

Abstract
The papillomavirus E2 proteins are indispensable for the viral life cycle, and their functions are subject to tight regulation. The E2 proteins undergo posttranslational modifications that regulate their properties and roles in viral transcription, replication, and genome maintenance. During persistent infection, the E2 proteins from many papillomaviruses act as molecular bridges that tether the viral genomes to host chromosomes to retain them within the host nucleus and to partition them to daughter cells. The betapapillomavirus E2 proteins bind to pericentromeric regions of host mitotic chromosomes, including the ribosomal DNA loci. We recently reported that two residues (arginine 250 and serine 253) within the chromosome binding region of the human papillomavirus type 8 (HPV8) E2 protein are required for this binding. In this study, we show that serine 253 is phosphorylated, most likely by protein kinase A, and this modulates the interaction of the E2 protein with cellular chromatin. Furthermore, we show that this phosphorylation occurs in S phase, increases the half-life of the E2 protein, and promotes chromatin binding from S phase through mitosis.
INTRODUCTION
Papillomaviruses are ubiquitous, small double-stranded DNA viruses that infect the mucosal and cutaneous epithelia of their natural hosts. They are the etiological agents of a wide spectrum of diseases that range from mild asymptomatic infections to malignant carcinomas. Human papillomavirus type 8 (HPV8) belongs to the betapapillomavirus (BPV) genus, which contains viruses that infect the cutaneous epithelium. In healthy individuals, these viruses are associated with asymptomatic infections (1), but in individuals with immune disorders, such as epidermodysplasia verruciformis, they cause lesions that can become cancerous after decades of infection (11). Members of this genus have also been implicated in nonmelanoma skin cancer (10).
One of the hallmarks of papillomavirus infection is the ability of the virus to establish persistent infection of the host. An essential feature of persistent infection is the ability of the viral E2 protein to tether viral genomes to host chromosomes during mitosis as a means of ensuring their nuclear retention and partitioning at the end of cell division (reviewed in reference 21). The E2 protein consists of two conserved domains: a carboxy-terminal DNA binding and dimerization domain (CTD) that binds to palindromic 12-bp target sequences on the viral genome and an amino-terminal transactivation domain that (along with the CTD) functions in viral replication and transcription. The two domains are separated by a highly flexible and nonconserved hinge region (13, 22). Initially, the hinge region was thought to function only as a flexible linker; however, several diverse functions have now been mapped to the hinge domains of different E2 proteins. Regions that regulate nuclear localization in HPV11 E2 (44), proteasomal degradation in BPV type 1 (BPV1) E2 (31), and transcriptional regulation and chromosome binding in HPV8 E2 (35, 39) have been defined.
The E2 proteins from different papillomaviruses have been shown to associate with distinct chromosomal targets (29). BPV1 and several other papillomavirus E2 proteins bind as small speckles over the arms of all mitotic chromosomes in association with the cellular protein Brd4 (25, 43). However, HPV8 E2 is primarily observed as large pericentromeric foci on mitotic chromosomes (25, 32). BPV1 E2 interacts with host chromosomes through the amino-terminal transactivation domain (2, 43), but the pericentromeric binding of HPV8 E2 does not require either the N-terminal transactivation domain or the Brd4 interaction to bind to mitotic chromosomes (32). The HPV8 E2 protein has been shown to associate with the repetitive ribosomal DNA (rDNA) loci on the short arms of human acrocentric chromosomes and to colocalize with the rDNA transcription factor upstream binding factor (UBF) (32). We have previously identified a 16-amino-acid region of the HPV8 E2 hinge that, when fused to the C-terminal DNA binding domain, is both essential and sufficient for this binding. Furthermore, two residues within this peptide, arginine 250 (R250) and serine 253 (S253), are critical for this interaction (35).
The regulatory mechanisms governing E2-mediated genome tethering and partitioning have not yet been completely elucidated. Posttranslational modifications regulate the functions of many viral proteins and modulate their interactions with cellular proteins. For example, posttranslational modifications regulate the chromatin binding function of the Epstein-Barr virus (EBV) tethering protein EBNA1. A glycine-arginine-rich chromatin binding region of EBNA1 is both phosphorylated, most likely by calmodulin-dependent kinase II, and methylated by protein arginine methyltransferases PRMT1 and PRMT5 (36). Phosphorylation of multiple serine residues within the glycine-arginine-rich region is important for the partitioning function of EBNA1 (36). EBNA1 is primarily a nuclear protein, with small amounts observed in the nucleolus. Inhibition of arginine methylation of EBNA1 alters its localization within the nucleolus from a diffuse pattern to a perinucleolar ring. This suggests that methylation affects the interaction of EBNA1 with either nucleolar RNA or proteins (36).
The papillomavirus E2 proteins are also subject to posttranslational modifications that regulate their functions and properties. In the case of BPV1 E2 protein, casein kinase 2 (CK2) phosphorylation of serine residue 301 within the hinge region triggers a conformational switch that targets the E2 protein for proteosomal degradation (30, 31). Mutation of E2 serine 301 in the background of the BPV1 genome results in expression of a highly stable E2 protein and a very high copy number of viral DNA (23). Furthermore, when the half-life of the E2 protein is increased by mutation of serine 301, significantly higher numbers of viral genomes are found tethered to the mitotic chromosomes, indicating that E2 phosphorylation could directly regulate viral genome copy number (37). Recent work has shown that nuclear receptor interaction protein (NRIP) activates the calcineurin phosphatase and promotes dephosphorylation of the HPV16 E2 protein, which in turn increases E2 stability and E2-mediated transcription (6). Conversely, phosphorylation of HPV16 E2 in the S phase of the cell cycle results in increased E2 stabilization (16); however, the exact site or functional significance of this E2 modification in the viral life cycle is not completely known. Studies have also reported that sumoylation influences E2 stability (41).
We have previously shown that the HPV8 E2 protein is highly phosphorylated (35). The two residues crucial for chromosomal association, R250 and S253, lie within a common kinase motif, RXXS, that is highly conserved in the E2 proteins of all members of the betapapillomavirus genus. We have also shown that S253 within this motif is phosphorylated (35). The fact that the residues required for HPV8 E2 chromosomal association and phosphorylation are identical raises the possibility that S253 phosphorylation regulates the E2 chromosomal association function. In addition, a comparison of the chromosome binding regions of the gamma herpesvirus proteins EBNA1 and LANA with the chromosome binding region of HPV8 E2 reveals that all three tethering proteins contain RXXS motifs within their respective chromosome binding regions (35). Thus, the chromosome binding function of these proteins might be regulated by a common mechanism. To gain further insight into regulation of the chromosome binding function of the HPV8 E2 protein, we investigated the role of phosphorylation of S253. We show that phosphorylation of S253, most likely by protein kinase A (PKA), stabilizes the E2 protein and regulates its interaction with host chromatin.
MATERIALS AND METHODS
Plasmids.
E2-derived proteins were expressed from an inducible metallothionein promoter in the pMEP4 expression vector, as described previously (35). All E2-derived proteins had an N-terminal FLAG epitope tag fused to the amino terminus. Plasmids encoding the HPV8 E2 CTD from amino acids 240 to 255 (240-255-CTD) and CTD from amino acids 216 to 255 (216-255-CTD) have been described previously (35). Plasmids expressing full-length HPV8 E2 with mutations R250A and S253A were generated by standard mutagenesis techniques.
Establishment of stably expressing E2 cell lines.
CV-1 cell lines containing the E2 expression vectors were generated by transfection of the pMEP-E2 plasmids using Fugene 6 (Roche) and selection of drug-resistant cells with 200 μg/ml hygromycin B (Roche). After 2 weeks of selection, drug-resistant colonies were pooled and expanded into a stable polyclonal cell line.
PKA inhibitor and enhancer treatments.
CV-1 cell lines containing E2 expression plasmids were plated onto 10-cm plates so as to achieve 80 to 85% confluence at harvest. On the following day, cells were pretreated for 2 h with the PKA inhibitor H-89 (371962; Calbiochem) at 10 μM or the PKA enhancer forskolin (344270; Calbiochem) at 10 μM or cholera toxin (CTX; 227036; Calbiochem) at 100 ng/ml. Following pretreatment, E2 expression was induced with 3 μM CdSO4 for 3 h in the presence or absence of PKA inhibitor or enhancer. Samples were prepared for immunoprecipitation and immunoblotting as described below.
Combined immunoprecipitation and immunoblotting.
Cell extracts were prepared in modified radioimmunoprecipitation assay (RIPA) buffer (20 mM HEPES, pH 7.0, 150 mM NaCl, 1 mM EDTA, 1% NP-40, 1% deoxycholate, 0.1% sodium dodecyl sulfate [SDS]) containing complete protease inhibitor cocktail (Roche) and PhosSTOP phosphatase inhibitor cocktail (Roche). Protein concentration was determined using a bicinchoninic acid (BCA) protein assay kit (Pierce). Equal amounts of total protein (about 300 μg) were immunoprecipitated using M2 anti-FLAG antibody beads (Sigma). Immune complexes were washed five times with RIPA buffer, and E2 proteins were eluted in 50 μl of lithium dodecyl sulfate (LDS) sample buffer and separated on a NuPage 4 to 12% gradient polyacrylamide gel (Invitrogen) in 1× MES (morpholineethanesulfonic acid) running buffer. Proteins were electrotransferred onto Immobilon-P membranes (Millipore) and detected by immunoblotting, as described below. E2 proteins were detected with rabbit anti-FLAG polyclonal antibody (F7425; Sigma) and phosphorylated E2 proteins with rabbit anti-RXXS motif antibodies (9624 and 9621; Cell Signaling Technology), followed by horseradish peroxidase-conjugated goat anti-rabbit immunoglobulin G (Pierce). Proteins were detected on the membrane with the chemiluminescent reagent SuperSignal Extended West Dura (Pierce). Kodak MI software was used to quantify protein bands.
Immunoblotting.
Cellular proteins were extracted in 2% SDS, 50 mM Tris-HCl, pH 6.8, 10% glycerol, complete protease inhibitor (Roche), and PhosSTOP (Roche). The protein concentration was determined using a BCA protein assay kit (Pierce) according to the manufacturer's instructions. For each sample, 12 to 20 μg protein was separated by SDS-polyacrylamide gel electrophoresis (PAGE) and electrotransferred onto Immobilon-P membranes (Millipore). E2 proteins were detected with M2 anti-FLAG monoclonal antibody (Sigma) or with rabbit anti-FLAG polyclonal antibody (F7425; Sigma). Phosphorylated E2 proteins were detected with rabbit anti-RXXS motif antibodies (9624 and 9621; Cell Signaling Technology) after anti-FLAG immunoprecipitation. Immune complexes were detected on the membrane with horseradish peroxidase-conjugated goat anti-rabbit or anti-mouse immunoglobulin G (Pierce) and the chemiluminescent reagent SuperSignal West Dura (ThermoScientific). Images were captured using a Kodak Image station system, and quantitation was performed using Kodak MI software.
Indirect immunofluorescence.
CV-1 cell lines containing E2 expression vectors were plated on Superfrost Plus slides (Daigger). For cell cycle experiments, cells were synchronized in 2 mM thymidine for 16 h. The thymidine was removed to allow progression of the cell cycle, and cells were fixed at the indicated time points. For mitotic synchronization, thymidine was removed and cells were cultured for an additional 9 h. E2 expression was induced by the addition of 3 μM CdSO4 during the last 3 h of each treatment. Cells were fixed in 4% paraformaldehyde–phosphate-buffered saline (PBS) for 20 min at room temperature and permeabilized with 0.1% Triton X-100 in PBS for 15 min at room temperature. The E2 proteins were detected with monoclonal mouse anti-FLAG M2 antibody (1:500; F3165; Sigma), UBF was detected with rabbit anti-UBF antiserum (1:100; sc-9131; Santa Cruz Biotechnologies), and centromeres were detected with human anti-ACA serum (1:500; HCT0100; Immunovision). Slides were stained with the appropriate DyLight secondary antibodies (Jackson ImmunoResearch). Slides were mounted in Prolong Gold (Invitrogen) containing 1 μg/ml DAPI (4′,6′-diamidino-2-phenylindole). Images were collected with a Leica TCS-SP5 laser scanning confocal imaging system.
E2 protein half-life determination.
CV-1 cell lines containing E2 expression plasmids were plated onto 10-cm plates so as to achieve 80 to 85% confluence at harvest. On the next day, E2-expressing cells were either untreated or pretreated with cholera toxin (100 ng/ml) for 4 h. E2 expression was induced for the last 2 h with 4 μM CdSO4, following which the cadmium was removed and fresh medium was added. At this point, protein synthesis was inhibited by the addition of 25 μM cycloheximide (C7698; Sigma) and 25 μM emetine (E2375; Sigma). At various time points, cells were extracted in 2% SDS, 50 mM Tris-HCl (pH 6.8), 10% glycerol containing complete protease inhibitor cocktail (Roche) and PhosSTOP (Roche).
Isolation of chromatin-bound and unbound proteins.
To isolate proteins that were not bound to chromatin, E2-expressing cells were first extracted in a Triton X-100 lysis buffer (0.5% Triton X-100, 0.1 M NaCl, 2 mM EDTA, 0.1 M Tris, pH 8) containing complete protease inhibitor cocktail (Roche) and PhosSTOP (Roche). To extract chromatin-bound fractions, cell debris was pelleted from the chromatin-unbound fraction and was further extracted in 2% SDS, 50 mM Tris-HCl (pH 6.8), 10% glycerol containing complete protease inhibitor cocktail (Roche) and PhosSTOP (Roche). Equivalent amounts of total protein were separated on a NuPage 4 to 12% gradient polyacrylamide gel and transferred to an Immobilon-P membrane, E2 protein was detected using M2 anti-FLAG antibody (F3165; Sigma), and alpha-tubulin was detected using anti-alpha-tubulin (T-5168; Sigma). Kodak MI software was used to quantify the signals.
RESULTS
Specific detection of HPV8 E2 protein phosphorylated at residue serine 253.
We have previously shown that a 16-amino-acid peptide from the hinge region of the HPV8 E2 protein, when fused to the C-terminal DNA binding domain, is sufficient for interaction with host mitotic chromosomes. Residues 250 and 253 within this peptide are specifically required for this interaction. These residues lie within a common kinase motif (RXXS) that is highly conserved among all betapapillomavirus E2 proteins, and we have previously reported that S253 is phosphorylated (35). To further investigate the role of S253 phosphorylation on the chromosome binding function of the E2 protein, we tested two commercially available phosphorylation-specific antibodies that could bind proteins phosphorylated at the serine residue within the RXXS motif. One of these antibodies detects phosphorylated serine within the context of an RRXS motif (9624; Cell Signaling), while the other recognizes phosphorylated serine within RXXS (9621; Cell Signaling). To determine whether these antibodies could specifically detect E2 protein phosphorylated on S253, cell extracts were prepared from CV-1 cell lines expressing wild-type and S253-mutated E2-derived proteins. The E2 proteins were immunoprecipitated using the FLAG epitope tag and analyzed for phosphorylation by immunoblotting. Both phospho-RXXS and phospho-RRXS motif antibodies detected the S253-phosphorylated E2 protein (Fig. 1) but did not bind to E2 proteins mutated in the S253 residue.
Fig 1.
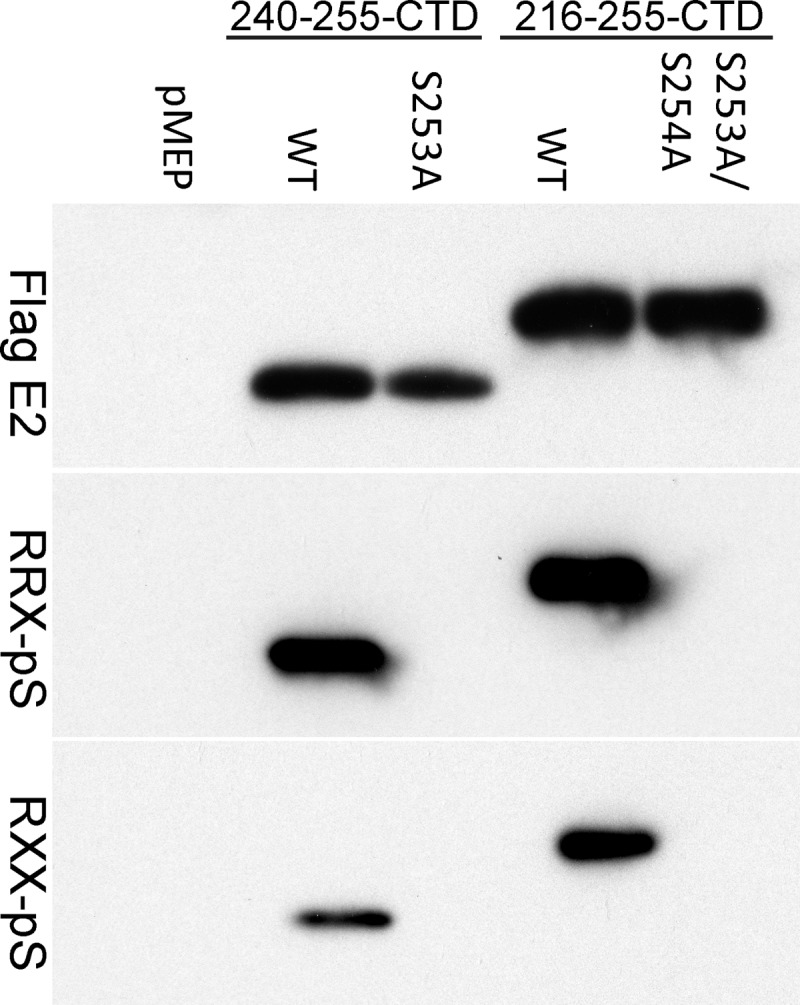
Phospho-RXXS-specific antibodies can recognize HPV8 E2 proteins phosphorylated at S253. Proteins extracted from CV-1 cell lines carrying control vector pMEP4 or plasmids expressing truncated E2 proteins were immunoprecipitated with M2 anti-FLAG antibody beads. Two versions of wild-type (WT) and mutated E2 proteins were used; one contained a 16-amino-acid region of the hinge fused to the C-terminal domain (240-255-CTD) and the other contained a 40-amino-acid region of the hinge fused to CTD (216-255-CTD). The mutated E2 protein in the 240-255-CTD background has an alanine substitution at residue 253 (S253A). The mutated E2 protein in the 216-255-CTD background has alanine substitutions at residues S253 and S254 (S253A/S254A). Phosphorylated E2 proteins were detected by phospho-RXXS (RRX-pS) and phospho-RRXS motif-specific antibodies. Total E2 protein was detected using rabbit anti-FLAG antibody.
To further prove that the phospho-RXXS antibodies were specific, antibodies against phospho-mitogen-activated protein kinase (MAPK)/cyclin-dependent kinase, phospho-AKT, and phospho-(Ser) protein kinase C substrates were also tested. None of these antibodies recognized the phosphorylated E2 proteins. Thus, the phospho-RXXS motif-specific antibodies are valuable tools to study HPV8 E2 S253 phosphorylation. All further studies used the phospho-RRXS motif antibody (9624; Cell Signaling).
HPV8 E2 protein is phosphorylated at the S253 residue by PKA.
The RXXS motif is a common kinase motif that is shared by a number of different cellular kinases, including protein kinase A and aurora kinases. To understand the role of S253 phosphorylation in regulating the chromosome binding function of the HPV8 E2 protein, we proceeded to first identify the cellular kinase responsible for S253 phosphorylation. Using the pkaPS predictor program, which predicts PKA phosphorylation sites on the basis of a simplified kinase substrate binding model (27), S253 was predicted to be a strong PKA substrate site (data not shown). Out of the 59 serine residues in the HPV8 E2 protein, serine 253 had the highest score. PKA is a cyclic AMP (cAMP)-dependent protein kinase that phosphorylates many different cellular proteins involved in regulating signal transduction pathways, immune regulation, and gene expression (33).
To further confirm the role of cellular PKA in S253 phosphorylation, we used different modulators that either activate or inhibit PKA activity and examined their effects on the phosphorylation status of the E2 protein. These experiments were carried out in the background of the 240-255-CTD E2 truncated protein to ensure that we were specifically analyzing residue 253. CV-1 cells carrying either control vector plasmid pMEP4 or plasmid expressing wild type 240-255-CTD E2 protein were pretreated for 2 h with the PKA inhibitor H-89 or the PKA enhancer forskolin or CTX. Pretreatment was followed by induction of E2 expression for 3 h in the presence of the PKA modulators. H-89 is a competitive antagonist that binds to the ATP site on the catalytic subunit of PKA and prevents phosphorylation of target proteins. The PKA enhancers forskolin and CTX, on the other hand, act by increasing the intracellular levels of cAMP, which result in increased PKA activity. Treatment of E2-expressing CV-1 cells with 10 μM H-89 resulted in reduced levels of S253 phosphorylation compared to those in untreated cells (Fig. 2). However, there is some residual amount of phosphorylated E2 protein observed even after the inhibitor treatment. This could be due to either incomplete inhibition of PKA at the concentrations used or redundancy with a secondary protein kinase that can phosphorylate E2 in the absence of PKA. However, reciprocally, treatment of E2-expressing cells with enhancers of PKA activity (10 μM forskolin or 100 ng/ml CTX) greatly increased the level of S253 phosphorylation compared to that in untreated cells (Fig. 2). Thus, modulation of cellular PKA activity alters the level of HPV8 E2 phosphorylation at S253, indicating that PKA likely phosphorylates residue S253.
Fig 2.

PKA phosphorylates HPV8 E2 protein at the serine 253 residue. CV-1 cell lines carrying control vector plasmid pMEP4 or plasmid expressing 240-255-CTD E2 protein were either untreated or pretreated for 2 h with the PKA inhibitor H-89 at 10 μM or the PKA enhancer forskolin at 10 μM or CTX at 100 ng/ml, followed by E2 induction for 3 h. (A) RIPA cell extracts were prepared, and equivalent amounts of total proteins were immunoprecipitated with M2 anti-FLAG antibody beads. Phosphorylated E2 protein was detected using phospho-RRXS motif-specific antibody, followed by reblotting for total E2 protein using rabbit anti-FLAG antibody. (B) Quantitative representation of the relative amounts of phosphorylated E2 protein to total E2 protein observed by immunoblotting in panel A. UT, untreated. Error bars represent standard deviations from three experimental repeats.
Modulation of cellular PKA activity affects the mitotic localization of the HPV8 E2 protein.
Since the two residues that are critical for the chromosomal association function of the HPV8 E2 protein (R250 and S253) are also important for E2 phosphorylation (35), we examined whether modulating the activity of cellular PKA would affect mitotic localization of the E2 protein. A CV-1 cell line expressing the E2-derived 240-255-CTD protein was treated with the different PKA modulators (as described above), and the localization of E2 in mitotic cells was examined by indirect immunofluorescence. Cells were also stained with ACA, an autoimmune antiserum that detects centromeric proteins.
Following treatment with the PKA inhibitor H-89, a reduction in the number of mitotic cells with E2 foci on mitotic chromosomes was observed compared to that for untreated cells (Fig. 3A and B). However, there were almost no mitotic cells observed with E2 protein excluded from the chromosomes. These observations raise the possibility that E2 protein which is not bound to mitotic chromosomes has a very short-half life and is therefore undetectable by immunofluorescence. The percentages of untreated (54%) or H-89-treated (46%) interphase cells expressing E2 were similar, and no change in the overall level of total E2 protein following treatment with the different PKA modulators was observed (Fig. 2A). Thus, the decrease in the number of mitotic cells with E2 foci was not due to an overall decrease in E2 protein.
Fig 3.

Mitotic localization of the HPV8 E2 protein is affected by modulators of cellular PKA activity. (A) CV-1 cell lines expressing the 240-255-CTD E2 protein were either untreated or pretreated for 2 h with 10 μM H-89, 10 μM forskolin, or 100 ng/ml CTX, followed by E2 induction for 3 h. Cells were visualized by indirect immunofluorescence to determine the location of E2 proteins on the mitotic chromosomes. E2 proteins were detected using anti-FLAG antibody (green), centromeres were detected with anti-ACA antibody (red), and cellular DNA was stained with DAPI (blue). (B) Quantitative representation of the relative proportion of E2-expressing mitotic cells with E2 foci on the mitotic chromosomes in the untreated or H-89- or CTX-treated samples. Error bars represent standard deviations from three experimental repeats.
In contrast, treatment with the PKA enhancer forskolin or CTX resulted in an increase in the number of mitotic cells with visible E2 foci compared to that for untreated cells (Fig. 3). However, no significant difference in the number of E2 foci per mitotic cell was observed. We believe that each pericentromeric focus consists of stretches of repetitive DNA and/or chromatin structure that become more and more occupied as the E2 concentration increases. Therefore, the number of foci does not significantly increase with increased E2 levels, but instead, each locus becomes more and more visible as E2 occupancy increases. Thus, modulating the activity of cellular PKA increases or decreases the mitotic localization of the HPV8 E2 protein, indicating that cellular PKA regulates the chromosomal association function of the HPV8 E2 protein.
Modulation of cellular PKA activity affects the interphase localization of the HPV8 E2 protein.
We have previously shown that both the full-length HPV8 E2 protein and the truncated 240-255-CTD E2 protein localize to distinct foci at the pericentromeric regions of mitotic chromosomes of both African green monkey CV-1 cells and human C33A cells (32, 35). In the case of C33A cells, these pericentromeric E2 foci localize to the rDNA loci on the acrocentric chromosomes and colocalize completely with the RNA polymerase I transcription factor UBF (32). However, in CV-1 cells there are two populations of E2-associated mitotic pericentromeric foci; one is UBF positive and one is UBF negative (Fig. 4A). We find that upregulation of PKA activity enhances E2 binding to both UBF-positive and UBF-negative regions.
Fig 4.

Localization of E2 to pericentromeric foci in mitotic and interphase cells. E2 protein expression was induced in CV-1 cell lines expressing the 240-255-CTD E2 protein for 3 h. (A) E2 proteins were detected in mitotic cells using anti-FLAG antibody (green), centromeres were detected with anti-ACA antibody (blue), rDNA loci were detected with anti-UBF (red), and cellular DNA was stained with DAPI (gray). (B) (Left) Wild-type 240-255-CTD E2 protein was detected using anti-FLAG antibody (green), and the rDNA loci were detected with anti-UBF (red); (center) wild-type 240-255-CTD E2 protein was detected using anti-FLAG antibody (green), and the centromeres were detected with anti-ACA antibody (red); (right) the S253A 240-255-CTD E2 was detected using anti-FLAG antibody (green), and the centromeres were detected with anti-ACA antibody (red). mt, S253A mutation in E2.
As shown in Fig. 4B (left and middle), the 240-255-CTD E2 protein forms a diffuse nuclear pattern with some nucleolar staining in interphase CV-1 cells. However, in a subset of cells, the 240-255-CTD E2 protein localizes in addition to distinct, bright foci. These foci are adjacent to the centromeres (stained with ACA antibody in Fig. 4B, middle) and represent the UBF-negative foci that are observed in mitotic CV-1 cells. As shown in Fig. 4B, left, they do not localize with the UBF-positive foci, which are sequestered in the nucleolus in interphase cells. The ACA-positive pericentromeric interphase foci are completely absent in C33A cells and in cells expressing the S253A-mutated 240-255 CTD E2 protein (Fig. 4B, right). The presence of the foci allowed us to study the association of the 240-255-CTD E2 protein with pericentromeric chromatin in interphase cells.
To analyze whether the appearance of the E2-associated interphase pericentromeric foci correlated with the phosphorylation status of the E2 protein, their localization was examined following treatment with the different PKA modulators. As shown in Fig. 5A, treatment of wild-type 240-255-CTD E2 protein-expressing cells with the PKA inhibitor H-89 almost completely abrogated the appearance of E2 bound to pericentromeric foci in interphase cells. Most interphase cells showed only a diffuse nuclear E2 staining pattern, with some minor nucleolar localization. However, in cells treated with the PKA enhancers, there was a dramatic increase in the number of interphase cells with E2 staining in pericentromeric foci (Fig. 5A and B). These results indicate that the localization of E2 adjacent to the centromere in CV-1 interphase cells is linked to the phosphorylation status of the E2 protein and that these foci most likely represent the UBF-negative pericentromeric E2 foci observed in mitotic CV-1 cells. Thus, enhanced levels of E2 phosphorylation result in an increase in the number of interphase cells with pericentromeric E2 foci, whereas inhibition of PKA results in an almost complete loss of these foci (Fig. 5B).
Fig 5.

Modulation of PKA activity alters the association of E2 with pericentromeric foci in interphase cells. (A) CV-1 cell lines carrying control vector plasmid or plasmid expressing E2 protein 240-255-CTD were either untreated or pretreated for 2 h with the PKA inhibitor H-89 at 10 μM or the PKA enhancer forskolin at 10 μM or CTX at 100 ng/ml, followed by E2 induction for 3 h. Cells were visualized by indirect immunofluorescence to determine the location of E2 proteins in interphase cells. E2 proteins were detected using anti-FLAG antibody (green), centromeres were detected with anti-ACA antibody (red), and cellular DNA was stained with DAPI (blue). (B) Quantitative representation of the relative proportion of E2-expressing interphase cells with E2-associated pericentromeric foci in untreated or H-89-, forskolin-, and CTX-treated samples. Error bars represent standard deviations from three experimental repeats.
S253 phosphorylation of HPV8 E2 increases during S phase and is high in G2 and M phases.
Only a subset of E2-expressing interphase cells contained distinct pericentromeric foci, and this indicated that they might be cell cycle dependent. To further understand the dynamics and regulation of the chromosomal association function of the HPV8 E2 protein, we examined the levels of E2 phosphorylation during different stages of the cell cycle. CV-1 cells containing either an E2 expression plasmid or the control vector pMEP were synchronized by a thymidine block, which arrests cells at the G1/S boundary and within S phase. After releasing the cells from thymidine block, they were allowed to progress through the cell cycle and proteins were extracted at various time points. Proteins were also extracted from E2-expressing mitotic cells that were collected by mitotic shake off at 9 h after thymidine release. E2 proteins were extracted in modified RIPA buffer at the indicated times and analyzed for phosphorylation by immunoprecipitation and immunoblotting. As shown in Fig. 6A, there was an increase in the level of E2 phosphorylation during S phase compared to that during G1 phase or to that in an asynchronous population of cells. The levels of phosphorylated E2 remained high throughout the late stages of S phase, G2 phase, and mitosis. The overall levels of E2 dropped in mitosis, but as shown in the quantitative data in Fig. 6B, the level of E2 phosphorylation was high, further indicating that phosphorylated E2 is bound to host chromatin, since only chromosome-bound E2 can be detected at this stage.
Fig 6.

HPV8 E2 phosphorylation at S253 increases in S phase and remains high until mitosis. (A) CV-1 cell lines carrying control vector plasmid or plasmid expressing 240-255-CTD E2 protein were synchronized with thymidine and proteins extracted at 0 h (G1/S), 3 h (S), 6 h (S/G2), and 9 h (G2/M) after thymidine release in RIPA extraction buffer following E2 induction for 3 h. Cell cycle stage was confirmed by flow cytometry. Mitotic cells (M) were collected by mitotic shake off at 9 h after thymidine release. Equivalent amounts of total protein were immunoprecipitated with M2 anti-FLAG antibody beads. Phosphorylated E2 protein (E2-P) was detected using phospho-RRXS motif-specific antibody, followed by reblotting for total E2 protein using rabbit anti-FLAG antibody. (B) Quantitative representation of the relative ratio of phosphorylated E2 protein to total E2 protein observed by immunoblotting in panel A. The error bars shown are a representation of the standard deviations of two experimental repeats. The asynchronous population (Asyn) has an arbitrary value of 1.
HPV8 E2 proteins phosphorylated at S253 have an increased half-life.
While examining the mitotic localization of E2 protein following H-89 inhibitor treatment, we observed that there were almost no mitotic cells with E2 protein excluding the chromosomes. The levels of E2 protein in mitotic cells without chromosome-bound E2 were undetectable. These observations lead us to hypothesize that E2 protein that is not bound to chromosomes is unstable and hence is not observed by immunofluorescence. To investigate this possibility further, we carried out a pulse-chase experiment and compared the half-life of the 240-255-CTD E2 protein with that of the S253A-mutated version. As shown in Fig. 7A and B, both wild-type and S235A 240-255-CTD E2 proteins had a very short half-life, and there was a dramatic decrease in the levels of E2 protein in the first 20 min following inhibition of protein synthesis. However, from 40 min to 3 h, the levels of wild-type protein remained stable. Thus, E2 decay was biphasic, indicating that there might be two populations of E2 protein. In contrast, the S253A-mutated E2 protein underwent a rapid degradation and by 40 min had almost disappeared. To determine whether enhancement of PKA phosphorylation would result in enhanced stability of the wild-type protein, E2-expressing cells were pretreated with CTX for 2 h, followed by induction of E2 expression, as described above. Pulse-chase analysis showed that the wild-type 240-255 E2 protein had an increased half-life in CTX-treated cells and about 25 to 40% remained stable for at least 3 h, while the stability of the S253A protein was unchanged by CTX treatment (Fig. 7A and B). Thus, PKA phosphorylation increased the half-life of the wild-type E2 protein.
Fig 7.

Wild-type E2 protein has a longer half-life than phosphorylation-defective E2 protein. (A) CV-1 cell lines carrying control vector plasmid or plasmids expressing 240-255-CTD E2 protein or S253-mutated E2 protein (S253A) were either untreated or treated with 100 ng/ml CTX for 2 h, followed by E2 induction for 2 h with 4 μM CdSO4. Thereafter, cells were treated with protein synthesis inhibitors cycloheximide (25 μM) and emetine (25 μM) for the indicated times. Cell extracts were prepared in SDS extraction buffer at the indicated time points, E2 protein was detected using M2 anti-FLAG antibody, and alpha-tubulin was detected using anti-alpha-tubulin. (B) Quantitative representation of the blot in panel A showing the percentage of E2 protein remaining plotted against time (in minutes).
In our previous study we had replaced S253 with the phosphomimetic S253D (35), but this negatively charged amino acid was not able to substitute for phosphoserine in E2 chromatin binding. We have not carried out an extensive analysis of the half-life of the 240-255-CTD S253D E2 protein, but the steady-state levels of this protein were reduced compared to those of the wild-type protein in our previous study (35). Thus, replacement of serine 253 with a negatively charged amino acid cannot stabilize the E2 protein, and the mutated protein cannot bind chromatin. This failure of a phosphomimetic to substitute for phosphoserine is similar to the findings of an earlier study from our laboratory showing that CKII phosphorylation of BPV1 E2 induced a conformational switch that triggers degradation of the papillomavirus E2 protein (30). Again, a phosphomimetic could not substitute for phosphoserine, demonstrating that a negative charge cannot substitute for the precise structure of phosphoserine in these situations.
Next, we wanted to investigate whether the observed increase in E2 phosphorylation and stability was related to chromatin binding. To this end, we compared the half-life of the E2 protein in chromatin-bound and unbound fractions. The unbound fraction was extracted using a standard lysis buffer (0.5% Triton X-100, 0.1 M NaCl, 2 mM EDTA, 0.1 M Tris, pH 8), and the chromatin-bound fraction was extracted from the insoluble pellet of the unbound fraction using SDS lysis buffer (2% SDS, 50 mM Tris-HCl, pH 6.8, 10% glycerol). As can be observed in Fig. 8, chromatin-bound wild-type 240-255 E2 protein is quite stable through the different time points following inhibition of protein synthesis. Conversely, almost no S253A-mutated protein could be detected in the bound fraction. This further supports the finding that S253A is defective in chromatin binding. Both wild-type and S253A E2 proteins were observed to be unstable in the unbound fraction, further supporting our hypothesis that there is a correlation between chromatin binding and protein stability.
Fig 8.

Chromatin-bound E2 protein has a longer half-life than unbound protein. (A) Chromatin-bound and chromatin-unbound fractions were prepared from CV-1 cell lines carrying control vector plasmid pMEP or plasmids expressing the 240-255-CTD E2 protein or the S253-mutated E2 protein (S253A) that were either untreated or treated, as described in the legend to Fig. 7A. E2 protein was detected using M2 anti-FLAG antibody; and alpha-tubulin (tub) was detected using anti-alpha-tubulin. (B) Quantitation of the experiment shown in panel A. The decrease in the wild-type E2 protein levels at time 40 min is a result of experimental variation and was not a consistent phenomenon in experimental repeats.
We also analyzed the difference in chromatin binding of E2 protein extracted from cells treated with CTX to enhance E2 phosphorylation. Increasing PKA phosphorylation with CTX resulted in an increase in the amount of wild-type E2 protein in the chromatin-bound fraction but had no effect on the distribution of the S253A E2 protein (Fig. 8). Therefore, the increase in E2 protein half-life observed after CTX treatment correlates perfectly with the increase in the proportion of E2 bound to chromatin after enhancement of phosphorylation. The E2 proteins in the non-chromatin-bound fraction were unstable and did not show much difference in half-life following CTX treatment. Taken together, these observations indicate that enhanced E2 phosphorylation correlates with increased binding to host chromatin and an increased half-life of the HPV8 E2 protein.
Residues arginine 250 and serine 253 are essential for the chromosome binding function of the full-length HPV8 E2 protein.
We have shown that residues R250 and S253 are essential for binding to mitotic chromosomes in the background of an E2-derived protein consisting of 16 amino acids from the hinge (amino acids 240 to 255) fused to the C-terminal DNA binding and dimerization domain (35). We wanted to ensure that mutating these residues in the background of the full-length protein also abrogated the chromosome binding function. We individually mutated each of these residues in the background of the full-length HPV8 E2 protein and examined the localization of the E2 proteins on the mitotic chromosomes by indirect immunofluorescence. As shown in Fig. 9, alanine substitutions of residue R250 or S253 abrogated the chromosome binding function of the mutated full-length E2 proteins. Furthermore, the wild-type E2 protein was bound to mitotic chromosomes in a large percentage of mitotic cells, whereas the mutated proteins were present (chromosome excluded) in only a much lower percentage of cells. This correlates well with the finding that binding to chromatin stabilizes the E2 protein.
Fig 9.

Residues arginine 250 and serine 253 are critical for the association of full-length HPV8 E2 with mitotic chromosomes. (A) CV-1 cell lines carrying control vector pMEP4 or plasmids expressing wild-type HPV8 E2 (WT), R250-mutated E2 (R250A), or S253-mutated E2 (S253A) were visualized by indirect immunofluorescence to determine the location of E2 proteins on the mitotic chromosomes. E2 proteins were detected using anti-FLAG antibody (green), and cellular DNA was stained with DAPI (blue). (B) The percentage of mitotic cells containing E2 bound or excluded from the chromosomes is shown for each protein. Results are from three independent experiments, and values were adjusted for the percentage of interphase cells expressing E2. The phenotype of the double mutation RSAA (R250A and S253A) is also shown.
DISCUSSION
We recently reported that residues R250 and S253, located within the chromosome binding region of the HPV8 E2 protein, are required for E2's chromosome binding function. Furthermore, S253, located within the highly conserved RXXS motif, is phosphorylated (35). In the current study, we have shown that phosphorylation of S253 regulates the chromosome binding function of the HPV8 E2 protein. RXXS is a common kinase motif shared by various cellular kinases, including PKA. Using modulators of cellular PKA activity, we have demonstrated that S253 is most likely phosphorylated by PKA.
PKA is a cAMP-dependent protein kinase that regulates many biological processes (40). PKA is involved in multiple functions during the different stages of the cell cycle, ranging from regulation of S-phase replication to mitotic progression (42). We report here that the level of E2 phosphorylation at residue S253 increases in S phase and remains high through mitosis. This finding is in line with the findings of previous studies that have shown that PKA is required for chromosomal DNA replication and so is active during the S phase of the cell cycle (9). PKA is ubiquitous and, hence, is under strict spatial and temporal regulation. The A kinase-anchoring proteins (AKAPs) play an important role in directing PKA to specific cellular sites so that they are in close proximity to their substrates (38). One such protein is AKAP95, which recruits the regulatory subunit of PKA to chromosomes during mitosis and plays a key role in chromatin condensation at mitosis (7, 19). Since E2 also binds host chromatin, it is possible that PKA anchored to chromatin by AKAP95 is readily available to phosphorylate E2 protein.
Previous studies have also implicated PKA in the HPV life cycle. PKA has been shown to phosphorylate the discs large protein (Dlg)/PDZ binding motif of high-risk HPV E6 (18) and regulate the E6-dependent degradation of the human Dlg. PKA also plays a role in HPV16 E7-mediated transformation by contributing to cellular alkalinization, a common characteristic of transformed cells (5). More recently, the HPV16 E5 protein has been reported to utilize the PKA pathway to inhibit apoptosis of transformed cervical epithelial cells by stimulating proteosome-mediated degradation of the Bax protein (28). Although the functional significance is not known, the HPV11 E1∧E4 protein has also been reported to be phosphorylated in vitro by PKA and MAPK (4). These findings, along with those of our study, highlight the importance of PKA in the HPV life cycle.
The HPV8 E2 protein binds to mitotic chromosomes as large distinct pericentromeric foci. In human cells, these foci colocalize with UBF and overlap the rDNA loci found on the short arm of acrocentric chromosomes (32). However, in CV-1 cells, two populations of E2-associated pericentromeric foci are observed on mitotic chromosomes. One population consists of UBF-positive E2 foci that colocalize with UBF at the rDNA loci on acrocentric chromosomes. The other consists of UBF-negative foci that are localized adjacent to the centromeres of nonacrocentric chromosomes (35). We believe that the latter regions consist of pericentromeric satellite DNA. Similar heterochromatic satellite regions are observed in mouse cells (15), and the gammaherpesvirus-tethering protein LANA has been shown to bind to these murine chromocenters (20). The E2-associated UBF-negative pericentromeric foci are not observed in human cells, and it is possible that in human cells similar repetitive regions are dispersed throughout the chromosomes and, hence, are difficult to detect. Although the HPV8 E2 protein has been shown to colocalize with the rDNA genes in human cells, the exact target has not been identified. It is possible that E2 is interacting with repetitive DNA sequences that are interspersed among the copies of rDNA genes and that these regions are similar to the monkey pericentromeric satellite DNA.
While the HPV8 E2 protein binds to different chromosomal locations in cell types from different species, the binding is regulated in a very similar manner. Mutation of the S253 phosphorylation site in E2 eliminates chromatin binding of both populations in human and monkey cells, as does the inhibition of PKA activity. There is no colocalization of E2 with UBF-positive foci in either monkey or human interphase cells, most likely because the rDNA loci are sequestered into the nucleoli. However, in monkey cells, the 240-255-CTD E2 protein can be observed to be binding to the UBF-negative pericentromeric regions in interphase as well as in mitosis, most likely because these targets are not sequestered into the nucleoli. This interphase localization provides a useful tool to elucidate the regulation of the E2-chromatin association throughout the cell cycle. Our analysis shows that E2 is bound to the UBF-negative foci during S phase and that this interaction is increased by PKA phosphorylation of E2.
There is a strong correlation between E2 phosphorylation, protein half-life, and chromatin binding. PKA phosphorylation of HPV8 E2 enhances both chromatin binding and protein half-life. However, it is not clear whether E2 phosphorylation directly regulates protein half-life, thus leaving only stable, phosphorylated E2 to bind to host chromatin, or whether phosphorylation regulates chromatin binding; and chromatin-bound E2 has a much longer half-life than unbound protein, which is rapidly degraded. A dramatic decrease in the amount of E2 protein is observed by both immunofluorescence and Western blotting as cells enter mitosis, and only chromosome-bound E2 can be detected in mitosis. Presumably, the labile population of unbound E2 protein rapidly disappears as cells cease transcription and translation at the onset of mitosis. It has also been shown that HPV16 E2 has both increased phosphorylation and an extended half-life in S phase (16).
The attachment of the E2 protein (with the viral genome) to host chromatin could be important for several viral functions. This could promote increased transcription and replication of viral DNA by bringing the genome into the vicinity of active host transcription and replication complexes and could also promote genome partitioning by ensuring that genomes are distributed to daughter cells during the process of cell division. We observed no significant difference in the ability of the full-length E2 S253A protein to support transient replication or activate transcription (data not shown). However, under these transient conditions the cells do not efficiently progress throughout the cell cycle, which we believe is necessary for E2 to become posttranslationally modified and attached to chromatin. A better experiment, especially if the role of E2 phosphorylation is to regulate partitioning, is to introduce these mutations into the viral genome and study replication and transcription over several cell divisions. Unfortunately, there is no faithful replication assay for studying genome replication of betapapillomaviruses. Geimanen et al. have recently shown that several papillomaviruses replicate in U2OS cells (12), and so we used this system to analyze the replication of the R250A and S253A mutations into the HPV8 genome. Unfortunately, in our hands, HPV8 replication in U2OS cells is not E2 dependent and so we could not use it to determine the phenotype of the R250A and S253A E2 mutations.
Our findings are in accordance with those of a recent study that showed that the chromosome binding glycine-arginine repeat regions of the EBV tethering protein EBNA1 could inhibit its proteasomal degradation and stabilize the protein (8). These chromosomal tethering regions are thought to resemble A-T hook motifs that bind with high avidity to A-T-rich sequences on cellular DNA (34). Another study demonstrated that EBV replicons replicate while bound to host chromatin and the daughter molecules remain paired at the site of DNA synthesis until they are partitioned in mitosis (26). HPV8 replication could follow a similar scenario in which viral genomes are tethered to host chromatin by the E2 protein in S phase and the daughter molecules remain bound throughout mitosis. The presence of RXXS motifs within the chromosome binding regions of EBNA1 and LANA further underscores the possibility of a common mechanism regulating the chromosome binding function of all three tethering proteins. It is well established that the tethering proteins EBNA, LANA, and E2 have common mechanisms and structural domains. For example, all three proteins have dimeric DNA binding domains that have been either shown or predicted to have very similar structures (3, 14) most likely due to convergent evolution. Similar to the HPV8 E2 protein, LANA has been shown to bind to pericentromeric and peritelomeric regions on mitotic chromosomes (17). In addition, the serine residues within the chromosome binding motifs of EBNA1 have been shown to be phosphorylated, and mutating these serine residues abrogates the partitioning function of EBNA1 (36).
The RXXS motifs are highly conserved in the hinge region of the betapapillomaviruses, but they are not conserved in this exact position in all papillomavirus E2 proteins. However, the papillomaviruses have coevolved along with their hosts and each genus has been distinct for millions of years, so it is not surprising that the E2 protein of each genus has acquired properties that may be similar to those of other nonpapillomaviruses but are not necessarily shared among all papillomaviruses. The E2 proteins fall into very different phylogenetic groups, especially with regard to tethering (24, 29). The betapapillomaviruses have distinct targets on mitotic chromosomes, and all betapapillomaviruses sequenced to date have conserved RXXS motifs.
In summary, we have shown that PKA phosphorylates the HPV8 E2 protein at S253. E2 phosphorylated at S253 can bind to specific, UBF-negative regions of host chromatin in interphase, and phosphorylated E2 has a much longer half-life than unphosphorylated E2. We propose a model in which E2 is phosphorylated primarily in S phase and binds to host chromatin. Phosphorylated, chromatin-bound E2 remains stabilized at these sites through mitosis. As cells transition from interphase to mitosis, additional phosphorylated E2 protein can also associate with UBF-positive regions of chromosomes that become available when the nucleolus disassembles. Both types of binding could be used to tether viral DNA to host chromosomes for viral genome partitioning. On the contrary, the population of E2 protein that is unphosphorylated and not bound to chromatin has a short half-life and disappears even more quickly as transcription and translation cease at the onset of mitosis. Finally, when infected cells exit from mitosis and reenter interphase, the E2 protein is likely dephosphorylated and degraded. Newly synthesized E2 protein would not be highly phosphorylated on serine 253 until the next S phase.
ACKNOWLEDGMENTS
We are grateful to members of the A. A. McBride laboratory for critical reading of the manuscript.
This work was funded by the Intramural Research Program of the NIAID, NIH.
Footnotes
Published ahead of print 11 July 2012
REFERENCES
- 1. Antonsson A, Forslund O, Ekberg H, Sterner G, Hansson BG. 2000. The ubiquity and impressive genomic diversity of human skin papillomaviruses suggest a commensalic nature of these viruses. J. Virol. 74:11636–11641 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2. Baxter MK, McPhillips MG, Ozato K, McBride AA. 2005. The mitotic chromosome binding activity of the papillomavirus E2 protein correlates with interaction with the cellular chromosomal protein, Brd4. J. Virol. 79:4806–4818 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3. Bochkarev A, et al. 1995. Crystal structure of the DNA-binding domain of the Epstein-Barr virus origin-binding protein EBNA 1. Cell 83:39–46 [DOI] [PubMed] [Google Scholar]
- 4. Bryan JT, Han A, Fife KH, Brown DR. 2000. The human papillomavirus type 11 E1∧E4 protein is phosphorylated in genital epithelium. Virology 268:430–439 [DOI] [PubMed] [Google Scholar]
- 5. Cardone RA, et al. 2008. HPV16 E7-dependent transformation activates NHE1 through a PKA-RhoA-induced inhibition of p38alpha. PLoS One 3:e3529 doi:10.1371/journal.pone.0003529 [DOI] [PMC free article] [PubMed] [Google Scholar] [Retracted]
- 6. Chang SW, Tsao YP, Lin CY, Chen SL. 2011. NRIP, a novel calmodulin binding protein, activates calcineurin to dephosphorylate human papillomavirus E2 protein. J. Virol. 85:6750–6763 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7. Collas P, Le GK, Tasken K. 1999. The A-kinase-anchoring protein AKAP95 is a multivalent protein with a key role in chromatin condensation at mitosis. J. Cell Biol. 147:1167–1180 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8. Coppotelli G, Mughal N, Marescotti D, Masucci MG. 2011. High avidity binding to DNA protects ubiquitylated substrates from proteasomal degradation. J. Biol. Chem. 286:19565–19575 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9. Costanzo V, Avvedimento EV, Gottesman ME, Gautier J, Grieco D. 1999. Protein kinase A is required for chromosomal DNA replication. Curr. Biol. 9:903–906 [DOI] [PubMed] [Google Scholar]
- 10. Feltkamp MC, et al. 2003. Seroreactivity to epidermodysplasia verruciformis-related human papillomavirus types is associated with nonmelanoma skin cancer. Cancer Res. 63:2695–2700 [PubMed] [Google Scholar]
- 11. Fuchs PG, Iftner T, Weninger J, Pfister H. 1986. Epidermodysplasia verruciformis-associated human papillomavirus 8: genomic sequence and comparative analysis. J. Virol. 58:626–634 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12. Geimanen J, et al. 2011. Development of a cellular assay system to study the genome replication of high- and low-risk mucosal and cutaneous human papillomaviruses. J. Virol. 85:3315–3329 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13. Giri I, Yaniv M. 1988. Structural and mutational analysis of E2 trans-activating proteins of papillomaviruses reveals three distinct functional domains. EMBO J. 7:2823–2829 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14. Grundhoff A, Ganem D. 2003. The latency-associated nuclear antigen of Kaposi's sarcoma-associated herpesvirus permits replication of terminal repeat-containing plasmids. J. Virol. 77:2779–2783 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15. Guenatri M, Bailly D, Maison C, Almouzni G. 2004. Mouse centric and pericentric satellite repeats form distinct functional heterochromatin. J. Cell Biol. 166:493–505 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16. Johansson C, Graham SV, Dornan ES, Morgan IM. 2009. The human papillomavirus 16 E2 protein is stabilised in S phase. Virology 394:194–199 [DOI] [PubMed] [Google Scholar]
- 17. Kelley-Clarke B, Ballestas ME, Komatsu T, Kaye KM. 2007. Kaposi's sarcoma herpesvirus C-terminal LANA concentrates at pericentromeric and peri-telomeric regions of a subset of mitotic chromosomes. Virology 357:149–157 [DOI] [PubMed] [Google Scholar]
- 18. Kuhne C, Gardiol D, Guarnaccia C, Amenitsch H, Banks L. 2000. Differential regulation of human papillomavirus E6 by protein kinase A: conditional degradation of human discs large protein by oncogenic E6. Oncogene 19:5884–5891 [DOI] [PubMed] [Google Scholar]
- 19. Landsverk HB, et al. 2001. Regulation of anchoring of the RIIalpha regulatory subunit of PKA to AKAP95 by threonine phosphorylation of RIIalpha: implications for chromosome dynamics at mitosis. J. Cell Sci. 114:3255–3264 [DOI] [PubMed] [Google Scholar]
- 20. Matsumura S, Persson LM, Wong L, Wilson AC. 2010. The latency-associated nuclear antigen interacts with MeCP2 and nucleosomes through separate domains. J. Virol. 84:2318–2330 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21. McBride AA. 2008. Replication and partitioning of papillomavirus genomes. Adv. Virus Res. 72:155–205 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22. McBride AA, Byrne JC, Howley PM. 1989. E2 polypeptides encoded by bovine papillomavirus type 1 form dimers through the common carboxyl-terminal domain: transactivation is mediated by the conserved amino-terminal domain. Proc. Natl. Acad. Sci. U. S. A. 86:510–514 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23. McBride AA, Howley PM. 1991. Bovine papillomavirus with a mutation in the E2 serine 301 phosphorylation site replicates at a high copy number. J. Virol. 65:6528–6534 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24. McPhillips MG, Oliveira JG, Spindler JE, Mitra R, McBride AA. 2006. Brd4 is required for e2-mediated transcriptional activation but not genome partitioning of all papillomaviruses. J. Virol. 80:9530–9543 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25. McPhillips MG, Ozato K, McBride AA. 2005. Interaction of bovine papillomavirus E2 protein with Brd4 stabilizes its association with chromatin. J. Virol. 79:8920–8932 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26. Nanbo A, Sugden A, Sugden B. 2007. The coupling of synthesis and partitioning of EBV's plasmid replicon is revealed in live cells. EMBO J. 26:4252–4262 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27. Neuberger G, Schneider G, Eisenhaber F. 2007. pkaPS: prediction of protein kinase A phosphorylation sites with the simplified kinase-substrate binding model. Biol. Direct 2:1 doi:10.1186/1745-6150-2-1 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28. Oh JM, et al. 2010. Human papillomavirus type 16 E5 protein inhibits hydrogen-peroxide-induced apoptosis by stimulating ubiquitin-proteasome-mediated degradation of Bax in human cervical cancer cells. Carcinogenesis 31:402–410 [DOI] [PubMed] [Google Scholar]
- 29. Oliveira JG, Colf LA, McBride AA. 2006. Variations in the association of papillomavirus E2 proteins with mitotic chromosomes. Proc. Natl. Acad. Sci. U. S. A. 103:1047–1052 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30. Penrose KJ, Garcia-Alai M, Prat-Gay G, McBride AA. 2004. CK2 phosphorylation-induced conformational switch triggers degradation of the papillomavirus E2 protein. J. Biol. Chem. 279:22430–22439 [DOI] [PubMed] [Google Scholar]
- 31. Penrose KJ, McBride AA. 2000. Proteasome-mediated degradation of the papillomavirus E2-TA protein is regulated by phosphorylation and can modulate viral genome copy number. J. Virol. 74:6031–6038 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32. Poddar A, Reed SC, McPhillips MG, Spindler JE, McBride AA. 2009. The human papillomavirus type 8 E2 tethering protein targets the ribosomal DNA loci of host mitotic chromosomes. J. Virol. 83:640–650 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33. Sassone-Corsi P. 1998. Coupling gene expression to cAMP signalling: role of CREB and CREM. Int. J. Biochem. Cell Biol. 30:27–38 [DOI] [PubMed] [Google Scholar]
- 34. Sears J, et al. 2004. The amino terminus of Epstein-Barr virus (EBV) nuclear antigen 1 contains AT hooks that facilitate the replication and partitioning of latent EBV genomes by tethering them to cellular chromosomes. J. Virol. 78:11487–11505 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35. Sekhar V, Reed SC, McBride AA. 2010. Interaction of the betapapillomavirus E2 tethering protein with mitotic chromosomes. J. Virol. 84:543–557 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36. Shire K, et al. 2006. Regulation of the EBNA1 Epstein-Barr virus protein by serine phosphorylation and arginine methylation. J. Virol. 80:5261–5272 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37. Skiadopoulos MH, McBride AA. 1998. Bovine papillomavirus type 1 genomes and the E2 transactivator protein are closely associated with mitotic chromatin. J. Virol. 72:2079–2088 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38. Skroblin P, Grossmann S, Schafer G, Rosenthal W, Klussmann E. 2010. Mechanisms of protein kinase A anchoring. Int. Rev. Cell Mol. Biol. 283:235–330 [DOI] [PubMed] [Google Scholar]
- 39. Steger G, Schnabel C, Schmidt HM. 2002. The hinge region of the human papillomavirus type 8 E2 protein activates the human p21(WAF1/CIP1) promoter via interaction with Sp1. J. Gen. Virol. 83:503–510 [DOI] [PubMed] [Google Scholar]
- 40. Taylor SS. 1989. cAMP-dependent protein kinase. Model for an enzyme family. J. Biol. Chem. 264:8443–8446 [PubMed] [Google Scholar]
- 41. Wu YC, Bian XL, Heaton PR, Deyrieux AF, Wilson VG. 2009. Host cell sumoylation level influences papillomavirus E2 protein stability. Virology 387:176–183 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42. Yanagida M, et al. 1999. Control of metaphase-anaphase progression by proteolysis: cyclosome function regulated by the protein kinase A pathway, ubiquitination and localization. Philos. Trans. R. Soc. Lond. B Biol. Sci. 354:1559–1569 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43. You J, Croyle JL, Nishimura A, Ozato K, Howley PM. 2004. Interaction of the bovine papillomavirus E2 protein with Brd4 tethers the viral DNA to host mitotic chromosomes. Cell 117:349–360 [DOI] [PubMed] [Google Scholar]
- 44. Zou N, et al. 2000. The hinge of the human papillomavirus type 11 E2 protein contains major determinants for nuclear localization and nuclear matrix association. J. Virol. 74:3761–3770 [DOI] [PMC free article] [PubMed] [Google Scholar]