

Abstract
Hepatocytes are the main source of hepatitis C virus (HCV) replication and contain the maximum viral load in an infected person. Chronic HCV infection is characterized by weak cellular immune responses to viral proteins. Cathepsin S is a lysosomal cysteine protease and controls HLA-DR–antigen complex presentation through the degradation of the invariant chain. In this study, we examined the effect of HCV proteins on cathepsin S expression and found it to be markedly decreased in dendritic cells (DCs) exposed to HCV or in hepatocytes expressing HCV proteins. The downregulation of cathepsin S was mediated by HCV core and NS5A proteins involving inhibition of the transcription factors interferon regulatory factor 1 (IRF-1) and upstream stimulatory factor 1 (USF-1) in gamma interferon (IFN-γ)-treated hepatocytes. Inhibition of cathepsin S by HCV proteins increased cell surface expression of the invariant chain. In addition, hepatocytes stably transfected with HCV core or NS5A inhibited HLA-DR expression. Together, these results suggested that HCV has an inhibitory role on cathepsin S-mediated major histocompatibility complex (MHC) class II maturation, which may contribute to weak immunogenicity of viral antigens in chronically infected humans.
INTRODUCTION
Hepatitis C virus (HCV) infection often causes progressive liver disease, including hepatic fibrosis, cirrhosis, and hepatocellular carcinoma. The virus efficiently escapes host immune responses and establishes chronic infection in >80% of infected humans. HCV infection induces an interferon (IFN) response but fails to clear virus. IFN therapy alone or together with a nucleoside analog (ribavirin) is efficient for virus clearance in ∼50% of infected patients (40). We have previously shown that human fibrosarcoma (HT1080) cells expressing HCV proteins display gamma interferon (IFN-γ)-mediated HLA-DR expression (37). Hepatocytes are the primary cell type to support HCV replication. Therefore, eradication of HCV can be achieved only by an effective antiviral cellular immune response within the liver.
The endocytic compartments of antigen-presenting cells (APCs) contain a complex mixture of individual proteases involved in antigen processing. Endosomal/lysosomal proteases control two key events in antigen presentation: the degradation of protein antigen and the generation of peptide-receptive major histocompatibility complex (MHC) class II molecules. Nascent HLA class II molecules in the endoplasmic reticulum associate with the chaperone invariant chain (Ii), and are guided to the endosomal compartment by a targeting motif in the invariant-chain cytoplasmic tail. Expression of HLA class II molecules at the cell surface does not occur until the invariant chain is cleaved by a cathepsin, with cathepsin S being the strongest candidate in dendritic cells (DCs) (7, 16). Cathepsin S therefore controls antigen presentation on CD4+ T cells by MHC class II molecules or on NK1.1+ T cells via CD1 molecules. Cathepsin S regulates the expression levels of several MHC class II antigen presentation-related proteins, including intracellular HLA-DR α/β dimer, HLA-DM, and the invariant chain (13, 41, 44).
Hepatocytes normally do not express MHC class II molecules, although in clinical hepatitis, aberrant MHC class II expression occurs (17, 19). The persistence of liver pathogens is often accompanied by a weak CD8+ T cell response to hepatocellular antigens resulting, in part, from impaired antigen presentation mechanisms (20). A high level of hepatitis B virus production has been shown to induce robust IFN-γ and tumor necrosis factor alpha (TNF-α) production in virus-specific CD8+ T cells, while limiting amounts of viral antigen, in both hepatocyte-like cells and naturally infected human hepatocytes, preferentially stimulate CD8+ T cell degranulation. The liver functions as an immune privilege organ, and various factors affect cleavage and loading of antigenic peptides onto MHC class I and class II molecules in hepatocytes and dendritic cells (33). Dendritic cells have a specialized capacity to process exogenous antigens into the MHC class I pathway. This function, known as cross-presentation, provides the immune system with an important mechanism for generating immunity to viruses and tolerance for self (22). Thus, a weak CD8+ T cell response can be augmented by CD4+ T cell help. MHC class II-expressing hepatocytes induce T helper 2 (Th2) cell differentiation of uncommitted CD4+ T cells and abrogate the ability of previously differentiated Th1 cells to secrete IFN-γ, even in the presence of proinflammatory microbial signals. In vivo, MHC class II expression by hepatocytes impaired IFN-γ production by lymphocytic choriomeningitis virus-specific CD4+ and CD8+ T cells and prolonged viral persistence (44). Thus, by instructing infiltrating CD4+ T cells to differentiate into a less inflammatory phenotype, MHC class II-expressing hepatocytes may impair antiviral CD8+ T cell response and viral clearance. On the other hand, MHC class II-expressing hepatocytes, as found in clinical hepatitis, can present antigen and activate CD4+ T cells (25). Further, inappropriate MHC class II expression on hepatocytes and mononuclear cell infiltration were suggested to affect the autoimmune nature of chronic Doberman hepatitis (18, 39).
Since HCV regulates dendritic cell maturation, as well as HLA-DR expression (37), we hypothesized that HCV may directly control cathepsin S to regulate antigen presentation on hepatocytes, which may lead to the escape of virus-infected cells from immune attack. Here, we have shown inhibition of cathepsin S synthesis by HCV proteins, which is consistent with the reduced level of interferon regulatory factor 1 (IRF-1) and upstream stimulatory factor 1 (USF-1) transcription factors. As a result, surface expression of the invariant chain on hepatocytes was enhanced, along with a reduction in HLA-DR expression. Perturbation of antigen presentation on hepatocytes by HCV may affect induction of a CD4+ T cell response in facilitating virus clearance.
MATERIALS AND METHODS
Cytokine and antibodies.
Commercially available cathepsin S inhibitor (EMD Chemicals, CA), IFN-γ and antibodies to cathepsin S (R&D Systems, MN), fluorescein isothiocyanate (FITC)-conjugated mouse anti-human HLA-DR and mouse IgG2a(κ) isotype control (BD Bioscience, CA), CD74 (Santa Cruz, CA), STAT3 and Tyr705phospho-STAT3 (Cell Signaling, MA), and actin (Sigma, MO) were procured.
Patient materials.
Liver biopsy specimens from patients with chronic HCV infection (n = 10) were used in this study. Clinical specimens were collected after approval of the research protocol by the Saint Louis University Institutional Review Board, as previously described (4). All subjects gave written informed consent. Patients were seropositive for anti-HCV and HCV RNA. The viral load was assessed and expressed as IU/ml, and HCV genotypes were determined by commercial assays. The liver biopsy specimens were read by an experienced liver pathologist, and the fibrosis stage was graded and staged according to a system described by Scheuer (38). Commercially available control liver RNAs were procured (Abcam, MA; Clonetics, CA; CloneTech, CA; and Lonza, NJ) and used in this study.
Hepatocytes, transduction, or transfection.
Immortalized human hepatocytes (IHH) and a human hepatoma cell line (Huh7) were used in this study. Hepatocytes were transduced with lentivirus expressing core, NS5A from genotype 1a, or lentivirus with a puromycin selection marker alone (as a vector control). Total RNA was prepared after 72 h and subjected to cDNA synthesis for real-time PCR. Hepatocytes were transfected with plasmid DNA from a mammalian expression vector (pcDNA3) carrying HCV genomic regions (core, NS2, NS3/4A, or NS5A) under the control of a cytomegalovirus promoter using Lipofectamine 2000 (Life Technologies, Inc., MD). For stable transfectants, cell colonies were selected using neomycin (800 μg/ml) and pooled for subsequent studies to avoid artifactual results from clonal variation. Parental cells transfected with empty-vector DNA were used in parallel as a control. Stable transfectants were maintained in Dulbecco's modified Eagle medium (DMEM) containing 10% fetal calf serum and a lower dose of the selection antibiotic (400 μg/ml of neomycin). Alternatively, cells transiently transfected with a specific HCV genomic region were used after 72 h.
Generation of cell culture-grown HCV and infection of cells.
HCV genotype 1a (clone H77) or 2a (clone JFH1) was grown in human hepatocytes, as previously described (28). Cell culture-grown HCV released in culture supernatant was filtered through a 0.45-μm cellulose acetate membrane (Nalgene, NY), aliquoted, and stored at −70°C for a single use. RNA was quantified (IU/ml) by real-time PCR (Prism 7500 real-time thermocycler; ABI) with the use of HCV analyte-specific reagents (ASR) (Abbott) in the Pathology Clinical Laboratory at Saint Louis University. Infectious virus titers from cell culture supernatants were measured in fluorescent focus-forming units (FFU) using an NS5A-specific monoclonal antibody. IHH or Huh7 cells were infected with cell culture-grown HCV at a multiplicity of infection (MOI) of ∼1 or as indicated.
Exposure of DCs to cell culture-grown HCV.
Immature DCs were prepared from human monocytes treated with granulocyte-macrophage colony-stimulating factor (GM-CSF) and interleukin 4 (IL-4) for 6 days (protocol approved by the Internal Review Board, Saint Louis University) and maintained as previously described (37). Briefly, immature DCs (105 cells/ml in RPMI 1640 supplemented with 10% fetal bovine serum and 2 mM l-glutamine) were seeded in plastic tubes. The next day, the DCs were exposed to 105 FFU of cell culture-grown HCV or conditioned medium (CM) from infected hepatocytes and a maturation cocktail (MC) containing 5 ng/ml IL-1β, 10 ng/ml IL-6 (BD Pharmingen, CA), 10 ng/ml TNF-α (Calbiochem, CA), and prostaglandin E2 (Sigma) for 48 h to induce maturation. The DCs were subjected to fluorescence-activated cell sorter (FACS) analysis for HLA-DR expression.
Flow cytometry.
Cell surface expression of HLA-DR or invariant chain (CD74) was analyzed by flow cytometry. DCs exposed to cell culture-grown HCV or hepatocytes expressing HCV core or NS5A protein were grown in DMEM supplemented with 10% fetal bovine serum (FBS) and antibiotics. Adherent cells were treated with 10 mM EDTA, neutralized with medium containing 10% FBS, and washed with phosphate-buffered saline (PBS). For staining, cells were incubated with fluorochrome-tagged mouse monoclonal antibody to HLA-DR (BD Biosciences, CA) in PBS containing 0.5% bovine serum albumin (BSA) for 30 min at room temperature. CD74 was stained with a specific primary antibody (Santa Cruz, CA). After washing, the cells were treated with FITC-conjugated secondary antibody for 30 min on ice and resuspended in PBS. Both HLA-DR- and CD74-stained cells were gated according to their size (forward light scatter) and granularity (side light scatter) using a flow cytometer (Beckon Dickinson). Surface marker expression on the gated cells was analyzed using CellQuest software (BD Immunocytometry Systems).
Real-time PCR.
A quantitative real-time PCR analysis was performed for IRF-1, USF-1, cathepsin S, or HLA-DRA mRNA using specific TaqMan primers and probes (Applied Biosystems, CA) as previously described (4). RNA was isolated from experimental hepatocytes or liver biopsy specimens using TRIzol (Invitrogen, CA). cDNA synthesis was done using random hexamers and the Superscript III first-strand synthesis kit (Invitrogen). IRF-1, USF-1, cathepsin S, or HLA-DRA mRNA was evaluated using specific oligonucleotide primers (IRF-1, Hs00971960_ml; USF-1, Hs00273038_m1; cathepsin S, Hs0017540-m1; HLA-DRA, Hs00219575_ml). The 6-carboxyfluorescein (FAM)-labeled 1× 18S gene expression assay (Hs03928990-g1) was used as an endogenous control. All reactions were performed in triplicate with an ABI Prism 7700 analyzer, and samples were analyzed at least 3 times.
Luciferase reporter assay.
Huh7 cells (1 × 106) were seeded on plastic plates 24 h before transfection. The cells were transfected together with a cathepsin S promoter-luciferase reporter construct and HCV protein expression plasmid or empty-vector DNA as a negative control. IFN-γ was added to the cells 24 h after transfection and incubated for 18 h. The cells were treated with lysis buffer (Promega, WI) for 30 min at room temperature. Cell lysates prepared after clarification by centrifugation were subjected to luciferase reporter assays using a luminometer (Opticomp II; MGM Instruments).
Western blot analysis.
Proteins in cell lysates were resolved by SDS-PAGE, transferred onto nitrocellulose membranes, and blotted with specific antibody. A positive signal for protein was detected using a peroxidase-conjugated secondary antibody. Protein bands were visualized by enhanced chemiluminescence with a Super Signal West Detection Kit (Thermo Chemical Company, IL). Cellular actin was detected similarly for comparison of protein loads in each lane.
Statistical analysis.
Statistical analysis was performed using a two-tailed unpaired Student t test or one-way analysis of variance (ANOVA) in GraphPad Prism 5 (GraphPad, La Jolla, CA). Results were expressed as the mean and standard deviation (SD), and a P value of <0.05 was considered statistically significant.
RESULTS
HCV inhibits dendritic cell maturation and reduces cathepsin S expression.
DCs undergo a process of maturation, migration, and relocation after antigen internalization, (11). During maturation, DCs upregulate MHC class II and a variety of costimulatory molecules. Exposure of DCs to HCV inhibited DC maturation, although HCV protein expression was observed in only a small percentage of cells (37). DCs exposed to cell culture-grown HCV were examined for cell surface HLA-DR expression by FACS analysis. As shown in a representative example, DCs expressed a higher mean fluorescence of HLA-DR staining when incubated with MC and CM from hepatocytes (Fig. 1A). In contrast, DCs exposed to cell culture-grown HCV expressed a much lower level of HLA-DR under identical experimental conditions. The mean fluorescence intensity of HLA-DR-positive cells was significantly reduced following incubation with cell culture-grown HCV.
Fig 1.
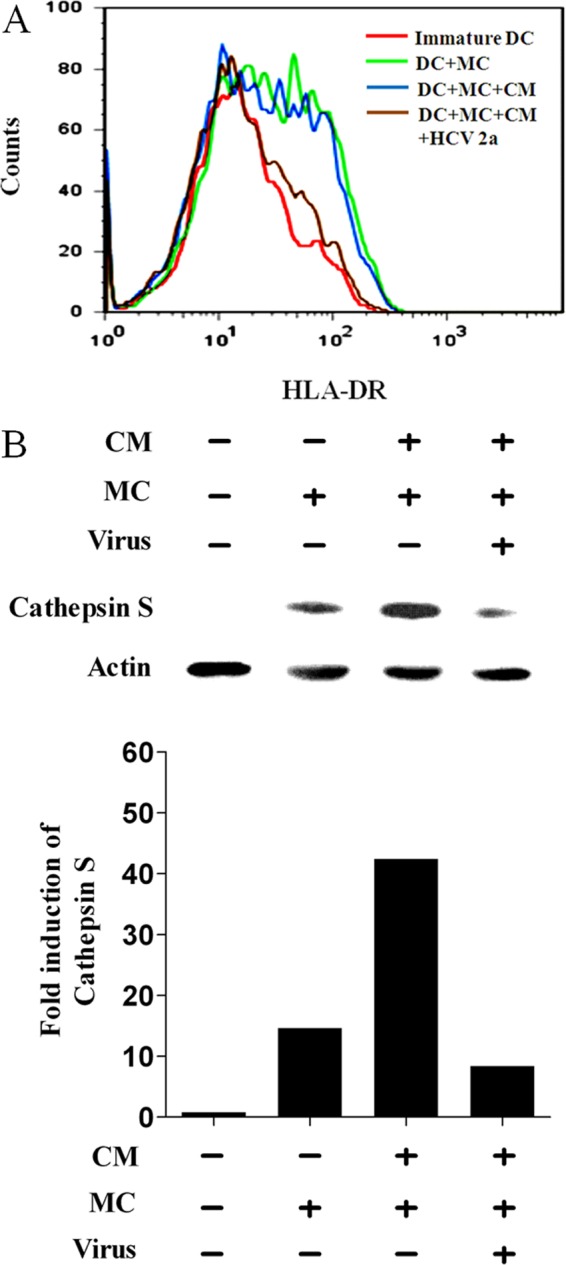
Analyses for HLA-DR and cathepsin S status in DCs following exposure to HCV. (A) FACS analysis of a typical expression profile of HLA-DR on the surfaces of monocyte-derived DCs from a healthy volunteer after stimulation with MC as a positive control. The DCs were treated separately with CM from hepatocytes as a mock control or exposed to cell culture-grown HCV genotype 2a. (B) Western blot analysis displaying cathepsin S expression levels in DCs following incubation with cell culture-grown HCV and MC. The DCs were also incubated with CM alone or together with MC from mock-infected cells. Densitometric analysis of the Western blot result displaying fold induction of cathepsin S is shown in the bar graph below.
Expression of HLA-DR on the cell surface does not occur until the invariant chain is cleaved by cathepsin S (16). The level of cathepsin S increased approximately 15-fold following incubation of immature DCs with MC. This increase using MC was enhanced 3-fold further in the presence of CM generated from uninfected hepatocytes used to mock treat DCs (Fig. 1B). Interestingly, when DCs were incubated with cell culture-grown HCV (already containing CM), an ∼8-fold decrease in the cathepsin S level compared to a mock-treated control was observed. This is paradoxical, as only a small fraction of cells generally display detectable HCV protein expression (37), and the observed effect may be due to interaction of HCV with DC surface molecules.
HCV core and NS5A downregulate cathepsin S expression in hepatocytes.
Since we observed inhibition of HLA-DR expression following exposure of DCs to cell culture-grown HCV, we examined the cathepsin S status in chronically HCV-infected liver biopsy specimens and in cell culture-grown HCV-infected or HCV gene-transfected hepatocytes by real-time PCR. The results suggested 6 out of 10 chronically HCV-infected liver biopsy specimens displayed an ∼5-fold reduction in mRNA expression of cathepsin S compared to three normal liver RNAs (Fig. 2A). The changes in the cathepsin S level did not correlate with the liver fibrosis stage. Similarly, an ∼2.5-fold decrease in the cathepsin S mRNA level in HCV genotype 2a-infected Huh7 cells was observed (Fig. 2B). HCV core- or NS5A-transfected hepatocytes also exhibited reductions of ∼2- and ∼2.5-fold in cathepsin S mRNA, respectively, compared to empty-vector-transfected cells (Fig. 2C).
Fig 2.

HCV inhibits cathepsin S production. (A, B, and C) Real-time PCR analysis for cathepsin mRNA status in chronically HCV-infected human liver biopsy specimens, in cell culture-grown HCV-infected human hepatocytes, and in HCV gene-transfected hepatocytes, respectively. mRNA expression levels in all experimental samples were normalized with 18S RNA. (D) Huh7 cells were transiently cotransfected with a pGL3/cathepsin S promoter-luciferase construct and FL HCV genotype 1a. HCV genotype 1b subgenomic replicon-harboring cells were also used in the promoter assay. Cell lysates were analyzed for cathepsin S promoter activity after 48 h. The promoter activity (%) was compared with that of vector-transfected or control Huh7 cells arbitrarily set at 100%. The P value (***, P < 0.001) was determined by using a two-tailed t test. (E) Huh7 cells were transiently cotransfected with vector or the HCV genomic region (core, NS2, NS3/4A, or NS5A) and a pGL3/cathepsin S promoter-luciferase construct. After 24 h of transfection, the cells were treated or not with IFN-γ for 18 h and examined for luciferase activity using a firefly luciferase assay system. The P value was determined by using a two-tailed t test. (A to C and F) P values were as follows: *, P < 0.05; **, P < 0.01. (D) The P value (***) was less than 0.001 compared to normal liver biopsy samples or vector control. (F) Cells were treated with increasing doses of IFN-γ (0, 500, 1,000, and 2,000 U/ml) for 18 h after transient cotransfection with pGL3/cathepsin S promoter-luciferase construct and core or NS5A plasmid to analyze IFN-γ-induced cathepsin S promoter activity. The results are shown as the means and standard deviations from triplicate experiments. The P value was determined by one-way ANOVA (P < 0.0293). (G and H) Cathepsin S protein expression was analyzed in HCV-infected or HCV gene-transfected cells by Western blot analyses. (I) Western blot analyses for HCV core or NS5A protein expression in transfected Huh7 cells.
To identify the effects of HCV proteins on cathepsin S promoter regulation, HCV full-length (FL) or subgenomic replicon-bearing cells from HCV genotype 1b (BB7) were separately transfected, along with the cathepsin S promoter construct (nucleotides −808 to +44) with a luciferase reporter gene. Cathepsin S promoter activation was measured as a function of luciferase activity and was found to be dramatically decreased in HCV FL transfected or subgenomic replicon-harboring (BB7) cells (Fig. 2D). To further verify the roles of HCV proteins, plasmids encoding HCV core, NS2, NS3/NS4A, or NS5A were separately transfected, together with the cathepsin S promoter construct, in Huh7 cells, and luciferase activity was measured with or without IFN-γ treatment. Hepatocytes transfected with HCV core- or NS5A-encoding plasmids reduced basal cathepsin S promoter activity compared to empty-vector-transfected control cells (Fig. 2E). Cathepsin S promoter activity is primarily regulated by IRF-1 upon IFN-γ treatment of epithelial cells (42). A similar effect was observed in IFN-γ-treated hepatocytes. However, transfection with NS2 and NS3/NS4A had no significant effect on cathepsin S promoter activation. Downregulation of IFN-γ-mediated cathepsin S promoter activation was also shown in the presence of HCV core or NS5A (Fig. 2F).
Since we observed a marked difference in cathepsin S protein expression levels in mock- and HCV-exposed DC lysates, similar analyses were performed using hepatocytes infected with two different HCV genotypes. Both HCV genotypes 1a and 2a induced a major reduction in cathepsin S expression in two separate human hepatocyte cell lines (IHH and Huh7), as determined by Western blot analysis (Fig. 2G). HCV infection of hepatocytes was measured by immunofluorescence using an NS5A-specific monoclonal antibody (data not shown). Next, we examined whether the modulation of cathepsin S expression is mediated by specific HCV proteins. Since cathepsin S promoter activity, as well as mRNA expression, was downregulated only in the presence of core and NS5A, we selected these two proteins for Western blot analysis. Huh7 cells expressing HCV core or NS5A significantly reduced cathepsin S protein expression (Fig. 2H). Expression of HCV proteins in virus-infected cells was also shown by Western blot analysis (Fig. 3I). These results underscore a significant inhibitory role of HCV core and NS5A proteins on cathepsin S.
Fig 3.

HCV infection or transfection of HCV core/NS5A inhibits IRF-1 and USF-1 expression in hepatocytes. (A) Real-time PCR analyses for the mRNA expression status of IRF-1 and USF-1 in HCV genotype 1a- or 2a-infected IHH or Huh7 cells. The results were normalized with 18S RNA and are presented as mean values and standard deviations from triplicate experiments. The asterisks indicate statistical significance (***, P < 0.001). (B) IHH and Huh 7 cells were infected with HCV genotype 1a or 2a. Whole-cell lysates were prepared 5 days after infection. The expression levels of transcription factors, USF-1, phospho-STAT3, IRF-1, and PU.1, were detected by Western blot analysis. (C) Real-time PCR analyses for the mRNA expression status of IRF-1 and USF-1 in IHH transduced with lentivirus vector expressing core or NS5A from HCV genotype 1a. (D) Similar analyses were performed using Huh-7 cells transiently transfected with mammalian expression vector encoding core or NS5A from HCV genotype 1a. The results in both panels C and D were analyzed as described for panel A. The asterisks indicate statistical significance (*, P < 0.05; ***, P < 0.001). (E) Huh7 cells were transiently transfected with the HCV core, NS2, NS3/4A, or NS5A genomic region, and expression of transcription factors was analyzed similarly by Western blotting after 48 h. (F) Huh7 cells were transiently transfected with HCV core or NS5A and treated or not with 1,000 U/ml of IFN-γ for 2 h, and expression of the transcription factors was analyzed by Western blotting. The blots were reprobed with an antibody to actin for comparison of the protein loads in each lane.
HCV infection or transfection of HCV core/NS5A modulates hepatocyte transcription factors.
Cathepsin S expression is regulated by IRF-1, PU.1, and phospho-STAT3 in antigen-presenting cells (12, 42, 43). In addition, the cathepsin S promoter contains an E-box region that is a binding site for the USF-1 transcription factor. We examined whether HCV infection or transfection of HCV core/NS5A alters the status of specific transcription factors with the potential for cathepsin S regulation. For this, hepatocytes were infected with HCV genotype 1a or 2a, and the expression status of transcription factors (USF-1, phospho-STAT3, IRF-1, and PU.1) was compared with that of mock-infected hepatocytes at the mRNA level by real-time PCR. The results indicated a significant reduction of IRF-1 and USF-1 mRNAs in hepatocytes infected with HCV genotype 1a or 2a (Fig. 3A). Western blot analysis for protein expression status was also performed (Fig. 3B), and the results suggested expression of both IRF-1 and USF-1 was significantly inhibited. In contrast, the phospho-STAT3 and PU.1 status remained unchanged as a result of HCV infection.
IHH transduced with lentivirus expressing core or NS5A from HCV genotype 1a displayed a significant reduction of IRF1 or USF-1 mRNA levels by real-time PCR (Fig. 3C). Similar reduction was also observed in Huh7 cells transiently transfected with HCV core- or NS5A-expressing plasmid DNA (Fig. 3D). Huh7 cells were transiently transfected with the empty vector, HCV core, NS2, NS3/4A, or NS5A genomic region, and the expression of USF-1 and IRF-1 transcription factors was analyzed after 48 h by Western blotting (Fig. 3E). The results suggested that only HCV core and NS5A proteins reduce USF-1 and IRF-1, while HCV NS2 or NS3/4A does not inhibit expression of the transcription factors. Huh7 cells transiently transfected with the HCV core or NS5A gene were also examined for a virus protein-specific effect on transcription factors. IFN-γ was used separately to stimulate the cells for 2 h in determining altered expression of the transcription factors (Fig. 3F). USF-1 expression was strongly inhibited in transiently transfected cells and, as expected, did not respond to IFN-γ stimulation. Further, expression of IRF-1 in the presence of IFN-γ is modulated in both core- and NS5A-transfected cells.
HLA-DR expression is reduced in hepatocytes transfected with HCV genomic regions or in chronically infected liver biopsy specimens.
We examined the expression level of HLA-DR by FACS in hepatocytes transiently transfected with HCV core or NS5A plasmid DNA. Empty-vector-transfected cells were used as controls. IFN-γ treatment increased HLA-DR expression in vector-transfected cells compared to untreated cells. Cells transfected with HCV core and NS5A reduced HLA-DR expression compared to IFN-γ-treated vector control (Fig. 4A). Subsequently, HLA-DR mRNA status was examined for IFN-γ-treated IHH transfected with HCV genomic regions by real-time PCR analysis. HCV core or NS5A transfection reduced HLA-DR expression to a significant degree (Fig. 4B). Thus, the results from this set of experiments suggested that IFN-γ-stimulated hepatocytes transfected with HCV core or NS5A protein suppress HLA-DR expression. HCV infection also decreased HLA-DR mRNA expression in liver biopsy specimens from chronically infected patients compared to healthy controls (Fig. 4C). The pattern of HLA-DR reduction in these HCV-infected liver biopsy specimens did not correlate with the liver fibrosis stage or the cathepsin S mRNA expression level, as shown in earlier results (Fig. 2A). However, HLA-DR is not dependent on cathepsin S production, and we did not observe regulation of HLA-DR expression by cathepsin S.
Fig 4.

Inhibition of HLA-DR expression on hepatocytes by HCV proteins. (A) HCV core- or NS5A-transfected Huh7 cells were stimulated with 500 U/ml of IFN-γ for 48 h and analyzed for HLA-DR cell surface expression after immunostaining with FITC-conjugated specific antibody. Cells stained with the FITC-mouse IgG2a(κ) isotype control are shown as the negative control. (B) Real-time PCR analysis for HLA-DR mRNA status in HCV gene-transfected IHH treated with IFN-γ. (C) HLA-DR mRNA status in chronically HCV-infected human liver biopsy specimens. The mRNA level was normalized with 18S RNA. The results are shown as means and standard deviations from triplicate assays. The P value was determined by using a two-tailed t test: *, P < 0.05; **, P < 0.01 compared to vector control or normal liver biopsy samples.
HCV infection or transfection with HCV core/NS5A genomic region inhibits invariant-chain degradation in hepatocytes.
Invariant chain (CD74) associates with the α and β chains of HLA-DR and stabilizes nascent HLA-DR α/β heterodimers (31). Since cathepsin S mediates proteolytic processing of the invariant chain and controls MHC class II antigen presentation; the reduction of HLA-DR and cathepsin S in hepatocytes may be correlated with the inhibition of CD74 degradation. We examined the expression status of CD74 in HCV-infected hepatocytes or hepatocytes transfected with a plasmid carrying the cloned HCV genomic region (core or NS5A) by Western blot analysis. CD74 expression was enhanced in Huh7 cells infected with cell culture-grown HCV genotype 1a or 2a (Fig. 5A) or transfected with the HCV core or NS5A genomic region (Fig. 5B).
Fig 5.

Invariant-chain degradation is inhibited by HCV infection or transfection with HCV core/NS5A. (A) Hepatocytes were infected with HCV genotype 1a or 2a. Cell lysates prepared 5 days after infection were analyzed for invariant-chain (CD74) expression by Western blotting. Mock-infected cells were treated as controls for comparison. (B) Huh7 cells transfected with HCV core or NS5A were analyzed similarly for expression of CD74 by Western blotting. (C) Cell surface expression of CD74 on HCV core- and NS5A-transfected Huh7 cells, stimulated or not with 500 U/ml IFN-γ for 48 h. The results of FACS analyses after immunostaining with anti-CD74 antibody, followed by FITC-conjugated goat anti-mouse treatment, are shown. (D) IHH were transfected with plasmids containing HBV X to enhance HLA-DR expression. The transfectants were stimulated with 500 U/ml IFN-γ, and CD74 cell surface expression was examined after 48 h by FACS. (E) Huh7 cells were treated or not with cathepsin S inhibitor (2.36 μM) and stimulated with 500 U/ml IFN-γ for 48 h. Cell surface expression of CD74 was examined by FACS. The result from cells stained with a secondary antibody is shown as the negative control.
Since hepatocytes express a low level of HLA-DR on their surfaces, induction of HLA-DR using IFN-γ (14) or HBV-X protein (26) expression was necessary to identify cell surface expression of CD74 associated with HLA-DR. To examine cell surface expression of CD74, hepatocytes were stimulated with IFN-γ for 48 h and analyzed by FACS using a monoclonal antibody to CD74, followed by staining with a FITC-conjugated secondary antibody. IFN-γ treatment modestly increased CD74 expression compared to negative-control or non-IFN-γ-treated cells. On the other hand, core- and NS5A-transfected hepatocytes displayed significantly enhanced CD74 expression, suggesting that CD74 degradation did not occur in HCV protein-expressing cells (Fig. 5C). In a similar experiment, CD74 expression was increased by core and NS5A in HBV X-expressing hepatocytes (Fig. 5D). Cell surface expression of HLA-DR was increased by HBV X (26), but HCV proteins did not alter HLA-DR expression on HBV X protein-expressing cells (data not shown). Together, these results suggested that IFN-γ and HBV X increase CD74 expression. Since CD74 expression is regulated by cathepsin S and HCV proteins inhibit cathepsin S expression, this may lead to enhanced CD74 cell surface expression. To distinguish between a difference in expression of CD74 independent of cathepsin S activity versus lysosomal enzymatic degradation, cells were incubated with a specific cathepsin S inhibitor. The inhibitor increased CD74 cell surface expression compared to untreated control cells, indicating that degradation of CD74 was regulated by cathepsin S (Fig. 5E). These results suggested that HCV core and NS5A inhibit dissociation of CD74 from HLA-DR through the inhibition of cathepsin S, which may lead to impairment of MHC class II maturation.
DISCUSSION
In this study, we have examined the role of HCV proteins in cathepsin S expression and related functions important for MHC class II maturation in human hepatocytes, the primary cell type infected by HCV. Our results suggest that cathepsin S expression is markedly decreased in hepatocytes infected with HCV or exposed to core or NS5A protein, resulting in inhibition of CD74 degradation and increased cell surface expression leading to a reduction in the maturation of MHC class II on the cell surface. DCs are important for initiation of adaptive immune responses to foreign antigens and T cell activation. DCs from patients with chronic HCV infection fail to stimulate allogeneic T cells and produce IFN-γ (30). HCV-exposed DCs switch their cytokine profile to the suppressive phenotype of IL-10 and transforming growth factor beta (TGF-β) predominance, which results in the prevention of DC maturation and allostimulation capacity. In addition, the TNF-α-stimulated MHC class II expression level is reduced considerably in DCs derived from patients with chronic HCV infection, indicating that immature DCs of patients with chronic HCV infection do not respond appropriately to stimulation with cytokine (2). Increasing evidence suggests that viral proteins normally found in the cytoplasm and exocytic compartments can be efficiently presented by MHC class II proteins. Conversely, viruses escape detection by CD4+ T cells by reducing the expression of MHC class II molecules and inhibiting MHC class II antigen presentation (23). Our results provide direct evidence that monocyte-derived DCs exposed to cell culture-grown HCV maintain an immature phenotype, secrete IL-10 (37), and display reduced levels of HLA-DR and cathepsin S, which are in agreement with the observations of chronically HCV-infected patients (2, 3). Two C-type lectin receptors found on dendritic cells, DC-SIGN and L-SIGN, have been shown to bind HCV (34). In addition, active replication of the HCV genome has been detected in dendritic cells derived from monocytes (3, 32). Virus-specific positive-strand HCV RNA and its replicative intermediate negative-strand HCV RNA were detected in peripheral blood dendritic cells, although viral replication occurs at a very low level (21, 32, 36). Therefore, binding of HCV to DCs or expression of HCV proteins from low-level virus genome replication may affect cathepsin S expression. Further studies should address the mechanistic aspect of these findings.
MHC class II-expressing hepatocytes in clinical hepatitis function as antigen-presenting cells and activate CD4+ T cells (25). HCV core protein inhibits priming of antigen-specific CD4+ and CD8+ T cell responses by downregulation of MHC molecules and costimulatory molecules on antigen-presenting cells and induces development of IL-10-producing T cells (45). We previously reported that HCV NS5A acts as an immunomodulator by inhibiting IFN-γ production in HCV NS5A transgenic mouse liver (29). In this study, HLA-DR-expressing hepatocytes were studied in the presence of IFN-γ, which acted as a stimulus to enhance HLA-DR production. In general, hepatocytes express a very low level of HLA-DR, so we introduced IFN-γ and/or HBV X to enhance cell surface expression of HLA-DR (14, 26). Interestingly, HCV core and NS5A, but not HBV X, inhibited IFN-γ-mediated HLA-DR expression. Since HCV proteins inhibit the IFN-γ-mediated STAT-1 signaling pathway (4), HLA-DR expression mediated by IFN-γ might be inhibited by a STAT-1-dependent mechanism. In addition, we have shown that HCV core or NS5A inhibits cathepsin S production, which results in an increase of invariant-chain expression and a reduction of mature MHC class II molecules on the cell surface. The inhibition of MHC class II maturation by HCV may impair antigen presentation on the hepatocyte cell surface and allows escape from recognition by CD4+ T cells.
Cathepsin S is known to increase the degradation of the invariant chain and to accelerate MHC class II antigen presentation on epithelial cells (7, 10). Therefore, our observations of cathepsin S downregulation by HCV suggest a regulatory mechanism for MHC class II maturation and antigen presentation in virus-infected hepatocytes. The cathepsin S expression level is significantly reduced in human DCs exposed to cell culture-grown HCV and in hepatocytes expressing core or NS5A. A reduced level of cathepsin S in virus-infected cells leads to failure of the degradation of the invariant chain, which results in an increase of HLA-DR–CD74 complex expression on the cell surface. This may influence the types of T cell epitopes generated in antigen-presenting cells and/or the rate of class II MHC peptide loading. CD74 may also be expressed on the cell surface in the absence of class II molecules, as was shown in a mutant class II-negative cell line (24). In most cases; however, CD74 is associated with HLA-DR on the cell surface when MHC class II is not matured with antigen, and the complex internalizes rapidly to the endosome (31). In this study, we observed increased cell surface expression of CD74 in core- or NS5A-transfected cells, indicating that cathepsin S activity is altered. Because surface CD74 is physically associated with HLA-DR, our results suggest that the expression of the HLA-DR–CD74 complex increased on the cell surface and that MHC class II maturation is inhibited by HCV proteins.
We have previously shown that HCV modulates the JAK-STAT1 pathway (9) and that inhibition of STAT1 activation reduces CIITA gene expression, important for induction of HLA-DR gene expression (37). In this study, we observed that the HLA-DR mRNA expression status is lower in liver biopsy specimens from patients chronically infected with HCV and in IFN-γ-stimulated hepatocytes transfected with HCV core or NS5A. The inhibition of HLA-DR mRNA might be caused by HCV-mediated CIITA gene inhibition. However, in 4 of the 10 liver biopsy specimens from hepatitis C patients, the expression of HLA-DR did not correlate with that of cathepsin S, suggesting cathepsin S-independent regulation of HLA-DR. Thus, HLA-DR expression is modulated outside cathepsin S, which introduces another variable and will be addressed in future studies.
We also found that HCV infection and HCV core or NS5A expression inhibit cathepsin S expression through the inhibition of IRF-1 and USF-1. However, IRF-1 levels were altered only in IFN-γ-treated Huh7 cells. Interestingly, USF-1 expression was completely inhibited in HCV core or NS5A stimulated or not with IFN-γ, indicating that cathepsin S may be regulated by USF-1 at the basal level and by IRF-1 in the presence of IFN-γ. These two HCV proteins have a wide range of regulatory activities on a number of diverse cellular genes and proteins and are involved in distinct signaling pathways for immunomodulatory (1, 4, 8, 37), metabolic (5, 6), and oncogenic (5) networks. We have shown that HCV impairs IRF-1 expression primarily at the transcriptional level. In our previous study (4), we also showed by real-time PCR that IRF-1 mRNA is significantly inhibited in liver biopsy specimens of HCV-infected patients. Pflugheber et al. (35) previously suggested that IRF-1 activation is blocked by HCV NS5A. Subsequent studies by Kanazawa et al. (27) have shown that suppression of IRF-1 mRNA expression occurs in HCV replicon-harboring cells, while Huh7 cells with the replicon eliminated (cured) display IRF-1 expression, implying replicon genome replication or viral protein expression in replicon-harboring cells suppresses IRF-1 expression. On the other hand, Ciccaglione et al. (15) reported that the expression levels of IRF-1 were decreased in clones harboring the full-length-HCV genome, and our results presented here are very similar. The minor differences in all these observations could be due to the expression levels of viral proteins in different cell types and the genotypes of virus used. IRF-1 binding to IFN-stimulated response element (ISRE) on the cathepsin S promoter and a role of IRF-1 in cathepsin S expression have already been shown (42). Thus, a significant decrease in IRF-1 at the transcriptional level likely impairs cathepsin S expression. Our data have addressed some of the basic mechanistic aspects of cathepsin S regulation by HCV proteins. Further work should help to unravel the complete mechanism of the observed results.
ACKNOWLEDGMENTS
We thank Aleem Siddiqui for providing the HBV X clone, Ratna B. Ray for providing lentivirus expressing HCV core or NS5A, Leonard Grosso for measuring HCV genome copy numbers in virus stock, and Lin Cowick for preparation of the manuscript.
This work was supported by research grants AI068769 and DK80812 from the National Institutes of Health.
Footnotes
Published ahead of print 3 July 2012
REFERENCES
- 1. Ait-Goughoulte M, et al. 2010. Hepatitis C virus core protein interacts with fibrinogen-beta and attenuates cytokine stimulated acute-phase response. Hepatology 51:1505–1513 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2. Auffermann-Gretzinger S, Keeffe EB, Levy S. 2001. Impaired dendritic cell maturation in patients with chronic, but not resolved, hepatitis C virus infection. Blood 97:3171–3176 [DOI] [PubMed] [Google Scholar]
- 3. Bain C, et al. 2001. Impaired allostimulatory function of dendritic cells in chronic hepatitis C infection. Gastroenterology 120:512–524 [DOI] [PubMed] [Google Scholar]
- 4. Banerjee A, et al. 2011. Transcriptional repression of C4 complement by hepatitis C virus proteins. J. Virol. 85:4157–4166 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5. Banerjee A, Meyer K, Mazumdar B, Ray RB, Ray R. 2010. Hepatitis C virus differentially modulates activation of forkhead transcription factors and insulin-induced metabolic gene expression. J. Virol. 84:5936–5946 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6. Banerjee S, et al. 2008. Hepatitis C virus core protein upregulates serine phosphorylation of insulin receptor substrate-1 and impairs the downstream Akt/protein kinase B signaling pathway for insulin resistance. J. Virol. 82:2606–2612 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7. Bania J, et al. 2003. Human cathepsin S, but not cathepsin L, degrades efficiently MHC class II-associated invariant chain in nonprofessional APCs. Proc. Natl. Acad. Sci. U. S. A. 100:6664–6669 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8. Basu A, et al. 2006. Microarray analyses and molecular profiling of Stat3 signaling pathway induced by hepatitis C virus core protein in human hepatocytes. Virology 349:347–358 [DOI] [PubMed] [Google Scholar]
- 9. Basu A, Meyer K, Ray RB, Ray R. 2001. Hepatitis C virus core protein modulates the interferon induced transacting factors of JAK/STAT signaling pathway but does not affect the activation of interferon-stimulated genes. Virology 288:379–390 [DOI] [PubMed] [Google Scholar]
- 10. Beers C, et al. 2005. Cathepsin S controls MHC class II-mediated antigen presentation by epithelial cells in vivo. J. Immunol. 174:1205–1212 [DOI] [PubMed] [Google Scholar]
- 11. Bell D, Young JW, Banchereau J. 1999. Dendritic cells. Adv. Immunol. 72:255–324 [DOI] [PubMed] [Google Scholar]
- 12. Chan LLY, Cheung BKW, Li JCB, Lau ASY. 2010. A role for STAT3 and cathepsin S in IL-10 down-regulation of IFN-γ-induced MHC class II molecules on primary human blood macrophages. J. Leukoc. Biol. 88:303–311 [DOI] [PubMed] [Google Scholar]
- 13. Chapman HA. 2006. Endosomal proteases in antigen presentation. Curr. Opin. Immunol. 18:78–84 [DOI] [PubMed] [Google Scholar]
- 14. Chiu JH, et al. 1997. Class I and class II major histocompatibility complex antigens expression on human hepatocytes and hepatoma cells: an approach with high sensitivity and specificity. Cytometry 30:317–323 [PubMed] [Google Scholar]
- 15. Ciccaglione AR, et al. 2007. Repression of interferon regulatory factor 1 by hepatitis C virus core protein results in inhibition of antiviral and immunomodulatory genes. J. Virol. 81:202–214 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16. Cresswell P. 1998. Proteases, processing, and thymic selection. Science 280:394–395 [DOI] [PubMed] [Google Scholar]
- 17. Dienes HP, Hütteroth T, Hess G, Meuer SC. 1987. Immunoelectron microscopic observations on the inflammatory infiltrates and HLA antigens in hepatitis B and non-A, non-B. Hepatology 7:1317–1325 [DOI] [PubMed] [Google Scholar]
- 18. Dyggve H, et al. 2011. Association of Doberman hepatitis to canine major histocompatibility complex II. Tissue Antigens 77:30–35 [DOI] [PubMed] [Google Scholar]
- 19. Franco A, et al. 1988. Expression of class I and class II major histocompatibility complex antigens on human hepatocytes. Hepatology 8:449–454 [DOI] [PubMed] [Google Scholar]
- 20. Gehring AJ, et al. 2007. The level of viral antigen presented by hepatocytes influences CD8 T-cell function. J. Virol. 81:2940–2949 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21. Goutagny N, et al. 2003. Evidence of viral replication in circulating dendritic cells during hepatitis C virus infection. J. Infect. Dis. 187:1951–1958 [DOI] [PubMed] [Google Scholar]
- 22. Heath WR, Carbone FR. 2001. Cross-presentation in viral immunity and self-tolerance. Nat. Rev. Immunol. 1:126–134 [DOI] [PubMed] [Google Scholar]
- 23. Hegde NR, Chevalier MS, Johnson DC. 2003. Viral inhibition of MHC class II antigen presentation. Trends Immunol. 24:278–285 [DOI] [PubMed] [Google Scholar]
- 24. Henne C, Schwenk F, Koch N, Moller P. 1995. Surface expression of the invariant chain (CD74) is independent of concomitant expression of major histocompatibility complex class II antigens. Immunology 84:177–182 [PMC free article] [PubMed] [Google Scholar]
- 25. Herkel J, et al. 2003. MHC class II-expressing hepatocytes function as antigen-presenting cells and activate specific CD4 T lymphocytes. Hepatology 37:1079–1085 [DOI] [PubMed] [Google Scholar]
- 26. Hu KQ, Vierling JM, Siddiqui A. 1990. Trans-activation of HLA-DR gene by hepatitis B virus X gene product. Proc. Natl. Acad. Sci. U. S. A. 87:7140–7144 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27. Kanazawa N, et al. 2004. Regulation of hepatitis C virus replication by interferon regulatory factor 1. J. Virol. 78:9713–9720 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28. Kanda T, et al. 2006. Generation of infectious hepatitis C virus in immortalized human hepatocytes. J. Virol. 80:4633–4639 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29. Kanda T, Steele R, Ray R, Ray RB. 2009. Inhibition of intrahepatic gamma interferon production by hepatitis C virus nonstructural protein 5A in transgenic mice. J. Virol. 83:8463–846919553305 [Google Scholar]
- 30. Kanto T, et al. 1999. Impaired allostimulatory capacity of peripheral blood dendritic cells recovered from hepatitis C virus-infected individuals. J. Immunol. 162:5584–5591 [PubMed] [Google Scholar]
- 31. Moldenhauer G, Henne C, Karhausen J, Möller P. 1999. Surface-expressed invariant chain (CD74) is required for internalization of human leucocyte antigen-DR molecules to early endosomal compartments. Immunology 96:473–484 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32. Navas MC, Fuchs AE, Schvoerer Bohbot A, Aubertin AM, Stoll-Keller F. 2002. Dendritic cell susceptibility to hepatitis C virus genotype 1 infection. J. Med. Virol. 67:152–161 [DOI] [PubMed] [Google Scholar]
- 33. Osna NA. 2009. Hepatitis C virus and ethanol alter antigen presentation in liver cells. World J. Gastroenterol. 15:1201–1208 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34. Pachiadakis I, Pollara G, Chain BM, Naoumov NV. 2005. Is hepatitis C virus infection of dendritic cells a mechanism facilitating viral persistence? Lancet Infect. Dis. 5:296–304 [DOI] [PubMed] [Google Scholar]
- 35. Pflugheber J, et al. 2002. Regulation of PKR and IRF-1 during hepatitis C virus RNA replication. Proc. Natl. Acad. Sci. U. S. A. 99:4650–4655 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36. Pham TN, et al. 2004. Hepatitis C virus persistence after spontaneous or treatment-induced resolution of hepatitis C. J. Virol. 78:5867–5874 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37. Saito K, et al. 2008. Hepatitis C virus inhibits cell surface expression of HLA-DR, prevents dendritic cell maturation and induces IL-10 production. J. Virol. 82:3320–3328 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38. Scheuer PJ. 1991. Classification of chronic viral hepatitis: a need for reassessment. J. Hepatol. 13:372–374 [DOI] [PubMed] [Google Scholar]
- 39. Speeti M, Ståhls A, Meri S, Westermarck E. 2003. Upregulation of major histocompatibility complex class II antigens in hepatocytes in Doberman hepatitis. Vet. Immunol. Immunopathol. 96:1–12 [DOI] [PubMed] [Google Scholar]
- 40. Stribling R, Sussman N, Vierling JM. 2006. Treatment of hepatitis C infection. Gastroenterol. Clin. North Am. 35:463–486 [DOI] [PubMed] [Google Scholar]
- 41. Thurmond RL, Sun S, Karlsson L, Edwards JP. 2005. Cathepsin S inhibitors as novel immunomodulators. Curr. Opin. Investig. Drugs 6:473–482 [PubMed] [Google Scholar]
- 42. van's Gravesande KS, et al. 2002. IFN regulatory factor-1 regulates IFN-γ-dependent cathepsin S expression. J. Immunol. 168:4488–4494 [DOI] [PubMed] [Google Scholar]
- 43. Wang Y, et al. 2006. PU. 1 regulates cathepsin S expression in professional APCs. J. Immunol. 176:275–283 [DOI] [PubMed] [Google Scholar]
- 44. Wiegard C, et al. 2007. Defective T helper response of hepatocytes-stimulated CD4T cells impairs antiviral CD8 response and viral clearance. Gastroenterology 133:2010–2018 [DOI] [PubMed] [Google Scholar]
- 45. Zimmermann M, et al. 2008. Hepatitis C virus core protein impairs in vitro priming of specific T cell responses by dendritic cells and hepatocytes. J. Hepatol. 48:51–61 [DOI] [PubMed] [Google Scholar]