

Abstract
The recently identified human metapneumovirus (HMPV) is a worldwide respiratory virus affecting all age groups and causing pneumonia and bronchiolitis in severe cases. Despite its clinical significance, no specific antiviral agents have been approved for treatment of HMPV infection. Unlike the case for most paramyxoviruses, the fusion proteins (F) of a number of strains, including the clinical isolate CAN97-83, can be triggered by low pH. We recently reported that residue H435 in the HRB linker domain acts as a pH sensor for HMPV CAN97-83 F, likely through electrostatic repulsion forces between a protonated H435 and its surrounding basic residues, K295, R396, and K438, at low pH. Through site-directed mutagenesis, we demonstrated that a positive charge at position 435 is required but not sufficient for F-mediated membrane fusion. Arginine or lysine substitution at position 435 resulted in a hyperfusogenic F protein, while replacement with aspartate or glutamate abolished fusion activity. Studies with recombinant viruses carrying mutations in this region confirmed its importance. Furthermore, a second region within the F2 domain identified as being rich in charged residues was found to modulate fusion activity of HMPV F. Loss of charge at residues E51, D54, and E56 altered local folding and overall stability of the F protein, with dramatic consequences for fusion activity. As a whole, these studies implicate charged residues and potential electrostatic interactions in function, pH sensing, and overall stability of HMPV F.
INTRODUCTION
Human metapneumovirus (HMPV) is a recently discovered paramyxovirus that is a major cause of upper and lower respiratory tract disease worldwide in all age groups (8, 9, 28, 41). Numerous studies have shown that in addition to exacerbating underlying diseases such as chronic obstructive pulmonary disease (COPD), HMPV can cause significant morbidity and some mortality, especially in infants, the elderly, and people with compromised immune systems (8, 15, 16). Indeed, studies have estimated that HMPV is the second or third most common cause of severe acute respiratory tract infection in children (28). Despite its widespread human pathogenicity, there is currently no approved antiviral medication or vaccine against HMPV.
HMPV expresses three surface glycoproteins: the putative attachment protein (G), the fusion protein (F), and the small hydrophobic protein (SH) (25). The F proteins of paramyxoviruses, including that of HMPV, are trimeric type I fusion proteins. The membrane fusion event is intimately linked to final formation of a six-helix bundle in the F protein after extensive conformational changes bring two heptad repeat (HR) regions—HRA and HRB—into close proximity (37). For paramyxoviruses, the triggering event that drives these conformational changes is hypothesized to occur following attachment of the virus to its receptor. This triggering must be regulated carefully, as the subsequent conformational change of the F protein from its metastable prefusion form to its stable postfusion form is irreversible. Before a paramyxovirus F protein can be triggered to promote membrane fusion, it must undergo one or more proteolytic cleavage events, which cut the precursor form of the protein (F0) into an active, disulfide-linked form (F1+F2) (25, 37). Unlike most paramyxovirus F proteins, which are processed by proteases present in the host cell, HMPV F requires exogenous protease cleavage to be fusogenically active. Thus, for infection to occur, HMPV requires proteolytic activation after budding. In vitro, the cleavage of HMPV F can be achieved by artificially adding a low concentration of trypsin to the system (35).
Despite being classified as a paramyxovirus, HMPV is novel in several aspects. While most other paramyxoviruses require the expression of an attachment protein to be infectious, HMPV, like its near relative respiratory syncytial virus (RSV), is infectious in vitro when devoid of the G protein (7, 23, 25). However, unlike RSV, HMPV lacking the G protein is also infectious in animals, including nonhuman primates (3, 7). Additionally, the HMPV F protein alone is able to promote cell-cell fusion in the absence of the G protein (35). These observations suggest that HMPV F can promote both attachment and efficient membrane fusion.
Another distinguishing feature of HMPV is that for at least some strains of the virus, low pH can trigger the fusion activity of the F protein (19, 35), while most paramyxovirus F proteins are triggered at neutral pH (25). Our laboratory has shown that low-pH treatment of Vero cells transfected with the F protein of HMPV strain CAN97-83 (clade A2) leads to F triggering and subsequent syncytium formation (35). Furthermore, inhibitors of endosomal acidification, such as bafilomycin A and concanamycin, were able to confer partial protection against infection with a recombinant green fluorescent protein (GFP)-expressing HMPV CAN97-83 strain as well as a B1 strain of the virus (27, 34). Certain strains of HMPV clade A1 also show low-pH-promoted fusion (19).
While low-pH triggering is unusual for paramyxoviruses, several viruses from other virus families require low pH for membrane fusion. A classic example of a type I fusion protein triggered by low pH is influenza virus hemagglutinin (HA), which has a triggering threshold of approximately pH 5.4 (30). Mechanistically, it is thought that electrostatic repulsive forces that arise between residues that become protonated at low pH and their neighboring residues are responsible for destabilizing the prefusion form and allowing it to trigger and refold into its postfusion conformation (18, 21). The effects of electrostatic interactions on the stability of a protein vary dramatically depending on the location of the interaction and the surrounding environment. For instance, modeling studies showed a strong attraction between the positively charged HA1 subunits and the negatively charged HA2 subunits of influenza virus HA. At low pH, however, the HA1 subunits become protonated, increasing the repulsive force between neighboring HA1 subunits in the trimeric protein. This results in protein destabilization, which aids in facilitating the transition to the postfusion structure (20).
Previous studies have emphasized the role of histidine residues in the influenza virus HA triggering event as well as in those for other fusion proteins (22). These moieties, with a side chain pKa value of 6.04 in solution, can undergo protonation under physiological conditions. Interestingly, sequence analysis of different influenza virus serotypes showed that a number of histidine residues are conserved across strains, highlighting the potential role of these residues in the function of the fusion protein (22). Analysis of a homology model of the prefusion form of the HMPV F protein showed the presence of a region in the HRB linker domain consisting of a central histidine, H435, that is surrounded by several basic residues, most notably K295, R396, and K438 (Fig. 1) (34). Previous studies indicated that the HRB linker domain is important for modulating fusion in the F protein of parainfluenza virus 5 (PIV5) (31). Mutagenesis studies confirmed the importance of this region for the low-pH-dependent triggering of HMPV F, as alanine substitutions for the above-mentioned basic residues resulted in a dramatic decrease in fusion activity (34). Thus, the electrostatic repulsion forces that arise upon protonation of H435 at low pH could be a driving force for fusion triggering of HMPV F.
Fig 1.
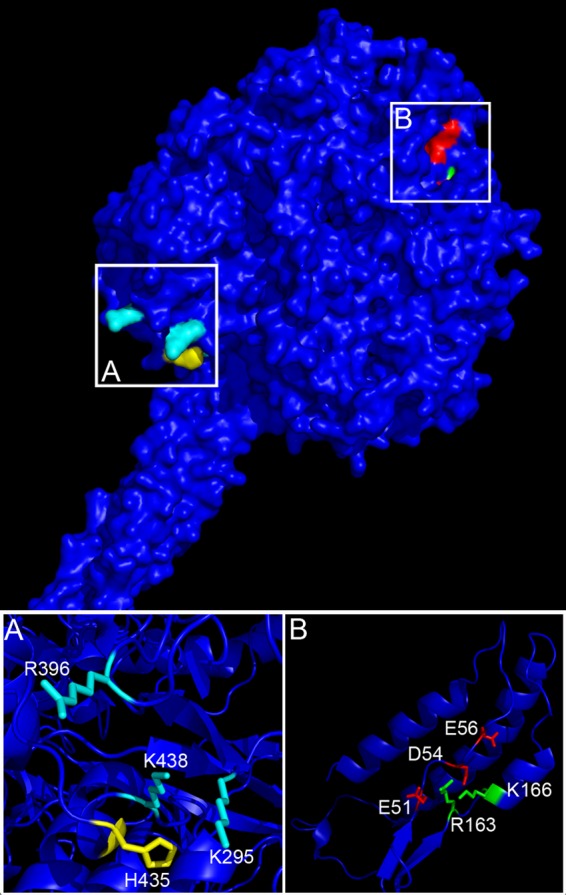
Homology model of HMPV F in its prefusion conformation, highlighting residues around H435 and the F2 region. The amino acid sequence of HMPV F was threaded onto the crystal structure of PIV5 F in its metastable prefusion form (42). (A) HRB linker domain showing H435 (yellow) in close proximity to K295, R396, and K438 (cyan). (B) Amino acid residues E51, D54, and E56 (red) lie in close proximity, allowing potential salt bridge interactions with R163 and K166 (green).
In this study, we tested the hypothesis that changes in the ionization state of H435 provide the driving force to trigger fusion. While the introduction of a negatively charged residue stabilized the protein, preventing fusion triggering, a positive charge at this position restored triggering at pH 5 but not at a higher pH. These results indicate that a positive charge in this region is required but not sufficient for triggering. Further analysis of the HMPV F protein revealed a second unique, highly charged region, located in F2. We therefore performed mutagenesis studies in this region and report that residues E51, D54, and E56 are also important for the triggering and stability of HMPV F. The results reported in this study shed new light on the potential roles of electrostatic interactions in HMPV F-mediated membrane fusion and suggest that interactions in multiple regions drive conformational changes in the F protein.
MATERIALS AND METHODS
Cell lines.
Vero and BSR T7/5 cells (provided by Karl-Klaus Conzelmann, Max Pettenkofer Institut) (10) were grown in Dulbecco's modified Eagle's medium (DMEM; Gibco Invitrogen, Carlsbad, CA) supplemented with 10% fetal bovine serum (FBS) and 1% penicillin and streptomycin (P/S). The medium of BSR cells was supplemented with 0.5 mg/ml G418 sulfate (Gibco Invitrogen, Carlsbad, CA) every third passage to select for T7 polymerase expression.
Plasmids and antibodies.
The HMPV F gene within the pGEM-3Zf(+) vector was described previously (34). The HMPV F H435R, H435K, H435D, H435E, E51A, D54A, E56A, R163A, K166A, E51K, E51K/H435R, D54A/E56A, D54A/E56A/H435R, D54A/H435R, E56A/H435R, D54N/E56Q, and D54N/E56Q/H435R protein mutants were created using QuikChange site-directed mutagenesis according to the manufacturer's protocol (Stratagene). The HMPV F wild-type (WT) and mutant genes were released from pGEM-3Zf(+), ligated into the pCAGGS mammalian expression vector, and sequenced in their entirety after ligation. Antipeptide antibodies against HMPV F (Genemed Synthesis, San Francisco, CA) were generated using amino acids 524 to 538 of HMPV F.
Viruses.
Recombinant GFP-expressing HMPV (rgHMPV) strain CAN97-83, a genotype group A2 virus with a codon-stabilized SH gene (2), was described previously (26). HMPV at a starting multiplicity of infection (MOI) of 0.01 was propagated in Vero cells incubated at 32°C with Opti-MEM supplemented with 200 mM L-glutamine and replenished with 0.3 μg/ml tosylsulfonyl phenylalanyl chloromethyl ketone (TPCK)-trypsin every day. On the seventh day, cells and medium were collected and frozen at −80°C. The virus-containing suspension was thawed to 37°C and subjected to centrifugation at 2,500 rpm for 10 min at 4°C in a Sorval RT7 tabletop centrifuge. The supernatant was then subjected to centrifugation over a 20% sucrose cushion for 2 h 15 min at 27,000 rpm and 4°C, using an SW28 swinging-bucket rotor on a Beckman Optima L90-K ultracentrifuge. The resulting pellet was resuspended in 500 μl of Opti-MEM, left at 4°C overnight, divided into aliquots the next morning, and stored at −80°C. Viral titers were determined by titration in a 96-well plate and by counting the number of GFP-expressing cells the following day. Recombinant GFP-expressing HMPVs with the F protein mutations K295A, R396A, H435A, H435N, and K438A were generated from cDNA. First, adapters containing the HMPV G-F and F-M2 intergenic regions were added to the specific mutant HMPV F genes by PCR; PCR fragments were cloned into pCAGGS and sequenced. Next, a 1,653-bp NheI/PacI DNA fragment containing the wild-type HMPV F gene was removed from the pgHMPV full-length cDNA plasmid and replaced by the individual mutant HMPV F genes. Recombinant GFP-expressing HMPVs with mutant F genes were recovered from cDNAs as described previously (6). rgHMPVs with the F protein mutations K295A, R396A, H435A, H435R, and K438A were propagated under the same conditions as those for wild-type virus; virus stocks were harvested either when cell death became prominent or when GFP expression was seen in all cells.
Syncytium assay of transfected cells.
Subconfluent monolayers of Vero cells plated in 6-well plates were transiently transfected with a total of 0.5 μg of DNA consisting of pCAGGS-HMPV F WT, pCAGGS-HMPV F mutants, or the empty pCAGGS vector (MCS control) by use of Lipofectamine and Plus reagents (Invitrogen) according to the manufacturer's instructions. The next morning, confluent cell monolayers were incubated at 37°C in 1 ml Opti-MEM with 0.3 μg/ml TPCK-trypsin for 1.5 h. Cells were then subjected to four 4-min pH pulses every 2 h with 1 ml of phosphate-buffered saline (PBS) at the indicated pH, buffered with 10 mM HEPES and 10 mM morpholineethanesulfonic acid (MES), at 37°C. After an overnight incubation at 32°C in order to allow final cellular rearrangements to take place, digital photographs of syncytia were taken with a Spot Insight Firewire digital camera mounted on a Carl Zeiss Axiovert 100 inverted microscope using a 10× objective (Thornwood, NY).
Reporter gene fusion assay.
Vero cells in 60-millimeter dishes were transfected with 0.55 μg pCAGGS-HMPV F WT or mutant F and 0.55 μg T7 control plasmid (Promega, Madison, WI) containing luciferase cDNA under the control of the T7 promoter by use of Lipofectamine and Plus reagents according to the manufacturer's instructions. The following day, Vero cells in one 60-millimeter dish were lifted from the plate surface and subjected to centrifugation at 1,300 rpm and 4°C in a Sorval RT7 tabletop centrifuge for 5 min. Cells were resuspended in 2 ml of DMEM plus 10% FBS and 1% P/S and overlaid onto two 35-millimeter dishes of confluent BSR cells (1 ml of Vero cells per 35-millimeter dish). Cells were incubated at 32°C for 1.5 h and then treated with 1 ml of PBS at the indicated pH, buffered with 10 mM HEPES and 10 mM MES, for 4 min at 37°C. Buffered PBS was replaced by 1 ml of Opti-MEM with 0.3 μg/ml TPCK-trypsin, and the pH pulse was repeated 1 h later. Cells were then incubated in 2 ml DMEM plus 10% FBS and 1% P/S at 37°C for 4 h to allow for luciferase production. Luciferase activity was assessed using a luciferase assay system (Promega) according to the manufacturer's protocol. Light emission was measured using an Lmax luminometer (Molecular Devices, Sunnyvale, CA) for 5 s, with a 1.6-s delay between measurements. Student t tests or analysis of variance (ANOVA) tests were used to determine statistically significant differences.
Protein expression, metabolic labeling, surface biotinylation, and immunoprecipitation.
Cells in 60-millimeter dishes were transfected with 1.10 μg pCAGGS-HMPV F WT or mutant F (with empty pCAGGS as the MCS control) by use of Lipofectamine and Plus reagents according to the manufacturer's protocol. At 18 to 24 h posttransfection, cells were starved in methionine- and cysteine-deficient DMEM for 1 h and then metabolically labeled with Tran[35S] label (100 μCi/ml; Perkin-Elmer) with 0.3 μg/ml TPCK-trypsin for 4 h. Following radiolabeling, cells were washed three times with ice-cold PBS, pH 8, and surface proteins were biotinylated with 1 mg/ml EZ-Link sulfo-NHS-biotin (Pierce, Rockford, IL) diluted in PBS, pH 8, for 30 min with rocking at 4°C and then for 20 min at room temperature. Cells were lysed in 1 ml RIPA buffer containing 1 kallikrein inhibitory unit of aprotinin (Calbiochem, San Diego, CA), 1 mM phenylmethylsulfonyl fluoride (Sigma, Saint Louis, MO), and 25 mM iodoacetamide (Sigma) after removal of the biotin solution. The lysates were subjected to centrifugation at 136,500 × g for 15 min at 4°C, and supernatants were collected. Antipeptide sera and protein A-conjugated Sepharose beads (Amersham, Piscataway, NJ) were used to immunoprecipitate the F proteins as previously described (29). Immunoprecipitated protein was boiled away from the beads by use of a total of 100 μl of 10% SDS (40 μl for the first boil and 60 μl for the second boil). Fifteen percent of the total protein collected was used for total expression analysis, and the remaining 85% was diluted in 500 μl biotinylation dilution buffer (20 mM Tris [pH 8], 150 mM NaCl, 5 mM EDTA, 1% Triton X-100, 0.2% bovine serum albumin) and incubated with immobilized streptavidin (Thermo Scientific, Rockford, IL) for 1 h at 4°C. Samples were washed, resolved by SDS-15% polyacrylamide gel electrophoresis (SDS-PAGE), and visualized using a Typhoon imaging system. The signal intensity for each sample was quantified using ImageQuant software, and Student t tests or ANOVA tests were used to determine statistically significant differences.
Homology modeling.
A model of the prefusion conformation of the HMPV F protein was generated from the molecular coordinates (mmdbld: 37132) determined from the crystal structure of the prefusion form of PIV5 F (42), using DeepView/Swiss-PdbViewer v4.0.1 (http://spdbv.vital-it.ch/) as previously described (34).
RESULTS
A positive charge at residue 435 is required but not sufficient for efficient triggering of HMPV F.
We previously reported that the histidine at position 435 (H435), located in the HRB linker domain (31), and the basic residues K295, R396, and K438 that surround it (Fig. 1A) are important for low-pH triggering of HMPV F (34). Because estimating the positions of the side chains from a homology model is less accurate than estimating that of the protein backbone, the distances between the β-carbons of H435 and residues K295, R396, and K438 were measured and were estimated to be less than 15 Å (Table 1). Therefore, the side chains of these residues could be positioned such that addition of another positive charge through the protonation of H435 at pH 5 could destabilize the metastable prefusion form, causing it to trigger the fusion process. To test this, a positive charge at position 435 was introduced by making arginine and lysine substitutions (HMPV F H435R and H435K). Additionally, HMPV F H435D and H435E mutants were created to introduce a negative charge in the region.
Table 1.
Estimated distances between β-carbons of H435 and surrounding basic residues in the HMPV F homology model
| Starting residue | Ending residue | Distance (Å) |
|---|---|---|
| H435 | K295 | 10 |
| R396 | 13 | |
| K438 | 6 |
Surface biotinylation was performed using an antibody against the cytoplasmic tail of HMPV F to examine surface expression and proteolytic cleavage of the mutant proteins. For all mutant proteins, the inactive F0 precursor form and the cleaved, active F1 form were expressed at the cell surface (Fig. 2A and B), suggesting that these proteins were properly folded. Total and surface expression levels of the F1 form of HMPV F H435D, H435K, and H435E mutants were decreased by 30 to 50% compared to WT levels, whereas the HMPV F H435R mutant was expressed at WT levels. The percentage of cleaved protein varied from 54 to 59% for the WT and mutant F proteins, with differences that were not statistically significant.
Fig 2.

A positive charge at position H435 is required but not sufficient for HMPV F-mediated membrane fusion. (A) Quantification of the total and surface expression of the F1 form in metabolically labeled Vero cells expressing HMPV F WT, H435R, H435K, H435D, and H435E in the presence of TPCK-trypsin. Data are presented as percentages of WT HMPV F expression, which was set to 100% (n = 4). (B) Representative gel of surface proteins used for quantification in panel A. (C) Representative images of syncytium formation of cells expressing the H435 mutants after pulses at pH 5 (n = 4). (D) Luciferase reporter gene assay of Vero cells transfected with mutant HMPV F that were used to overlay BSR cells and subjected to two pH pulses. Data are presented as percentages of WT luminosity (fusion) at pH 5 (n = 5). Error bars show standard errors of the means (SEM).
Fusogenic activity of these mutants was determined using a syncytium assay in which cells expressing the mutant proteins were exposed to four 4-min low-pH pulses (Fig. 2C). Both the HMPV F H435R and H435K mutants were capable of promoting cell-to-cell fusion when exposed to pH 5 pulses, and they formed syncytia at levels equivalent to or slightly higher than the WT level. Like the WT, however, these mutants did not show significant fusion activity at pH 6 or pH 7 (data not shown). These data indicate that a positive charge at position 435 is not sufficient to drive membrane fusion, though our results suggest that a positive charge at this position is necessary for triggering. Similar to the previously published results for HMPV F H435A and H435N protein mutants (34) and in agreement with results shown by Mas et al. (27), fusion activity was not detected for the HMPV F H435D and H435E mutants, potentially because the positive charge involved in triggering was replaced by a negative charge that stabilized the prefusion form of the protein.
To confirm the findings from the syncytium assay and to quantify fusogenic activity, a firefly luciferase reporter gene assay was employed. Briefly, Vero cells cotransfected with the different HMPV F proteins and a T7-luciferase plasmid were overlaid onto T7 polymerase-expressing BSR cells. Upon membrane fusion of the two cell lines, luciferase production occurred, allowing quantification of fusion. Consistent with the syncytium assay results, the HMPV F H435R and H435K mutants were fusogenically active at pH 5 but not at pH 6 or pH 7 (Fig. 2D). Interestingly, luciferase production of cells expressing the HMPV F H435R mutant increased by 25% compared to that of cells expressing the WT. Despite a 45% decrease in surface expression for the HMPV F H435K mutant, cells transfected with this mutant F protein retained 84% of the fusion activity of the WT, suggesting that the HMPV F H435K mutant may have a hyperfusogenic phenotype, as previous studies with other paramyxovirus F proteins have indicated that fusogenic activity correlates with the level of surface expression of F (13, 31, 36). Fusion levels above background at pH 6 or 7 were not detected for any of the mutants tested, and as seen in the syncytium assay, HMPV F H435D and H435E mutants did not show any fusion above background at pH 5 (Fig. 2D) (P > 0.05; ANOVA). Together, these results suggest that a positive charge at position 435, likely present after protonation at low pH, is necessary but not sufficient for the triggering of HMPV F.
Mutations in H435 and the surrounding basic residues impair viral growth.
Our previous studies showed that H435 and its surrounding basic residues are important for low-pH-dependent cell-cell fusion activity of HMPV F (34). To determine if these changes in fusion activity translated into changes in viral growth, recombinant GFP-expressing HMPVs carrying the HMPV F mutations H435A, H435N, K295A, R396A, and K438A were created by replacing the HMPV F WT gene in the rgHMPV full-length cDNA plasmid (6) with the mutant HMPV F genes. Mutant viruses were recovered by transfection of BSR cells (6), sequenced to confirm the presence of the desired mutation and the absence of other mutations in the F protein, and used subsequently to generate later passages. Viruses were then titrated to ensure that cells were infected at an MOI of 0.01. Compared to the WT, all mutant viruses exhibited at least a 100-fold reduction in viral growth. On day 6 after infection with WT rgHMPV, 100% of the cells in a plate were infected, resulting in high GFP expression levels and a titer between 106 and 107 PFU/ml, while the rates of infection and growth of the mutant viruses were dramatically lower (Fig. 3). Consequently, propagation of these viruses required a much longer incubation period to achieve workable viral titers (data not shown). Interestingly, viruses carrying the K295A and R396A mutations were more impaired than HMPVs carrying the H435A and H435N mutations in their F proteins, producing titers of <102 PFU/ml. These results confirm that alterations of H435 and the region surrounding it are deleterious for HMPV growth, verifying that this region is important for the fusion activity of HMPV F and for the overall fitness of the virus.
Fig 3.
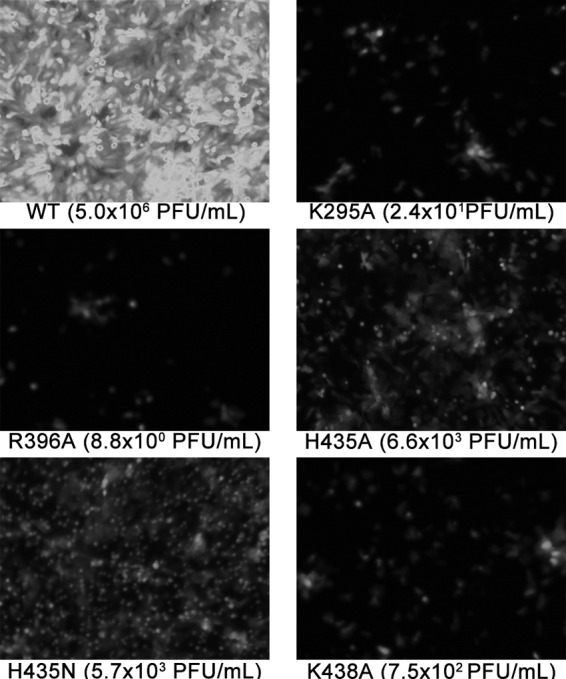
Recombinant HMPVs carrying mutations in the HRB linker domain of HMPV F show drastic decreases in infectivity. GFP expression is shown for WT and mutant HMPV-infected plates 6 days after infection with passage 2 virus grown in Opti-MEM with 200 mM l-glutamine and 0.3 μg/ml TPCK-trypsin. Values in parenthesis are representative titers obtained from supernatant and cell lysates after a 6-day incubation.
An electrostatic-rich region in F2 modulates HMPV F low-pH-dependent fusion.
Since our results suggested that a positive charge at position 435 was necessary but not sufficient to trigger HMPV F, the homology model was further examined for additional regions where changes in charge-charge interactions could potentially contribute to low-pH triggering. When the amino acid sequence of HMPV F was aligned with those of other paramyxovirus fusion proteins, a distinct stretch of negatively charged residues (including E51, D54, and E56) that was not present in other paramyxovirus F proteins was identified in the HMPV F2 domain. This region is homologous to the CBF2 region shown to modulate fusion of PIV5 F (17). Structural analysis of PIV5 F indicated that a portion of the CBF2 region interacts with sections of heptad repeat A (HRA) to form a three-strand β-sheet that is hypothesized to stabilize the F protein structure in its prefusion conformation (17, 42). Interestingly, the homology model of HMPV F predicts these CBF2 residues to be positioned in proximity to two positively charged moieties from the HRA region—R163 and K166 (Fig. 1B). Measurements from our homology model estimate the distance between β-carbons of these residues to be between 4 and 11 Å (Table 2), a distance permissible for the formation of salt bridges between side chains at neutral pH, potentially stabilizing the overall structure of HMPV F. For salt bridges to form, the charged groups of the side chain need to be less than 4 Å apart (1, 24). The highly charged local environment in this region could theoretically alter the pKa of the side chains such that a drop in pH from 7 to 5 could lead to changes in the protonation state of E51, D54, and/or E56, resulting in destabilization and facilitating triggering. To determine if these amino acids play a role in low-pH-dependent fusion activity, HMPV F E51A, D54A, and E56A mutants (F2 mutants), as well as R163A and K166A mutants (HRA mutants), were created and tested for proper expression and fusogenic activity at various pHs.
Table 2.
Estimated distances between β-carbons of selected amino acid residues in F2 in the HMPV F homology model
| Starting residue | Ending residue | Distance (Å) |
|---|---|---|
| E51 | R163 | 4 |
| D54 | R163 | 5 |
| E56 | R163 | 10 |
| E51 | K166 | 8 |
| D54 | K166 | 5 |
| E56 | K166 | 10 |
With the exception of the E51A mutant, for which surface F1 expression was 66% of the WT level, all F2 and HRA mutants were expressed close to or above WT levels on the surfaces of transfected Vero cells (Fig. 4A and B). The fusogenic activity of the mutant proteins was determined by syncytium and reporter gene fusion assays. All of the mutants were able to promote efficient syncytium formation at pH 5, except for HMPV F E51A (Fig. 4C). Cells transfected with HMPV F D54A, E56A, R163A, and K166A mutants produced more numerous and larger syncytia than those transfected with the WT at pH 5. In contrast, syncytium formation was not detected in cells transfected with HMPV F E51A, despite a significant amount of protein being expressed at the cell surface (Fig. 4A to C). To our surprise, all of the mutants except for HMPV F E51A also showed syncytia at pH 6, though they were smaller (Fig. 4C). However, compared to that at pH 5, syncytium formation was significantly lower at pH 6 and undetectable at pH 7 (data not shown).
Fig 4.

Charged residues in F2 and HRA regions of HMPV F are important for the stability and fusogenic activity of the protein. (A) Quantification of total and surface expression levels of the cleaved F1 form of mutant proteins HMPV F E51A, D54A, E56A, R163A, and K166A in Vero cells (n = 4). Data are presented as percentages of WT HMPV F expression, which was set to 100%. The asterisk indicates a significant difference from the WT control at the same pH (P < 0.05). Error bars show SEM. (B) Representative surface biotinylation of mutant HMPV F used for quantification in panel A. (C) Representative pictures of syncytium assay of HMPV F mutants at pH 5 and pH 6 (n = 4). Arrows point to small syncytia seen at pH 6. The areas delimited by the black squares for the D54A and E56A mutants at pH 6 were enlarged and are shown below (D54A 2× and E56A 2×, respectively). (D) Luciferase reporter gene assay of mutant F proteins tested for panels A, B, and C (n = 3). Data are presented as percentages of WT luminosity (fusion) at pH 5. Asterisks indicate significant differences compared to the WT control (P < 0.05). Error bars show SEM.
In agreement with the syncytium assay results, all of the mutant HMPV F proteins, with the exception of the E51A mutant, were able to efficiently promote fusion at pH 5 in the reporter gene assay (Fig. 4D). In fact, the D54A, E56A, and K166A mutants promoted fusion levels that were at least 25% above the WT level. The HMPV F R163A mutant exhibited similar fusogenic activity to that of the WT, while fusion activity for HMPV F E51A was diminished to near background levels. The HMPV F D54A, E56A, and K166A mutants also promoted fusion above WT levels at pH 5.5, although as seen with the WT, the fusogenic activity was reduced more than 50% compared to that at pH 5. Fusion levels for HMPV F R163A were similar to that of the WT, and no fusion above background was observed for HMPV F E51A at pH 5.5. Interestingly, no fusion activity above background was detected at pH 6 or above for any of the mutants. The disparate results between the syncytium and reporter gene assays at pH 6 may be due to differences in the kinetics of the fusion process triggered at this pH as well as to different amounts of activated F present at the surface. While luciferase production was assessed after two pulses spaced 1 h apart, syncytium formation was induced by a total of four pH pulses given every 2 h. Additionally, for the luciferase reporter to be synthesized, a pore large enough to transfer plasmid DNA or the T7 polymerase must be formed between the time of the pH pulse and cell lysis (4 h). Syncytium formation, on the other hand, was assessed after an overnight incubation, which allowed sufficient time for slower proteins to undergo the conformational changes needed to enlarge the fusion pore (14).
E51 is required for proper local folding and cleavage activation of HMPV F.
The results for the F2 and HRA mutants suggest that this region plays two important yet different roles in the stability and triggering of HMPV F. E51 appears to be involved in the overall fusion activity of HMPV F and could potentially serve as another pH sensor. Therefore, to test whether E51 acts similarly to H435, expression and fusion of the HMPV F E51K mutant were assessed.
Replacement of E51 with a lysine (E51K) decreased the surface expression levels of the F1 form of HMPV F by nearly 50% (Fig. 5A and B). The reduction in surface expression observed for both the E51A and E51K mutants suggests that the negative charge at this position is important for stability and expression of the protein at the cell surface. The importance of this residue is underscored by the fact that the portion of the HMPV F E51K mutant that remained on the surface also failed to promote cell-to-cell membrane fusion at any pH, as assessed by our reporter gene and syncytium formation assays.
Fig 5.

Different charged residues in F2 are responsible for the stability and local folding of HMPV F. (A) Quantification of total and surface expression of the F1 form of HMPV F E51K, E51K/H435R, D54A/H435R, E56A/H435R, D54A/E56A, D54A/E56A/H435R, D54N/E56Q, and D54N/E56Q/H435R mutant proteins as previously described (n = 5). Data are presented as percentages of WT HMPV F expression, which was set to 100%. Error bars show SEM. (B) Representative gel used for quantification in panel A. (C) Luciferase reporter gene assay of HMPV F mutant proteins shown in panel A. Data are presented as percentages of WT luminosity (fusion) at pH 5 (n = 6). Error bars show SEM. (D) Representative photographs of syncytium formation of Vero cells transfected with HMPV F mutants at pH 5 (n = 4). Arrows point to small syncytia.
To test whether there is cross talk between E51 and H435, the E51K/H435R mutant was also made and analyzed. Like that of HMPV F E51K, surface expression of the E51K/H435R mutant was reduced. Furthermore, cell-to-cell fusion activity was not detected at any pH, despite the introduction of the hyperfusogenic mutation H435R (Fig. 5C and D). These results suggest that the E51K mutation is dominant over H435R and confirm that residue E51 plays a key role in the overall function of F.
Even though the HMPV F E51A, E51K, and E51K/H435R mutants showed less surface expression, a significant amount of mutant F reached the cell surface, suggesting that the effects of these mutations on the overall structure are not severe enough to result in complete endoplasmic reticulum (ER) retention due to gross misfolding. Since E51 is near the cleavage site of HMPV F, electrostatic interactions involving this moiety may be important for the proper folding and positioning of the proteolytic processing site. To test this, surface expression of HMPV F E51A and E51K mutants was examined in the absence of trypsin. Although HMPV F strains or mutants have been identified in which some processing to the mature F1+F2 form is observed in the absence of exogenous trypsin (4, 33), WT HMPV CAN97-83 F protein cleavage in vitro has been shown in our laboratory and others to require trypsin treatment (5, 35, 40). Fitting with this, the WT HMPV F protein was observed in the mature, cleaved F1 form only after addition of trypsin (Fig. 6A and B). Interestingly, more than 30% of the E51A or E51K mutant was cleaved in the absence of trypsin (Fig. 6A and B), suggesting that the removal of the negative charge at position 51 caused local misfolding that exposed a cleavage site normally not accessible to an endogenous protease. Indeed, analysis of the sequence of HMPV F revealed the presence of an arginine residue located two residues upstream of the putative cleavage site that could be utilized by an endogenous trypsin-like protease. This aberrant cleavage event, however, did not result in a fusogenically active F protein (Fig. 5C and D). These results therefore suggest that E51 is important for positioning of the fusion peptide for proper proteolytic processing.
Fig 6.
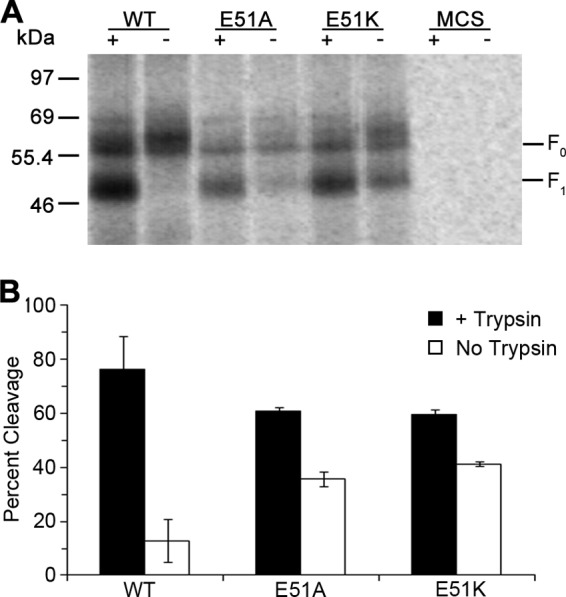
E51 is important for proper folding at the HMPV F cleavage site. (A) Representative gel showing surface expression levels of HMPV F WT and HMPV F E51A and E51K mutants in the presence or absence of 0.3 μg/ml of TPCK-trypsin. (B) Quantification of percent cleavage for HMPV F WT and HMPV F E51A and E51K mutants, calculated from the gel shown in panel A as follows: percent cleavage = F1/(F1 + F0) (n = 3). Error bars show SEM.
Charged residues in F2 are important for HMPV F prefusion and postfusion stability.
The hyperfusogenic phenotype of the HMPV F D54A and E56A mutants suggests that the D54 and E56 residues are involved in modulating the stability of the protein, potentially through salt bridge interactions with R163 and K166 in the prefusion state. At low pH, neutralization of D54 and/or E56 could disrupt these salt bridges, allowing F fusion to trigger. HMPV F D54A/E56A and D54N/E56Q double mutants were created and analyzed to determine the effects of disrupting these potential interactions. The mutations in the latter mutant, i.e., D54N and E56Q, are more conservative, isosteric substitutions that remove the charged residues while maintaining polarity at these positions. Analysis of the R163 and K166 mutations was not pursued further due to the location of these residues in HRA, as their potential role in six-helix-bundle formation means that fusion changes may not be attributed directly to triggering. Additionally, HMPV F D54A/H435R, E56A/H435R, D54A/E56A/H435R, and D54N/E56Q/H435R mutants were made to test whether changes in one region would influence the other.
Though the HMPV F D54A and E56A mutants were expressed at or above WT levels (Fig. 4A), surface expression of the F1 form of the double alanine mutant (D54A/E56A) decreased by more than 40%. Consistent with a decrease in surface expression levels, a reduction in fusion activity was also observed for the HMPV F D54A/E56A mutant. The more conservative polar substitutions (D54N/E56Q), however, restored the surface expression to WT levels (Fig. 5A and B), suggesting that either polar residues or steric similarity is needed to stabilize the prefusion form of the protein.
According to our reporter gene fusion assay, fusion activity was significantly enhanced for HMPV F D54N/E56Q at pH 5 (Fig. 5C), to levels equivalent to those for the E56A mutant. However, the effect of the mutations on the stability and fusogenic activity of the protein was different. The D54N/E56Q mutant was more hyperfusogenic at pH 5.5 than the E56A mutant, and while the addition of the H435R mutation to the D54N/E56Q mutant (D54N/E56Q/H435R) had a modest effect on fusion at pH 5.5, the E56A/H435R mutant exhibited a dramatic increase in fusion activity at the same pH. More importantly, analysis of the syncytium assay revealed that cells expressing the HMPV F D54N/E56Q and D54N/E56Q/H435R mutants produced numerous syncytia that were significantly smaller than those for the WT and the E56A and E56A/H435R mutants at pH 5 (Fig. 5D). The larger amount of smaller syncytia agrees with the hyperfusogenic phenotype observed in the reporter gene fusion assay, as this would result in more cells fusing and in increased luciferase production. No syncytium formation was detected at pH 6 or pH 7 (data not shown). Thus, our data indicate that replacement of the negative charges at positions 54 and 56 leads to changes in the stability of the fusion protein that can be seen by changes in surface expression and fusogenic activity.
The addition of the hyperfusogenic mutation H435R (D54A/H435R, E56A/H435R, D54A/E56A/H435R, and D54N/E56Q/H435R) did not significantly alter the fusion levels of the mutant proteins at pH 5 (Fig. 4A and D and 5C and D). However, the presence of the H435R mutation led to a modest increase in fusion activity at pH 5.5, with the notable exceptions of the D54A/H435R and E65A/H435R mutants, for which the increase in fusion was significantly more pronounced. Together with a minor decrease in F1 surface expression (Fig. 4A and 5A), our data suggest that the H435R mutation also affects the stability of the protein, allowing it to trigger fusion at a higher pH. The destabilizing force of adding a basic residue to an already positively charged region may indeed be responsible for the hyperfusogenic phenotype of the H435R mutant, as disruptions of charge-charge interactions have been shown to alter the triggering threshold of influenza virus HA (12, 30). The pH dependency of HMPV F-mediated membrane fusion, however, was not abolished by mutations at positions 54 and/or 56. Therefore, while H435 mediates the stability of the HRB linker region to behave as a pH sensor, the stability effect conferred by residues D54 and E56 is not involved in sensing low pH.
DISCUSSION
The results presented in this study provide new insights into the multiple roles that electrostatic interactions play in the folding, stability, and triggering of HMPV F. We demonstrated that a positive charge at position 435 is necessary but not sufficient to trigger HMPV F, with this charge most likely arising through protonation of H435 upon its exposure to a low-pH environment. This conclusion is supported by a recent report in which this histidine residue was mutated in the F proteins of phylogenetically distinct HMPVs, with similar results (27). Furthermore, our mutagenesis studies revealed that the novel enrichment of charged residues present in the F2 and HRA domains of HMPV F is important for proper local folding and the overall stability of the protein. Removal of the negative charge at position 51 affected proteolytic processing and led to an inactive protein on the cell surface, consistent with an alteration in the local conformation. Loss of charge at positions 54, 56, 163, and/or 166 also affected stability and fusogenic activity of HMPV F.
Several reports have shown important differences between members of the Pneumovirinae subfamily and their Paramyxovirinae counterparts. While viruses in the Paramyxovirinae subfamily require the expression of the attachment protein for membrane fusion and infection to occur, both RSV and HMPV remain infectious in vitro in the absence of their putative attachment proteins (7, 23). Furthermore, our laboratory has shown that triggering of HMPV F can occur in the absence of the HMPV G protein (35). Indeed, recent studies suggested that the protein responsible for the primary attachment step for both of these viruses is F (11, 38). The possibility of triggering the F proteins of certain clades of HMPV by low pH further distinguishes this virus from the rest of the family (19, 35). Close examination of the sequence alignment of HMPV F and other paramyxovirus F proteins and of a homology model based on the prefusion form of PIV5 F (42) reveals areas rich in charged residues that are absent in other F proteins. Therefore, changes in the ionization state of these residues could explain the triggering of HMPV F by low pH, as seen in influenza virus HA (30, 32, 39).
The first highly charged region identified is located at the HRB linker domain and is centered at H435 (34). Based on mutagenesis studies, we and others determined that H435 and the neighboring K295, R396, and K438 residues, in addition to G294, are important for low-pH-mediated fusion (19, 34), and the severe attenuation of recombinant HMPV bearing an H435A, H435N, K295A, R396A, or K438A mutation in the F protein (Fig. 3) highlights the importance of this region for the virus. Furthermore, analyses of HMPV F H435R and H435K mutants presented here and elsewhere strongly suggest that a positive charge in this region is required though not sufficient for low-pH triggering of HMPV F. Both the H435D and H435E mutants were not fusogenically active, likely due to the presence of a negative charge. Such a phenotype, we hypothesize, is due to stabilization of the prefusion form via electrostatic attraction, in contrast to the hyperfusogenic profile observed when a positive charge is present at position 435.
Despite the dramatic reduction in cell-to-cell fusion observed when the charged residues around HMPV F H435 were removed (34), we were able to rescue infectious HMPV particles bearing mutations in these residues (Fig. 3). Interestingly, while viral titers of all mutant viruses were reduced by three or more logarithmic units compared to the wild-type titer, titers of recombinant viruses expressing the H435A and H435N mutations were higher than those of viruses bearing the K295A and R396A mutations. Our homology model of the HMPV F protein in its prefusion conformation indicates that K295 and R396 are freely exposed at the surface of the protein (Fig. 1). It is possible that these residues have other important roles in the life cycle of the virus, thereby increasing the selective pressure and making these mutations more difficult to overcome. We have shown that HMPV entry is initially mediated by interactions between the F protein and heparan sulfate (11). Therefore, one possibility is that the positively charged residues K295 and R396 are needed for interactions with the negatively charged heparan sulfate during virus entry. Even though the residues around H435 are located at the base of the head domain, interactions could take place at an intermediate step or with longer branches of heparan sulfate.
Protonation of H368 was recently suggested by Mas et al. to also be important for low-pH-dependent F proteins (27). Our homology model predicts this residue to be more than 20 Å from H435 (data not shown), and it is predicted to lie adjacent to and interacting with the fusion peptide (34). Alanine and asparagine mutations of H368 in HMPV F CAN97-83 suggest that protonation of this residue is not essential for low-pH-mediated fusion of this F protein (34). Furthermore, with the exception of the H368A mutant, for which fusion activity was not detected, none of the mutations of H368 reported for CAN97-83 (34) or NL/1/00 (27) showed a pH-independent triggering unless it was paired with a positively charged residue at position 435. These results further emphasize the importance of a positive charge at position 435 and support our previous hypothesis that side chain packing around the fusion peptide modulates the fusion efficiency and stability of the F protein (34, 36).
Another unique region in HMPV F CAN97-83 where charged residues are predicted to be concentrated in our homology model is the interface between the F2 and HRA domains of the protein. While the charged residues are novel relative to other paramyxovirus F proteins, sequence alignments indicate that this enrichment is conserved across different HMPV clades (data not shown). This region has been shown to be important in modulating fusion for PIV5 F (17), and structural analysis of PIV5 F in its prefusion conformation (42) indicated that residues in this area form a three-strand β-sheet, with two of the three strands derived from HRA residues and the remaining strand derived from F2 residues. Though HMPV and PIV5 belong to different subfamilies, the importance of this region for the proper folding of F and the promotion of membrane fusion is preserved. Interestingly, there are striking differences between the F2 region of PIV5 F and that of HMPV F. This region in PIV5 F is composed primarily of nonpolar residues, while polar and charged moieties are more predominant in this region of HMPV F. Mechanistically, the way that this region modulates folding and fusion in PIV5 F is still unclear. However, because these charged residues in the F2 domain of HMPV F are predicted to lie near those in HRA, we suggest that potential electrostatic interactions between HRA and this region in F2 are involved in the folding and fusion of HMPV F.
The results shown in this study identify several areas where electrostatic interactions, including salt bridges, may play important roles in HMPV F function. We found that a positive charge at the previously identified residue H435 (34) results in a hyperfusogenic state but is not sufficient alone to cause pH-independent fusion, while a negative charge at this position blocks the F protein's fusogenic activity. Furthermore, we showed a charged region in F2 to be important for proper proteolytic activation and for the overall stability of HMPV F. We have previously shown that HMPV infection can be inhibited partially by agents that raise endosomal pH. Additionally, HMPV infection was greatly reduced after inhibition of clathrin-mediated endocytosis and dynamin by chlorpromazine and dynasore, respectively (34). A recent report found variable inhibition of the infectivity of other strains of HMPV by concanamycin A, without a clear link between a pH-triggered F protein and inhibition by the drug, and no inhibition by ammonium chloride of any tested HMPV strain (27). While our previous results using chlorpromazine and dynasore suggest a role for endocytosis in HMPV entry, the correlation between viral entry by endocytosis and the requirement for low pH remains unclear. Electrostatic interactions may also play a role in viral binding, as we have recently shown that an interaction between HMPV F and heparan sulfate plays a critical role in initial binding (7, 11), suggesting that a cascade of electrostatic interactions occurs to drive HMPV F binding and fusion promotion.
ACKNOWLEDGMENTS
We thank Rachel Schowalter (NCI, NIH) for help in generating some of the HMPV F mutant constructs. We are also grateful to Trevor Creamer and members of the Dutch lab for helpful discussions and for critical reviews of the manuscript.
This work was supported by NIH grants R01AI051517 and 2P20 RR020171 from the National Center for Research Resources to R.E.D., by AHA Great Rivers Affiliate predoctoral fellowship 10PRE4230022 to A.C., and by funds from the Intramural Research Program of the NIH, NIAID Division, to U.J.B. and C.C.W.
Footnotes
Published ahead of print 3 July 2012
REFERENCES
- 1. Barlow DJ, Thornton JM. 1983. Ion-pairs in proteins. J. Mol. Biol. 168:867–885 [DOI] [PubMed] [Google Scholar]
- 2. Biacchesi S, Murphy BR, Collins PL, Buchholz UJ. 2007. Frequent frameshift and point mutations in the SH gene of human metapneumovirus passaged in vitro. J. Virol. 81:6057–6067 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3. Biacchesi S, et al. 2005. Infection of nonhuman primates with recombinant human metapneumovirus lacking the SH, G, or M2-2 protein categorizes each as a nonessential accessory protein and identifies vaccine candidates. J. Virol. 79:12608–12613 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4. Biacchesi S, et al. 2006. Modification of the trypsin-dependent cleavage activation site of the human metapneumovirus fusion protein to be trypsin independent does not increase replication or spread in rodents or nonhuman primates. J. Virol. 80:5798–5806 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5. Biacchesi S, et al. 2003. Genetic diversity between human metapneumovirus subgroups. Virology 315:1–9 [DOI] [PubMed] [Google Scholar]
- 6. Biacchesi S, et al. 2004. Recovery of human metapneumovirus from cDNA: optimization of growth in vitro and expression of additional genes. Virology 321:247–259 [DOI] [PubMed] [Google Scholar]
- 7. Biacchesi S, et al. 2004. Recombinant human metapneumovirus lacking the small hydrophobic SH and/or attachment G glycoprotein: deletion of G yields a promising vaccine candidate. J. Virol. 78:12877–12887 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8. Boivin G, et al. 2002. Virological features and clinical manifestations associated with human metapneumovirus: a new paramyxovirus responsible for acute respiratory-tract infections in all age groups. J. Infect. Dis. 186:1330–1334 [DOI] [PubMed] [Google Scholar]
- 9. Boivin G, et al. 2003. Human metapneumovirus infections in hospitalized children. Emerg. Infect. Dis. 9:634–640 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10. Buchholz UJ, Finke S, Conzelmann KK. 1999. Generation of bovine respiratory syncytial virus (BRSV) from cDNA: BRSV NS2 is not essential for virus replication in tissue culture, and the human RSV leader region acts as a functional BRSV genome promoter. J. Virol. 73:251–259 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11. Chang A, Masante C, Buchholz UJ, Dutch RE. 2012. Human metapneumovirus (HMPV) binding and infection are mediated by interactions between the HMPV fusion protein and heparan sulfate. J. Virol. 86:3230–3243 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12. Daniels RS, et al. 1985. Fusion mutants of the influenza virus hemagglutinin glycoprotein. Cell 40:431–439 [DOI] [PubMed] [Google Scholar]
- 13. Dutch RE, Joshi SB, Lamb RA. 1998. Membrane fusion promoted by increasing surface densities of the paramyxovirus F and HN proteins: comparison of fusion reactions mediated by simian virus 5 F, human parainfluenza virus type 3 F, and influenza virus HA. J. Virol. 72:7745–7753 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14. Dutch RE, Lamb RA. 2001. Deletion of the cytoplasmic tail of the fusion (F) protein of the paramyxovirus simian virus 5 (SV5) affects fusion pore enlargement. J. Virol. 75:5363–5369 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15. Falsey AR, Erdman D, Anderson LJ, Walsh EE. 2003. Human metapneumovirus infections in young and elderly adults. J. Infect. Dis. 187:785–790 [DOI] [PubMed] [Google Scholar]
- 16. Falsey ARMD. 2008. Human metapneumovirus infection in adults. Pediatr. Infect. Dis. J. 27:S80–S83 [DOI] [PubMed] [Google Scholar]
- 17. Gardner AE, Dutch RE. 2007. A conserved region in the F(2) subunit of paramyxovirus fusion proteins is involved in fusion regulation. J. Virol. 81:8303–8314 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18. Harrison SC. 2008. Viral membrane fusion. Nat. Struct. Mol. Biol. 15:690–698 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19. Herfst S, et al. 2008. Low-pH-induced membrane fusion mediated by human metapneumovirus F protein is a rare, strain-dependent phenomenon. J. Virol. 82:8891–8895 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20. Huang Q, Opitz R, Knapp E-W, Herrmann A. 2002. Protonation and stability of the globular domain of influenza virus hemagglutinin. Biophys. J. 82:1050–1058 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21. Huang Q, et al. 2003. Early steps of the conformational change of influenza virus hemagglutinin to a fusion active state: stability and energetics of the hemagglutinin. Biochim. Biophys. Acta 1614:3–13 [DOI] [PubMed] [Google Scholar]
- 22. Kampmann T, Mueller DS, Mark AE, Young PR, Kobe B. 2006. The role of histidine residues in low-pH-mediated viral membrane fusion. Structure 14:1481–1487 [DOI] [PubMed] [Google Scholar]
- 23. Karron RA, et al. 1997. Respiratory syncytial virus (RSV) SH and G proteins are not essential for viral replication in vitro: clinical evaluation and molecular characterization of a cold-passaged, attenuated RSV subgroup B-mutant. Proc. Natl. Acad. Sci. U. S. A. 94:13961–13966 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24. Kumar S, Nussinov R. 2002. Close-range electrostatic interactions in proteins. ChemBioChem 3:604–617 [DOI] [PubMed] [Google Scholar]
- 25. Lamb RA, Parks GD. 2007. Paramyxoviridae: the viruses and their replication, p 1449–1646 In Knipe DM, et al. (ed), Fields virology, 5th ed, vol 1 Lippincott Williams & Wilkins, Philadelphia, PA [Google Scholar]
- 26. Le Nouën C, et al. 2009. Infection and maturation of monocyte-derived human dendritic cells by human respiratory syncytial virus, human metapneumovirus, and human parainfluenza virus type 3. Virology 385:169–182 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27. Mas V, Herfst S, Osterhaus ADME, Fouchier RAM, Melero JA. 2011. Residues of the human metapneumovirus fusion (F) protein critical for its strain-related fusion phenotype: implications for the virus replication cycle. J. Virol. 85:12650–12661 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28. Papenburg J, Boivin G. 2010. The distinguishing features of human metapneumovirus and respiratory syncytial virus. Rev. Med. Virol. 20:245–260 [DOI] [PubMed] [Google Scholar]
- 29. Paterson RG, Lamb RA. 1993. The molecular biology of influenza viruses and paramyxoviruses, p 35–73 In Davidson A, Elliott RM. (ed), Molecular virology: a practical approach. IRL Oxford University Press, Oxford, United Kingdom [Google Scholar]
- 30. Rachakonda PS, et al. 2007. The relevance of salt bridges for the stability of the influenza virus hemagglutinin. FASEB J. 21:995–1002 [DOI] [PubMed] [Google Scholar]
- 31. Russell CJ, Kantor KL, Jardetzky TS, Lamb RA. 2003. A dual-functional paramyxovirus F protein regulatory switch segment: activation and membrane fusion. J. Cell Biol. 163:363–374 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32. Russell RJ, et al. 2004. H1 and H7 influenza haemagglutinin structures extend a structural classification of haemagglutinin subtypes. Virology 325:287–296 [DOI] [PubMed] [Google Scholar]
- 33. Schickli JH, Kaur J, Ulbrandt N, Spaete RR, Tang RS. 2005. An S101P substitution in the putative cleavage motif of the human metapneumovirus fusion protein is a major determinant for trypsin-independent growth in Vero cells and does not alter tissue tropism in hamsters. J. Virol. 79:10678–10689 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34. Schowalter RM, Chang A, Robach JG, Buchholz UJ, Dutch RE. 2009. Low-pH triggering of human metapneumovirus fusion: essential residues and importance in entry. J. Virol. 83:1511–1522 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35. Schowalter RM, Smith SE, Dutch RE. 2006. Characterization of human metapneumovirus F protein-promoted membrane fusion: critical roles for proteolytic processing and low pH. J. Virol. 80:10931–10941 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36. Smith EC, Dutch RE. 2010. Side chain packing below the fusion peptide strongly modulates triggering of the Hendra virus F protein. J. Virol. 84:10928–10932 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37. Smith EC, Popa A, Chang A, Masante C, Dutch RE. 2009. Viral entry mechanisms: the increasing diversity of paramyxovirus entry. FEBS J. 276:7217–7227 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38. Tayyari F, et al. 2011. Identification of nucleolin as a cellular receptor for human respiratory syncytial virus. Nat. Med. 17:1132–1135 [DOI] [PubMed] [Google Scholar]
- 39. Thoennes S, et al. 2008. Analysis of residues near the fusion peptide in the influenza hemagglutinin structure for roles in triggering membrane fusion. Virology 370:403–414 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40. van den Hoogen BG, et al. 2001. A newly discovered human pneumovirus isolated from young children with respiratory tract disease. Nat. Med. 7:719–724 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41. Walsh EE, Peterson DR, Falsey AR. 2008. Human metapneumovirus infections in adults: another piece of the puzzle. Arch. Intern. Med. 168:2489–2496 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42. Yin HS, Wen X, Paterson RG, Lamb RA, Jardetzky TS. 2006. Structure of the parainfluenza virus 5 F protein in its metastable, prefusion conformation. Nature 439:38–44 [DOI] [PMC free article] [PubMed] [Google Scholar]