

Abstract
Idiopathic inflammatory myopathies (IIMs) comprise a group of autoimmune diseases that are characterized by symmetrical skeletal muscle weakness and muscle inflammation with no known cause. Like other autoimmune diseases, IIMs are treated with either glucocorticoids or immunosuppressive drugs. However, many patients with an IIM are frequently resistant to immunosuppressive treatments, and there is compelling evidence to indicate that not only adaptive immune but also several non-immune mechanisms play a role in the pathogenesis of these disorders. Here, we focus on some of the evidence related to pathologic mechanisms, such as the innate immune response, endoplasmic reticulum stress, non-immune consequences of MHC class I overexpression, metabolic disturbances, and hypoxia. These mechanisms may explain how IIM-related pathologic processes can continue even in the face of immunosuppressive therapies. These data indicate that therapeutic strategies in IIMs should be directed at both immune and non-immune mechanisms of muscle damage.
Introduction
The term 'idiopathic inflammatory myopathy' (IIM) usually refers to one of three related diseases: polymyositis (PM), dermatomyositis (DM), and inclusion body myositis (IBM) . Each variation of the disease has defining characteristics (for example, the heliotrope rash of DM or the intramuscular rimmed vacuoles of IBM), but there are some common characteristics. In PM and DM, patients typically present clinically with diffuse muscle weakness that is worse in proximal muscles than in distal muscles, whereas in IBM profound weakness may be present in both proximal and distal muscles. Histologically, patients show muscle inflammation, fiber degeneration, and overexpression of the MHC class I molecule [1-3]. This muscle pathology is not always uniform, and many patients display isolated patches of inflammation and degeneration in otherwise healthy muscle tissue. In affected patients, significant causes of morbidity and mortality include difficulty in performing daily activities, dysphagia, and respiratory failure [4,5]. Immunosuppressive therapies such as prednisolone and methotrexate are common first-line treatments, with cyclosporine and other powerful immunosuppressants as second-line agents [6-8]. Unfortunately, these current therapies have produced mixed results; for example, patients with DM are most likely to benefit from glucocorticoid therapy (with a reduction in inflammation, and improvement in muscle function), and most PM patients typically respond to immunosuppressive therapy [9-11]. On the other hand, patients with IBM (and a subset of PM patients) do not typically respond to either glucocorticoids or other immunosuppressant therapies [7]. Studies have shown that the therapeutic response to glucocorticoid treatment varies significantly; furthermore, the degrees of inflammatory infiltration and muscle function are dissociated, suggesting a role for other mechanisms in muscle dysfunction in these diseases [12,13]. Here we review some of the non-adaptive immune mechanisms that may be responsible for muscle weakness in the absence of overt inflammation, and we provide evidence that skeletal muscle, but not inflammation, is an active participant in the progression of muscle disease in these disorders.
Toll-like receptors on skeletal muscle
It is generally thought that skeletal muscle is merely a target of immune-mediated damage and that removal of immune cells from the milieu could result in preservation of skeletal muscle and improvement in skeletal muscle function. However, the fact that disease progression occurs in the absence of significant inflammation suggests that skeletal muscle itself contributes to the disease process. How the skeletal muscle senses signals from the injured microenvironment clearly requires further study. Mammalian cells are able to rapidly and efficiently recognize not only foreign invaders such as microbes but also damage- or injury-associated self-proteins via germline-encoded pattern recognition receptors. These receptors recognize damage-associated molecular patterns (DAMPs), which are either derived from pathogens (pathogen-associated molecular patterns, or PAMPs) or are self-proteins (stress or danger-associated molecular patterns, or SAMPs) [14,15]. Recognition of damage-associated molecular patterns by Toll-like receptors (TLRs) initiates a signaling cascade, leading to the activation or inhibition of genes that control the inflammatory response.
In chronic inflammatory conditions such as myositis, some of the self-proteins released from damaged cells may act as SAMPs. TLRs are known to sense several of these endogenous ligands and activate inflammatory signaling pathways [16]. It is important to consider this mechanism of inflammation since some IIM patients can continue to experience muscle fiber degeneration even after successful suppression of autoreactive lymphocytes. In IIMs, several endogenous inflammatory molecules (such as high mobility group box 1 (HMGB1), which is discussed in more detail later) have been proposed to play a role in disease pathogenesis [17,18]. Two separate investigations of patient biopsies have identified TLR-3 and TLR-7 (receptors for double-stranded RNA and GU-rich single-stranded RNA, respectively) on the surface of regenerating muscle fibers in patient biopsies [19,20]. TLRs have also been observed on skeletal muscle cells in other myopathies, where the activation of the innate immune pathway caused the skeletal muscle cells to secrete the inflammatory cytokine IL-1β [21]. Interestingly, the U1-snRNP molecule, which is capable of activating both TLR-3 and TLR-7, has been previously reported to be associated with several autoimmune rheumatoid diseases, and a small number of IIM patients develop anti-U1 autoantibodies [22,23]. Both HMGB1 and U1-snRNP are nuclear proteins that are not found in the extracellular space in healthy tissue, thereby suggesting that these endogenous TLR ligands may be released from inflamed or necrotic fibers, which then induce inflammation in neighboring fibers.
Skeletal muscle cells secrete several cytokines and chemokines
Cytokines play a central role in the immune response by promoting the activation of antigen-specific and non-specific effector mechanisms and tissue repair. Most discussions about cytokines revolve around their production from lymphocytes, but it is important to consider cytokines that are produced from the skeletal muscle itself. Human skeletal muscle cells constitutively produce the cytokine IL-6, and its expression can be increased by stimulation with IL-1α, IL-1β, TNFα and IFNγ in a dose-dependent manner [24,25]. IL-6 has a role in regulating metabolic rates in skeletal muscle, but it can also act as an inflammatory molecule. Excessive secretion of IL-6 has previously been linked to other autoimmune diseases (for example, rheumatoid arthritis) and is known to promote the development of potentially autoreactive TH-17 cells [26,27]. The pathogenic role of IL-6 in myositis has also been supported by IL-6 blockade in one mouse model of C-protein-induced myositis [28]. In IL-6 knockout mice, both the incidence and the severity of myositis were reduced compared to C57BL/6 controls. It should also be noted that previous examinations of muscle biopsies from IIM patients have found fibers that were positive for IL-1α, TNFα, and IL-1β staining, suggesting that lymphocytes may not be necessary to induce overexpression of IL-6 [29,30]. Furthermore, it has been suggested that a feedback loop linking IL-6 production and TLR-3 stimulation exists in inflamed skeletal muscle, and may play a role in the pathogenesis of IIMs [19].
Apart from IL -6, recent investigations have confirmed that IFNα plays a prominent role in the pathogenesis of IIMs [31-35]. Inquiries into the sources of some of the type I IFNs have indicated that dendritic cells and mast cells are the major producers of IFNα and IFNβ in DM patients. These same cells are also known to be major producers of IL-1β [36,37]. IFNα is known to upregulate a number of antiviral target genes, such as MXA, IFIT2, and ISG15. Strong intramuscular staining for these IFNα targets has previously been observed in biopsies from DM and PM patients [34]. One such target, ISG15, is known to disrupt the normal ubiquitination process. During viral infections, ISG15 is substituted for ubiquitin and interferes with the maturation and function of viral proteins, such as the HIV Gag and Ebola VP40 proteins. Histological examination of muscle biopsies has revealed that ISG15 is absent from healthy muscle but is detectable in patients with PM, IBM, and DM [35]. Further experiments are still needed to establish the functional consequences of ISG15 expression in muscle cells. The clinical significance of IFNα was underscored in a small study in which IIM patients failed to respond to infliximab, and a worsening in symptoms was correlated with an increase in IFNα serum levels [31].
In addition to classical cytokines, non-traditional cytokines/chemokines also appear to play a significant role in controlling the inflammatory process. Two such proteins, histidyl tRNA synthetase (HisRS) and HMGB1, have been associated with myositis [18,38,39]. The inflammatory properties of the HMGB1 protein have previously been demonstrated in other inflammatory diseases [40-42]. This non-histone nuclear protein acts as a potent inflammatory molecule when released from necrotic cells. HMGB1-mediated inflammation is signaled through either TLR-4 or receptor for advanced glycation endproducts (RAGE) [43]. Examination of human muscle biopsies has revealed that cytoplasmic HMGB1 expression is widespread in PM and DM patients but absent from healthy controls. Furthermore, when isolated primary muscles fibers are exposed to extracellular HMGB1 in vitro, the fibers begin expressing MHC class I antigens on their surface and demonstrate an accelerated, dose-dependent efflux of calcium ions [17], suggesting a role for HMGB1 in the perpetuation of inflammation and muscle fiber degeneration even in the absence of infiltrating lymphocytes.
Role of MHC class I upregulation on skeletal muscle fibers
MHC class I proteins are essential for immune surveillance. They are expressed on nearly all cells, with few exceptions (for example, mature neurons, oligodendrocytes, pancreatic islet cells, and skeletal muscle fibers). In these few cases, constitutive expression is either low or absent. As mentioned previously, one of the histological hallmarks of skeletal muscle in IIM is the widespread and constitutive presence of MHC class I protein [44-47]. Even though the immunological roles of these proteins (for example, antigen presentation) are well-studied, their non-immunological roles are not well defined. It is worthwhile to mention that other autoimmune diseases (for example, type I diabetes or multiple sclerosis) can be initiated by forced expression of the MHC class I in the few cell types (that is, pancreatic islet or oligodendrocyte cells) where it is normally absent [48,49]. It is not clear why MHC class I expression is restricted in these cell types, but one possible explanation can be found in recent research into the formation and maintenance of the neuromuscular junction (NMJ). Recent studies of neurons in the central nervous system have indicated that MHC class I expression is dynamic (as is that of its secondary receptors, CD3z and PirB) and is involved in communication across the synaptic junction and the maintenance of synaptic plasticity [50,51]. These studies have indicated that classical MHC class I proteins are expressed in motor neurons, with predominant expression occurring at the presynaptic end of the NMJ. In another recent study, it was reported that a knockout of the MHC class I gene resulted in abnormally high densities of NMJs during development, and abnormal formation of NMJs after denervation and re-innervation [52]. Together, these reports suggest that MHC class I acts as a signaling molecule to promote pruning NMJs during development and potentially to force the remodeling of established NMJs. This newly discovered role for MHC class I may explain why its expression is so tightly controlled by the cells surrounding the NMJ (neurons, oligodendrocytes, and muscle fibers). Under this model, the constitutive expression of MHC class I from the muscle may have a disruptive effect on the structure and/or function of the NMJ, though this hypothesis has not yet been tested.
The endoplasmic reticulum stress response pathway
Endoplasmic reticulum (ER) stress is a known cause of pathology in certain conditions, including viral infections and neurodegenerative disorders, and there is emerging evidence that ER stress within the muscle cells contributes to pathology in IIMs [53]. The ER is known as the site of protein synthesis, folding, post-translational modification, lipid biosynthesis, and intracellular calcium storage within the cell. Since muscle contractions are signaled through calcium release, the regulation of intra-cellular calcium concentrations is of paramount importance in muscle cells. Within muscle cells, the ER is a specially modified organelle that is also known as the sarcoplasmic reticulum. Excessive changes in the load of proteins, composition of lipids, or calcium concentration within the ER can trigger multiple stress response path-ways acting through ATF4 (activating transcription factor 4), ATF6 (activating transcription factor 6), and PERK (protein kinase RNA-like endoplasmic reticulum kinase). If the ER stress cannot be resolved, then the cell will initiate an apoptotic response via the expression of DDIT3 (DNA-damage-inducible transcript; also called CHOP) and activation of caspase 4 (or caspase-11 and caspase-12 in mice) [54-56]. Interestingly, there is some evidence to suggest that mature skeletal muscle, like pancreatic islet cells, may tax the sarcoplasmic reticulum to near its limits, even under conditions of normal protein synthesis [57].
We and others have suggested that, in myositis, over-expression of the MHC class I molecule in skeletal muscle can induce ER stress. This hypothesis is based upon observations made in the transgenic mouse model of myositis [45,46,58] and in human patients with IIMs [47,59,60]. As described previously, skeletal muscle cells do not express detectable levels of MHC class I molecules under normal conditions, although transient expression can be observed following injury or viral infections. In the mouse model of myositis, the forced expression of MHC class I molecules is sufficient to precipitate the onset of myositis in mice. Histological and biochemical examinations of the mouse muscle tissue revealed ER stress in muscle fibers, as evidenced by the upregulation of multiple ER chaperone proteins (for example, Grp78, Grp94, calnexin, calreticulin) and activation of the NF-κB pathway. Very similar changes were found in patients in whom muscle cells that were positive for MHC class I were also found to show increased expression of Grp78, Grp94, and calreticulin [47,61-63]. It should also be noted that lymphocytic infiltration was not correlated with staining for ER stress markers [47].
Several laboratories have examined the potential role of ER stress and ER stress-associated degradation in the accumulation of misfolded proteins in IBM patients. Histological analysis of patients with IBM has shown scattered inclusion bodies (thought to be protein aggregates) that are not seen in PM or DM. The appearance of inclusion bodies can be hypothesized to be indicative of defective protein folding or defective degradation of misfolded proteins, although the available evidence seems to favor defective degradation. Examination of patients with IBM has revealed that the mRNA and protein expression of amyloid-B protein and BACE1 (β-site amyloid-β precursor protein (AβPP)-cleaving enzyme 1) are increased in IBM muscle. However, it should be noted that the potential pathogenic role of β-amyloid in IBM is a controversial topic [64,65]. In a separate investigation, patients with IBM were found to exhibit an increased expression of autophagosomes, a common ER stress response. Furthermore, the proteases cathepsin B and cathepsin D in patients with IBM showed diminished protease activity when compared to those in controls. These data indicate that increased ER stress and protein aggregation may be the result of impaired protein degradation in IIMs [66].
Metabolic disturbances
Another potential mechanistic explanation for the persistent muscle weakness seen in IIMs comes from the observation that there is an apparent disturbance in patients' metabolism and ATP production. Muscle fibers are generally composed of type I (slow-twitch) and type II (fast-twitch) fibers. Muscles composed of slow fibers depend on oxidative phosphorylation and are responsible for long-term, low-force muscle activity. Conversely, muscles composed of fast-twitch fibers often depend on glycolysis in order to carry out rapid, strenuous movements. However, in the case of IIM patients, there are indications that the fast-twitch fibers are more prone to degenerate, and that these patients' ability to produce ATP in the skeletal muscle may be impaired. Mass spectrometry analysis of myositis biopsies has shown a significant loss of type II (fast-twitch)-specific proteins (for example, myosin heavy chain 1 (MYH1), troponin T3 (TNNT3), and actinin 3 (ACTN3)) and a modest increase in type I (slow-twitch) muscle proteins (for example, myosin heavy chain 7 (MYH7), troponin T1 (TNNTL1), and actinin 2 (ACNT2)) [64]. Similarly, IIM patients have shown a modest decrease in enzymes required for glycolysis in the muscle (for example, glycogen debranching enzyme (AGL), muscle phosphofructokinase (PFKM), fructose-1,6-bis-phosphatase isozyme 2 (FBP2), and phosphoglycerate mutase 2 (PGAM)), with the most significant decreases being found in patients with IBM [64].
In recent decades, the results of multiple studies have indicated that the loss of another type II-specific muscle enzyme, AMPD1, is potentially responsible for disturbances in ATP metabolism and muscle weakness [67,68]. These findings have been corroborated by magnetic resonance spectroscopy in patients with juvenile-onset DM; in that study, the stable isotope 31P was used to compare the levels of ATP, ADP, AMP, and phosphocreatine in the quadriceps of DM patients and healthy controls. The DM patients were found to have roughly 4.03 ± 0.26 mmoles/kg of total ATP, compared to 6.26 ± 0.23 mmoles/kg in the controls. The concentrations of ADP, AMP, and phosphocreatine were similarly decreased in the DM patients [69]. Interestingly, there is some evidence that chronic ER stress can alter the metabolism of muscle fibers: activation of the unfolded protein response pathway in skeletal muscle has been reported to occur during exercise, with repeated activation of this pathway being linked to a gradual shift in muscle metabolism to adapt to exercise training. In addition, the transcriptional coactivator PGC-1α (peroxisome proliferator-activated receptor gamma coactivator 1-alpha) has been found to interact with ATF6; prior investigations have linked PGC-1α to a role in promoting type I muscle fiber genes, and ATF6 has been shown to be activated by ER stress. It is interesting to note that mice deficient in ATF6 have an impaired ability to recover from exercise, while mice lacking PGC-1α experience exercise intolerance that can be partially reversed by inhibitors of CHOP [70]. Taken together, these results suggest that ER stress has important consequences for metabolic homeostasis and force generation in skeletal muscle cells.
Role of hypoxia
Histological analysis of muscle from some IIM patients has shown that these patients have fewer capillaries per unit area in their skeletal muscle than do healthy patients [71,72]. This observation has led investigators to propose that the resulting lower oxygen levels are responsible for the muscle weakness seen in myositis patients. Gene profiling of patient biopsies has revealed that both promoters of angiogenesis (for example, hypoxia inducible factor (HIF)-1α, CD146, and fibronectin) and inhibitors of angiogenesis (for example, CXCL10, transforming growth factor-β, and angiopoietin 2) are expressed simultaneously, indicating that capillaries are being actively remodeled in patients with myositis [73]. The transcription factor HIF-1α is especially noteworthy because it is a direct sensor of hypoxia and helps to maintain oxygen homeostasis. A recent publication has corroborated these expression profiling data by demonstrating that HIF-1 proteins accumulate in IIM patients [71,74]. This accumulation is possible only under low-oxygen conditions, indicating that the skeletal muscle of all IIM patients are very likely subjected to hypoxic conditions. This hypoxia hypothesis is supported by the reported benefits of physical exercise in myositis patients, suggesting a role for vascular endothelial cell dysfunction and hypoxia in the pathogenesis of IIMs [72,75].
Conclusion
The evidence to support a role for non-immune mechanisms in the pathogenesis of IIMs is growing. These data place the myofiber at the center of disease pathobiology, with an initial insult to the myofibers (whether viral, damage, or a genetic predisposition) leading to myofiber TLR signaling, which in and of itself can induce weakness via type 1 IFN pathways by interfering with energy production and mitochondrial respiration. Infiltrating leukocytes augment and exacerbate this signaling. The relationship between all of these pathologic mechanisms is shown in Figure 1, with a special emphasis on their convergence on the production of inflammatory cytokines. These data also explain why immune suppression is variable in terms of its efficacy in restoring muscle strength, because the innate dysfunction of myofibers remains despite the achievement of successful immune suppression.
Figure 1.
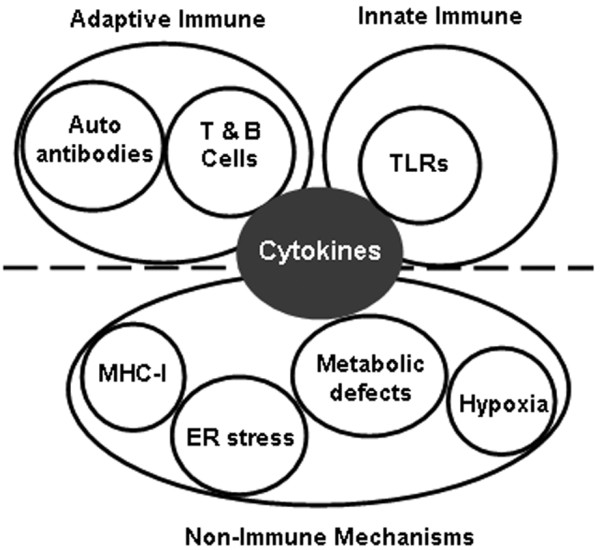
Relationship between the possible pathologic mechanisms leading to idiopathic inflammatory myopathies.ER, endoplasmic reticulum; MHC, major histocompatibility complex; TLR, Toll-like receptor.
Abbreviations
DM: dermatomyositis; ER: endoplasmic reticulum; HIF: hypoxia inducible factor; HMGB1: high mobility group box 1; IBM: inclusion body myositis; IFN: interferon; IIM: idiopathic inflammatory myopathy; IL: interleukin; MHC: major histocompatibility complex; NF: nuclear factor; NMJ: neuromuscular junction; PM: polymyositis; TLR: Toll-like receptor; TNF: tumor necrosis factor.
Competing interests
The authors declare that they have no competing interests.
Note
Autoimmune Basis of Rheumatic Diseases
This article is part of a series on Myositis, edited by Ingrid Lundberg, which can be found online at http://arthritis-research.com/series/myositis
This series forms part of a special collection of reviews covering major autoimmune rheumatic diseases, available at: http://arthritis-research.com/series/abrd
Contributor Information
William Coley, Email: wcoley@cnmcresearch.org.
Sree Rayavarapu, Email: SRayavarapu@cnmcresearch.org.
Kanneboyina Nagaraju, Email: KNagaraju@cnmcresearch.org.
Acknowledgements
KN is supported by the National Institutes of Health (RO1-AR050478 and 5U54HD053177); the myositis association; and the US Department of Defense (W81XWH-05-1-0616).
References
- Mastaglia FL, Garlepp MJ, Phillips BA, Zilko PJ. Inflammatory myopathies: clinical, diagnostic and therapeutic aspects. Muscle Nerve. 2003;27:407–425. doi: 10.1002/mus.10313. [DOI] [PubMed] [Google Scholar]
- Tanimoto K, Nakano K, Kano S, Mori S, Ueki H, Nishitani H, Sato T, Kiuchi T, Ohashi Y. Classification criteria for polymyositis and dermatomyositis. J Rheumatol. 1995;22:668–674. [PubMed] [Google Scholar]
- Miller F, Rider L, Plotz P, Oddis C, Isenberg D. Diagnostic criteria for polymyositis and dermatomyositis. Lancet. 2003;362:1762–1763. doi: 10.1016/S0140-6736(03)14862-3. [DOI] [PubMed] [Google Scholar]
- Santmyire-Rosenberger B, Dugan EM. Skin involvement in dermatomyositis. Curr Opin Rheumatol. 2003;15:714–722. doi: 10.1097/00002281-200311000-00006. [DOI] [PubMed] [Google Scholar]
- Ramanan AV, Feldman BM. Clinical features and outcomes of juvenile dermatomyositis and other childhood onset myositis syndromes. Rheum Dis Clin North Am. 2002;28:833–857. doi: 10.1016/S0889-857X(02)00024-8. [DOI] [PubMed] [Google Scholar]
- Mastaglia FL, Zilko PJ. Inflammatory myopathies: how to treat the difficult cases. J Clin Neurosci. 2003;10:99–101. doi: 10.1016/S0967-5868(02)00271-0. [DOI] [PubMed] [Google Scholar]
- Dalakas MC. Immunotherapy of myositis: issues, concerns and future prospects. Nat Rev Rheumatol. 2010;6:129–137. doi: 10.1038/nrrheum.2010.2. [DOI] [PubMed] [Google Scholar]
- Miller FW, Rider LG, Chung YL, Cooper R, Danko K, Farewell V, Lundberg I, Morrison C, Oakley L, Oakley I, Pilkington C, Vencovsky J, Vincent K, Scott DL, Isenberg DA. International Myositis Outcome Assessment Collaborative Study Group: Proposed preliminary core set measures for disease outcome assessment in adult and juvenile idiopathic inflammatory myopathies. Rheumatology (Oxford) 2001;40:1262–1273. doi: 10.1093/rheumatology/40.11.1262. [DOI] [PubMed] [Google Scholar]
- Nagaraju K. Update on immunopathogenesis in inflammatory myopathies. Curr Opin Rheumatol. 2001;13:461–468. doi: 10.1097/00002281-200111000-00002. [DOI] [PubMed] [Google Scholar]
- Bunch TW. Prednisone and azathioprine for polymyositis: long-term followup. Arthritis Rheum. 1981;24:45–48. doi: 10.1002/art.1780240107. [DOI] [PubMed] [Google Scholar]
- Kissel JT, Halterman RK, Rammohan KW, Mendell JR. The relationship of complement-mediated microvasculopathy to the histologic features and clinical duration of disease in dermatomyositis. Arch Neurol. 1991;48:26–30. doi: 10.1001/archneur.1991.00530130034016. [DOI] [PubMed] [Google Scholar]
- Imai T, Hirayama K, Osumi E. [Muscle histopathology and responsiveness to steroid therapy in polymyositis and dermatomyositis] Rinsho Shinkeigaku. 1995;35:243–246. [PubMed] [Google Scholar]
- Lundberg I, Kratz AK, Alexanderson H, Patarroyo M. Decreased expression of interleukin-1alpha, interleukin-1beta, and cell adhesion molecules in muscle tissue following corticosteroid treatment in patients with polymyositis and dermatomyositis. Arthritis Rheum. 2000;43:336–348. doi: 10.1002/1529-0131(200002)43:2<336::AID-ANR13>3.0.CO;2-V. [DOI] [PubMed] [Google Scholar]
- Seong SY, Matzinger P. Hydrophobicity: an ancient damage-associated molecular pattern that initiates innate immune responses. Nat Rev Immunol. 2004;4:469–478. doi: 10.1038/nri1372. [DOI] [PubMed] [Google Scholar]
- Takeda K, Kaisho T, Akira S. Toll-like receptors. Annu Rev Immunol. 2003;21:335–376. doi: 10.1146/annurev.immunol.21.120601.141126. [DOI] [PubMed] [Google Scholar]
- Barton GM, Medzhitov R. Toll-like receptor signaling pathways. Science. 2003;300:1524–1525. doi: 10.1126/science.1085536. [DOI] [PubMed] [Google Scholar]
- Grundtman C, Bruton J, Yamada T, Ostberg T, Pisetsky DS, Harris HE, Andersson U, Lundberg IE, Westerblad H. Effects of HMGB1 on in vitro responses of isolated muscle fibers and functional aspects in skeletal muscles of idiopathic inflammatory myopathies. FASEB J. 2010;24:570–578. doi: 10.1096/fj.09-144782. [DOI] [PubMed] [Google Scholar]
- Howard OM, Dong HF, Yang D, Raben N, Nagaraju K, Rosen A, Casciola-Rosen L, Härtlein M, Kron M, Yang D, Yiadom K, Dwivedi S, Plotz PH, Oppenheim JJ. Histidyl-tRNA synthetase and asparaginyl-tRNA synthetase, autoantigens in myositis, activate chemokine receptors on T lymphocytes and immature dendritic cells. J Exp Med. 2002;196:781–791. doi: 10.1084/jem.20020186. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Tournadre A, Lenief V, Miossec P. Expression of Toll-like receptor 3 and Toll-like receptor 7 in muscle is characteristic of inflammatory myopathy and is differentially regulated by Th1 and Th17 cytokines. Arthritis Rheum. 2010;62:2144–2151. doi: 10.1002/art.27465. [DOI] [PubMed] [Google Scholar]
- Cappelletti C, Baggi F, Zolezzi F, Biancolini D, Beretta O, Severa M, Coccia EM, Confalonieri P, Morandi L, Mora M, Mantegazza R, Bernasconi P. Type I interferon and Toll-like receptor expression characterizes inflammatory myopathies. Neurology. 2011;76:2079–2088. doi: 10.1212/WNL.0b013e31821f440a. [DOI] [PubMed] [Google Scholar]
- Rawat R, Cohen TV, Ampong B, Francia D, Henriques-Pons A, Hoffman EP, Nagaraju K. Inflammasome up-regulation and activation in dysferlin-deficient skeletal muscle. Am J Pathol. 2010;176:2891–2900. doi: 10.2353/ajpath.2010.090058. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Brouwer R, Hengstman GJ, Vree Egberts W, Ehrfeld H, Bozic B, Ghirardello A, Grøndal G, Hietarinta M, Isenberg D, Kalden JR, Lundberg I, Moutsopoulos H, Roux-Lombard P, Vencovsky J, Wikman A, Seelig HP, van Engelen BG, van Venrooij WJ. Autoantibody profiles in the sera of European patients with myositis. Ann Rheum Dis. 2001;60:116–123. doi: 10.1136/ard.60.2.116. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kattah NH, Kattah MG, Utz PJ. The U1-snRNP complex: structural properties relating to autoimmune pathogenesis in rheumatic diseases. Immunol Rev. 2010;233:126–145. doi: 10.1111/j.0105-2896.2009.00863.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bartoccioni E, Michaelis D, Hohlfeld R. Constitutive and cytokine-induced production of interleukin-6 by human myoblasts. Immunol Lett. 1994;42:135–138. doi: 10.1016/0165-2478(94)90076-0. [DOI] [PubMed] [Google Scholar]
- Nagaraju K, Raben N, Merritt G, Loeffler L, Kirk K, Plotz P. A variety of cytokines and immunologically relevant surface molecules are expressed by normal human skeletal muscle cells under proinflammatory stimuli. Clin Exp Immunol. 1998;113:407–414. doi: 10.1046/j.1365-2249.1998.00664.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bettelli E, Oukka M, Kuchroo VK. T(H)-17 cells in the circle of immunity and autoimmunity. Nat Immunol. 2007;8:345–350. doi: 10.1038/ni0407-345. [DOI] [PubMed] [Google Scholar]
- Nishimoto N, Yoshizaki K, Miyasaka N, Yamamoto K, Kawai S, Takeuchi T, Hashimoto J, Azuma J, Kishimoto T. Treatment of rheumatoid arthritis with humanized anti-interleukin-6 receptor antibody: a multicenter, double-blind, placebo-controlled trial. Arthritis Rheum. 2004;50:1761–1769. doi: 10.1002/art.20303. [DOI] [PubMed] [Google Scholar]
- Okiyama N, Sugihara T, Iwakura Y, Yokozeki H, Miyasaka N, Kohsaka H. Therapeutic effects of interleukin-6 blockade in a murine model of polymyositis that does not require interleukin-17A. Arthritis Rheum. 2009;60:2505–2512. doi: 10.1002/art.24689. [DOI] [PubMed] [Google Scholar]
- Goebels N, Michaelis D, Engelhardt M, Huber S, Bender A, Pongratz D, Johnson MA, Wekerle H, Tschopp J, Jenne D, Hohlfeld R. Differential expression of perforin in muscle-infiltrating T cells in polymyositis and dermatomyositis. J Clin Invest. 1996;97:2905–2910. doi: 10.1172/JCI118749. [DOI] [PMC free article] [PubMed] [Google Scholar]
- De Bleecker JL, Meire VI, Declercq W, Van Aken EH. Immunolocalization of tumor necrosis factor-alpha and its receptors in inflammatory myopathies. Neuromuscul Disord. 1999;9:239–246. doi: 10.1016/S0960-8966(98)00126-6. [DOI] [PubMed] [Google Scholar]
- Dastmalchi M, Grundtman C, Alexanderson H, Mavragani CP, Einarsdottir H, Helmers SB, Elvin K, Crow MK, Nennesmo I, Lundberg IE. A high incidence of disease flares in an open pilot study of infliximab in patients with refractory inflammatory myopathies. Ann Rheum Dis. 2008;67:1670–1677. doi: 10.1136/ard.2007.077974. [DOI] [PubMed] [Google Scholar]
- Eloranta ML, Barbasso Helmers S, Ulfgren AK, Ronnblom L, Alm GV, Lundberg IE. A possible mechanism for endogenous activation of the type I interferon system in myositis patients with anti-Jo-1 or anti-Ro 52/anti-Ro 60 autoantibodies. Arthritis Rheum. 2007;56:3112–3124. doi: 10.1002/art.22860. [DOI] [PubMed] [Google Scholar]
- Walsh RJ, Kong SW, Yao Y, Jallal B, Kiener PA, Pinkus JL, Beggs AH, Amato AA, Greenberg SA. Type I interferon-inducible gene expression in blood is present and reflects disease activity in dermatomyositis and polymyositis. Arthritis Rheum. 2007;56:3784–3792. doi: 10.1002/art.22928. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Greenberg SA, Pinkus JL, Pinkus GS, Burleson T, Sanoudou D, Tawil R, Barohn RJ, Saperstein DS, Briemberg HR, Ericsson M, Park P, Amato AA. Interferon-alpha/beta-mediated innate immune mechanisms in dermatomyositis. Ann Neurol. 2005;57:664–678. doi: 10.1002/ana.20464. [DOI] [PubMed] [Google Scholar]
- Salajegheh M, Kong SW, Pinkus JL, Walsh RJ, Liao A, Nazareno R, Amato AA, Krastins B, Morehouse C, Higgs BW, Jallal B, Yao Y, Sarracino DA, Parker KC, Greenberg SA. Interferon-stimulated gene 15 (ISG15) conjugates proteins in dermatomyositis muscle with perifascicular atrophy. Ann Neurol. 2010;67:53–63. doi: 10.1002/ana.21805. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Greenberg SA, Pinkus GS, Amato AA, Pinkus JL. Myeloid dendritic cells in inclusion-body myositis and polymyositis. Muscle Nerve. 2007;35:17–23. doi: 10.1002/mus.20649. [DOI] [PubMed] [Google Scholar]
- Rider LG, Miller FW. Mast cells and type I interferon responses in the skin of patients with juvenile dermatomyositis: are current therapies just scratching the surface? Arthritis Rheum. 2010;62:2619–2622. doi: 10.1002/art.27525. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Soejima M, Kang EH, Gu X, Katsumata Y, Clemens PR, Ascherman DP. Role of innate immunity in a murine model of histidyl-transfer RNA synthetase (Jo-1)-mediated myositis. Arthritis Rheum. 2011;63:479–487. doi: 10.1002/art.30113. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Howard OM, Dong HF, Su SB, Caspi RR, Chen X, Plotz P, Oppenheim JJ. Autoantigens signal through chemokine receptors: uveitis antigens induce CXCR3- and CXCR5-expressing lymphocytes and immature dendritic cells to migrate. Blood. 2005;105:4207–4214. doi: 10.1182/blood-2004-07-2697. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wang H, Bloom O, Zhang M, Vishnubhakat JM, Ombrellino M, Che J, Frazier A, Yang H, Ivanova S, Borovikova L, Manogue KR, Faist E, Abraham E, Andersson J, Andersson U, Molina PE, Abumrad NN, Sama A, Tracey KJ. HMG-1 as a late mediator of endotoxin lethality in mice. Science. 1999;285:248–251. doi: 10.1126/science.285.5425.248. [DOI] [PubMed] [Google Scholar]
- Yang H, Ochani M, Li J, Qiang X, Tanovic M, Harris HE, Susarla SM, Ulloa L, Wang H, DiRaimo R, Czura CJ, Wang H, Roth J, Warren HS, Fink MP, Fenton MJ, Andersson U, Tracey KJ. Reversing established sepsis with antagonists of endogenous high-mobility group box 1. Proc Natl Acad Sci USA. 2004;101:296–301. doi: 10.1073/pnas.2434651100. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Li J, Xie H, Wen T, Liu H, Zhu W, Chen X. Expression of high mobility group box chromosomal protein 1 and its modulating effects on downstream cytokines in systemic lupus erythematosus. J Rheumatol. 2010;37:766–775. doi: 10.3899/jrheum.090663. [DOI] [PubMed] [Google Scholar]
- Park JS, Gamboni-Robertson F, He Q, Svetkauskaite D, Kim JY, Strassheim D, Sohn JW, Yamada S, Maruyama I, Banerjee A, Ishizaka A, Abraham E. High mobility group box 1 protein interacts with multiple Toll-like receptors. Am J Physiol Cell Physiol. 2006;290:C917–924. doi: 10.1152/ajpcell.00401.2005. [DOI] [PubMed] [Google Scholar]
- Choi JH, Park YE, Kim SI, Kim JI, Lee CH, Park KH, Kim DS. Differential immunohistological features of inflammatory myopathies and dysferlinopathy. J Korean Med Sci. 2009;24:1015–1023. doi: 10.3346/jkms.2009.24.6.1015. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Nagaraju K, Raben N, Loeffler L, Parker T, Rochon PJ, Lee E, Danning C, Wada R, Thompson C, Bahtiyar G, Craft J, Hooft Van Huijsduijnen R, Plotz P. Conditional up-regulation of MHC class I in skeletal muscle leads to self-sustaining autoimmune myositis and myositis-specific autoantibodies. Proc Natl Acad Sci USA. 2000;97:9209–9214. doi: 10.1073/pnas.97.16.9209. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Nagaraju K, Casciola-Rosen L, Lundberg I, Rawat R, Cutting S, Thapliyal R, Chang J, Dwivedi S, Mitsak M, Chen YW, Plotz P, Rosen A, Hoffman E, Raben N. Activation of the endoplasmic reticulum stress response in autoimmune myositis: potential role in muscle fiber damage and dysfunction. Arthritis Rheum. 2005;52:1824–1835. doi: 10.1002/art.21103. [DOI] [PubMed] [Google Scholar]
- Vitadello M, Doria A, Tarricone E, Ghirardello A, Gorza L. Myofiber stress-response in myositis: parallel investigations on patients and experimental animal models of muscle regeneration and systemic inflammation. Arthritis Res Ther. 2010;12:R52. doi: 10.1186/ar2963. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Allison J, Campbell IL, Morahan G, Mandel TE, Harrison LC, Miller JF. Diabetes in transgenic mice resulting from over-expression of class I histocompatibility molecules in pancreatic beta cells. Nature. 1988;333:529–533. doi: 10.1038/333529a0. [DOI] [PubMed] [Google Scholar]
- Turnley AM, Morahan G, Okano H, Bernard O, Mikoshiba K, Allison J, Bartlett PF, Miller JF. Dysmyelination in transgenic mice resulting from expression of class I histocompatibility molecules in oligodendrocytes. Nature. 1991;353:566–569. doi: 10.1038/353566a0. [DOI] [PubMed] [Google Scholar]
- Oliveira AL, Thams S, Lidman O, Piehl F, Hokfelt T, Karre K, Linda H, Cullheim S. A role for MHC class I molecules in synaptic plasticity and regeneration of neurons after axotomy. Proc Natl Acad Sci USA. 2004;101:17843–17848. doi: 10.1073/pnas.0408154101. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Shatz CJ. MHC class I: an unexpected role in neuronal plasticity. Neuron. 2009;64:40–45. doi: 10.1016/j.neuron.2009.09.044. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Thams S, Brodin P, Plantman S, Saxelin R, Karre K, Cullheim S. Classical major histocompatibility complex class I molecules in motoneurons: new actors at the neuromuscular junction. J Neurosci. 2009;29:13503–13515. doi: 10.1523/JNEUROSCI.0981-09.2009. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Yoshida H. ER stress and diseases. FEBS J. 2007;274:630–658. doi: 10.1111/j.1742-4658.2007.05639.x. [DOI] [PubMed] [Google Scholar]
- Zinszner H, Kuroda M, Wang X, Batchvarova N, Lightfoot RT, Remotti H, Stevens JL, Ron D. CHOP is implicated in programmed cell death in response to impaired function of the endoplasmic reticulum. Genes Dev. 1998;12:982–995. doi: 10.1101/gad.12.7.982. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kalai M, Lamkanfi M, Denecker G, Boogmans M, Lippens S, Meeus A, Declercq W, Vandenabeele P. Regulation of the expression and processing of caspase-12. J Cell Biol. 2003;162:457–467. doi: 10.1083/jcb.200303157. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Rao RV, Hermel E, Castro-Obregon S, del Rio G, Ellerby LM, Ellerby HM, Bredesen DE. Coupling endoplasmic reticulum stress to the cell death program. Mechanism of caspase activation. J Biol Chem. 2001;276:33869–33874. doi: 10.1074/jbc.M102225200. [DOI] [PubMed] [Google Scholar]
- Iwawaki T, Akai R, Kohno K, Miura M. A transgenic mouse model for monitoring endoplasmic reticulum stress. Nat Med. 2004;10:98–102. doi: 10.1038/nm970. [DOI] [PubMed] [Google Scholar]
- Alger HM, Rayavarapu S, Nagaraju K. Measurement of activation of the endoplasmic reticulum stress response in autoimmune myositis. Methods Enzymol. 2011;489:207–225. doi: 10.1016/B978-0-12-385116-1.00012-1. [DOI] [PubMed] [Google Scholar]
- Vattemi G, Engel WK, McFerrin J, Askanas V. Endoplasmic reticulum stress and unfolded protein response in inclusion body myositis muscle. Am J Pathol. 2004;164:1–7. doi: 10.1016/S0002-9440(10)63089-1. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Nogalska A, Engel WK, McFerrin J, Kokame K, Komano H, Askanas V. Homocysteine-induced endoplasmic reticulum protein (Herp) is up-regulated in sporadic inclusion-body myositis and in endoplasmic reticulum stress-induced cultured human muscle fibers. J Neurochem. 2006;96:1491–1499. doi: 10.1111/j.1471-4159.2006.03668.x. [DOI] [PubMed] [Google Scholar]
- Li CK, Knopp P, Moncrieffe H, Singh B, Shah S, Nagaraju K, Varsani H, Gao B, Wedderburn LR. Overexpression of MHC class I heavy chain protein in young skeletal muscle leads to severe myositis. Implications for juvenile myositis. Am J Pathol. 2009;175:1030–1040. doi: 10.2353/ajpath.2009.090196. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Salomonsson S, Grundtman C, Zhang SJ, Lanner JT, Li C, Katz A, Wedderburn LR, Nagaraju K, Lundberg IE, Westerblad H. Upregulation of MHC class I in transgenic mice results in reduced force-generating capacity in slow-twitch muscle. Muscle Nerve. 2009;39:674–682. doi: 10.1002/mus.21129. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Nogalska A, Wojcik S, Engel WK, McFerrin J, Askanas V. Endoplasmic reticulum stress induces myostatin precursor protein and NF-kappaB in cultured human muscle fibers: relevance to inclusion body myositis. Exp Neurol. 2007;204:610–618. doi: 10.1016/j.expneurol.2006.12.014. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Parker KC, Walsh RJ, Salajegheh M, Amato AA, Krastins B, Sarracino DA, Greenberg SA. Characterization of human skeletal muscle biopsy samples using shotgun proteomics. J Proteome Res. 2009;8:3265–3277. doi: 10.1021/pr800873q. [DOI] [PubMed] [Google Scholar]
- Fratta P, Engel WK, McFerrin J, Davies KJ, Lin SW, Askanas V. Proteasome inhibition and aggresome formation in sporadic inclusion-body myositis and in amyloid-beta precursor protein-overexpressing cultured human muscle fibers. Am J Pathol. 2005;167:517–526. doi: 10.1016/S0002-9440(10)62994-X. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Nogalska A, D'Agostino C, Terracciano C, Engel WK, Askanas V. Impaired autophagy in sporadic inclusion-body myositis and in endoplasmic reticulum stress-provoked cultured human muscle fibers. Am J Pathol. 2010;177:1377–1387. doi: 10.2353/ajpath.2010.100050. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Norman B, Nygren AT, Nowak J, Sabina RL. The effect of AMPD1 genotype on blood flow response to sprint exercise. Eur J Appl Physiol. 2008;103:173–180. doi: 10.1007/s00421-008-0683-0. [DOI] [PubMed] [Google Scholar]
- Fischer H, Esbjornsson M, Sabina RL, Stromberg A, Peyrard-Janvid M, Norman B. AMP deaminase deficiency is associated with lower sprint cycling performance in healthy subjects. J Appl Physiol. 2007;103:315–322. doi: 10.1152/japplphysiol.00185.2007. [DOI] [PubMed] [Google Scholar]
- Park JH, Olsen NJ, King L Jr, Vital T, Buse R, Kari S, Hernanz-Schulman M, Price RR. Use of magnetic resonance imaging and P-31 magnetic resonance spectroscopy to detect and quantify muscle dysfunction in the amyopathic and myopathic variants of dermatomyositis. Arthritis Rheum. 1995;38:68–77. doi: 10.1002/art.1780380111. [DOI] [PubMed] [Google Scholar]
- Wu J, Ruas JL, Estall JL, Rasbach KA, Choi JH, Ye L, Boström P, Tyra HM, Crawford RW, Campbell KP, Rutkowski DT, Kaufman RJ, Spiegelman BM. The unfolded protein response mediates adaptation to exercise in skeletal muscle through a PGC-1alpha/ATF6alpha complex. Cell Metab. 2011;13:160–169. doi: 10.1016/j.cmet.2011.01.003. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Konttinen YT, Mackiewicz Z, Povilenaite D, Sukura A, Hukkanen M, Virtanen I. Disease-associated increased HIF-1, alphavbeta3 integrin, and Flt-1 do not suffice to compensate the damage-inducing loss of blood vessels in inflammatory myopathies. Rheumatol Int. 2004;24:333–339. doi: 10.1007/s00296-003-0379-z. [DOI] [PubMed] [Google Scholar]
- Grundtman C, Tham E, Ulfgren AK, Lundberg IE. Vascular endothelial growth factor is highly expressed in muscle tissue of patients with polymyositis and patients with dermatomyositis. Arthritis Rheum. 2008;58:3224–3238. doi: 10.1002/art.23884. [DOI] [PubMed] [Google Scholar]
- Nagaraju K, Rider LG, Fan C, Chen YW, Mitsak M, Rawat R, Patterson K, Grundtman C, Miller FW, Plotz PH, Hoffman E, Lundberg IE. Endothelial cell activation and neovascularization are prominent in dermatomyositis. J Autoimmune Dis. 2006;3:2. doi: 10.1186/1740-2557-3-2. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Probst-Cousin S, Neundorfer B, Heuss D. Microvasculopathic neuromuscular diseases: lessons from hypoxia-inducible factors. Neuromuscul Disord. 2010;20:192–197. doi: 10.1016/j.nmd.2010.01.005. [DOI] [PubMed] [Google Scholar]
- Dastmalchi M, Alexanderson H, Loell I, Stahlberg M, Borg K, Lundberg IE, Esbjornsson M. Effect of physical training on the proportion of slow-twitch type I muscle fibers, a novel nonimmune-mediated mechanism for muscle impairment in polymyositis or dermatomyositis. Arthritis Rheum. 2007;57:1303–1310. doi: 10.1002/art.22996. [DOI] [PubMed] [Google Scholar]