

Abstract
MVA is an attenuated strain of vaccinia virus (VACV) that is a popular vaccine vector. MVA infection activates NF-κB. For 293T cells, it is known that MVA early gene expression activates extracellular signal-regulated kinase 2 (ERK2), resulting in NF-κB activation. However, other viral and cellular mechanisms responsible for this event are ill defined. The data presented here show that the epidermal growth factor receptor (EGFR) is at least one apical trigger in this pathway: ERK2 and NF-κB activation was diminished when MVA infections occurred in cells devoid of the EGFR (CHO K1 cells) or in the presence of a drug that inhibits EGFR activation (AG1478) in 293T cells. The expression of dominant negative Ras or Raf proteins still permitted NF-κB activation, suggesting that a nonclassical EGFR-based signal transduction pathway triggered ERK2–NF-κB activation. C11R is an early gene present in MVA and other orthopoxviruses. It encodes the soluble, secreted vaccinia virus growth factor (VGF), a protein that binds to and stimulates the EGFR. Here it was observed that NF-κB was activated in 293T cells transfected with a plasmid encoding the C11R gene. Silencing by small interfering RNA (siRNA) or deletion of the C11R gene (MVAΔC11R) reduced both MVA-induced ERK2 and NF-κB activation in 293T cells or the keratinocyte line Hacat, suggesting that this mechanism of MVA-induced NF-κB activation may be common for several cell types.
INTRODUCTION
MVA is an attenuated vaccinia virus (VACV) that was created by serially passaging wild-type VACV in chicken embryo fibroblasts (CEFs) >500 times (34). MVA was used safely as a smallpox vaccine in the 1970s (25) and is more advantageous than wild-type VACV because it is replication incompetent in all human cells tested (3, 8). However, a drawback for its use as a smallpox vaccine is that higher or multiple doses of MVA are required to achieve the anti-VACV antibody response obtained with a single dose of wild-type VACV (55). More recently, MVA has been used as a vector for vaccines against a variety of infectious diseases of humans (AIDS, malaria) and wildlife (rabies) (26).
Wild-type VACV strains, such as WR, encode immunoevasion genes, whose products inhibit NF-κB, whereas these genes are missing from MVA (2, 38). As a result, WR infection inhibits NF-κB, whereas MVA infection activates NF-κB (41). While these data would suggest that NF-κB activation diminishes the immunogenicity of MVA, one cannot regard this model as proven, because other profound differences exist between WR and MVA. In comparison to WR, MVA is replication incompetent (8), and MVA lacks myriad immunoevasion genes whose products dampen immune responses other than NF-κB activation (2).
To more accurately evaluate the relationship between NF-κB and the immunogenicity of MVA, our rationale is to identify the viral component(s) responsible for MVA-induced NF-κB activation and to use this information to create MVA constructs in which NF-κB-activating capacity is compromised. Some of these components are already known. For example, in 293T human fibroblasts, MVA-induced NF-κB activation requires the expression of the early class of poxviral genes (33). Next, an early gene product(s) either directly or indirectly stimulates activation of the MEK1 mitogen-activated protein kinase (MAPK), resulting in the activation of extracellular signal-related kinase 2 (ERK2), an event that is upstream of and necessary for MVA-induced NF-κB activation (18, 33).
The epidermal growth factor receptor (EGFR), when interacting with its cognate EGF, triggers receptor activation to induce events, including MEK/ERK activation, proliferation, cell survival, and NF-κB activation (44). The EGFR stimulates these myriad events via distinct upstream signal transduction events. For example, EGFR-Sos-Ras-Raf-MEK1 activation activates the ERK1 and ERK2 (ERK1/2) proteins, resulting in cellular proliferation (44). EGFR autophosphorylation can also stimulate NF-κB-inducing kinase (NIK) (23), and it has been reported that EGFR-induced NIK activation results in NF-κB activation (23).
The goal of this study was to identify a viral protein(s) that is an upstream activator of this ERK2–NF-κB activation pathway, using 293T cells as a model system. The data indicate that the EGFR is partially responsible for initiating this pathway during MVA infection. While the EGFR classically is known to activate ERK2 via the Sos-Ras-Raf-MEK1 pathway, we show here that a Ras- and Raf-independent pathway triggers EGFR-induced NF-κB activation (44). MVA, like all other orthopoxviruses, encodes vaccinia virus growth factor (VGF), a homolog to EGF (49). We observed that either silencing or removal of the gene encoding VGF (C11R) resulted in an MVA virus with reduced ability to activate ERK2 and NF-κB. This phenotype was observed during infection of 293T cells and Hacat keratinocytes, suggesting that MVA infection activates NF-κB via similar pathways in multiple cell types.
MATERIALS AND METHODS
Cells, viruses, and plasmids.
The cell lines used in this study were HEK293T human kidney fibroblasts (293T cells), Chinese hamster ovary cells (CHO K1), and transformed human keratinocytes (Hacat). Cell lines were cultivated in Eagle's minimal essential medium (EMEM) supplemented with 10% fetal calf serum (FCS; HyClone), referred to as complete medium. MVA is an attenuated strain of vaccinia virus that was created by serially passaging a wild-type strain of vaccinia virus through primary chicken embryo fibroblasts (34). The WR strain of vaccinia virus and a recombinant strain, MVA/5.2kb, were used as controls because NF-κB is inactive in cells infected with either virus.
An MVA virus in which the C11R gene was replaced by a gene encoding green fluorescent protein (GFP) (MVAΔC11R) was created by homologous recombination in primary CEFs (24). MVAΔC11R progeny viruses were selected on the basis of their ability to stably express GFP and were plaque purified four times. After plaque purification, the absence of contaminating parental virus DNA and the presence of GFP in the C11R region were verified by PCR analysis of viral genomes, using primers that specifically bind to each of the regions flanking the C11R gene. Stocks of pure recombinant MVAΔC11R were propagated in CEFs.
Plasmid pC11R, in which the VACV C11R gene was inserted into pcDNA3.1, has been described previously (48). Plasmids encoding dominant negative (DN) forms of Raf (pRafDN) and Ras (pRasDN) were a gift from Richard Jove (City of Hope, Duarte, CA). pNIKDN is a plasmid encoding a dominant negative NIK protein (pNIKDN). Plasmid p53 was a gift from Kim Kemper (University of Illinois).
Measurement of NF-κB activation by use of a firefly luciferase reporter assay.
A luciferase reporter assay was utilized to quantitate NF-κB activation in infected or transfected cells, as described previously (18). For assays involving virus infection, subconfluent 293T, Hacat, or CHO K1 cellular monolayers in 12-well plates were transfected with 50 ng pRL-null (Promega) and 450 ng pNF-κBluc (Stratagene) by using the FuGene 6 transfection reagent (Roche) according to the manufacturer's instructions. Transfections proceeded for 24 h prior to virus infection. To evaluate the role of the EGFR during MVA infection, transfected cells were incubated in regular medium or dimethyl sulfoxide (DMSO)-containing medium either lacking or containing AG1478 (50 μM; Sigma-Aldrich) for 30 min prior to infection, with the compounds remaining present in the medium throughout the infection. To evaluate the roles of Raf and Ras in MVA-induced NF-κB activation, cells were cotransfected with 50 ng pRL-null, 450 ng pNF-κBluc, and 1,000 ng either pRafDN, pRasDN, pNIKDN, or pcDNA3.1 for 24 h prior to virus infection. Regardless of these differences, for all luciferase assays involving virus infections, cells were either mock infected or infected with MVA, MVAΔC11R, or WR at a multiplicity of infection (MOI) of 10 PFU/cell, unless stated otherwise. At 6 h postinfection (p.i.), unless stated differently, cellular monolayers were lysed in passive lysis buffer (PLB; Promega) for 15 min according to the manufacturer's suggestions.
To test the effect of ectopic expression of the viral gene C11R on NF-κB-controlled luciferase activity, subconfluent 293T cells were cotransfected with 50 ng pRL-null, 450 ng pNF-κBluc, and either 1,000 ng pcDNA3.1 or 1,000 ng pC11R. At the times posttransfection indicated in Fig. 7, cellular monolayers were lysed in PLB.
Fig 7.
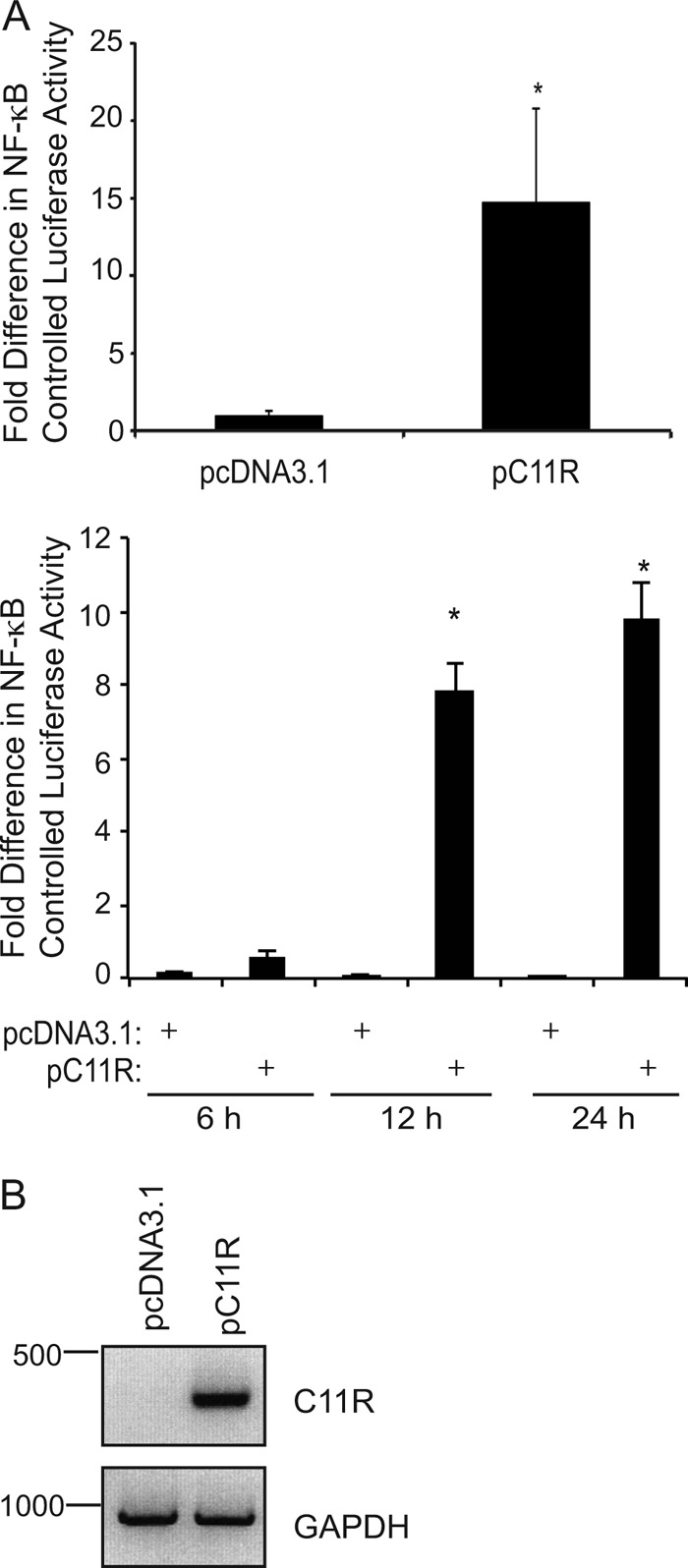
Ectopic expression of C11R induces NF-κB activation. Subconfluent 293T cellular monolayers were cotransfected with pNF-κBluc, pRL-null, and either pcDNA3.1 or pC11R. (A) At 24 h (top) or at 6, 12, or 24 h (bottom) posttransfection, cells were lysed in PLB, and luciferase activities were measured. An asterisk indicates statistical significance (P < 0.05). The ratios of firefly to sea pansy luciferase activities were normalized to that of mock-infected cells, whose value was set to 1. (B) Cells were lysed, and total RNA was isolated from cellular lysates. When total RNA isolated from transfected cells was used, 0.5 μg of total RNA per sample was reverse transcribed using oligo d(T) as a primer to generate cDNA. Then primers specific for either C11R or GAPDH were used in subsequent PCRs. A portion of each PCR product was analyzed by electrophoresis in a 1% agarose gel, and amplicons were visualized by ethidium bromide staining.
For all luciferase assays, the resultant lysates were assessed for luciferase activities as described previously (46). All assays were performed in triplicate. For each experimental point, firefly luciferase activity was divided by sea pansy luciferase activity. The resultant ratios were averaged and normalized to the appropriate control cells (mock-infected cells or cells cotransfected with pcDNA3.1). The value for the control cells was set to 1. Results were displayed as the relative fold change in luciferase activity from activity in control cells. Statistical significance was determined by using the Student t test. P values of <0.05 were considered statistically significant.
Western immunoblotting.
A previously published protocol was used to extract cytoplasmic proteins from virus-infected or transfected cells (46). In some experiments that evaluate the role of Raf and Ras signaling in MVA-induced NF-κB activation, 293T cells were transfected with 1,000 ng of pRafDN, pRasDN, pNIKDN, or pcDNA3.1 for 24 h prior to virus infection. Alternatively, cells were cotransfected with 1,000 ng pcDNA3 or FLAG-p53 and 1,000 ng of pcDNA3.1, pRafDN, or pRasDN and were incubated for 24 h. For infections of either untransfected or transfected cells, 293T, CHO K1, or Hacat cells were either mock infected or infected with MVA, MVAΔC11R, or WR. Infections proceeded for the indicated times, and cells were harvested, collected by centrifugation, and then lysed in CE buffer (41). In an experiment to evaluate the role of the EGFR in ERK1/2 activation, 293T cells were incubated with a DMSO-containing medium that either lacked or contained AG1478 (50 μM; Sigma-Aldrich) for 30 min prior to and throughout infection.
In some experiments, the transcriptional silencing of the C11R gene was used to study ERK1/2 activation in the absence of the VGF protein. In this case, subconfluent monolayers of 293T cells were plated onto 24-well plates. Twenty-four hours later, cells were transfected with a solution containing small interfering RNA (siRNA) molecules specific for 4 different regions of a targeted viral gene transcript. The final concentration of each siRNA in cellular medium was either 25 or 50 nM, such that the total final concentration of siRNA duplexes was either 100 or 200 nM, respectively. siRNA duplexes were transfected into cells using Dharmafect 1 (Thermo Fisher Scientific, Dharmacon Products) according to the manufacturer's directions. In downregulating the expression of the C11R gene, the following siRNA-duplexed oligonucleotides were utilized (only the sense strand is shown): 5′-CUAGAGAUAUCGACGGUAU-3′, 5′-CGGUAUUAUGCUUGUAUUA-3′, 5′-AGUGGUAACGCUAUCGAAA-3′, and 5′-GCUAUCAGAUUAUGCGGUC-3′. Several known (E3L, J2R, H5R, B18R, E5R) or suspected (C ORF B, F ORF A) VACV early genes were silenced in parallel as controls. The siRNA sequences used to downregulate the expression of the C ORF B gene were 5′-AGUCAUAACCGUAAUCCAA-3′, 5′-ACGCUAACGGUAUCAAUAA-3′, 5′-GCAUCGUGCUUUAACAUCA-3′, and 5′-CAUAUUCGUCCCUACUAUA-3′. The siRNA sequences used to downregulate the expression of the J2R gene were 5′-CGAGUUAGACGUUAUCAAA-3′, 5′-GACUAUGGACGCAUGAUAA-3′, 5′-GCGGACAUAUUCAGUUGAU-3′, and 5′-UAUAGUAGCCGCACUCGAU-3′. The siRNA sequences used to downregulate the expression of the H5R gene were 5′-CUGUAGAGCUCGAUGAUAG-3′, 5′-CGACGGUUCUAGAGGAUGU-3′, 5′-AAAGGUGGCUACCGACAAU-3′, and 5′-UUACAACACUAUCUGAUCU-3′. The siRNA sequences used to downregulate the expression of the B18R gene were 5′-ACACGUAGCAUGUGAAUAU-3′, 5′-CGGCAGAUUCGAUAACUUA-3′, 5′-AUAGAUGAGCGUCGUAUAA-3′, and 5′-GCACUACACUGCUAUUUGU-3′. The siRNA sequences used to downregulate the expression of the E5R gene were 5′-UGAUAUAGUUGAACCAUGU-3′, 5′-GGUUAUACGGCUACAAUUU-3′, 5′-UGACUUUGUUAGUGAAUAG-3′, and 5′-GAGAAUCCGCUACAUAUUA-3′. The siRNA sequences used to downregulate the expression of the F ORF A gene were 5′-GACCUCCUACUACGUAUAA-3′, 5′-UUUCAGAAGACUCGGCAUA-3′, 5′-GGAAUCCAAUUGUUUGAUA-3′, and 5′-AACCAACGCUCAACAGAUG-3′. For silencing of the expression of the E3L gene, cells were instead transfected with 50 nM double-stranded synthetic oligonucleotides specific for one region of the E3L mRNA. In this case, the siRNA sequence used was 5′-AAUAUCGUCGGAGCUGUACAC-3′. A nonspecific siRNA (NC) was also used, in which the siRNA sequence was predicted not to bind to any known poxviral or cellular mRNA. For all experiments involving siRNA silencing, the medium was removed at 24 h posttransfection. Next, cells were infected with MVA (MOI, 5) and were finally harvested at 6 h postinfection.
Regardless of whether cells were transfected or infected, cells were harvested and collected by using centrifugation (30 s at 10,000 × g). Supernatants were removed, and cellular pellets were suspended and lysed in CE buffer (41) under conditions known not to disrupt the nuclei. Clarified supernatants containing the cytoplasmic proteins were transferred to new centrifuge tubes, and the protein concentration of each sample was determined by using the bicinchoninic acid (BCA) assay (Pierce). An equivalent amount of protein from each sample was loaded into separate lanes of sodium dodecyl sulfate (SDS)-12% polyacrylamide gels. Proteins were separated electrophoretically and were then transferred to a polyvinyl difluoride (PVDF) membrane (Millipore). Membranes were incubated in blocking buffer (Tris-buffered saline [TBS; pH 7.4] containing 5% milk and 0.1% Tween 20) for at least 1 h. Next, membranes were incubated at 4°C overnight in TBS-T (TBS containing 0.1% Tween 20 and 0.5% milk) containing one of the primary antibodies listed below, diluted in TBS-T. After membranes were washed 3 times in TBS-T, blots were incubated with a secondary antibody consisting of either horseradish peroxidase (HRP)-conjugated goat anti-rabbit IgG (dilution, 1:10,000; Fisher Scientific) or HRP-conjugated goat anti-mouse IgG (1:5,000; Fisher Scientific) diluted in TBS-T. The antigen-antibody reactions on each immunoblot were detected by using chemiluminescence and autoradiography. The GE SuperSignal West Pico substrate was utilized except when blots were probed with anti-IκBα antiserum, according to the manufacturer's directions. Primary antibodies included rabbit anti-phospho-ERK1/2 antiserum (1:1,000; Santa Cruz Biotechnology), which interacts with the phosphorylated form of ERK2 and, to a lesser extent, the highly similar ERK1 protein; rabbit anti-ERK1/2 antiserum (1:1,000; Santa Cruz Biotechnology), which recognizes ERK2 and the highly similar ERK1 protein. For immunoblots detecting IκBα, rabbit anti-IκBα (1:5,000; Cell Signaling) was used, and immunoblots were reprobed with anti-actin antiserum (1:1,000). In some cases, a separate immunoblot was incubated with mouse monoclonal anti-E3 antiserum, which interacts with the VACV E3 protein (52). A polyclonal anti-NIK antibody (1:500; Pierce) was used to detect endogenous and dominant negative forms of NIK. Anti-hemagglutinin (anti-HA) (1:1,000; Sigma), anti-FLAG (1:1,000; Sigma), and anti-Ras (1:1,000; Santa Cruz Biotechnology) antisera were used to detect HA-tagged Raf, FLAG-tagged p53, and Ras, respectively.
Detection of mRNA transcripts by RT-PCR.
Reverse transcriptase PCR (RT-PCR) was used to evaluate the extent of siRNA-mediated silencing of several VACV genes during an MVA infection: C11R, E3L, F ORFA, and C ORF B. In this case, cells previously transfected with siRNAs and infected with MVA (MOI, 5) were harvested at 6 h p.i. For experiments using pC11R to express the VGF protein ectopically, the expression level of C11R transcription was evaluated by RT-PCR at 24 h posttransfection. In another experiment, RT-PCR was used to detect the transcription of the cellular tumor necrosis factor (TNF) gene in infected cells. In this case, 293T cells were infected (MOI, 10), and infections proceeded for the indicated times before cells were harvested. Regardless of these differences, cells were lysed in RLT buffer (Qiagen), and total RNA was extracted using the Qiagen RNeasy kit. To produce cDNA from the collected RNA, 0.5 μg of RNA was reverse transcribed into single-stranded cDNA with SuperScript II reverse transcriptase (Invitrogen) and oligo(dT) primers at 42°C for 50 min. For all PCRs, 1 μl of template cDNA was used, and the primers used to amplify cDNA are as follows. Primers for C11R were 5′-ATCAGATTATGCGGTCCAGAGGGA-3′ and antisense oligonucleotide 5′-ACAAGCATAATACCGGGAGATGGG-3′ and were expected to yield a 260-bp product. Primers for glyceraldehyde-3-phosphate dehydrogenase (GAPDH) were 5′-AAGGTCGGAGTCAACGGATTTGGT-3′ and antisense oligonucleotide 5′-ACAAAGTGGTCGTTGAGGGCAATG-3′, yielding a 911-bp product. Amplification of cDNAs from RNA from virus-infected cells was performed with the following oligonucleotides. Primers for E3L were 5′-GCAGAGATTGTGTGTGCGGCTATT-3′ and antisense oligonucleotide 5′-GGTGACAGGGTTAGCATCTTTCCA-3′ and were expected to yield a 327-bp product (11). Primers for C ORF B were 5′-CCTCGCTTTGTGTATCATATTCGTCCC-3′ and antisense oligonucleotide 5′-CGGTTATGACTAAATGGTGTGCTCCG-3′, yielding a 114-bp product. Primers for F ORF A were 5′-GGATGCCACTGCTGGATTACATCT-3′ and reverse primer 5′-TGG ATT CCA ATT CCT CCG ATG-3′, yielding a 186-bp product. Primers for J2R were 5′-TACGGAACGGGACTATGGAC-3′ and reverse primer 5′-GTTTGCCATACGCTCACAGA-3′. Primers for H5R were 5′-CTCCATCTCCTGGAGTCAGC-3′ and reverse primer 5′-CGGTAGCCACCTTTAGGTCA-3′. Primers for B18R were 5′-TGTCGTTTGATAAATACGATGG-3′ and reverse primer 5′-CTTTTTCGAATTATGGCATCTT-3′. For all experiments, GoTaq polymerase (Promega) was used for PCR. PCR conditions were 25 amplification rounds consisting of a denaturation step at 95°C for 45 s, an annealing step of 58°C (C11R, E3L, GAPDH, C ORF B, J2R, H5R, and B18R primer sets) or 55°C (F ORF A primer sets) for 30 s, and an extension step of 72°C for 90 s. An additional extension step of 72°C lasted for 5 min. For amplification of the TNF cDNA, primers and PCR conditions published previously were used (46). A portion of each PCR product was analyzed by 1% agarose gel electrophoresis, and amplicons were visualized by ethidium bromide staining.
Electrophoretic mobility shift assays (EMSAs) of nuclearly extracted proteins to detect NF-κB nuclear translocation and DNA binding.
For EMSAs, 293T cells were either mock infected or infected with MVA or MVAΔC11R. For experiments involving siRNAs, cells were transfected with siRNAs targeting a specific VACV gene and were then infected with MVA (MOI, 5) as described above. At 6 h postinfection, or at the indicated times postinfection, cells were collected by scraping; the method used for isolating nuclear proteins from virus-infected cells has been described previously (18). The concentration of nuclear protein in each sample was determined by using the BCA protein assay kit (Pierce). Five micrograms of each extract was incubated with 0.35 pmol of 32P-labeled double-stranded oligonucleotides containing binding sites for the NF-κB transcription factor in Gel Shift Assay System Binding buffer (Promega) according to the manufacturer's directions. Some reaction mixtures also included excess molar amounts of nonradiolabeled oligonucleotides that either contained NF-κB binding sites or lacked NF-κB binding sites, such as the AP-1 oligonucleotide, or 1 μg monoclonal anti-p65 antiserum (Santa Cruz Biotechnology). Following incubation, reaction products were resolved electrophoretically in a 6% acrylamide gel (Invitrogen) under nondenaturing conditions. Afterward, the gels were dried on filter paper and were exposed to a phosphorimager plate (Molecular Devices), and images were developed and analyzed using the ImageGauge and ImageReader programs, respectively (Fuji).
RESULTS
MVA-induced NF-κB and ERK2 activation is triggered by the EGFR.
MVA infection activates NF-κB in the human fibroblast line 293T and in other primary and transformed fibroblasts of human or murine origin (12, 18, 32, 33). Previously, we showed that ERK2 activation occurs prior to, and is necessary for, MVA-induced NF-κB activation in 293T cells (18, 33). However, the upstream proteins responsible for MVA-induced ERK2 activation were unknown. To identify these, we asked whether the inhibition of signal transduction pathways known to activate ERK2 also prevented MVA-induced NF-κB activation.
One such pathway involves the EGFR; the autophosphorylation of the cytoplasmic tail of the EGFR results in the downstream activation of the ERK1 and ERK2 proteins (44). It is also known that EGFR activation and ERK1/2 activation can each stimulate NF-κB (23, 42, 45). Thus, our working hypothesis was that the EGFR activated NF-κB via ERK2 activation. While the EGFR is present on the surfaces of 293T cells, it is absent from the surfaces of CHO K1 cells (4, 27). However, both cell lines permit poxviral uptake and early gene expression during infection (17, 32). To probe the role of the EGFR in MVA-induced NF-κB activation, CHO K1 cells were infected with MVA, and an NF-κB-dependent luciferase reporter assay was used to evaluate NF-κB activation (Fig. 1A). The results were compared to those for infections that occurred in 293T cells in parallel, because it is known that NF-κB nuclear translocation and activation peak at 3 to 8 h postinfection in these cells, as measured by gel mobility shift assays (33). For this study, NF-κB activation was detected via luciferase reporter assays at 6 h postinfection, a time when NF-κB activation is likely to be high. As reported previously (33, 41), MVA infection of 293T cells activated NF-κB, as indicated by the increased levels of luciferase activity in comparison to luciferase activity from mock-infected 293T cells. In contrast, luciferase activity remained low in mock- or MVA-infected CHO K1 cells, suggesting that the EGFR was necessary for MVA-induced NF-κB activation.
Fig 1.
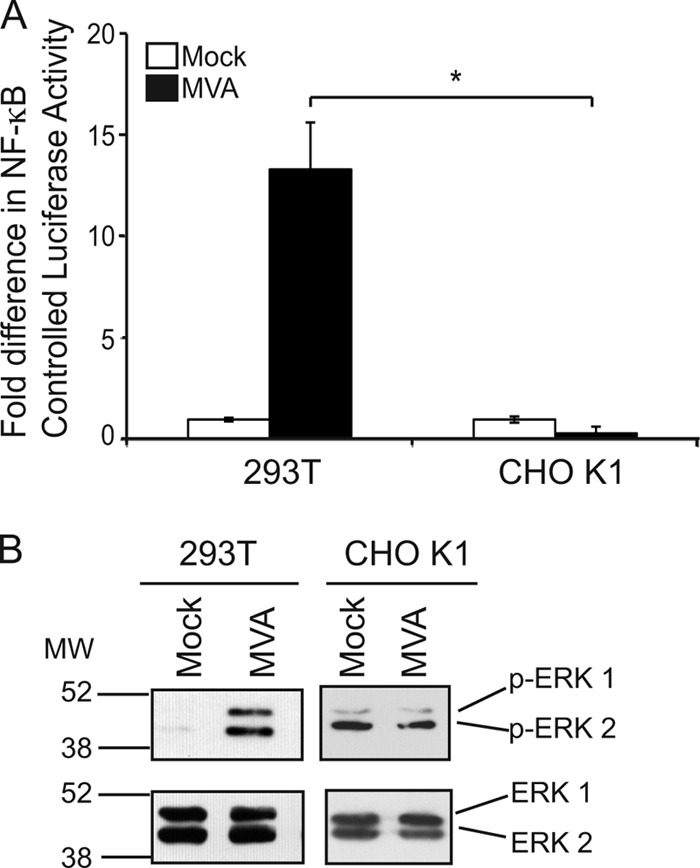
MVA does not activate NF-κB in cells lacking the EGFR. (A) Subconfluent monolayers of 293T or CHO-K1 cells were transfected with pNF-κBluc and pRL-null. Twenty-four hours later, cells were either mock infected or infected with MVA (MOI, 10). Cells were lysed in PLB at 6 h p.i., and the ratio of firefly to sea pansy luciferase activities was normalized to that of the corresponding mock-infected cells, whose value was set to 1. The asterisk indicates statistical significance (P < 0.05). (B) 293T or CHO K1 cells were either mock infected or infected with MVA (MOI, 10). At 4 h p.i., cells were harvested and lysed in CE buffer. Fifteen micrograms of each prepared cytoplasmic extract was electrophoretically separated in SDS-12% polyacrylamide gels and was transferred to PVDF membranes. Membranes were incubated with antibodies recognizing the phosphorylated forms of ERK1 and ERK 2 (p-ERK1/2) and were then reprobed with antibodies recognizing the unmodified forms of ERK1 and ERK2. This blot is representative of results observed in at least three independent experiments.
ERK2 activation precedes and is required for MVA-induced NF-κB activation, and it peaks at 3 to 6 h postinfection in 293T cells (18) (33). We chose to examine phospho-ERK levels at 4 h postinfection, since strong ERK1/2 activation should occur by then. In mock-infected 293T cells, ERK1/2 proteins remained inert; the phosphorylated forms of ERK1/2 were not visibly present (Fig. 1B). In contrast, MVA infection increased MEK/ERK activation, as detected by the presence of the phosphorylated form of the 42-kDa ERK2 protein. The anti-phospho-ERK2 antibody also cross-reacts with the highly similar phospho-ERK1 protein, as evidenced by the presence of a higher-molecular-size (44-kDa) band in most samples. MVA infection of EGFR-deficient CHO K1 cells resulted in a different ERK activation pattern: while ERK2 was activated in uninfected cells, MVA infection did not visibly increase ERK2 activation (Fig. 1B). The immunoblot in Fig. 1B shows a result representative of those of multiple experiments (data not shown).
AG1478 (18) is a compound that selectively inhibits EGFR autophosphorylation (10). Its presence has been reported to reduce MVA-induced NF-κB activation by approximately 20% at 8 h postinfection (18), suggesting that the EGFR may be one of many cellular proteins that facilitate MVA-induced NF-κB activation. When the effects of AG1478 on MVA-induced NF-κB activation were observed at earlier times postinfection, a similar trend occurred: MVA-induced luciferase activity was diminished at 4 and 6 h postinfection (Fig. 2A). Compared to those for infections occurring in a medium containing DMSO, AG1478 treatment reduced luciferase activities approximately 60 and 70% at 4 and 6 h postinfection, respectively. No differences in luciferase activities between a medium containing DMSO alone and one containing DMSO plus AG1478 were statistically significant. At an earlier time postinfection (2 h), luciferase activities were lower in MVA-infected cells than in mock-infected cells, likely reflecting the fact that substantial amounts of NF-κB-controlled luciferase had not yet been expressed.
Fig 2.

The drug AG1478 reduces ERK2 and NF-κB activation in MVA-infected cells. (A) Subconfluent 293T cellular monolayers were transfected with plasmids pNF-κBluc and pRL-null. At 24 h posttransfection, cells were either mock infected or infected with MVA (MOI, 10) in a regular medium or in a DMSO-containing medium in which AG1478 (50 μM) was present or absent for 30 min prior to and throughout infection. At 2, 4, and 6 h p.i., cells were lysed in PLB, and luciferase activities were measured. The ratio of firefly to sea pansy luciferase activities was normalized to that of mock-infected cells, whose value was set to 1. (B) 293T cellular monolayers were pretreated with a DMSO-containing medium either lacking or containing AG1478 (50 μM) for 30 min prior to infection. The respective compounds remained present in the medium for the duration of infection. Cells were either mock infected or infected with MVA (MOI, 10). Cells were harvested at 4 or 6 h p.i., and 20 μg of each of the prepared cytoplasmic extracts was separated by SDS-12% PAGE and was transferred to PVDF membranes. (Top) Membranes were analyzed for the activated (phosphorylated) forms of the ERK1 and ERK2 proteins via immunoblotting. (Bottom) Blots were reprobed with antibodies recognizing the unmodified ERK1 and ERK2 proteins.
The effect of AG1478 on MVA-induced MEK/ERK activation was studied at 4 and 6 h postinfection, times at which NF-κB activation was inhibited in luciferase assays (Fig. 2A). AG1478 also decreased MVA-induced ERK2 activation in 293T cells; the intensity of the 42-kDa phospho-ERK2-containing band was reduced when infections proceeded in the presence versus the absence of AG1478 (Fig. 2B). This band was no longer visible when lysates from cells harvested at 6 h postinfection were used. Unmodified ERK1 and ERK2 protein levels were detected in similar amounts in MVA-infected cells, regardless of the presence or absence of the drug, indicating that AG1478 did not cause its effects by decreasing ERK2 protein levels.
The Raf and Ras proteins are dispensable for MVA-induced ERK2 and NF-κB activation.
It is well known that EGFR phosphorylation activates Ras GTPase, which in turn binds to and phosphorylates Raf (44). Raf then phosphorylates MEK proteins, the upstream kinases that activate ERK1/2 proteins (44). To evaluate the requirement for the Ras and Raf proteins in the activation of NF-κB by MVA, dominant negative (DN) forms of Raf or Ras, which were previously shown to inhibit ERK1/2 activation (16, 47), were overexpressed in 293T cells. Next, cells were infected with MVA, and NF-κB-controlled luciferase activity was measured. As shown in Fig. 3A, MVA infection of pcDNA3.1-transfected cells activated NF-κB. MVA retained its ability to activate NF-κB in cells transfected with pRasDN, suggesting that Ras was dispensable for MVA-induced ERK2 activation. The overexpression of DN Raf itself activated NF-κB; luciferase activity levels were 45-fold higher in mock-infected cells transfected with pRafDN versus pcDNA3.1. Luciferase activity increased further when cells were infected with MVA, indicating that Raf was not required for NF-κB activation. The IκB kinase (IKK) complex is a central upstream activator of NF-κB, and it is known that MEK/ERK activation results in downstream IKK activation (40). We previously showed that IKK inhibition, by overexpression of DN IKKβ, dramatically decreases MVA-induced NF-κB activation (18). Since NIK directly stimulates IKK (13), we transfected a set of cells with plasmids encoding a dominant negative NIK protein as a control. In contrast to DN Ras and DN Raf, DN NIK expression inhibited MVA-induced NF-κB activation (Fig. 3A). Both the DN Raf and DN Ras proteins inhibit p53-induced MEK/ERK activation (29). Figure 3A shows that the pRafDN and pRasDN plasmids expressed functional dominant negative proteins, because p53-induced ERK1/2 activation was inhibited in pRasDN- or pRafDN-transfected cells.
Fig 3.

The effect of DN Ras, Raf, or NIK on MVA-induced NF-κB or ERK2 activation. (A) 293T cells were cotransfected with pNF-κBluc and pRL-null and either pcDNA3.1, pRafDN, pRasDN, or pNIKDN. At 24 h posttransfection, cells were either mock infected or infected with MVA (MOI, 10). Cells were harvested at 6 h p.i. and were evaluated for luciferase activities. The ratio of firefly to sea pansy luciferase activities for each sample was normalized to that of mock-infected cells, whose value was set to 1. A similar set of 293T cells was cotransfected with 1,000 ng pcDNA3.1, pRafDN, pRasDN, or pNIKDN and 1,000 ng of pcDNA3.1 or FLAG-p53. At 24 h posttransfection, cells were lysed, and lysates were probed with an antiserum recognizing the activated form of ERK1/2, inactive ERK1/2, Ras, HA-Raf, or FLAG-p53. (B) 293T cells were transfected with 1,000 ng of pcDNA3.1, pRafDN, pRasDN, or pNIKDN. Twenty-four hours later, cells were either mock infected or infected with MVA (MOI, 10). At 4 h p.i., cells were harvested and lysed, and 15 μg of each of the cellular lysates was analyzed by immunoblotting with antibodies against phosphorylated ERK1/2 (p-ERK1/2) and was then developed by using chemiluminescence. The blot was reprobed with antibodies against ERK1/2 and was developed by using chemiluminescence. Lysates from cells transfected with either pcDNA3.1 or pNIKDN were probed for NIK expression by immunoblotting using an anti-NIK antiserum.
Based on the data from Fig. 3A, it is hypothesized that MVA-induced ERK2 activation would still occur in cells expressing DN Ras or DN Raf but not in cells expressing DN NIK. As shown in Fig. 3B, the phospho-ERK2-containing bands were more intense in cells infected with MVA than in mock-infected cells, regardless of the presence of DN Raf, DN Ras, or DN NIK. These data suggest that MVA-induced ERK2 activation was independent of the upstream Ras and Raf proteins. Notably, MEK/ERK activation occurred in DN NIK-expressing cells. These data suggest that NIK functions either downstream of ERK2 activation or via an ERK2-independent NF-κB activation pathway.
Silencing of the C11R gene reduces ERK2 and NF-κB activation.
The C11R gene of orthopoxviruses encodes VGF, a soluble, secreted viral homolog of epidermal growth factor (EGF) (5, 49, 50). VGF binds to the extracellular domain of the EGFR, resulting in EGFR activation (51). To test if the C11R gene was responsible for MVA-induced ERK2 or NF-κB activation, we infected cells with MVA under conditions in which the C11R gene was transcriptionally silenced, and we asked if ERK2 remained activated (Fig. 4A). When cells were transfected with siRNAs directed against C11R, MVA-induced phospho-ERK2 levels also were greatly reduced from those in mock-transfected cells (Fig. 4A). In this case, phospho-ERK1 proteins were no longer detected, because a smaller amount of protein from each sample (7 μg) was used for immunoblotting than in other experiments. This effect was specific for the silencing of the C11R gene, because MVA still activated ERK2 when cells were transfected with siRNAs specific for two other predicted MVA genes (F ORF A, C ORF B) or two genes known to be present in MVA (J2R and H5R), or when cells were transfected with siRNAs that were predicted not to target any known cellular or viral gene (NC siRNA) (35). As would be expected, unmodified ERK1 and ERK2 protein levels remained similar in cells regardless of the infection state or the presence or absence of transfected siRNAs. As shown in Fig. 4A, the transfection of siRNAs specific for either C11R, C ORF B, F ORF A, J2R, or H5R greatly reduced the production of the mRNAs for those genes, but not for GAPDH. Another indicator of the specificity of the siRNAs was the fact that siRNA silencing of the E5R gene did not affect the expression of the B18R gene. NC siRNA transfection also did not alter the transcription of the C11R gene.
Fig 4.

siRNA silencing of the C11R gene diminishes MVA-induced ERK2 and NF-κB activation. Subconfluent 293T cellular monolayers either were transfected with chemically synthesized siRNAs against the indicated VACV genes or were mock transfected. A 100 nM concentration of siRNA was used for transfection when the C11R, C ORF B, or F ORF A gene was silenced; 50 nM siRNA was used for transfection to silence E3L; 200 nM siRNA was used to silence J2R, H5R, B18R, or E5R gene expression or as a control (NC). Twenty-four hours later, cells were either mock infected or infected with MVA (MOI, 5), and infected cells were harvested 6 h later. Cytoplasmic (A) or nuclear (B) proteins were extracted. For both experiments, total RNA was isolated from an identical set of cells that were transfected and infected in parallel, and RNA was reverse transcribed to cDNA for further PCR amplification. (A) Seven micrograms of prepared cytoplasmic extracts was analyzed by immunoblotting. The blots were probed with antibodies against the phosphorylated form of ERK2 (p-ERK2). The blots were then reprobed with an antiserum against ERK1/2. From the RNA isolated from an identical set of cells, mRNA was reverse transcribed into cDNA. C11R, C ORF B, F ORF A, J2R, H5R, E5R, B18R, or GAPDH cDNAs were amplified by using PCR. In another experiment to prove the specificity of the siRNA silencing, cDNA generated from samples of infected cells treated with siRNA against B18R or E5R was incubated with primers specific for B18R. A portion of each reaction product was analyzed by electrophoresis in a 1% agarose gel. Ethidium bromide was used to visualize the presence of PCR amplicons. (B) (Top) Nuclear extracts from infected cells were incubated with [32P]ATP-labeled oligonucleotides containing a consensus NF-κB binding site. In some reaction mixtures, nonradiolabeled oligonucleotides containing either NF-κB binding sites (NF-κB) or AP-1 binding sites (AP-1) were included. An antibody against the p65 subunit of NF-κB was also included where indicated (p65 Ab), and a plus sign indicates the supershifted band containing p65, NF-κB, and oligonucleotides. The reaction mixture was analyzed by electrophoresis in a 6% gel under nondenaturing conditions. An asterisk indicates the NF-κB-bound probe. (Bottom) C11R, E3L, and GAPDH cDNAs were PCR amplified. A portion of each reaction product was analyzed by electrophoresis in a 1% agarose gel. Ethidium bromide was used to visualize the presence of PCR amplicons, and the image was inverted.
Next, siRNA-transfected cells were infected and evaluated for their NF-κB activation state by using electrophoretic mobility shift assays (EMSAs), which detect both NF-κB nuclear translocation and the ability of nuclearly translocated NF-κB to bind to its promoter sequence (18). If extracted nuclear proteins shift the mobility of radiolabeled oligonucleotides that possess NF-κB binding sequences, then NF-κB is considered to be present in the nucleus and activated (able to bind to its target DNA) (18). Luciferase assays were not used to measure NF-κB activation because of technical difficulties encountered when cells were transfected sequentially with luciferase reporter plasmids and siRNAs prior to infections. As shown in Fig. 4B (lanes 4, 5, 11, and 12), the intensity of an NF-κB-containing band (marked by an asterisk) was greater in nuclear extracts from MVA-infected cells than in those from mock-infected cells, indicating that MVA infection activated NF-κB. The mobility of this band was supershifted (marked by a plus sign) when an anti-NF-κB antibody (p65 Ab) was present in reaction mixtures, proving that NF-κB was indeed present in the complex. The band disappeared when an excess of unlabeled oligonucleotides containing NF-κB binding sites, but not AP-1 binding sites, was present in the reaction mixture, showing that the interaction between NF-κB and its target oligonucleotide was specific. The C11R gene was partially responsible for MVA-induced NF-κB activation, because the intensity of the NF-κB-containing band was reduced when C11R was silenced (Fig. 4B, lane 7), but not when E3L was silenced (Fig. 4B, lanes 6 and 14), or in the absence of siRNA oligonucleotides (Fig. 4B, lane 5), or in the presence of siRNAs that were predicted not to bind to any known cellular or poxviral transcript (lane 13). The effectiveness of the siRNA silencing of C11R was assessed by RT-PCR. The intensities of the C11R- or E3L-containing amplicons were much lower in cells transfected with C11R or E3L siRNA than in mock-transfected cells. NC or E3L siRNA did not affect the transcription of the poxvirus C11R gene (data not shown), indicating that the siRNA silencing of a viral gene was specific. GAPDH mRNA levels remained similar, illustrating that the decrease in C11R or E3L gene expression did not result in a general decrease in transcription.
Deletion of the C11R gene from MVA reduces ERK2 and NF-κB activation in 293T cells.
To continue to test the hypothesis that the C11R gene product was responsible for MVA-induced NF-κB activation, we created a mutant MVA virus lacking the C11R gene (MVAΔC11R). While deletion of C11R from wild-type VACV results in a mutant virus with a smaller plaque phenotype and a diminished replication efficiency (6, 7), MVAΔC11R and MVA foci were similar in size and replicated to similar titers in CEF monolayers (data not shown). The vaccinia virus early protein E3 was also detected to similar levels in both 293T and Hacat cells infected with MVAΔC11R or MVA, indicating that early gene expression still occurred, despite the absence of C11R (data not shown).
The NF-κB activation states of cells infected with MVA versus MVAΔC11R were compared by using gel mobility shift assays to detect nuclearly translocated and activated NF-κB. The results of a representative assay are shown in Fig. 5A. No differences in NF-κB-containing bands were obvious between MVA- and MVAΔC11R-infected cells at 1 and 2 h postinfection. However, the band was less intense in MVAΔC11R-infected cells at 3 h postinfection. This trend was also observed at 5 and 6 h postinfection. Since gel mobility shift assays are not quantitative, NF-κB activation was also assessed by a luciferase reporter assay. In this case, cells were examined at 6 h postinfection to measure luciferase activity that culminated during virus infection (33). NF-κB-controlled luciferase activity in MVAΔC11R-infected cells was decreased by 30% from that in MVA-infected cells (Fig. 5B), suggesting that the C11R gene product does indeed contribute to MVA-induced NF-κB activation. Levels of TNF mRNA in infected 293T cells were detected by using semiquantitative RT-PCR as another means of evaluating NF-κB activation (Fig. 5C). At 2 h p.i., TNF cDNA levels were increased by infection with either MVA or MVAΔC11R, indicating that C11R did not affect NF-κB activation at this time point. At 4 h postinfection, while TNF gene transcription was decreased in cells infected with a virus that inhibits NF-κB (MVA/5.2kb), the TNF amplicons in samples from MVA- and MVAΔC11R-infected cells were similar. However, differences in the amplicon intensities were apparent at 6 h postinfection, when the TNF cDNA levels in MVAΔC11R- and MVA/5.2kb-infected cells were lower than those in mock- or MVA-infected cells.
Fig 5.
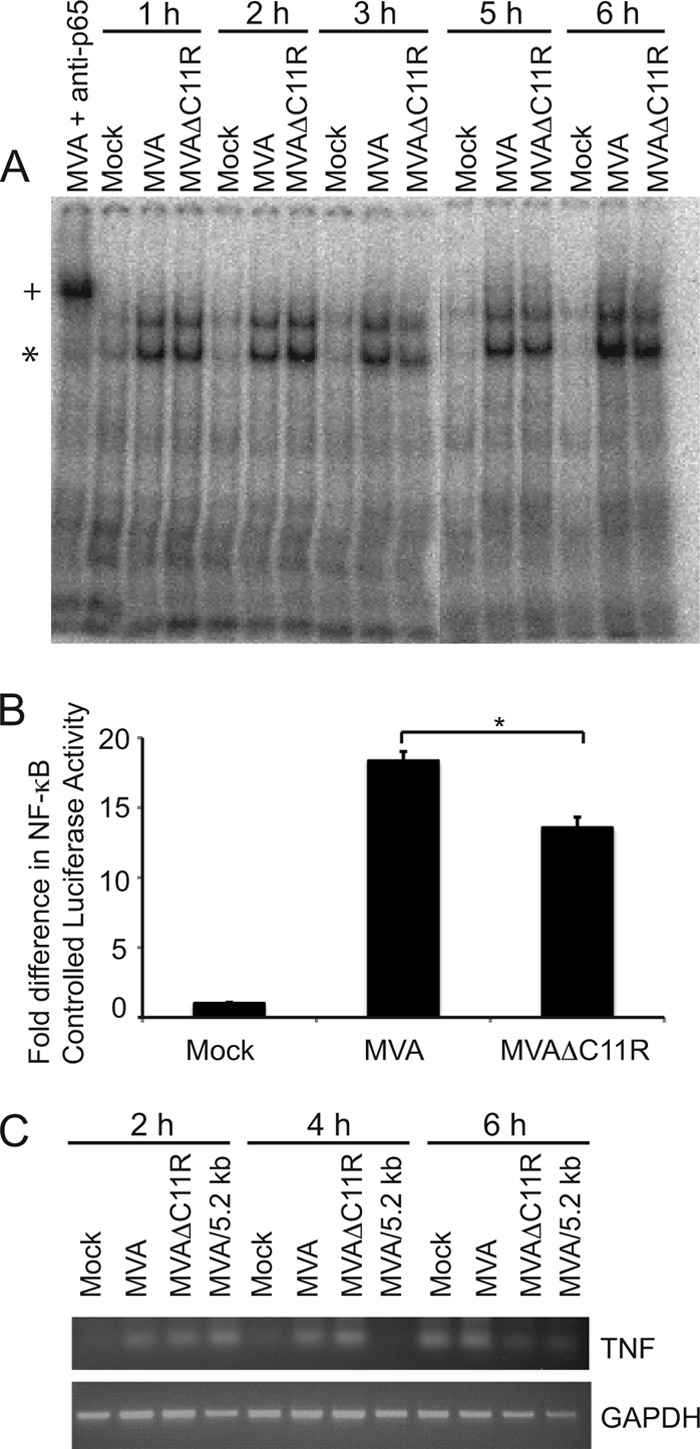
An MVA virus lacking C11R (MVAΔC11R) reduces NF-κB activation. (A) 293T cellular monolayers were either mock infected or infected with MVA or MVAΔC11R (MOI, 10). Infected cells were harvested at the indicated time points. Cells were lysed, and nuclear proteins were extracted. Nuclear extracts from infected cells were incubated with [32P]ATP-labeled oligonucleotides containing a consensus NF-κB binding site. An asterisk indicates the NF-κB-bound probe. An antibody against the p65 subunit of NF-κB (p65 Ab) was also included with extracts from MVA-infected cells (6 h p.i.) where indicated, and a plus sign indicates the supershifted band containing p65, NF-κB, and oligonucleotides. The reaction mixture was analyzed by electrophoresis in a 6% gel under nondenaturing conditions. (B) 293T cells were transfected with luciferase reporter plasmids (pNF-κBluc and pRL-null) 24 h before infection. Cells were either mock infected or infected with the indicated viruses (MOI, 5). At 6 h p.i., cells were lysed, and firefly and sea pansy luciferase activities were measured. The ratio of firefly to sea pansy luciferase activities was normalized to that of mock-infected cells, whose value was set to 1. An asterisk indicates statistical significance (P < 0.05). (C) 293T cells were infected with the indicated viruses (MOI, 10). At the times indicated on the figure, infected cells were harvested and lysed. Total RNA was first isolated and then reverse transcribed to cDNA for further PCR amplification. TNF or GAPDH cDNAs were amplified by using PCR. A portion of each reaction product was analyzed by electrophoresis in a 1% agarose gel. Ethidium bromide was used to visualize the presence of PCR amplicons.
Since ERK2 activation is required for MVA-induced NF-κB activation (18), phospho-ERK2 levels were expected to be diminished in cells infected with MVAΔC11R. As shown in Fig. 6, phosphorylated ERK2 levels were lower in MVAΔC11R-infected cells than in MVA-infected cells. This preceded NF-κB activation, was observed as early as 1 h postinfection, and continued until 6 h postinfection. Levels of unmodified ERK1/2 proteins remained constant in mock- or virus-infected cells, ruling out the possibility that a decrease in ERK2 activation was due to decreased ERK2 protein levels.
Fig 6.

An MVA virus lacking C11R (MVAΔC11R) reduces ERK2 activation. 293T cellular monolayers were either mock infected or infected with either MVA or MVAΔC11R (MOI, 10). At either 1, 2, 3, 4, 5, or 6 h p.i., cells were lysed, and 20 μg of cytoplasmic extracts from each sample was analyzed by immunoblotting, using antibodies recognizing the phosphorylated form of ERK2 (p-ERK1/2). The blot was reprobed with antibodies recognizing ERK1/2.
Ectopic expression of the C11R gene is sufficient to activate NF-κB.
To test if the C11R product induced NF-κB activation independently of virus infection, 293T cells were transfected with a plasmid encoding the C11R gene, and NF-κB activation was assessed by an NF-κB-dependent luciferase reporter assay. As shown in Fig. 7A, luciferase activity was 14-fold higher in cells transfected with pC11R than in cells transfected with a plasmid lacking C11R (pcDNA3.1). During a time course assay, luciferase activity was noticeably higher than that in pCI-transfected cells when the C11R plasmid had been expressed for either 12 or 24 h (data not shown). Unfortunately, antibodies against the orthologous variola virus C11 product (SPGF) (28) did not detect the VGF in cellular extracts or in concentrated supernatants (data not shown). Other strategies to detect C11 proteins by engineering epitope tags onto the C terminus of the C11R gene also proved fruitless: the epitope tags were not detectable by immunoblotting (data not shown). While not indicative of protein levels, RT-PCR was used to detect C11R mRNA so as to demonstrate gene transcription (Fig. 6B). Following reverse transcription of total RNA from pcDNA3.1- or pC11R-transfected cells, the resultant C11R cDNA was amplified by using PCR with primers specific for the C11R gene. As shown in Fig. 7B, the C11R-containing amplicon was visible by use of genetic material from pC11R-transfected cells but not from vector-transfected cells. Amplicons containing GAPDH cDNA were detected in similar amounts from both samples.
C11R is partially responsible for MVA-induced NF-κB and ERK2 activation in the human keratinocyte line Hacat.
The effect of MVAΔC11R infection in cells of the transformed keratinocyte line Hacat was examined to determine whether the MVAΔC11R effect occurred in other cell types. Hacat cells were studied specifically because keratinocytes are infected soon after inoculation with the smallpox vaccine (31, 56). In this case, NF-κB activation was measured by luciferase reporter assays (Fig. 8A) and detection of IκBα protein levels (Fig. 8B). Infection of Hacat cells with MVA activated NF-κB more than infection with MVAΔC11R; the greatest difference in NF-κB activation was observed at 6 h postinfection. IκBα levels were lower in lysates from MVA-infected cells than in those from MVAΔC11R-infected cells, supporting the data in Fig. 8A. In both assays, WR infection inhibited NF-κB-driven luciferase activity and IκBα degradation, as expected. As with 293T cells, MVA infection activated ERK2 at all time points tested (Fig. 8C). MVAΔC11R infection of Hacat cells resulted in visibly lower levels of phospho-ERK2 at 4, 6, and 8 h postinfection.
Fig 8.

C11R contributes to MVA-induced NF-κB and ERK2 activation in human keratinocytes. (A) Hacat cells were transfected with luciferase reporter plasmids (pNF-κBluc and pRL-null) 24 h before infection. Cells were either mock infected or infected with one of the indicated viruses (MOI, 10). At 4, 6, or 8 h p.i., cells were lysed, and firefly and sea pansy luciferase activities were measured. The ratio of firefly to sea pansy luciferase activities was normalized to that of mock-infected cells, whose value was set to 1. (B and C) Hacat cells were either mock infected or infected with MVA or MVAΔC11R (MOI, 10) or with WR. At 6 h p.i.(B) or 4, 6, or 8 h later (C), cells were lysed, and 10 μg of prepared cytoplasmic extracts from each sample was analyzed via immunoblotting. (B) Extracts were probed with antibodies against IκBα and were then reprobed with an antiserum against actin. (C) Extracts were probed with antibodies against phospho-ERK1/2 and were then reprobed with an antiserum against unmodified ERK1/2.
DISCUSSION
MVA is an attenuated VACV lacking many immune evasion genes, including several genes encoding NF-κB-inhibitory proteins that are otherwise present in its parental genome (2). As a result, MVA activates NF-κB in a variety of cell types (12, 32, 41, 53). For 293T cells, virus entry and early gene expression are required for NF-κB activation (33). The purpose of this study was to identify cellular and viral factors responsible for MVA-induced NF-κB activation. Here we show that chemical inhibition of the EGFR results in reduced NF-κB activation in response to MVA infection. The data presented here show that expression of the early C11R gene plays a role in NF-κB activation in 293T and Hacat cells; MVA viruses devoid of the C11R gene have a reduced ability to activate NF-κB. Moreover, ectopic expression of the C11R gene also activated NF-κB. Since the C11R gene encodes VGF, the current model is that extracellular VGF binding to the EGFR results in EGFR activation, triggering ERK2 activation and NF-κB activation.
In the past, treatment of cells with AG1478 diminished MVA-induced NF-κB activation by 20% at 8 h postinfection (18), indicating that the EGFR pathway may be a minor activator of NF-κB. An increased level of inhibition was observed at earlier times postinfection here. Recently, other cellular molecules were identified as transducing MVA-induced NF-κB activation, including protein kinase R (PKR) in mouse embryonic fibroblasts (MEFs) and HeLa cells, and Toll-like receptors 2 and 6 (TLR2/6) in THP-1 cells (12, 32, 53, 54). These data likely exemplify the complexity of virus infection in vivo, in which MVA may trigger different signal transduction events in different cell types. Thus, the successful neutralization of viral infections may require the simultaneous presence of small molecules that target myriad signal transduction pathways. It was reported previously that inhibition of MEK/ERK activity resulted in an approximately 80% decrease in NF-κB-driven luciferase activity (18). Thus, our current model is that MEK/ERK activation occurs as a downstream event common to the PKR, TLR2/6, and EGFR signal transduction pathways, ultimately resulting in NF-κB activation.
It was observed that the DN NIK protein inhibited MVA-induced NF-κB activation yet allowed MVA-induced MEK/ERK activation. How do these data fit within the model above? One hypothesis is that NIK functions immediately downstream of MEK/ERK. In support of this are multiple publications showing that NIK binds to IKK (either directly or indirectly) (30). Thus, MVA infection may stimulate events that eventually result in MEK/ERK activation, resulting in downstream NIK and IKK activation and NF-κB activation. We attempted to detect (activated) phospho-NIK proteins during MVA infection, using lysates from MVA-infected cells either expressing DN ERK1/2 or treated with U0126. However, inconsistent results made it difficult to draw any conclusions (data not shown).
There are several known effects of VGF or EGFR activation during a poxvirus infection. Wild-type VACV activates the EGFR via extracellular interactions to stimulate macropinocytosis of VACV (37). The VGF protein binds to the EGFR to suppress apoptosis in infected host cells (43), stimulate the expression of the mitogenic egr-1 gene in host cells (1), and induce proliferation in uninfected cells (7). This report describes a fourth outcome of VGF-EGFR activation, stimulating NF-κB during MVA infection. In contrast to the effects of VGF-EGFR interactions discussed above, NF-κB activation could present deleterious effects during virus infection: stimulating the transcription of myriad immune molecules with antiviral functions (22). We hypothesize that VGF binds to the EGFR to induce “proviral” events despite the fact that such interactions also activate NF-κB. To counteract this undesired activity, wild-type VACV expresses immune evasion proteins, such as B14 and K1 (9, 46), to specifically disarm EGFR-induced NF-κB activation while permitting proviral events. This exemplifies a sophisticated approach that poxviruses employ to differentially manipulate EGFR-triggered events for their benefit. Proper EGFR signal transduction is critical for the entry of several virus types into cells (14, 36), and aberrant EGFR expression or signaling is associated with certain types of cancers (39). Thus, identifying how VACV modulates EGFR-mediated signal transduction pathways could likely yield new approaches and targets for antiviral or anticancer therapies.
Activation of the EGFR stimulates myriad signaling pathways that trigger cellular proliferation, survival, and cell cycle progression (44). For example, phosphorylated EGFR triggers Sos, stimulating the Ras-Raf-MEK1/2-ERK1/2 pathway to result in cellular proliferation (15). The data presented here show that EGFR-induced NF-κB activation is distinct from this pathway, since DN Ras and Raf proteins permitted MVA-induced NF-κB and ERK2 activation. Identification of the upstream cellular molecules that are responsible for MEK/ERK activation will be the topic of future investigations.
MVA was used safely as a smallpox vaccine in Germany in the 1970s (25) and is now a promising vaccine vector for use against human and veterinary diseases (26). One shortcoming of the current MVA vector is its relatively low immunogenicity compared to that of its parent, VACV. Thus, many MVA-based vaccine protocols use multiple inoculations to generate a robust antibody response. NF-κB controls the important process of inflammation and cell survival (19), making it a prime candidate for controlling viral immunogenicity. Yet it is not clear how altering NF-κB would affect the immunogenicity of MVA. One hypothesis is that activated NF-κB decreases immunogenicity: NF-κB would stimulate the expression of intrinsic or extracellular antiviral immune response proteins to lyse cells before an appropriate acquired immune response would develop. A competing hypothesis is that NF-κB activation would stimulate the synthesis of prosurvival genes in infected cells, which would allow host cells to survive longer, giving prolonged signals to stimulate the immune response to increase immunogenicity. An MVA that no longer activates NF-κB is required in order to test these models. Here we demonstrate that MVAΔC11R is one such virus, in that it has a diminished ability to activate NF-κB and MEK/ERK. How this virus affects early events in virus-host interactions will help solve the question of the role of NF-κB activation in viral immunogenicity.
ACKNOWLEDGMENTS
We thank Amanda Birmingham and Jon Karpilow for reagents and discussions for siRNA experiments. We thank Amy MacNeill, Gail Scherba, and Dong-wan Yoo for helpful discussions.
S.M. was supported in part by NIH Cellular and Molecular Biology training grant T32 GM007283 and NIH/NIAID fellowship F21 AI082947. J.S. was supported by grant AI055530.
Footnotes
Published ahead of print 27 June 2012
REFERENCES
- 1. Andrade AA, et al. 2004. The vaccinia virus-stimulated mitogen-activated protein kinase (MAPK) pathway is required for virus multiplication. Biochem. J. 381:437–446 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2. Antoine G, Scheiflinger F, Dorner F, Falkner FG. 1998. The complete genomic sequence of the modified vaccinia Ankara strain: comparison with other orthopoxviruses. Virology 244:365–396 [DOI] [PubMed] [Google Scholar]
- 3. Blanchard TJ, Alcami A, Andrea P, Smith GL. 1998. Modified vaccinia virus Ankara undergoes limited replication in human cells and lacks several immunomodulatory proteins: implications for use as a human vaccine. J. Gen. Virol. 79(Pt 5):1159–1167 [DOI] [PubMed] [Google Scholar]
- 4. Bringman TS, Lindquist PB, Derynck R. 1987. Different transforming growth factor-alpha species are derived from a glycosylated and palmitoylated transmembrane precursor. Cell 48:429–440 [DOI] [PubMed] [Google Scholar]
- 5. Brown JP, Twardzik DR, Marquardt H, Todaro GJ. 1985. Vaccinia virus encodes a polypeptide homologous to epidermal growth factor and transforming growth factor. Nature 313:491–492 [DOI] [PubMed] [Google Scholar]
- 6. Buller RM, Chakrabarti S, Cooper JA, Twardzik DR, Moss B. 1988. Deletion of the vaccinia virus growth factor gene reduces virus virulence. J. Virol. 62:866–874 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7. Buller RM, Chakrabarti S, Moss B, Fredrickson T. 1988. Cell proliferative response to vaccinia virus is mediated by VGF. Virology 164:182–192 [DOI] [PubMed] [Google Scholar]
- 8. Carroll MW, Moss B. 1997. Host range and cytopathogenicity of the highly attenuated MVA strain of vaccinia virus: propagation and generation of recombinant viruses in a nonhuman mammalian cell line. Virology 238:198–211 [DOI] [PubMed] [Google Scholar]
- 9. Chen RA, Ryzhakov G, Cooray S, Randow F, Smith GL. 2008. Inhibition of IκB kinase by vaccinia virus virulence factor B14. PLoS Pathog. 4:e22 doi:10.1371/journal.ppat.0040022 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10. Daub H, Weiss FU, Wallasch C, Ullrich A. 1996. Role of transactivation of the EGF receptor in signalling by G-protein-coupled receptors. Nature 379:557–560 [DOI] [PubMed] [Google Scholar]
- 11. Dave RS, et al. 2006. siRNA targeting vaccinia virus double-stranded RNA binding protein [E3L] exerts potent antiviral effects. Virology 348:489–497 [DOI] [PubMed] [Google Scholar]
- 12. Delaloye J, et al. 2009. Innate immune sensing of modified vaccinia virus Ankara (MVA) is mediated by TLR2-TLR6, MDA-5 and the NALP3 inflammasome. PLoS Pathog. 5:e1000480 doi:10.1371/journal.ppat.1000480 [DOI] [PMC free article] [PubMed] [Google Scholar] [Retracted]
- 13. Dhawan P, Richmond A. 2002. A novel NF-κB-inducing kinase-MAPK signaling pathway up-regulates NF-κB activity in melanoma cells. J. Biol. Chem. 277:7920–7928 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14. Eierhoff T, Hrincius ER, Rescher U, Ludwig S, Ehrhardt C. 2010. The epidermal growth factor receptor (EGFR) promotes uptake of influenza A viruses (IAV) into host cells. PLoS Pathog. 6:e1001099 doi:10.1371/journal.ppat.1001099 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15. Fan PD, Goff SP. 2000. Abl interactor 1 binds to Sos and inhibits epidermal growth factor- and v-Abl-induced activation of extracellular signal-regulated kinases. Mol. Cell. Biol. 20:7591–7601 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16. Fiordalisi JJ, Holly SP, Johnson RL, II, Parise LV, Cox AD. 2002. A distinct class of dominant negative Ras mutants: cytosolic GTP-bound Ras effector domain mutants that inhibit Ras signaling and transformation and enhance cell adhesion. J. Biol. Chem. 277:10813–10823 [DOI] [PubMed] [Google Scholar]
- 17. Franke CA, Roseman NA, Hruby DE. 1985. Expression and regulation of the vaccinia virus thymidine kinase gene in non-permissive cells. Virus Res. 3:13–17 [DOI] [PubMed] [Google Scholar]
- 18. Gedey R, Jin XL, Hinthong O, Shisler JL. 2006. Poxviral regulation of the host NF-κB response: the vaccinia virus M2L protein inhibits induction of NF-κB activation via an ERK2 pathway in virus-infected human embryonic kidney cells. J. Virol. 80:8676–8685 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19. Ghosh S, May MJ, Kopp EB. 1998. NF-κB and Rel proteins: evolutionarily conserved mediators of immune responses. Annu. Rev. Immunol. 16:225–260 [DOI] [PubMed] [Google Scholar]
- 20. Reference deleted.
- 21. Reference deleted.
- 22. Guerra S, et al. 2004. Microarray analysis reveals characteristic changes of host cell gene expression in response to attenuated modified vaccinia virus Ankara infection of human HeLa cells. J. Virol. 78:5820–5834 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23. Habib AA, et al. 2001. The epidermal growth factor receptor engages receptor interacting protein and nuclear factor-κB (NF-κB)-inducing kinase to activate NF-κB. Identification of a novel receptor-tyrosine kinase signalosome. J. Biol. Chem. 276:8865–8874 [DOI] [PubMed] [Google Scholar]
- 24. Hinthong O, Jin XL, Shisler JL. 2008. Characterization of wild-type and mutant vaccinia virus M2L proteins' abilities to localize to the endoplasmic reticulum and to inhibit NF-κB activation during infection. Virology 373:248–262 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25. Hochstein-Mintzel V, Huber HC, Stickl H. 1972. Virulence and immunogenicity of a modified vaccinia virus (strain MVA). Z. Immunitatsforsch. Exp. Klin. Immunol. 144:104–156 (In German.) [PubMed] [Google Scholar]
- 26. Jacobs BL, et al. 2009. Vaccinia virus vaccines: past, present and future. Antiviral Res. 84:1–13 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27. Jo M, Thomas KS, O'Donnell DM, Gonias SL. 2003. Epidermal growth factor receptor-dependent and -independent cell-signaling pathways originating from the urokinase receptor. J. Biol. Chem. 278:1642–1646 [DOI] [PubMed] [Google Scholar]
- 28. Kim M, et al. 2004. Biochemical and functional analysis of smallpox growth factor (SPGF) and anti-SPGF monoclonal antibodies. J. Biol. Chem. 279:25838–25848 [DOI] [PubMed] [Google Scholar]
- 29. Lee SW, et al. 2000. Sustained activation of Ras/Raf/mitogen-activated protein kinase cascade by the tumor suppressor p53. Proc. Natl. Acad. Sci. U. S. A. 97:8302–8305 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30. Ling L, Cao Z, Goeddel DV. 1998. NF-κB-inducing kinase activates IKK-α by phosphorylation of Ser-176. Proc. Natl. Acad. Sci. U. S. A. 95:3792–3797 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31. Liu L, et al. 2010. Epidermal injury and infection during poxvirus immunization is crucial for the generation of highly protective T cell-mediated immunity. Nat. Med. 16:224–227 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32. Lynch HE, et al. 2009. Modified vaccinia virus Ankara can activate NF-κB transcription factors through a double-stranded RNA-activated protein kinase (PKR)-dependent pathway during the early phase of virus replication. Virology 391:177–186 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33. Martin S, Shisler JL. 2009. Early viral protein synthesis is necessary for NF-κB activation in modified vaccinia Ankara (MVA)-infected 293 T fibroblast cells. Virology 390:298–306 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34. Mayr A, Hochstein-Mintzel V, Stickl H. 1975. Creation of an attenuated strain of Ankara, MVA. Infection 3:6–14 [Google Scholar]
- 35. McCraith S, Holtzman T, Moss B, Fields S. 2000. Genome-wide analysis of vaccinia virus protein-protein interactions. Proc. Natl. Acad. Sci. U. S. A. 97:4879–4884 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36. Melnick M, Abichaker G, Htet K, Sedghizadeh P, Jaskoll T. 2011. Small molecule inhibitors of the host cell COX/AREG/EGFR/ERK pathway attenuate cytomegalovirus-induced pathogenesis. Exp. Mol. Pathol. 91:400–410 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37. Mercer J, et al. 2010. Vaccinia virus strains use distinct forms of macropinocytosis for host-cell entry. Proc. Natl. Acad. Sci. U. S. A. 107:9346–9351 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38. Mohamed MR, McFadden G. 2009. NF-κB inhibitors: strategies from poxviruses. Cell Cycle 8:3125–3132 [DOI] [PubMed] [Google Scholar]
- 39. Navolanic PM, Steelman LS, McCubrey JA. 2003. EGFR family signaling and its association with breast cancer development and resistance to chemotherapy. Int. J. Oncol. 22:237–252 [PubMed] [Google Scholar]
- 40. Nemoto S, DiDonato JA, Lin A. 1998. Coordinate regulation of IκB kinases by mitogen-activated protein kinase kinase kinase 1 and NF-κB-inducing kinase. Mol. Cell. Biol. 18:7336–7343 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41. Oie KL, Pickup DJ. 2001. Cowpox virus and other members of the orthopoxvirus genus interfere with the regulation of NF-κB activation. Virology 288:175–187 [DOI] [PubMed] [Google Scholar]
- 42. Pearson G, English JM, White MA, Cobb MH. 2001. ERK5 and ERK2 cooperate to regulate NF-κB and cell transformation. J. Biol. Chem. 276:7927–7931 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43. Postigo A, Martin MC, Dodding MP, Way M. 2009. Vaccinia-induced epidermal growth factor receptor-MEK signalling and the anti-apoptotic protein F1L synergize to suppress cell death during infection. Cell. Microbiol. 11:1208–1218 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44. Prenzel N, Fischer OM, Streit S, Hart S, Ullrich A. 2001. The epidermal growth factor receptor family as a central element for cellular signal transduction and diversification. Endocr. Relat. Cancer 8:11–31 [DOI] [PubMed] [Google Scholar]
- 45. Sethi G, Ahn KS, Chaturvedi MM, Aggarwal BB. 2007. Epidermal growth factor (EGF) activates nuclear factor-κB through IκBα kinase-independent but EGF receptor-kinase dependent tyrosine 42 phosphorylation of IκBα. Oncogene 26:7324–7332 [DOI] [PubMed] [Google Scholar]
- 46. Shisler JL, Jin XL. 2004. The vaccinia virus K1L gene product inhibits host NF-κB activation by preventing IκBα degradation. J. Virol. 78:3553–3560 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 47. Sutor SL, Vroman BT, Armstrong EA, Abraham RT, Karnitz LM. 1999. A phosphatidylinositol 3-kinase-dependent pathway that differentially regulates c-Raf and A-Raf. J. Biol. Chem. 274:7002–7010 [DOI] [PubMed] [Google Scholar]
- 48. Tscharke DC, et al. 2006. Poxvirus CD8+ T-cell determinants and cross-reactivity in BALB/c mice. J. Virol. 80:6318–6323 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 49. Twardzik DR, Brown JP, Ranchalis JE, Todaro GJ, Moss B. 1985. Vaccinia virus-infected cells release a novel polypeptide functionally related to transforming and epidermal growth factors. Proc. Natl. Acad. Sci. U. S. A. 82:5300–5304 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 50. Twardzik DR, Ranchalis JE, Moss B, Todaro GJ. 1987. Vaccinia growth factor: newest member of the family of growth modulators which utilize the membrane receptor for EGF. Acta Neurochir. Suppl. (Wien) 41:104–109 [DOI] [PubMed] [Google Scholar]
- 51. Tzahar E, et al. 1998. Pathogenic poxviruses reveal viral strategies to exploit the ErbB signaling network. EMBO J. 17:5948–5963 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 52. Weaver JR, et al. 2007. The identification and characterization of a monoclonal antibody to the vaccinia virus E3 protein. Virus Res. 130:269–274 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 53. Willis KL, Langland JO, Shisler JL. 2011. Viral double-stranded RNAs from vaccinia virus early or intermediate gene transcripts possess PKR activating function, resulting in NF-κB activation, when the K1 protein is absent or mutated. J. Biol. Chem. 286:7765–7778 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 54. Willis KL, Patel S, Xiang Y, Shisler JL. 2009. The effect of the vaccinia K1 protein on the PKR-eIF2α pathway in RK13 and HeLa cells. Virology 394:73–81 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 55. Wyatt LS, Earl PL, Eller LA, Moss B. 2004. Highly attenuated smallpox vaccine protects mice with and without immune deficiencies against pathogenic vaccinia virus challenge. Proc. Natl. Acad. Sci. U. S. A. 101:4590–4595 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 56. Yu Q, Hu N, Ostrowski M. 2009. Poxvirus tropism for primary human leukocytes and hematopoietic cells. Methods Mol. Biol. 515:309–328 [DOI] [PubMed] [Google Scholar]