

Abstract
Human cytomegalovirus (HCMV) is a herpesvirus that establishes a lifelong, latent infection within a host. At times when the immune system is compromised, the virus undergoes a lytic reactivation producing infectious progeny. The identification and understanding of the biological mechanisms underlying HCMV latency and reactivation are not completely defined. To this end, we have developed a tractable in vitro model system to investigate these phases of viral infection using a clonal population of myeloid progenitor cells (Kasumi-3 cells). Infection of these cells results in maintenance of the viral genome with restricted viral RNA expression that is reversed with the addition of the phorbol ester 12-O-tetradecanoylphorbol-13-acetate (TPA, also known as PMA). Additionally, a latent viral transcript (LUNA) is expressed at times where viral lytic transcription is suppressed. Infected Kasumi-3 cells initiate production of infectious virus following TPA treatment, which requires cell-to-cell contact for efficient transfer of virus to other cell types. Importantly, lytically infected fibroblast, endothelial, or epithelial cells can transfer virus to Kasumi-3 cells, which fail to initiate lytic replication until stimulated with TPA. Finally, inflammatory cytokines, in addition to the pharmacological agent TPA, are sufficient for transcription of immediate-early (IE) genes following latent infection. Taken together, our findings argue that the Kasumi-3 cell line is a tractable in vitro model system with which to study HCMV latency and reactivation.
INTRODUCTION
Human cytomegalovirus (HCMV), a ubiquitous pathogen found in >50% of the general population by 40 years of age, is the most common cause of congenital birth defects yet rarely induces severe disease in immunocompetent hosts (6). Primary infection in healthy individuals results in mild mononucleosis-type symptoms in conjunction with a low-level viremia (68). Within a host, HCMV is detected in a wide range of tissues and cell types, including, but not limited to, epithelial, endothelial, fibroblast, and myeloid cells (22, 49). Primary infection is often resolved by a strong HCMV-specific adaptive immune response. However, HCMV, like all human herpesviruses, establishes a lifelong, latent infection within its host that is likely coupled with a subclinical persistent/latent infection (6, 23). During this phase of the viral life cycle, HCMV infection remains asymptomatic in immunocompetent individuals; however, upon immunosuppression, such as that which occurs in solid organ transplant recipients, bone marrow recipients, and AIDS patients, reactivation of the virus leads to severe morbidity and mortality (48). HCMV-associated disease in adults is predominantly due to reactivation of latent virus as opposed to primary infection, and therefore, understanding latent infection and reactivation is critical.
The reservoir for latent HCMV is commonly accepted to reside within hematopoietic stem cells within the bone marrow, particularly in undifferentiated cells of the myeloid lineage and monocytes (15, 26, 28, 32, 39, 55). Several hallmarks define herpesvirus latency, including long-term maintenance of the viral genome coupled with limited viral transcript expression and a lack of detectable productive viral replication (reviewed in reference 8). In vivo, latently infected cells represent only a small population of an individual's hematopoietic compartment. Typically, only 1 in 10,000 to 1 in 25,000 peripheral blood mononuclear cells (PBMCs) harbor viral genomes with a low viral copy number (2 to 13 viral genomes present per cell) (53). Considerable efforts have been made to identify specific viral transcripts that are expressed during stages of latency from naturally infected cells, including the identification of UL138 (13), viral interleukin-10 (vIL-10) (24), US28 (5), ORF94 (where ORF is open reading frame) (29), and a transcript that runs antisense to the UL81-UL82 locus called LUNA (4). The biological requirement of these viral factors for the establishment and maintenance of latency is the focus of ongoing investigations. It is generally established that virus reactivates from latency within either CD34+ hematopoietic progenitors or CD14+ monocytes as the cells mature into macrophages or dendritic cells, resulting in a lytic infection (reviewed in reference 44).
Previous studies using several model systems have laid the foundation for establishing paradigms of key components of HCMV latency and critical aspects of reactivation. While these studies have proven instrumental, these model systems possess characteristics that render studies assessing complete reactivation from latency problematic. In vitro HCMV latency models using either THP-1 cells (a monocyte cell line) (10) or NTera2 cells (embryonic carcinoma cell line) (2) have been extensively employed to study HCMV latency (5, 10, 11, 20, 25, 30, 31, 34, 45, 62, 65, 69). While these cell lines are valuable tools for identifying cellular factors that modulate viral latency, the cell types do not maintain the viral genome for extended periods of time, resulting in no clear demarcation between latency and reactivation. Importantly, these model systems lack the ability to recapitulate the critical defining characteristic of reactivation: the production of infectious virus progeny. Thus, these systems represent only a snapshot of the complete viral life cycle.
Ex vivo latency models that utilize primary CD34+/CD38− hematopoietic progenitor cells (HPCs) isolated from bone marrow or umbilical cord blood (13, 14, 26, 35, 42), as well as peripheral blood monocytes (7, 16, 27, 33, 50, 52, 56, 58, 67), represent perhaps a more complete assessment of HCMV latency. These model systems support HCMV latent infection, and importantly, the latent virus can be reactivated, producing infectious progeny (12, 14, 16, 50, 55, 56). However, these primary cell systems are hampered by finite cell numbers coupled with lower infectivity rates, limited life span ex vivo, donor variation, the complexity of a heterogeneous population, and the inability to exploit current molecular biology tools to manipulate gene expression. For example, since these primary cells have short life spans, it is not possible to generate stable cell lines that either repress or overexpress a gene of interest. Donor variation between samples and highly heterogeneous populations, coupled with reduced infectivity rates, renders global analyses by deep sequencing and/or commercial microarray platforms for transcript assessment problematic. Thus, although such ex vivo systems most closely represent a complete model of latency, it is difficult to further identify and define the biological roles of cellular and viral factors that are involved in latency.
In order to develop a model system that combines the positive attributes of current systems, we have defined a novel system for HCMV latency in vitro utilizing Kasumi-3 cells. Kasumi-3 cells are a clonal cell line, derived from a patient suffering from myeloperoxidase-negative acute leukemia (3). This patient harbored a chromosomal rearrangement, including a breakpoint that disrupts the normal repression of the EVI1 gene promoter. EVI1 is a nuclear activator of the cell cycle, and stem cell growth and expression of EVI1 aids in the transformation of these cells (37). This cell line is attractive as a potential resource for a latency model, as these cells are negative for HCMV and express cell surface markers indicative of myeloid progenitors, including CD13, CD33, CD34, HLA-DR, and c-Kit (3). Furthermore, Kasumi-3 cells maintain the ability to differentiate down the myeloid pathway and, specifically, are directed toward the monocyte lineage by the addition of the phorbol ester, 12-O-tetradecanoylphorbol-13-acetate (TPA, also known as PMA) (3). Therefore, we hypothesized that Kasumi-3 cells are a tractable cell line that could be used to study HCMV latency and reactivation. We report that the Kasumi-3 myeloid-derived cell line is well suited to study in vitro HCMV latency and recapitulates all of the aspects of viral dormancy and reactivation. We have observed key defining characteristics of HCMV latency using the Kasumi-3 cells, including initial infection, genome maintenance in the absence of viral lytic transcription, reactivation due to an external stimulus, and, finally, production of infectious progeny virions. Thus, the novel in vitro system described herein using Kasumi-3 cells serves as an attractive model system for investigations into mechanisms of HCMV latency and reactivation, providing a valuable resource to complement current ex vivo model systems.
MATERIALS AND METHODS
Cells and viruses.
Kasumi-3 cells (ATCC catalog no. CRL-2725) were maintained in RPMI medium containing 20% fetal bovine serum (FBS) and 100 U/ml each of penicillin and streptomycin at a concentration between 3 × 105 and 3 × 106 cells/ml. Primary human foreskin fibroblasts (HFFs; passages 9 to 20) were cultured in Dulbecco's modified Eagle medium (DMEM) containing 10% FBS, 1 mM sodium pyruvate, 10 mM HEPES, 2 mM l-glutamine, 0.1 mM nonessential amino acids, and 100 U/ml each of penicillin and streptomycin. Primary human retinal pigment epithelial cells (ARPE19 [ATCC catalog no. CRL-2302]; passages 27 to 36) were cultured in 1:1 DMEM-HAM's F-12 medium with 10% FBS, 2.5 mM l-glutamine, 0.5 mM sodium pyruvate, 15 mM HEPES, 1.2 g/liter NaHCO3, and 100 U/ml each of penicillin and streptomycin. Primary human umbilical vascular endothelial cells (HUVECs; passages 3 to 6) were cultured on tissue culture-treated plates coated with 3% porcine gelatin (Sigma) in EBM-2 medium containing 2% FBS and the EGM-2 supplements (Lonza). All cells were propagated with 5% CO2 at 37°C.
The bacterial artificial chromosome (BAC)-derived clinical strain TB40/E (clone 4) was used in this study (51). For infections, either TB40/Ewt-mCherry (38) or TB40/E expressing enhanced green fluorescent protein (eGFP) was used where indicated. Using bacterial recombineering methods (63), we generated TB40/Ewt-GFP, which expresses eGFP from the simian virus 40 (SV40) early promoter. In brief, galK was inserted between the US34 and TRS1 genes of TB40/E by homologous recombination using the following primers: forward, 5′-TGTATTTGTGACTATACTATGTGCAGTCGTGTGTCGATGTTCCTATTGGGCCTGTTGACAATTAATCATCGGCA-3′, and reverse, 5′-GATGTCTTCCTGCGTCCCACCATTCTTTATACCTCCTACATTCACACCCTTTCAGCACTGTCCTGCTCCTT-3′, where the underlined sequences correspond to galK. galK-positive clones were selected as described previously (63). A cassette containing eGFP driven by the SV40 promoter along with the bovine growth hormone poly(A) sequence was then PCR amplified using the following primers: forward, 5′-TTGTATTTGTGACTATACTATGTGCAGTCGTGTGTCGATGTTCCTATTGGGATCTGCGCAGCACCATGGCCTGAAATAACCTCTGAAAG-3′, and reverse, 5′-GATGTCTTCCTGCGTCCCACCATTCTTTATACCTCCTACATTCACACCCTTCTGCCCCAGCTGGTTCTTTCCGCCTCAGAAGCCATAGA-3′, where the underlined sequences corresponds to the intergenic region between HCMV US34 and TRS1. The resulting PCR product was transformed into electrocompetent SW105 cells and finally counterselected for galK to generate TB40/Ewt-GFP. All clones were sequenced to validate proper positional insertion and to validate the sequence of the cassette (data not shown).
Virus stocks were prepared as described previously (66). In brief, low-passage fibroblasts were transfected with BAC DNA along with 1 μg of pCGNpp71 plasmid, and virus was harvested from cells when they reached complete cytopathic effect by centrifugation through a 20% sorbitol cushion. Virus stocks were stored in DMEM containing 10% FBS and 1.5% bovine serum albumin (BSA) at −80°C. Viral stock titers were calculated using 50% tissue culture infectious dose (TCID50) assays on fibroblasts.
Kasumi-3 infection assay.
Kasumi-3 cells were infected with TB40/Ewt-GFP or TB40/Ewt-mCherry, where indicated, at a multiplicity of 10 PFU/cell by centrifugal enhancement at 1,000 × g for 30 min at room temperature. Infected cultures were incubated overnight at 37°C and 5% CO2. To remove debris, infected cells were cushioned onto Ficoll-Paque Plus (GE Healthcare) by low-speed centrifugation (450 × g) for 35 min at room temperature without the brake. The following day, GFP-positive/propidium iodide (PI)-negative cells indicative of viable infected cells were isolated at room temperature by fluorescence-activated cell sorting (FACS) using a FACSDiva (Becton, Dickinson). eGFP-positive cells were then returned to culture to establish a latent infection. For each time point, 20 nM 12-O-tetradecanoylphorbol-13-acetate (TPA, also known as PMA; Sigma), 20 μg/ml tumor necrosis factor alpha (TNF-α; Pepro Tech, Inc.), or interleukin-1β (IL-1β; Pepro Tech, Inc.) was added 48 h prior to harvest in order to induce viral lytic reactivation. Where indicated, cells were cultured with 300 μg/ml phosphonoacetic acid (PAA; Sigma). For these cultures, PAA-containing media were changed every 5 days.
Analysis of viral RNA and DNA.
Cell-associated and cell-free viral DNA was isolated as described previously by lysis with 40 μg/ml proteinase K (PK) and 0.16% sodium dodecyl sulfate (SDS) treatment, followed by phenol-chloroform-isoamyl alcohol extraction and ethanol precipitation (14). To ensure the measurement of encapsidated viral DNA, cell-free viral particles were pretreated with DNase prior to the addition of PK and SDS. The levels of cell-associated viral DNA were normalized to the cellular gene MDM2 using the following primers: forward, 5′-CCCCTTCCATCACATTGCA-3′, and reverse, 5′-AGTTTGGCTTTCTCAGAGATTTCC-3′. All samples were analyzed in triplicate by SYBR green (Applied Biosystems) using an Eppendorf Mastercycler RealPlex2 real-time PCR machine.
Intracellular viral RNA was quantified as described previously (61). In brief, RNA was isolated using TRI Reagent (Sigma) according to the manufacturer's instructions and precipitated with isopropanol. Samples were treated with DNase using the DNA-free kit (Ambion) according to the manufacturer's instructions. The concentration of RNA was determined, and 0.5 μg was used for the reverse transcriptase (RT) reaction. cDNA was synthesized using the TaqMan reverse transcription kit with random hexamers according to the manufacturer's protocol (Applied Biosystems). Equal amounts of samples were analyzed by quantitative PCR (qPCR) in triplicate using an Eppendorf Mastercycler RealPlex2 real-time PCR machine. RNA was normalized to cellular GAPDH: forward, 5′-ACCCACTCCTCCACCTTTGAC-3′, and reverse, 5′-CTGTTGCTGTAGCCAAATTCGT-3′.
Primer sets used for viral RNA and DNA are as follows: UL123 forward, 5′-GCCTTCCCTAAGACCACCAAT-3′, and reverse, 5′-ATTTTCTGGGCATAAGCCATAATC-3′; UL44 forward, 5′-TACAACAGCGTGTCGTGCTCCG-3′, and reverse, 5′-GGCGTGAAAAACATGCGTATCAAC-3′; UL99 forward, 5′-GTGTCCCATTCCCGACTCG-3′, and reverse, 5′-TTCACAACGTCCACCCACC-3′; UL122 forward, 5′-ATGGTTTTGCAGGCTTTGATG-3′, and reverse, 5′-ACCTGCCCTTCACGATTCC-3′; UL54 forward, 5-CCCTCGGCTTCTCACAACAAT-3′, and reverse, 5′-CGAGGTAGTCTTGGCCATGCAT-3′; and UL83 forward, 5′-CGTGGAAGAGGACCTGACGATGAC-3′, and reverse, 5′-GGGACACAACACCGTAAAGCCG-3′.
To amplify the latent transcript LUNA, a strand-specific RT reaction was performed on RNA prepared as described above. The TaqMan reverse transcription kit (Applied Biosystems) was used to generate LUNA-specific cDNA, with the exception that the random hexamers were replaced with the following strand-specific primer for LUNA: 5′-ATGACCTCTCATCCACACC-3′. As a negative control, a primer upstream of the LUNA coding region was used to ensure the amplification of LUNA-specific cDNA: 5′-CCTCCGCGTCACGCTGACCGG-3′. cDNA was then quantified by qPCR as describe above using the following primer set: forward, 5′-GAGCCTTGACGACTTGGTAC-3′, and reverse, 5′-GGAAAACACGCGGGGGA-3′.
Immunofluorescence detection of pUL99.
Kasumi-3 cells were infected with TB40/Ewt-mCherry at a multiplicity of 1.0 PFU/ml by low-speed centrifugation. Following 24 h at 37°C/5% CO2, cells were cushioned onto Ficoll-Paque Plus (GE Healthcare) and incubated at 37°C/5% CO2 for an additional 24 h, after which the cells were sorted for mCherry. Sorted cells were returned to culture for 18 days and then cultured an additional 2 days in the presence or absence of 20 nM TPA (Sigma). Nonadherent cells were removed from culture, and cells that had adhered were removed by trypsin treatment. For each treatment condition, adhered and nonadhered cells were combined, washed twice with phosphate-buffered saline (PBS), and fixed in 4% formaldehyde (Thermo Scientific) for 10 min at room temperature. The fixative was diluted with 5 times the volume of permeabilization buffer (1% BSA, 2 mM EDTA, 0.1% sodium azide, 0.1% saponin in PBS) and pelleted at 0.5 × g for 7 min. Cells were washed and pelleted an additional two times, after which the cells were incubated for 20 min at room temperature with a monoclonal antibody for pUL99 (47), diluted 1:10 in permeabilization buffer. Cells were diluted and washed with permeabilization buffer as described above and then incubated with Alexa Fluor 488 anti-mouse secondary antibody (Molecular Probes) diluted 1:1,000 in permeabilization buffer for 15 min in the dark at room temperature. Cells were diluted and washed as described above. Cells were washed an additional two times in buffer lacking saponin. Cells were then resuspended in Vectashield mounting medium (Vector Labs) containing 4′,6′-diamidino-2-phenylindole (DAPI) and mounted on Superfrost Plus microscope slides (Cardinal Health). Images were captured by using a Leica CTR5000 microscope equipped with tile scanning and outfitted with a Retiga-SRV camera (QImaging). Tiled images were collected at a ×10 magnification using Image Pro Plus, version 6.1 (MediaCybernetics) with an Oasis tile scanning module.
Assay for reactivation of infectious progeny virions.
Kasumi-3 cells were infected at a multiplicity of 10 PFU/cell by low-speed centrifugation, followed by overnight incubation at 37°C/5% CO2. Infected cells were treated with trypsin to remove virions that had not entered the cells and then cushioned onto Ficoll-Paque Plus (GE Healthcare) as described above. Infected Kasumi-3 cells were maintained in culture for an additional 6 days in the presence or absence of 20 nM TPA (Sigma). Cells were then washed with 1× PBS prior to coculture with primary HFFs to remove the TPA. Infected cells were cocultured with HFFs with or without cell-to-cell contact using transwells (0.4 μm; Corning). After 2 days, the infected Kasumi-3 cells were removed, and the HFFs were washed twice with PBS and then returned to culture with fresh media for an additional 4 days. Over the complete course of infection, HFFs were monitored for GFP-positive plaques, and HFF cell-associated DNA was analyzed for viral genomes by qPCR as described above.
Individual cultures of primary HUVECs, ARPE19 cells, or HFFs were infected at a multiplicity of 1.0 PFU/cell for 2 days and then trypsinized to remove any virus that had not entered the cells. Uninfected Kasumi-3 cells were cocultured with each cell type in the presence or absence of a transwell (0.4 μm; Corning) for 5 days, after which the Kasumi-3 cells were removed and replated in the presence or absence of 20 nM TPA (Sigma) for 2 days. Kasumi-3 cell-associated DNA was harvested to quantify viral DNA by qPCR as described above.
Limiting dilution assay.
An HCMV limiting dilution assay was performed as described previously (12), with minor modifications. Kasumi-3 cells were infected at a multiplicity of 10 PFU/cell, cushioned by Ficoll-Paque Plus (GE Healthcare) after 24 h to remove unattached virus and cell debris, and finally sorted by FACS the following day as described above. Cells were then cultured for 8 days to favor a latent infection. Infected cells were then cultured for an additional 2 days in the presence or absence of 20 nM TPA (Sigma). TPA and any residual cell-free virus was removed by trypsin treatment and subsequently washed twice with 1× PBS. The Kasumi-3 cells were then plated directly onto primary HFFs in a 96-well plate, where each well contained 1.0 × 104 HFF/well. The Kasumi-3 cells were plated in a 2-fold dilution beginning with 1,600 cells/well. In a parallel experiment, we plated infected Kasumi-3 cells that were sonicated prior to TPA treatment to control for residual cell-associated virus that might be infectious prior to reactivation. HFFs were monitored for plaque formation. Positive wells were scored and quantified with a 90% confidence interval using the extreme limiting dilution analysis (ELDA) Web tool interface (17). If no wells were positive for plaque formation across the wells with the most number of Kasumi-3 cells, the assumption was made that reactivation occurred in 1 in 38,400 cells or at a frequency of 2.6 × 10−5.
RESULTS
Infected Kasumi-3 cells maintain HCMV genomes while restricting viral gene transcription.
To determine if Kasumi-3 cells could support HCMV infection, we infected these cells by centrifugal enhancement at a multiplicity of 10 PFU/cell with TB40/Ewt-GFP and isolated viable eGFP-positive cells by FACS. We observed no detectable eGFP-positive cells for the viable mock-infected cells (Fig. 1A). Interestingly, we observed roughly 11.4% eGFP-positive cells from the viable infected Kasumi-3 cultures 48 h postinfection (hpi) (Fig. 1B), indicating that the virus was able to enter the Kasumi-3 cells and express a reporter gene that is incorporated into the viral genome. A common hallmark of herpesviral latency is the maintenance of the viral genome in the absence of robust lytic viral transcription. In order to determine the status of HCMV transcription in Kasumi-3 cells, we collected infected cells over a 10-day time course and isolated both RNA and DNA to quantify viral transcripts and genomes, respectively. To monitor viral DNA maintenance and levels of viral transcription, we evaluated the expression (RNA) and quantity (DNA) of the viral open reading frame (ORF) UL123. We found that although viral genomes increase over the time course, viral RNA increases slightly at 5 days postinfection (dpi) but then decreases significantly by 10 dpi (Fig. 2). In support of this finding, viral gene expression of UL123 in the ex vivo primary cell systems is activated at early times and is then repressed (14), suggesting that our Kasumi-3 latency model system mimics the findings in these other model systems. Taken together, these data suggest that infected Kasumi-3 cells maintain viral genomes yet restrict the transcription of viral genes.
Fig 1.
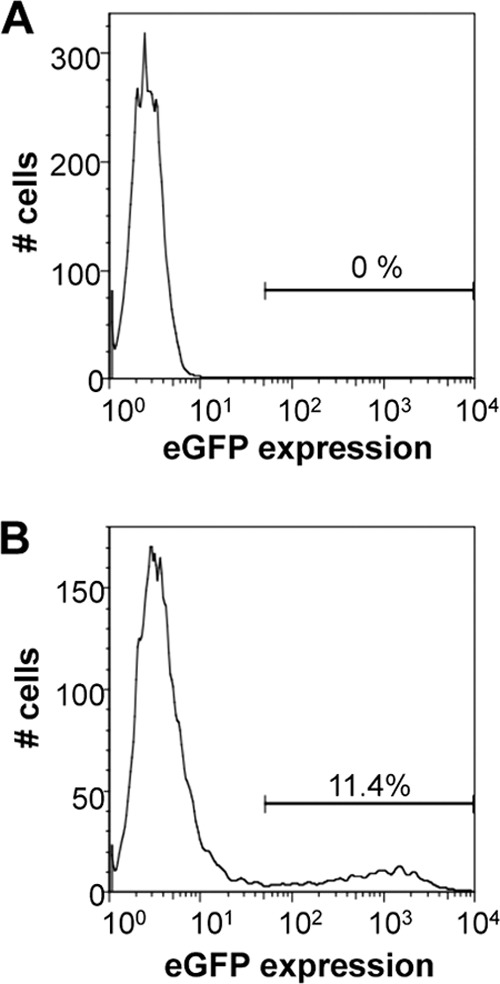
HCMV infection of Kasumi-3 cells results in approximately 11% infectivity rate. Kasumi-3 cells were mock (A) or HCMV (B) infected with TB40/Ewt-GFP at an MOI of 10 PFU/cell. Infected cells were gated first on viability and then gated for eGFP expression.
Fig 2.
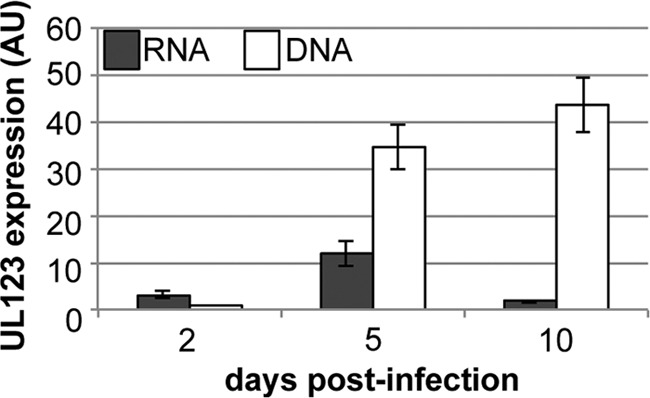
HCMV-infected Kasumi-3 cells maintain viral genomes with restricted viral gene expression. Viral RNA (black bars) and viral DNA (white bars) were assayed over the indicated times for UL123. RNA transcripts were normalized to cellular GAPDH (glyceraldehyde-3-phosphate dehydrogenase), and DNA was normalized to cellular MDM2. Each sample was performed in triplicate. AU, arbitrary units.
Viral transcription is induced in infected Kasumi-3 cells with the addition of TPA.
We next asked if the phorbol ester TPA could induce viral transcription in the infected Kasumi-3 cells, thereby relieving the repression of these transcripts. TPA is commonly used in other well-established in vitro herpesviral latency model systems, including Epstein-Barr virus (EBV) (36) and Kaposi's sarcoma-associated herpesvirus (KSHV) (46), to reactivate viral transcription, thereby inducing lytic replication (36). Additionally, partial HCMV reactivation is observed in THP-1 (31, 62, 64) and NTera2 (34) model systems after treatment with TPA. Finally, treatment of Kasumi-3 cells with TPA promotes their maturation to terminally differentiated cells of the myeloid lineage (3), an important cell type in HCMV reactivation (54, 60, 67). To determine if TPA is sufficient to induce viral gene transcription in our Kasumi-3 model system, we harvested total RNA over a time course from infected cells cultured in the presence or absence of this phorbol ester. We assessed representative viral transcripts from each class of viral genes (i.e., immediate early [IE], early [E], and late [L]). HCMV, like other herpesviruses, transcribes its genes in a coordinated cascade (59). IE genes are transcribed first and predominantly encode transcriptional activators and immune evasion proteins. The major immediate-early promoter (MIEP)-driven transcripts encode proteins (IE1 and IE2) that function to initiate E gene transcription and subsequent protein synthesis. The E genes encode DNA replication machinery and facilitate HCMV genome amplification, after which L genes are transcribed and function mainly in viral assembly and egress. Interestingly, TPA treatment induces IE (UL122 and UL123), E (UL44 and UL54), and L (UL83 and UL99) gene transcription. It is important to note that the IE, E, and L designations, while named on a temporal schedule, were originally defined by protein and DNA replication inhibitor treatments. As such, the accumulation of specific transcripts is more closely related to stages of viral replication as opposed to a defined time frame. Yet several of the transcripts are commonly used to monitor timing of lytic replication (e.g., UL123, UL44, and UL99). We observed a similar pattern of gene expression in our TPA-treated Kasumi-3 cells, as we see in lytic infections of fibroblasts. We observed maximal detection of UL123 IE gene expression at 5 dpi, followed by UL44 E gene expression at 6 to 7 dpi, and, finally, UL99 L gene expression at 8 dpi (Fig. 3). We did not observe robust HCMV transcription within infected cells that were untreated, suggesting the requirement of the phorbol ester on promoting reactivation. This indicates that TPA is sufficient for inducing lytic gene transcription in infected Kasumi-3 cells and that transcription occurs in a cascade similar to that observed in lytically infected cells.
Fig 3.

Repression of HCMV transcripts is released by TPA treatment. Kasumi-3 cells were infected with TB40/Ewt-GFP in the presence or absence of TPA. RNA was harvested at the indicated time points, and representative transcripts from each class of viral gene expression were assessed. UL123 and UL122 (IE, top panel), UL44 and UL54 (E, middle panel), and UL99 and UL83 (L, bottom panel) transcripts were increased with the addition of TPA to the infected cultures. Each sample was performed in triplicate and normalized to cellular GAPDH. AU, arbitrary units.
The HCMV latency transcript LUNA is expressed during viral transcriptional repression in infected Kasumi-3 cells.
Five viral transcripts are expressed in ex vivo HCMV latent model systems, including LUNA (4). Both the LUNA transcript and protein are expressed in peripheral blood of seropositive individuals (4). In order to determine if LUNA is expressed in our model system, we utilized a modified RT-qPCR assay, in which we used a strand-specific primer that yields a cDNA for LUNA but not the two viral transcripts expressed on the opposite strand from LUNA (i.e., UL81 and UL82). We also utilized a reverse transcriptase primer complementary to the region just upstream of the LUNA transcription start site as a negative control. Kasumi-3 cells infected with HCMV were FACs sorted and allowed to incubate for 20 days. The cells were either mock treated or treated with TPA for 2 days prior to harvesting for total RNA. We observe significantly more LUNA transcripts compared to UL123 transcripts in Kasumi-3 cells in the absence of TPA (Fig. 4). Upon stimulation by TPA, IE gene expression increased in relation to LUNA expression, consistent with activation of the lytic life cycle (Fig. 4). As expected, we were not able to detect amplification of LUNA when we utilized the upstream primer, thereby showing specificity of the assay (data not shown). These results suggest that a viral latency transcript is expressed at times during which IE gene expression is restricted. Further, this mimics the findings observed in natural and ex vivo HCMV latency models (40).
Fig 4.
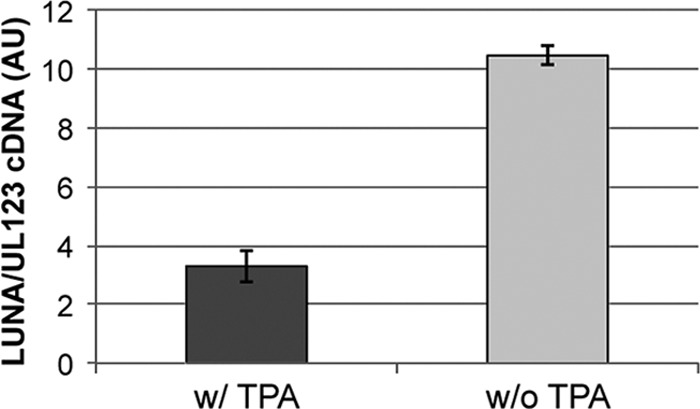
The latent transcript LUNA is expressed while UL123 transcription is repressed, which is reversed with the addition of TPA. Infected Kasumi-3 cells were sorted 2 dpi and cultured for an additional 18 days, at which point cells were cultured for an additional 2 days with or without TPA. Cells were then harvested, and total RNA was collected. LUNA-specific primers were used in the RT reaction to subsequently quantify the LUNA transcript. To quantify UL123, total cDNA was reverse transcribed using random hexamers. Each sample was analyzed in triplicate. AU, arbitrary units.
TPA induction of infected Kasumi-3 cells promotes the release of infectious progeny virions.
We have shown that Kasumi-3 cells are capable of supporting a latent HCMV infection and that transcription of lytic genes is induced by TPA treatment. We next asked if these cells could produce extracellular virions following TPA treatment. To this end, we infected Kasumi-3 cells and assessed their ability to produce cell-free virus in the presence or absence of TPA. We collected cell-free supernatants over time for use in quantitative analysis to determine the amount of released viral genomes. To ensure that we were quantifying encapsidated viral DNA and not free DNA arising from cellular necrosis or lysis, we first treated the cell-free supernatants with DNase prior to the release of encapsidated viral genomes by proteinase K and SDS. HCMV-infected Kasumi-3 cells treated with TPA indeed produced extracellular particles as determined by qPCR for viral genomes, whereas there were significantly fewer particles produced from infected cells that were untreated (Fig. 5A). This result suggests that TPA treatment of infected Kasumi-3 cells is sufficient for producing viral progeny.
Fig 5.

TPA-induced reactivation of HCMV-infected Kasumi-3 cells results in the production of infectious viral particles. (A) Extracellular viral production of HCMV-infected Kasumi-3 cells is increased with TPA treatment. Cell-free viral particles were harvested from TB40/Ewt-GFP-infected cells cultured in the presence or absence of TPA. Encapsidated viral DNA was assessed by qPCR using primers directed at UL123. (B and C) To determine if the viral particles were infectious, infected Kasumi-3 cells were cultured in the presence or absence of TPA for 6 days, after which the cells were washed with PBS and cocultured with HFFs for 2 days. Kasumi-3 cells were then removed from the cocultures, the HFFs were washed twice with PBS, and the HFFs were cultured for an additional 4 days. (B) Fluorescence microscopy reveals eGFP-positive plaques in the HFF monolayer cocultured with TPA-reactivated Kasumi-3-infected cells. (C) HFF-associated DNA was assayed for viral genomes using primers directed at UL44. Each sample was normalized to cellular MDM2 and assessed in triplicate. AU, arbitrary units.
Although informative, this assay does not yield information regarding the infectivity of these particles. A defining attribute of a suitable in vitro model for herpesvirus latency is the ability of the cells to produce infectious progeny upon lytic induction. To determine if infectious viral progeny are produced in TPA-reactivated, HCMV-infected Kasumi-3 cells, we assessed the ability of these cells to transfer virus to HFFs. Primary fibroblasts (e.g., HFFs and MRC5 cells) are commonly used to study the HCMV lytic life cycle in tissue culture, as they are highly permissive for viral replication and thus serve as a suitable cell type with which to determine the infectivity of the reactivated virus. We infected Kasumi-3 cells with TB40/Ewt-GFP as described above, in the presence or absence of TPA. To ensure that TPA treatment would not impact infection of the fibroblasts, we cleared the Kasumi-3 cells of TPA prior to coculturing. We cocultured these cells with HFFs by allowing direct physical contact or separated them with transwells to determine if cell-to-cell contact is necessary for the transfer of infectious virus. After 2 days of coculture between the two cell types, we removed the nonadherent Kasumi-3 cells and monitored the HFFs for plaque formation for an additional 4 days by fluorescence microscopy. Fibroblasts that were in contact (i.e., in the absence of a transwell) with infected, TPA-treated Kasumi-3 cells displayed distinct plaques that were eGFP positive (Fig. 5B). Importantly, we did not observe plaque formation within the fibroblast monolayer in cells cocultured with infected Kasumi-3 cells that were untreated, indicating that phorbol ester treatment is required to produce infectious virus within Kasumi-3 cells. The requirement of TPA treatment suggests that the Kasumi-3 cells are not merely functioning as a carrier of infectious particles from the original infection. We next qualitatively measured the infectivity rate of the mock- and TPA-treated Kasumi-3 cells on cocultured HFFs by monitoring the amplification of viral DNA within the HFFs by qPCR. Fibroblasts cocultured with TPA-treated Kasumi-3 cells displayed increased viral genome levels over background compared to either mock-infected cells treated with TPA or infected cells that were not treated with TPA (Fig. 5C). Infected Kasumi-3 cells that were treated with TPA but cocultured with HFFs in transwells failed to transmit virus to the fibroblasts (data not shown), which suggests that cell-to-cell contact is necessary for transferring virus to the fibroblasts, similar to the mode of viral transfer seen in ex vivo models (41). Taken together, these data suggest that infected Kasumi-3 cells treated with TPA produce viral particles that can establish a lytic infection in permissive fibroblasts.
We next sought to determine the efficiency of viral reactivation by extreme limiting dilution assay (ELDA) (17). Following initial infection of Kasumi-3 cells at a multiplicity of 10 PFU/cell and FACS for eGFP expression, we cultured the cells for 8 days, after which we treated the cells for an additional 2 days in the presence or absence of TPA to induce viral reactivation. We removed residual TPA by washing the Kasmui-3 cells so as to ensure this pharmacological agent would not influence infection of the fibroblasts. We then seeded the Kasmumi-3 cells in a 2-fold dilution from 1,600 to 12.5 cells per well of a 96-well plate seeded with a monolayer of primary fibroblasts to determine the number of reactivated Kasumi-3 cells required to form an infectious foci. To control for any infectious intracellular particles within the Kasumi-3 cells prior to reactivation, we included a lysate control, whereby the cells were disrupted by bath sonication (12). We monitored and quantified plaque formation in the HFFs 8 days post coculture with the Kasumi-3 cells using the web tool interface developed by Hu and Smyth (17) for analyses. We observed that the Kasumi-3 cells that were sonicated (i.e., the lysate control) failed to produce eGFP-positive wells in our assay, further confirming that the Kasumi-3 cells are not simply a carrier of preexisting virus left from the primary infection. We observed 1 eGFP-positive plaque for every 131 infected Kasumi-3 cells treated with TPA or a frequency of 7.6 × 10−3 compared to that of the lysate control, which was 1 in 38,400 cells or a frequency of 2.6 × 10−5 (data not shown). In cultures that were not treated with TPA to induce viral reactivation, the frequency of reactivation was only 2.8 × 10−3 or 1 eGPF-positive plaque per 349 cells. A log fraction plot of the limiting dilution model derived from our assay shows distinct slopes with nonoverlapping, 90% confidence interval plots for latently infected Kasumi-3 cell reactivation in TPA-treated versus untreated cultures, demonstrating that the addition of TPA significantly increases viral reactivation (Fig. 6). Although there is a low level of viral replication in the non-TPA-treated cultures, TPA significantly reduces the number of Kasumi-3 cells required to form plaques. Together, these results suggest that reactivation of HCMV in infected Kasumi-3 cells produces infectious virus following the treatment of these cells with TPA.
Fig 6.
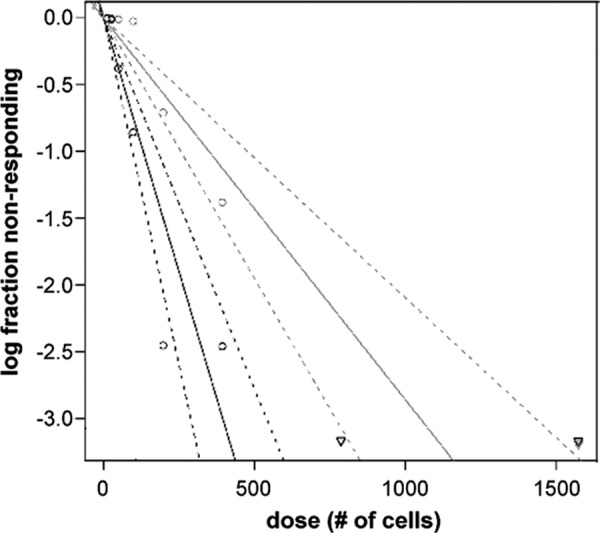
Frequency of reactivation in infected Kasumi-3 cultures in the presence and absence of TPA. Limiting dilution (ELDA) using fibroblasts reveals 131 infected Kasumi-3 cells treated with TPA are required for 1 plaque compared to untreated infected cells, which require 349 cells to generate 1 plaque (P < 0.0005). Black lines represent cultures treated with TPA; gray lines represent cultures not treated with TPA. Solid lines represent the trend line estimating the active cell frequency; dashed lines represent the 90% confidence interval. Circles represent data points of the log proportion of negative cultures; upside-down triangles represent data with zero negative response.
PAA treatment of infected Kasumi-3 cells limits spontaneous reactivation of HCMV.
We observed a low-level accumulation of viral DNA in the absence of reactivation by TPA treatment (Fig. 5A). Additionally, our limiting dilution assays suggest that infected Kasumi-3 cells can produce infectious virus (although inefficiently) in the absence of TPA stimulation (Fig. 6). There are three possible scenarios that could account for these observations: (i) infection with HCMV establishes a low-level, persistent infection that is stimulated by TPA treatment, (ii) a small population of infected Kasumi-3 cells fails to enter a repressed state upon infection, or (iii) latency is established following infection that demonstrates low levels of spontaneous lytic reactivation. In order to differentiate between these three possibilities, we monitored the expression of a viral late protein, pUL99, by immunofluorescence in Kasumi-3 cells infected for 20 days. If cells are persistently infected or fail to initially undergo a quiescent state, the relative intensity of pUL99 staining would increase with TPA treatment but the number of positive cells would not significantly change. However, if a latent infection is established, the number of pUL99 cells would increase in response to TPA treatment but the relative intensity of antigen staining would not vary significantly. HCMV mCherry-positive Kasumi-3 cells were sorted and maintained for 18 dpi, at which point cells were cultured for an additional 2 days in the presence or absence of TPA. These cells were then harvested and fixed for an immunofluorescence assay (IFA) using an antibody to pUL99 and DAPI to identify nuclei. Forty random fields of stained cells from three different experiments were counted for expression of pUL99. A representative field of view is shown in Fig. 7A. We observed ∼15% pUL99-positive cells in the infected Kasumi-3 cells that were not treated with TPA (Fig. 7B). Interestingly, we observe ∼40% pUL99 staining in cells that are treated with TPA, indicating that the number of cells, but not the staining intensity, has increased upon TPA treatment. This observation is consistent with a latent infection versus a persistent infection or incomplete quiescence.
Fig 7.

PAA treatment of infected Kasumi-3 cells limits spontaneous reactivation of HCMV. (A) Infected Kasumi-3 cells were sorted 2 dpi and cultured for 18 days, at which time the cells were cultured for an additional 2 days in the presence or absence of TPA. Cells were then harvested for IFA using an antibody to the late protein pUL99 and DAPI. (B) Infected Kasumi-3 cells that did not receive TPA treatment show a significantly less number of pUL99-positive cells than those that received TPA (P < 0.05). Additionally, the fluorescent intensity of pUL99 observed in the infected cells that were not treated with TPA did not vary between the infected cultures treated with TPA (panel A). (C) Kasumi-3 cells were infected and sorted as described above and then cultured for 18 days in the presence or absence of PAA. These cells were then cultured for an additional 2 days in the presence or absence of TPA. Total RNA was harvested and quantified by RT-qPCR for UL123 expression. Each sample was normalized to cellular GAPDH and analyzed in triplicate. AU, arbitrary units.
We next determined if background levels of viral transcripts in the absence of TPA is the result of spontaneous reactivation of HCMV and/or the result of cells that could not establish a complete latent state. We maintained infected, FACS-sorted Kasumi-3 cells in the presence or absence of phosphonoacetic acid (PAA) for 20 days. PAA is a nucleoside analogue that, in the presence of HCMV UL97, functions as an inhibitor for both viral and cellular DNA polymerases (1). Thus, cells undergoing an active lytic replication would be cleared from the population in the presence of PAA. In the absence of PAA, we can detect UL123 gene expression that is increased upon treatment with TPA (Fig. 7C). However, infected Kasumi-3 cells maintained in the presence of PAA display near-undetectable levels of UL123 gene expression. This suggests that PAA treatment is sufficient to eliminate cells that harbor a transcriptionally active viral genome.
Kasumi-3 cells require cell-to-cell contact with HCMV infected cells to establish an infection.
Although myeloid progenitor cells are an important cell type for HCMV pathogenesis in vivo, this cell type is not likely the first to come in contact with the virus. Cells at the epithelium such as endothelial and epithelial cells likely play an important role in primary HCMV infection in the host (6). Thus, we asked if endothelial and epithelial cells, as well as fibroblasts, could transfer HCMV to Kasumi-3 cells. We infected HUVECs (endothelial cells), ARPE19s (epithelial cells) or HFFs with TB40/Ewt-GFP at a multiplicity of 1.0 PFU/cell for 2 days, after which we removed virus that had bound but not entered by trypsin treatment so as to neutralize any remaining extracellular virus that may influence our results. We then cocultured these cells with uninfected Kasumi-3 cells either with direct contact or separated by transwells for 5 days. The nonadherent Kasumi-3 cells were then collected and cultured in the presence or absence of TPA. We assessed whole-cell DNA content of the Kasumi-3 cells and found that the TPA-treated cells that were cultured in the absence of the transwells harbored a significantly increased quantity of viral genomes compared to cocultured cells that were mock-treated with the phorbol ester (Fig. 8). In addition, our results indicate that cell-to-cell contact is required for HCMV to transfer from fibroblasts, endothelial, or epithelial cells to Kasumi-3 cells, as Kasumi-3 cells cocultured in transwells with these various cell types did not yield cell-associated viral DNA, even following TPA treatment (Fig. 8). Taken together, these results suggest that we are able to infect cells that likely represent sites of primary infection in vivo and transfer virus to Kasumi-3 cells, where the virus requires TPA for reactivation. These data further support our findings shown in Fig. 5, whereby transfer of virus between Kasumi-3 cells and fibroblasts is necessitated by cell-to-cell contact.
Fig 8.

Cell-to-cell contact is required for infected endothelial, epithelial, and fibroblast cells to transfer virus to Kasumi-3 cells. Primary HUVECs (endothelial cells) (A), ARPE19 cells (epithelial cells) (B), or HFFs (fibroblasts) (C) were infected and then cocultured with Kasumi-3 cells for 5 days. Kasumi-3 cells were removed from the coculture and cultured in the presence or absence of TPA for 2 days. Kasumi-3 cell-associated DNA was harvested to quantify viral genomes by qPCR using primers recognizing UL99. Samples were normalized to cellular MDM2 and analyzed in triplicate. AU, arbitrary units.
Inflammatory cytokines reactivate IE gene expression in Kasumi-3 cells.
Although TPA is sufficient to induce lytic reactivation in our in vitro model system, phorbol esters are not responsible for HCMV reactivation within a host. As tumor necrosis factor alpha (TNF-α) or interleukin-1β (IL-1β) promote reactivation of murine cytomegalovirus (MCMV) in vivo, we sought to determine if these inflammatory cytokines are sufficient for inducing lytic viral gene expression from the MIEP. The MIEP drives expression of both UL122 and UL123 viral mRNAs (59), and activation of this promoter is considered the initial step in the switch from a latent infection to a lytic infection (11). We observed that TNF-α or IL-1β treatment of infected Kasumi-3 cells resulted in the induction of UL123 IE gene expression by 48 h posttreatment, where TNF-α treatment was more efficient at inducing viral transcriptional activation (Fig. 9A). Although each of these inflammatory cytokines, as well as the pharmacological agent TPA, are sufficient for inducing UL123 expression, it is possible that the differentiation of these progenitor cells by these factors plays a role in viral reactivation. This is not without precedent: ex vivo systems utilizing monocytes display increased viral production, as these cells differentiate down the myeloid pathway following induction by specific growth factor treatments (16, 55–57). We therefore assessed IE gene expression within the first 30 h posttreatment with either TPA, TNF-α, or IL-1β treatment and found that TNF-α induces the transcription of UL123 mRNA beginning as early as 2 h posttreatment, whereas IL-1β-induced UL123 expression does not increase until 30 h posttreatment (Fig. 9B). The different kinetics of promoter activation by TNF-α compared to IL-1β suggest that the mechanisms of activation by these inflammatory cytokines may differ; however, each are sufficient for activating HCMV transcription within our Kasumi-3 model system.
Fig 9.
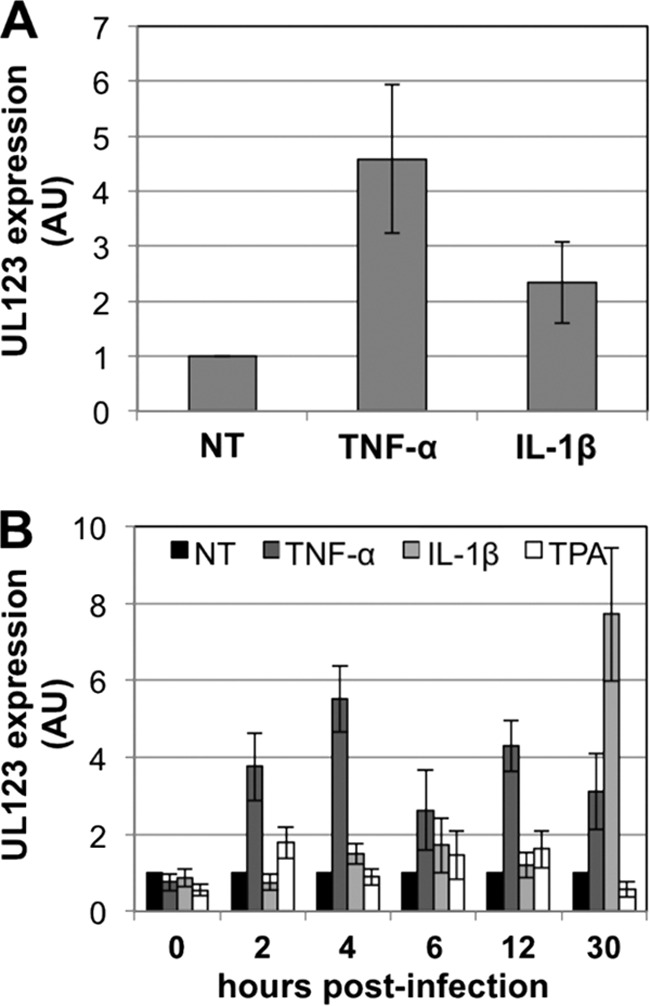
Inflammatory cytokines increase HCMV IE transcription. (A) Either TNF-α or IL-1β treatment induces UL123 gene expression in infected Kasumi-3 cells. Cells were treated with either cytokine for 48 h, after which cells were harvested for RT-qPCR for UL123. (B) TNF-α-induced IE gene expression as early as 2 h posttreatment. Infected Kasumi-3 cells (10 dpi) were treated with TNF-α, IL-1β, or TPA for the indicated times. Cells were collected for RT-qPCR for UL123. In all cases, samples were analyzed in triplicate with primers toward UL123. Each sample was normalized to cellular GAPDH and then normalized to the NT control. NT, no treatment; AU, arbitrary units.
DISCUSSION
In the present study, we have established a novel in vitro system with which to investigate HCMV latency. Our system utilizes a readily available clonal cell line termed Kasumi-3. We have shown that the Kasumi-3 cell line is capable of harboring a nonproductive HCMV infection that can be reactivated to produce infectious viral progeny. Importantly, we have shown that HCMV-infected Kasumi-3 cells recapitulate the key hallmarks of latency. These criteria include initial infection, minimal expression of viral transcripts coupled with viral genome maintenance, reactivation due to stimuli resulting in viral lytic transcription, and, importantly, the production of infectious viral progeny. The development of this system serves as a significant vertical advancement to the field of HCMV biology that will function as a powerful tool to complement current ex vivo model systems.
This is the first description of a myeloid cell line that meets all of the criteria for HCMV latency. To our knowledge, the only other cell line capable of full permissive infection by HCMV is the human glioblastoma-astrocytoma cell line U373 MG cells (43), which are no longer available from the ATCC, as there are questions about their authenticity (21). Using the homogenous population of Kasumi-3 cells, we were able to infect over 11% of viable cells, which, when cultured long-term, were capable of maintaining the viral genome in an environment of repressed viral gene transcription. Interestingly, when we treated these cells with the phorbol ester TPA to induce viral gene expression, we found that several of the representative viral genes of the IE, E, and L gene classes were transcribed in the temporal order observed during traditional lytic gene expression. These findings suggest that TPA-reactivated HCMV in the Kasumi-3 system results in lytic gene expression, consistent with that observed during de novo lytic infection with HCMV in culture. Further strengthening our model is the finding that Kasumi-3 cells infected with HCMV and subsequently treated with TPA are capable of transferring virus to primary fibroblasts by a cell-to-cell contact mechanism. While we observe robust transcriptional activation upon TPA treatment, production of infectious virus is not as equally robust. The efficiency of virus assembly is often cell-type specific, as one observes different particle-to-PFU ratios for HCMV depending on the source of the infected cell. However, we are able to detect a significant increase in the amount of viral production upon TPA stimulation, a defining hallmark of herpesviral reactivation. Thus, this model system represents a significant advancement, as Kasumi-3 cells are a suitable platform for use in investigations of the complete reactivation life cycle of HCMV, a prospect that was only available in ex vivo model systems.
In support of the idea that Kasumi-3 cells allow for the establishment of latency as opposed to a smoldering, persistent infection, we removed lytically infected cells from our population by the addition of PAA. In cells treated with this antiviral factor, we did not see background expression from the MIEP. Upon treatment with TPA, we observed a significant activation of the MIEP in this population of cells, suggesting that latency was established in the Kasumi-3 cells. If the Kasumi-3 cells were persistently infected, we would not observe an increase in UL123 expression with TPA treatment, as these cells would have been cleared from the population. Taken together, these experiments suggest that Kasumi-3 cells establish a latent infection.
Additionally, we have found that our Kasumi-3 model system is well suited for monitoring mechanisms of viral transmission between cell lines important during in vivo infections. While myeloid cells in vivo reactivate and transmit virus to other cell types and can be infected with virus ex vivo, it is likely that cells of the endothelium and epithelium come in contact and are infected first during a primary infection of the host (6). We tested this mode of infection within our Kasumi-3 system by infecting endothelial and epithelial cells, as well as fibroblasts, and asking if these cell types could transmit virus to naïve Kasumi-3 cells. We found that indeed this was the case and that this transmission of virus required cell-to-cell contact. Additionally treatment with TPA was required for viral replication within these cells. There is a possibility that infected, nonadherent HUVECs, ARPE19s, or HFFs could contribute to the source of viral DNA we observed; however, under visual inspection, the GFP-positive cells we analyzed were consistent in size and shape with Kasumi-3 cells. These data suggest that HCMV favors a latent infection in Kasumi-3 cells, whether the mode of infection is primary (i.e., direct infection of the Kasumi-3 cells with HCMV) or secondary (i.e., viral transmission to the Kasumi-3 cells from other cell types).
Immunologically stressed individuals who are latently infected with HCMV undergo lytic reactivation of the virus, leading to a productive infection and subsequent disease. Although the direct factors that impact reactivation of HCMV in vivo remain to be elucidated, an increase in inflammatory cytokines due to immune stress has been strongly implicated, at least in part, in HCMV reactivation. Evidence supporting this comes from studies utilizing ex vivo hematopoietic cells from healthy, seropositive patients, where inflammatory cytokines function to impact HCMV reactivation (18, 19). Additionally, in vitro systems that utilize latently infected THP-1 cells, for example, respond to inflammatory cytokines by inducing IE gene expression (31). Strong evidence for the involvement of inflammatory cytokines comes from the murine model for CMV reactivation. Mice latently infected with murine cytomegalovirus (MCMV) undergo viral reactivation following TNF-α or IL-1β treatment (9). Finally, in transgenic mice that express a HCMV MIEP-driven reporter construct, the MIEP is transcriptionally activated upon treatment with these cytokines either directly or by the addition of lipopolysaccharides (19). When we evaluated the ability of either TNF-α or IL-1β to induce HCMV IE gene expression in our model system, we discovered that both of these inflammatory cytokines induced IE gene expression, although TNF-α caused a more robust response than IL-1-β. This difference reflects perhaps distinct pathways influenced by these two inflammatory cytokines that, in the end, independently induce transcription of the IE genes. Alternatively, our observations may be due to differential cell surface expression of the receptors for these cytokines, a prospect we are actively pursuing. It is enticing to speculate that perhaps HCMV uses two independent yet redundant mechanisms to ensure reactivation in its host.
To further characterize the differences observed in the cytokine treatment protocols, we monitored the timing of MIEP activation upon treatment. There have been several studies that have described the production of infectious HCMV, as latently infected ex vivo cells are promoted to differentiate toward mature cells of the myeloid lineage (12, 56, 60). We assessed IE gene activation within the first 30 h following inflammatory cytokine treatment to begin to distinguish whether the viral reactivation in our system is due to differentiation caused by the various treatments or due to activation of specific biological factors (e.g., transcription factor/signaling pathway activation). IE gene expression due to TNF-α begins at least 2 h posttreatment, whereas IL-1β confers its effects after 24 h of treatment. This suggests that at least TNF-α alone is sufficient for at least “jump-starting” lytic replication. Additionally, this further supports the possibility that each of these inflammatory cytokines functions through a distinct mechanism to induce the IE genes.
The in vitro latency system we have described herein provides a critical resource that will function to complement current ex vivo systems. Our novel Kasumi-3 model system is well suited for high-throughput molecular techniques amenable for identification of both cellular factors critical for reactivation as well as pharmacological agents that inhibit viral reactivation. Additionally, the Kasumi-3 model for HCMV latency affords one the ability to generate stable cell lines for analysis of targeted manipulation of cellular and viral components to assess their role in viral reactivation. Investigators can extrapolate and extend findings from the HCMV Kasumi-3 latency model system into ex vivo systems. We favor the clonal Kasumi-3 model described herein as a suitable in vitro model system to investigate key aspects of HCMV latency and reactivation.
ACKNOWLEDGMENTS
We thank the Cleveland Clinic Flow Cytometry Core Facility for their assistance with cell sorting and Judy Drazba and John Peterson of the Cleveland Clinic Image Core for their assistance with the immunofluorescence assays.
C.M.O. was supported by a postdoctoral fellowship, award 119028-PF-10-164-01-MPC, from the American Cancer Society.
Footnotes
Published ahead of print 3 July 2012
REFERENCES
- 1. Alain S, et al. 1993. Rapid detection of cytomegalovirus strains resistant to ganciclovir through mutations within the gene UL97. Mol. Cell. Probes 7:487–495 [DOI] [PubMed] [Google Scholar]
- 2. Andrews PW, et al. 1984. Pluripotent embryonal carcinoma clones derived from the human teratocarcinoma cell line Tera-2. Differentiation in vivo and in vitro. Lab. Invest. 50:147–162 [PubMed] [Google Scholar]
- 3. Asou H, et al. 1996. Establishment of an undifferentiated leukemia cell line (Kasumi-3) with t(3;7)(q27;q22) and activation of the EVI1 gene. Jpn. J. Cancer Res. 87:269–274 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4. Bego MG, Keyes LR, Maciejewski J, St Jeor SC. 2011. Human cytomegalovirus latency-associated protein LUNA is expressed during HCMV infections in vivo. Arch. Virol. 156:1847–1851 [DOI] [PubMed] [Google Scholar]
- 5. Beisser PS, Laurent L, Virelizier JL, Michelson S. 2001. Human cytomegalovirus chemokine receptor gene US28 is transcribed in latently infected THP-1 monocytes. J. Virol. 75:5949–5957 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6. Britt W. 2008. Manifestations of human cytomegalovirus infection: proposed mechanisms of acute and chronic disease. Curr. Top. Microbiol. Immunol. 325:417–470 [DOI] [PubMed] [Google Scholar]
- 7. Chan G, Bivins-Smith ER, Smith MS, Smith PM, Yurochko AD. 2008. Transcriptome analysis reveals human cytomegalovirus reprograms monocyte differentiation toward an M1 macrophage. J. Immunol. 181:698–711 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8. Cohrs RJ, Gilden DH. 2001. Human herpesvirus latency. Brain Pathol. 11:465–474 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9. Cook CH, Trgovcich J, Zimmerman PD, Zhang Y, Sedmak DD. 2006. Lipopolysaccharide, tumor necrosis factor alpha, or interleukin-1beta triggers reactivation of latent cytomegalovirus in immunocompetent mice. J. Virol. 80:9151–9158 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10. Delannoy AS, Hober D, Bouzidi A, Wattre P. 1997. A macrophage-like cell model for testing anti-CMV drugs. Pathol. Biol. (Paris) 45:394–399 [PubMed] [Google Scholar]
- 11. Dósa R, Burian K, Gonczol E. 2005. Human cytomegalovirus latency is associated with the state of differentiation of the host cells: an in vitro model in teratocarcinoma cells. Acta Microbiol. Immunol. Hung. 52:397–406 [DOI] [PubMed] [Google Scholar]
- 12. Goodrum F, Jordan CT, Terhune SS, High K, Shenk T. 2004. Differential outcomes of human cytomegalovirus infection in primitive hematopoietic cell subpopulations. Blood 104:687–695 [DOI] [PubMed] [Google Scholar]
- 13. Goodrum F, Reeves M, Sinclair J, High K, Shenk T. 2007. Human cytomegalovirus sequences expressed in latently infected individuals promote a latent infection in vitro. Blood 110:937–945 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14. Goodrum FD, Jordan CT, High K, Shenk T. 2002. Human cytomegalovirus gene expression during infection of primary hematopoietic progenitor cells: a model for latency. Proc. Natl. Acad. Sci. U. S. A. 99:16255–16260 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15. Hahn G, Jores R, Mocarski ES. 1998. Cytomegalovirus remains latent in a common precursor of dendritic and myeloid cells. Proc. Natl. Acad. Sci. U. S. A. 95:3937–3942 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16. Hargett D, Shenk TE. 2010. Experimental human cytomegalovirus latency in CD14+ monocytes. Proc. Natl. Acad. Sci. U. S. A. 107:20039–20044 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17. Hu Y, Smyth GK. 2009. ELDA: extreme limiting dilution analysis for comparing depleted and enriched populations in stem cell and other assays. J. Immunol. Methods 347:70–78 [DOI] [PubMed] [Google Scholar]
- 18. Hummel M, Abecassis MM. 2002. A model for reactivation of CMV from latency. J. Clin. Virol. 25(Suppl 2):S123–S136 [DOI] [PubMed] [Google Scholar]
- 19. Hummel M, et al. 2001. Allogeneic transplantation induces expression of cytomegalovirus immediate-early genes in vivo: a model for reactivation from latency. J. Virol. 75:4814–4822 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20. Ioudinkova E, et al. 2006. Control of human cytomegalovirus gene expression by differential histone modifications during lytic and latent infection of a monocytic cell line. Gene 384:120–128 [DOI] [PubMed] [Google Scholar]
- 21. Ishii N, et al. 1999. Frequent co-alterations of TP53, p16/CDKN2A, p14ARF, PTEN tumor suppressor genes in human glioma cell lines. Brain Pathol. 9:469–479 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22. Jarvis MA, Nelson JA. 2007. Human cytomegalovirus tropism for endothelial cells: not all endothelial cells are created equal. J. Virol. 81:2095–2101 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23. Jarvis MA, Nelson JA. 2007. Molecular basis of persistence and latency. In Arvin A, Campadelli-Fiume G, Mocarski E, Moore PS, Roizman B, Whitley R, Yamanishi K. (ed), Human herpesviruses: biology, therapy, and immunoprophylaxis. Cambridge University Press, Cambridge, United Kingdom: [PubMed] [Google Scholar]
- 24. Jenkins C, Abendroth A, Slobedman B. 2004. A novel viral transcript with homology to human interleukin-10 is expressed during latent human cytomegalovirus infection. J. Virol. 78:1440–1447 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25. Keller MJ, et al. 2007. Reversal of human cytomegalovirus major immediate-early enhancer/promoter silencing in quiescently infected cells via the cyclic AMP signaling pathway. J. Virol. 81:6669–6681 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26. Khaiboullina SF, et al. 2004. Human cytomegalovirus persists in myeloid progenitors and is passed to the myeloid progeny in a latent form. Br. J. Haematol. 126:410–417 [DOI] [PubMed] [Google Scholar]
- 27. Khan KA, Coaquette A, Davrinche C, Herbein G. 2009. Bcl-3-regulated transcription from major immediate-early promoter of human cytomegalovirus in monocyte-derived macrophages. J. Immunol. 182:7784–7794 [DOI] [PubMed] [Google Scholar]
- 28. Kondo K, Kaneshima H, Mocarski ES. 1994. Human cytomegalovirus latent infection of granulocyte-macrophage progenitors. Proc. Natl. Acad. Sci. U. S. A. 91:11879–11883 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29. Kondo K, Mocarski ES. 1995. Cytomegalovirus latency and latency-specific transcription in hematopoietic progenitors. Scand. J. Infect. Dis. Suppl. 99:63–67 [PubMed] [Google Scholar]
- 30. Lashmit PE, Stinski MF, Murphy EA, Bullock GC. 1998. A cis repression sequence adjacent to the transcription start site of the human cytomegalovirus US3 gene is required to down regulate gene expression at early and late times after infection. J. Virol. 72:9575–9584 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31. Lee CH, et al. 1999. Factors affecting human cytomegalovirus gene expression in human monocyte cell lines. Mol. Cells 9:37–44 [PubMed] [Google Scholar]
- 32. Maciejewski JP, et al. 1992. Infection of hematopoietic progenitor cells by human cytomegalovirus. Blood 80:170–178 [PubMed] [Google Scholar]
- 33. McCormick AL, Roback L, Livingston-Rosanoff D, St Clair C. 2010. The human cytomegalovirus UL36 gene controls caspase-dependent and -independent cell death programs activated by infection of monocytes differentiating to macrophages. J. Virol. 84:5108–5123 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34. Meier JL. 2001. Reactivation of the human cytomegalovirus major immediate-early regulatory region and viral replication in embryonal NTera2 cells: role of trichostatin A, retinoic acid, and deletion of the 21-base-pair repeats and modulator. J. Virol. 75:1581–1593 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35. Meyer-König U, Hufert FT, von Laer DM. 1997. Infection of blood and bone marrow cells with the human cytomegalovirus in vivo. Leuk. Lymphoma 25:445–454 [DOI] [PubMed] [Google Scholar]
- 36. Miller G, et al. 1997. Selective switch between latency and lytic replication of Kaposi's sarcoma herpesvirus and Epstein-Barr virus in dually infected body cavity lymphoma cells. J. Virol. 71:314–324 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37. Nucifora G, Laricchia-Robbio L, Senyuk V. 2006. EVI1 and hematopoietic disorders: history and perspectives. Gene 368:1–11 [DOI] [PubMed] [Google Scholar]
- 38. O'Connor CM, Shenk T. 2011. Human cytomegalovirus pUS27 G protein-coupled receptor homologue is required for efficient spread by the extracellular route but not for direct cell-to-cell spread. J. Virol. 85:3700–3707 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39. Prösch S, Docke WD, Reinke P, Volk HD, Kruger DH. 1999. Human cytomegalovirus reactivation in bone-marrow-derived granulocyte/monocyte progenitor cells and mature monocytes. Intervirology 42:308–313 [DOI] [PubMed] [Google Scholar]
- 40. Reeves M, Woodhall D, Compton T, Sinclair J. 2010. Human cytomegalovirus IE72 protein interacts with the transcriptional repressor hDaxx to regulate LUNA gene expression during lytic infection. J. Virol. 84:7185–7194 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41. Reeves MB, Lehner PJ, Sissons JG, Sinclair JH. 2005. An in vitro model for the regulation of human cytomegalovirus latency and reactivation in dendritic cells by chromatin remodelling. J. Gen. Virol. 86:2949–2954 [DOI] [PubMed] [Google Scholar]
- 42. Reeves MB, Sinclair JH. 2010. Analysis of latent viral gene expression in natural and experimental latency models of human cytomegalovirus and its correlation with histone modifications at a latent promoter. J. Gen. Virol. 91:599–604 [DOI] [PubMed] [Google Scholar]
- 43. Reis B, et al. 1993. Stable constitutive expression of glycoprotein B (gpUL55) of human cytomegalovirus in permissive astrocytoma cells. J. Gen. Virol. 74(Pt 7):1371–1379 [DOI] [PubMed] [Google Scholar]
- 44. Sacher T, et al. 2012. The role of cell types in cytomegalovirus infection in vivo. Eur. J. Cell Biol. 91:70–77 [DOI] [PubMed] [Google Scholar]
- 45. Sanchez V, Dong JJ. 2010. Alteration of lipid metabolism in cells infected with human cytomegalovirus. Virology 404:71–77 [DOI] [PubMed] [Google Scholar]
- 46. Sarid R, Flore O, Bohenzky RA, Chang Y, Moore PS. 1998. Transcription mapping of the Kaposi's sarcoma-associated herpesvirus (human herpesvirus 8) genome in a body cavity-based lymphoma cell line (BC-1). J. Virol. 72:1005–1012 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 47. Silva MC, Yu QC, Enquist L, Shenk T. 2003. Human cytomegalovirus UL99-encoded pp28 is required for the cytoplasmic envelopment of tegument-associated capsids. J. Virol. 77:10594–10605 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 48. Sing GK, Ruscetti FW. 1995. The role of human cytomegalovirus in haematological diseases. Baillieres Clin. Haematol. 8:149–163 [DOI] [PubMed] [Google Scholar]
- 49. Sinzger C, Digel M, Jahn G. 2008. Cytomegalovirus cell tropism. Curr. Top. Microbiol. Immunol. 325:63–83 [DOI] [PubMed] [Google Scholar]
- 50. Sinzger C, et al. 2006. Macrophage cultures are susceptible to lytic productive infection by endothelial-cell-propagated human cytomegalovirus strains and present viral IE1 protein to CD4+ T cells despite late downregulation of MHC class II molecules. J. Gen. Virol. 87:1853–1862 [DOI] [PubMed] [Google Scholar]
- 51. Sinzger C, et al. 2008. Cloning and sequencing of a highly productive, endotheliotropic virus strain derived from human cytomegalovirus TB40/E. J. Gen. Virol. 89:359–368 [DOI] [PubMed] [Google Scholar]
- 52. Slobedman B, Cheung AK. 2008. Microarrays for the study of viral gene expression during human cytomegalovirus latent infection. Methods Mol. Med. 141:153–175 [DOI] [PubMed] [Google Scholar]
- 53. Slobedman B, Mocarski ES. 1999. Quantitative analysis of latent human cytomegalovirus. J. Virol. 73:4806–4812 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 54. Smith MS, Bentz GL, Alexander JS, Yurochko AD. 2004. Human cytomegalovirus induces monocyte differentiation and migration as a strategy for dissemination and persistence. J. Virol. 78:4444–4453 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 55. Söderberg-Nauclér C, Fish KN, Nelson JA. 1997. Reactivation of latent human cytomegalovirus by allogeneic stimulation of blood cells from healthy donors. Cell 91:119–126 [DOI] [PubMed] [Google Scholar]
- 56. Söderberg-Nauclér C, et al. 2001. Reactivation of latent human cytomegalovirus in CD14(+) monocytes is differentiation dependent. J. Virol. 75:7543–7554 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 57. Stein J, Volk HD, Liebenthal C, Kruger DH, Prosch S. 1993. Tumour necrosis factor alpha stimulates the activity of the human cytomegalovirus major immediate early enhancer/promoter in immature monocytic cells. J. Gen. Virol. 74(Pt 11):2333–2338 [DOI] [PubMed] [Google Scholar]
- 58. Stern JL, Slobedman B. 2008. Human cytomegalovirus latent infection of myeloid cells directs monocyte migration by up-regulating monocyte chemotactic protein-1. J. Immunol. 180:6577–6585 [DOI] [PubMed] [Google Scholar]
- 59. Stinski MF, Thomsen DR, Stenberg RM, Goldstein LC. 1983. Organization and expression of the immediate early genes of human cytomegalovirus. J. Virol. 46:1–14 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 60. Taylor-Wiedeman J, Sissons P, Sinclair J. 1994. Induction of endogenous human cytomegalovirus gene expression after differentiation of monocytes from healthy carriers. J. Virol. 68:1597–1604 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 61. Terhune S, et al. 2007. Human cytomegalovirus UL38 protein blocks apoptosis. J. Virol. 81:3109–3123 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 62. Turtinen LW, Seufzer BJ. 1994. Selective permissiveness of TPA differentiated THP-1 myelomonocytic cells for human cytomegalovirus strains AD169 and Towne. Microb. Pathog. 16:373–378 [DOI] [PubMed] [Google Scholar]
- 63. Warming S, Costantino N, Court DL, Jenkins NA, Copeland NG. 2005. Simple and highly efficient BAC recombineering using galK selection. Nucleic Acids Res. 33:e36 doi:10.1093/nar/gni035 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 64. Weinshenker BG, Wilton S, Rice GP. 1988. Phorbol ester-induced differentiation permits productive human cytomegalovirus infection in a monocytic cell line. J. Immunol. 140:1625–1631 [PubMed] [Google Scholar]
- 65. Yee LF, Lin PL, Stinski MF. 2007. Ectopic expression of HCMV IE72 and IE86 proteins is sufficient to induce early gene expression but not production of infectious virus in undifferentiated promonocytic THP-1 cells. Virology 363:174–188 [DOI] [PubMed] [Google Scholar]
- 66. Yu D, Smith GA, Enquist LW, Shenk T. 2002. Construction of a self-excisable bacterial artificial chromosome containing the human cytomegalovirus genome and mutagenesis of the diploid TRL/IRL13 gene. J. Virol. 76:2316–2328 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 67. Yurochko AD, Huang ES. 1999. Human cytomegalovirus binding to human monocytes induces immunoregulatory gene expression. J. Immunol. 162:4806–4816 [PubMed] [Google Scholar]
- 68. Zaia JA. 1990. Epidemiology and pathogenesis of cytomegalovirus disease. Semin. Hematol. 27:5–10 [PubMed] [Google Scholar]
- 69. Zydek M, Hagemeier C, Wiebusch L. 2010. Cyclin-dependent kinase activity controls the onset of the HCMV lytic cycle. PLoS Pathog. 6:e1001096 doi:10.1371/journal.ppat.1001096 [DOI] [PMC free article] [PubMed] [Google Scholar]