Grace AA, Vidal-Puig A, Huang CL. Deletion of the metabolic transcriptional coactivator PGC1β induces cardiac arrhythmia. Cardiovasc Res. 2011;92:29–38. doi: 10.1093/cvr/cvr155. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lengqvist J, Mata De Urquiza A, Bergman AC, Willson TM, Sjovall J, Perlmann T, Griffiths WJ. Polyunsaturated fatty acids including docosahexaenoic and arachidonic acid bind to the retinoid × receptor alpha ligand-binding domain. Mol Cell Proteomics. 2004;3:692–703. doi: 10.1074/mcp.M400003-MCP200. [DOI] [PubMed] [Google Scholar]
- Keller H, Dreyer C, Medin J, Mahfoudi A, Ozato K, Wahli W. Fatty acids and retinoids control lipid metabolism through activation of peroxisome proliferator-activated receptor-retinoid × receptor heterodimers. Proc Natl Acad Sci USA. 1993;90:2160–2164. doi: 10.1073/pnas.90.6.2160. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Na H-K, Surh Y-J. Peroxisome proliferator-activated receptor gamma (PPARgamma) ligands as bifunctional regulators of cell proliferation. Biochem Pharmacol. 2003;66:1381–1391. doi: 10.1016/S0006-2952(03)00488-X. [DOI] [PubMed] [Google Scholar]
- Hoggatt J, Pelus LM. Eicosanoid regulation of hematopoiesis and hematopoietic stem and progenitor trafficking. Leukemia. 2010;24:1993–2002. doi: 10.1038/leu.2010.216. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Poudyal H, Panchal SK, Diwan V, Brown L. Omega-3 fatty acids and metabolic syndrome: effects and emerging mechanisms of action. Prog Lipid Res. 2011;50:372–387. doi: 10.1016/j.plipres.2011.06.003. [DOI] [PubMed] [Google Scholar]
- Tontonoz P, Spiegelman BM. Fat and beyond: the diverse biology of PPARgamma. Annu Rev Biochem. 2008;77:289–312. doi: 10.1146/annurev.biochem.77.061307.091829. [DOI] [PubMed] [Google Scholar]
- Kliewer SA, Lenhard JM, Willson TM, Patel I, Morris DC, Lehmann JM. A prostaglandin J2 metabolite binds peroxisome proliferator-activated receptor gamma and promotes adipocyte differentiation. Cell. 1995;83:813–819. doi: 10.1016/0092-8674(95)90194-9. [DOI] [PubMed] [Google Scholar]
- Lim H, Gupta RA, Ma WG, Paria BC, Moller DE, Morrow JD, DuBois RN, Trzaskos JM, Dey SK. Cyclo-oxygenase-2-derived prostacyclin mediates embryo implantation in the mouse via PPARdelta. Genes Dev. 1999;13:1561–1574. doi: 10.1101/gad.13.12.1561. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Yu K, Bayona W, Kallen CB, Harding HP, Ravera CP, McMahon G, Brown M, Lazar MA. Differential activation of peroxisome proliferator-activated receptors by eicosanoids. J Biol Chem. 1995;270:23975–23983. doi: 10.1074/jbc.270.41.23975. [DOI] [PubMed] [Google Scholar]
- Huang JT, Welch JS, Ricote M, Binder CJ, Willson TM, Kelly C, Witztum JL, Funk CD, Conrad D, Glass CK. Interleukin-4-dependent production of PPAR-gamma ligands in macrophages by 12/15-lipoxygenase. Nature. 1999;400:378–382. doi: 10.1038/22572. [DOI] [PubMed] [Google Scholar]
- Nagy L, Tontonoz P, Alvarez JG, Chen H, Evans RM. Oxidized LDL regulates macrophage gene expression through ligand activation of PPARgamma. Cell. 1998;93:229–240. doi: 10.1016/S0092-8674(00)81574-3. [DOI] [PubMed] [Google Scholar]
- Zhao Y, Calon F, Julien C, Winkler JW, Petasis NA, Lukiw WJ, Bazan NG. Docosahexaenoic acid-derived neuroprotectin D1 induces neuronal survival via secretase- and PPARgamma-mediated mechanisms in Alzheimer's disease models. PLoS One. p. e15816. [DOI] [PMC free article] [PubMed]
- Kim YG, Lou AC, Saghatelian A. A metabolomics strategy for detecting protein-metabolite interactions to identify natural nuclear receptor ligands. Mol Biosyst. 2011;7:1046–1049. doi: 10.1039/c0mb00324g. [DOI] [PubMed] [Google Scholar]
- Masoodi M, Eiden M, Koulman A, Spaner D, Volmer DA. Comprehensive lipidomics analysis of bioactive lipids in complex regulatory networks. Anal Chem. 2010;82:8176–8185. doi: 10.1021/ac1015563. [DOI] [PubMed] [Google Scholar]
- Cohen G, Riahi Y, Shamni O, Guichardant M, Chatgilialoglu C, Ferreri C, Kaiser N, Sasson S. Role of lipid peroxidation and PPAR-δ in amplifying glucose-stimulated insulin secretion. Diabetes. 2011;60:2830–2842. doi: 10.2337/db11-0347. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Roberts RA, James NH, Hasmall SC, Holden PR, Lambe K, Macdonald N, West D, Woodyatt NJ, Whitcome D. Apoptosis and proliferation in nongenotoxic carcinogenesis: species differences and role of PPARalpha. Toxicol Lett. 2000;112-113:49–57. doi: 10.1016/s0378-4274(99)00243-x. [DOI] [PubMed] [Google Scholar]
- Gonzalez FJ, Shah YM. PPARalpha: mechanism of species differences and hepatocarcinogenesis of peroxisome proliferators. Toxicology. 2008;246:2–8. doi: 10.1016/j.tox.2007.09.030. [DOI] [PubMed] [Google Scholar]
- Roberts RA. Non-genotoxic hepatocarcinogenesis: suppression of apoptosis by peroxisome proliferators. Ann N Y Acad Sci. 1996;804:588–611. doi: 10.1111/j.1749-6632.1996.tb18647.x. [DOI] [PubMed] [Google Scholar]
- Peters JM, Cattley RC, Gonzalez FJ. Role of PPAR alpha in the mechanism of action of the nongenotoxic carcinogen and peroxisome proliferator Wy-14,643. Carcinogenesis. 1997;18:2029–2033. doi: 10.1093/carcin/18.11.2029. [DOI] [PubMed] [Google Scholar]
- Bayly AC, French NJ, Dive C, Roberts RA. Non-genotoxic hepatocarcinogenesis in vitro: the FaO hepatoma line responds to peroxisome proliferators and retains the ability to undergo apoptosis. J Cell Sci. 1993;104:307–315. doi: 10.1242/jcs.104.2.307. [DOI] [PubMed] [Google Scholar]
- Delaney J, Hodson MP, Thakkar H, Connor SC, Sweatman BC, Kenny SP, McGill PJ, Holder JC, Hutton KA, Haselden JN, Waterfield CJ. Tryptophan-NAD+ pathway metabolites as putative biomarkers and predictors of peroxisome proliferation. Arch Toxicol. 2004;79:208–223. doi: 10.1007/s00204-004-0625-5. [DOI] [PubMed] [Google Scholar]
- Connor SC, Hodson MP, Ringeissen S, Sweatman BC, McGill PJ, Waterfield CJ, Haselden JN. Development of a multivariate statistical model to predict peroxisome proliferation in the rat, based on urinary 1 H-NMR spectral patterns. Biomarkers. 2004;9:364–385. doi: 10.1080/13547500400006005. [DOI] [PubMed] [Google Scholar]
- Pogribny IP, Tryndyak VP, Boureiko A, Melnyk S, Bagnyukova TV, Montgomery B, Rusyn I. Mechanisms of peroxisome proliferator-induced DNA hypomethylation in rat liver. Mut Res. 2008;644:17–23. doi: 10.1016/j.mrfmmm.2008.06.009. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Podo F. Tumour phospholipid metabolism. NMR Biomed. 1999;12:413–439. doi: 10.1002/(SICI)1099-1492(199911)12:7<413::AID-NBM587>3.0.CO;2-U. [DOI] [PubMed] [Google Scholar]
- Klaunig JE, Kamendulis LM. The role of oxidative stress in carcinogenesis. Annu Rev Pharmacol Toxicol. 2004;44:239–267. doi: 10.1146/annurev.pharmtox.44.101802.121851. [DOI] [PubMed] [Google Scholar]
- Angermüller S, Islinger M, Völkl A. Peroxisomes and reactive oxygen species, a lasting challenge. Histochem Cell Biol. 2009;131:459–463. doi: 10.1007/s00418-009-0563-7. [DOI] [PubMed] [Google Scholar]
- Reddy JK, Hashimoto T. Peroxisomal β-oxidation and peroxisome proliferator-activated receptor α: an adaptive metabolic system. Annu Rev Nutr. 2001;21:193–230. doi: 10.1146/annurev.nutr.21.1.193. [DOI] [PubMed] [Google Scholar]
- Lake BG, Gray TJB, Stubberfield CR, Beamand JA, Gangolli SD. Induction of lauric acid hydroxylation and maintenance of cytochrome P-450 content by clofibrate in primary cultures of rat hepatocytes. Life Sci. 1983;33:249–254. doi: 10.1016/0024-3205(83)90383-1. [DOI] [PubMed] [Google Scholar]
- Giera DD, van Lier RBL. A convenient method for the determination of hepatic lauric acid ω-oxidation based on solvent partition. Fundam Appl Toxicol. 1991;16:348–355. doi: 10.1016/0272-0590(91)90119-O. [DOI] [PubMed] [Google Scholar]
- Tang Z, Guengerich FP. Dansylation of unactivated alcohols for improved mass spectral sensitivity and application to analysis of cytochrome P450 oxidation products in tissue extracts. Anal Chem. 2010;82:7706–7712. doi: 10.1021/ac1015497. [DOI] [PMC free article] [PubMed] [Google Scholar]