

Abstract
Hepatitis B virus (HBV) is a hepatotropic virus that can cause severe liver diseases. By conducting studies using four different transgenic mouse lines that carry either the wild-type HBV genome or the HBV genome incapable of expressing the X gene, we found that liver injury and regeneration induced by a partial hepatectomy (PHx) could have different effects on HBV replication depending on the mouse lines. Further studies using hydrodynamic injection to introduce different amounts of the HBV genomic DNA into the mouse liver revealed that liver injury and regeneration induced by PHx enhanced HBV replication when viral load was low and suppressed HBV replication when viral load was high. These effects of liver injury and regeneration on HBV were independent of the HBV X protein and apparently due to alpha and beta interferons (IFN-α/β), as the effects could be abolished by the injection of anti-IFN-α/β antibodies. Further analysis indicated that PHx could induce the expression of hepatocyte nuclear factor 3 gamma (HNF3γ) when viral load was low and activate the signal transducer and activator of transcription 3 (Stat3) and suppress the expression of the suppressor of cytokine signaling 3 (SOCS3) irrespective of viral load. As both HNF3γ and Stat3 are required to activate the HBV enhancer I to stimulate HBV gene expression and replication, these results provided an explanation to the viral-load-dependent effect of liver injury and regeneration on HBV replication. Our studies thus revealed a novel interaction between HBV and its host and provided important information for understanding HBV replication and pathogenesis during liver injury.
INTRODUCTION
Hepatitis B virus (HBV) is an important human pathogen that chronically infects approximately 350 million people worldwide. This virus is a hepatotropic virus and can cause acute and chronic hepatitis, liver cirrhosis, and hepatocellular carcinoma. HBV is a small DNA virus with a genome size of about 3.2 kb. This genome is partially double stranded and contains four genes. The S gene codes for the three viral envelope proteins collectively called surface antigens (HBsAgs). The C gene codes for the core protein and a related protein termed the precore protein, which is the precursor of the e antigen (HBeAg) detected in the sera of HBV patients. The P gene codes for the viral DNA polymerase, which is also a reverse transcriptase, and the X gene codes for a regulatory protein. After the infection of hepatocytes, the HBV genome is delivered into the nucleus, where it is converted to a covalently closed circular DNA (cccDNA). This cccDNA then directs the transcription of viral RNAs, which is controlled by two enhancer elements and four different promoters. The HBV core protein mRNA is larger than the genome size. This RNA, which is also termed the pregenomic RNA (pgRNA), is packaged by the core protein to form the core particle. The pgRNA is subsequently converted to the partially double-stranded genome by the viral DNA polymerase that is also packaged. The core particle subsequently interacts with surface antigens in intracellular membranes for the maturation of the virion, which is then released from cells (for a review, see reference 15).
HBV is not cytopathic, but its infection of hepatocytes can induce immune responses to cause liver injury and regeneration. The liver regeneration is a dynamic volume recovery process that can be triggered by a volume loss caused by immune responses to viral infections, partial hepatectomy (PHx), partial-graft liver transplantation, drug injury, or steatosis (3, 4, 10). During liver regeneration, multiple cytokines, including tumor necrosis factor alpha (TNF-α), lymphotoxin, and interleukin 6 (IL-6), are induced, leading to activation of the signal transducer and activator of transcription 3 (Stat3), a transcription factor, and the entry of quiescent hepatocytes into cell cycles (13). Stat3 can also be activated by interferons and their downstream Janus kinases (Jaks) (14). Its activation is followed by the induction of its negative regulator, the suppressor of cytokine signaling 3 (SOCS3) (1, 2, 9, 11, 12, 18). The liver regeneration process ceases when the liver regains its volume.
The studies on HBV replication in vivo have been hampered by the lack of a convenient animal model. However, the development of HBV transgenic mice that carry a 1.3-mer overlength HBV genome and the use of hydrodynamic injection, which is a procedure to transfect hepatocytes with the HBV DNA in vivo, have partially overcome this problem. HBV transgenic mice carrying the overlength genome express viral genes preferentially in the liver. This results in the replication of HBV DNA and the release of mature HBV particles into the blood. Due to immune tolerance, HBV transgenic mice do not have liver inflammation and thus resemble asymptomatic HBV carriers (7). In the hydrodynamic injection, a large volume of saline containing the HBV genomic DNA is injected into mice in a relatively short period of time. This results in the transfection of hepatocytes by the HBV DNA, which will then direct HBV gene expression and viral replication in vivo (21). Thus, the hydrodynamic injection creates a scenario that resembles acute HBV infection.
Previous studies indicated that the liver injury and regeneration induced by a PHx can suppress HBV replication in transgenic mice via a posttranscriptional mechanism (6). We had conducted similar studies using HBV transgenic mice that carried the 1.3-mer overlength HBV genome with or without the ability to express the HBV X protein (HBx) to investigate how liver injury and regeneration may affect HBV replication. To our surprise, we found that the effect of liver injury and regeneration on HBV replication was mouse line dependent. Our further analyses using hydrodynamic injection indicated that, depending on viral load, liver injury and regeneration induced by PHx could induce the expression of the transcription factor hepatocyte nuclear factor 3 gamma (HNF3γ) and positively or negatively regulate HBV replication. In addition, we found that the activation of Stat3 in the liver after a PHx was prolonged in HBV transgenic mice in a viral-load-independent manner, possibly due to the inhibition of expression of SOCS3 by HBV. Interestingly, in spite of the prolonged activation of Stat3, the liver regeneration rate after a PHx was not affected by HBV.
MATERIALS AND METHODS
HBV transgenic mice and DNA plasmids.
The HBV transgenic mouse lines Tg05, Tg08, Tg31, and Tg38 have been previously described (17, 20). The Tg05 and Tg08 mouse lines carried the 1.3-mer overlength wild-type HBV genome, and Tg31 and Tg38 also carried the 1.3-mer HBV genome, with the exception that the expression of the X protein (HBx) was abolished by an A-to-C missense mutation at nucleotide (nt) 1377 that removed the translation initiation codon and a C-to-T nonsense mutation at nt 1398 that disrupted the HBx coding sequence. All of the experiments were conducted using age-matched male mice with the C57BL/6 genetic background. The plasmid p1.3×HBV, which contains the 1.3-mer overlength HBV genome, has been described before (22).
Antibodies.
Rabbit anti-mouse alpha interferon (IFN-α) (PBL, New Jersey) and hamster anti-mouse IFN-β (Biolegend, San Diego) antibodies were used in this study. Purified rabbit IgG (Cell Signaling Technology) and hamster IgG (Abcam) were used as the control antibodies. Rabbit anti-HNF3α (Abcam), anti-HNF3β (Cell Signaling Technology), anti-HNF3γ (Sigma-Aldrich), anti-Stat3 (Cell Signaling Technology), anti-phosph-Stat3 (Tyr705) (Cell Signaling Technology), anti-SOCS3 (Cell Signaling Technology), anti-α-actin (Abcam), and anti-lamin-β (Abcam) antibodies were used for Western blot analyses. The rabbit anti-PCNA antibody (Abcam) was used for immunohistochemistry staining.
Partial hepatectomy and poly(I·C) injection.
A 70% partial hepatectomy (PHx) was performed on 9-week-old male mice as described by Greene and Puder (5). Briefly, male HBV transgenic mice with matched HBeAg levels were weighed prior to the surgery. The midline laparotomy was then performed, and right medial, left medial, and left lateral lobes of the liver were removed. The resected-liver weight was also measured. Mice were sacrificed at multiple time points after surgery, and the regenerating liver was harvested and used for the weight measurement, histological analyses, and other studies. The mouse serum was collected prior to the PHx and at the time when mice were sacrificed. For the injection of poly(I·C), HBV mice with matched HBeAg levels or age-matched male naïve mice were injected with saline with or without 200 μg poly(I·C) and sacrificed a day later for the isolation of the liver for a Western blot analysis.
Hydrodynamic injection of HBV DNA.
The 1.3-mer overlength HBV genomic DNA in the pUC19 vector was hydrodynamically injected into the tail vein of 9-week-old male C57BL/6 mice using our previous procedures (17). Two sets of mice were used for injection, with one set subjected to a PHx and the other set subjected to a sham operation to serve as the control. In all of the injections, the control vector pUC19 was included if necessary to ensure that the total amounts of DNA used for injection were identical among different mice. The Gaussia luciferase (Gluc) reporter pCMV-Glu was also included in the injection to monitor the injection efficiency. Twenty-four hours after the hydrodynamic injection, the serum was collected and Gluc and HBeAg were assayed by an enzyme-linked immunosorbent assay (ELISA). Mice with matched Gluc and HBeAg levels were used as a pair for the sham operation and PHx.
Immunohistochemistry.
Paraffin-embedded liver tissue sections were stained for the proliferating cell nuclear antigen (PCNA). Briefly, liver tissue sections were treated with 0.01 M sodium citrate in a microwave oven for 10 min for epitope retrieval and then blocked using the goat serum. Tissue sections were incubated with the primary rabbit anti-PCNA antibody and the secondary biotinylated affinity-purified goat anti-rabbit IgG, and the staining was then developed using the avidin-conjugated horseradish peroxidase (HRP) with diaminobenzidine (DAB) as the substrate (ultrasensitive ABC peroxidase staining kit; Thermo).
Southern, Northern, and Western blot analyses.
Total liver DNA and RNA were isolated as described before (17). The HBV DNA replicative intermediates (RI) in the core particles were isolated from liver homogenates after the treatment with micrococcal nuclease and DNase I using our previous protocol (22). Both Southern and Northern blot analyses were conducted using the 32P-labeled HBV DNA probe. A Western blot was performed using our previous procedures (16).
Real-time PCR analysis of serum HBV DNA.
The HBV DNA in the mouse serum was measured by real-time PCR as previously described (19). Briefly, 10 μl serum was treated with 10 μg DNase I and micrococcal nuclease for 30 min at 37°C for the removal of free DNA. The virion-associated HBV DNA was then isolated by proteinase K digestion, followed by the phenol-chloroform extraction (19). For HBV DNA real-time PCR analysis, the following primers were used: forward primer, 1552-CCGTCTGTGCCTTCTCATCTG-1572; reverse primer, 1667-AGTCCTCTTATGTAAGACCTT-1646. The TaqMan probe used was 1578-CCGTGTGCACTTCGCTTCACCTCTGC-1603. The thermal cycling was conducted using the ABI7500 system (Applied Biosystems) with the following parameters: 2 min at 50°C, 10 min at 95°C, and 40 cycles of 15 s at 95°C and 60 s at 60°C.
RESULTS
Mouse line-dependent effect of liver regeneration on HBV replication.
To investigate the effect of liver injury and regeneration on the replication of HBV, we performed a 70% PHx on four different HBV transgenic mouse lines that carried either the wild-type HBV genome or the HBV genome incapable of expressing HBx. Mice were sacrificed at different time points after the PHx for the isolation of the liver. The mouse liver resected during the surgery was used as the day 0 control. The HBV DNA replicative intermediates (RI) were isolated from the liver and analyzed by a Southern blot. As shown in Fig. 1A, the level of the HBV RI DNA in Tg05 mice reduced between day 2 and day 7 after the PHx, most notably on day 3 and day 5. Similar results were obtained when HBV titers in the serum were quantified by real-time PCR, with the HBV titer reduced to the lowest level on day 5, and the HBV titer then returned to approximately the presurgery level on day 10 when the liver was fully regenerated (see below). The analysis of the HBV core protein level in the mouse liver by a Western blot revealed a similar reduction of the core protein level between day 2 and day 7. However, the analysis of HBV RNAs revealed no reduction of their levels throughout the studied period. These results were consistent with the previous report, which suggested that the suppressive effect of liver regeneration on HBV DNA replication was a posttranscriptional event (6).
Fig 1.

Effect of a PHx on HBV replication in transgenic mice. Nine-week-old, serum HBeAg-matched male mice were subjected to a 70% PHx and sacrificed at the time points indicated. The liver resected from an individual mouse served as the day 0 (D0) control of that particular mouse. HBV RI DNA, RNAs, and the core protein in the mouse liver were then analyzed by a Southern blot (top panel), a Northern blot (second panel from the top), and a Western blot (fourth panel from the top), respectively. The HBV transgene in the mouse genome was used as the loading control for the Southern blot analysis. 28S and 18S rRNAs were used as the loading controls for the Northern blot analysis (third panel from the top), and α-actin was used as the control for the Western blot (bottom panel). The HBV titers in the serum before (gray bars) and after (white bars) the PHx were measured by real-time PCR and are shown in the histograms. (A) Tg05 mouse line; (B) Tg38 mouse line; (C) Tg08 mouse line; (D) Tg31 mouse line.
When the same experiments were conducted on Tg38 mice, there was a slight reduction of the HBV RI DNA level in the liver between day 2 and day 7 (Fig. 1B). However, there was no apparent difference in serum HBV titers and the liver HBV RNA and core protein levels at any time point after the PHx (Fig. 1B). These results indicated that the liver regeneration had only a marginal effect on HBV in this mouse line. Interestingly, when the same experiments were conducted on the Tg08 and Tg31 mouse lines, liver regeneration increased the HBV RI DNA, RNA, and core protein levels in the liver and the HBV DNA level in the serum between day 2 and day 7 after the PHx (Fig. 1C and D). Thus, the results shown in Fig. 1 indicate that PHx and liver regeneration may have different effects on HBV depending on the mouse lines. They also indicated that these effects of PHx and liver regeneration on HBV were not caused by the HBx protein, as Tg05 and Tg08 mice both contained the wild-type HBV genome but HBV in these two mouse lines had opposite responses to the PHx. Similarly, both the Tg31 and Tg38 mouse lines contained the X-null HBV genome but HBV in these two mouse lines also responded differently to the PHx.
Viral-load-dependent effect of partial hepatectomy on HBV replication.
The HBV transgenic mouse lines Tg05, Tg08, Tg31, and Tg38 produced approximately 5.7 × 108, 5.2 × 105, 2.7 × 106, and 3.4 × 107 genome copies of HBV per ml of serum, respectively (Fig. 2) (also see reference 17). Since a PHx had a strong suppressive effect on HBV in Tg05 mice, which produced the highest level of HBV, a marginal suppressive effect on HBV in Tg38 mice, which produced a moderate level of HBV, and a strong stimulating effect on HBV in Tg08 and Tg31 mice, which produced low levels of HBV, the effect of a PHx on HBV replication appeared to be inversely correlated with the viral titers. To test this possibility, we decided to perform the hydrodynamic injection to introduce different amounts of the 1.3-mer overlength HBV genomic DNA into the liver of naïve mice using our previous procedures (17). Mice injected with the HBV DNA were then subjected to a PHx 1 day after DNA injection and sacrificed 3 days after the PHx for the isolation of the liver for the analysis of HBV RI DNA, RNAs, and the core protein. As shown in Fig. 2, the PHx enhanced HBV DNA replication when the amount of HBV genomic DNA used for the injection was 14 μg or less. However, the PHx suppressed HBV DNA replication when the amount of the HBV genomic DNA used for injection was 20 μg or higher. Similar results were observed when HBV RNAs and the core protein were analyzed: the PHx increased HBV RNA and core protein levels when the amount of HBV DNA used for injection was 14 μg or lower, but it decreased HBV RNA levels, particularly the C gene transcripts, and the core protein level when the amount of HBV DNA used for injection was 20 μg or higher (Fig. 2). Note that the injection of 32 μg HBV DNA did not increase and, rather, reduced HBV RI DNA and RNA levels in the mouse liver compared to levels following the injection of 24 μg HBV DNA. This observation is consistent with our previous result and might be due to the reduction of the transfection efficiency when too much HBV DNA was used in the hydrodynamic injection (17). In any case, the results shown in Fig. 2 demonstrated that the effect of a PHx on HBV replication was indeed dependent on viral load.
Fig 2.
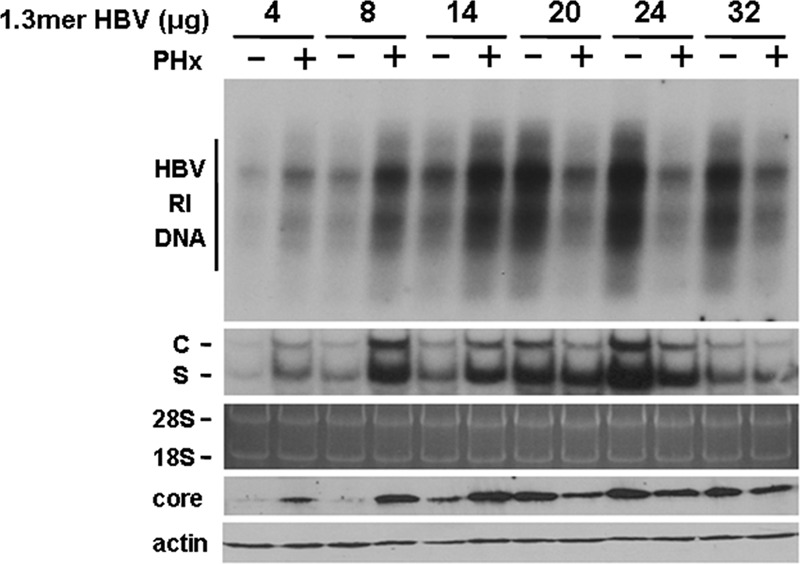
Viral-load-dependent effects of a PHx on HBV in mice. The hydrodynamic injection was used to introduce different amounts of the 1.3-mer HBV genomic DNA into the mouse liver. A PHx was performed on the mice 1 day after the DNA injection, and all of the mice were sacrificed 3 days after the PHx for the isolation of the liver for the analysis of the HBV RI DNA, RNAs, and the core protein as described in the Fig. 1 legend. −, with sham operation; +, with PHx.
Effects of a PHx on HBV, mediated by IFN-α/β.
The observation that a PHx enhanced HBV replication when viral load was low and suppressed HBV replication when viral load was high was reminiscent of our recent finding that alpha and beta interferons (IFN-α/β) could suppress or enhance HBV replication depending on the viral load. As liver injury may induce the expression of cytokines, including interferons, we first determined whether a PHx can also induce the interferon response in the liver of our HBV transgenic mice. As shown in Fig. 3A, an increase of the RNA level of 2′,5′-oligoadenylate synthetase (2′,5′-OAS), an interferon-stimulated gene, was observed in the liver 1 day after the PHx, indicating the induction of the interferon response. We next determined whether the effects of the PHx on HBV were mediated by IFN-α/β. Three transgenic mouse lines, Tg05, Tg08, and Tg31, which generated different HBV responses to the PHx, were injected with the control IgG or the anti-IFN-α/β antibodies. As shown in Fig. 3B, the suppressive effect of the PHx on HBV DNA replication in Tg05 mice, which produced a high level of HBV, was not affected by the control IgG (lane 1 versus lane 2), but it was abolished by the anti-IFN-α/β antibodies (lane 3 versus lane 4). Similar to the results shown in Fig. 1A, the PHx had only a marginal effect on HBV RNA but significantly reduced the HBV core protein level in Tg05 mice (Fig. 3B, lane 1 versus lane 2). These effects on HBV RNA and the core protein were also abolished by the anti-IFN-α/β antibodies. These results indicated that the effect of the PHx on HBV in Tg05 mice was indeed mediated by IFN-α/β. We also conducted the same study using Tg08 and Tg31 mice, which expressed low levels of HBV. As shown in the same figure, the control IgG did not prevent the PHx from increasing HBV DNA, RNA, and core protein levels in these two mouse lines (Fig. 3B, lane 5 versus lane 6 and lane 11 versus lane 12). However, this increase was abolished by the anti-IFN-α/β antibodies (Fig. 3B, lane 7 versus lane 8 and lane 9 versus lane 10). These results again demonstrated that the enhancement effect of a PHx on HBV in Tg08 and Tg31 mice was mediated by IFN-α/β.
Fig 3.

Effects of a PHx on HBV, mediated by IFN-α/β. (A) Induction of 2′,5′-OAS expression by a PHx. Total liver RNA was extracted from mice 1 day (D1) after the PHx for the analysis of the 2′,5′-OAS mRNA by semiquantitative reverse transcription-PCR (RT-PCR). The resected liver was used as the day 0 (D0) control. GAPDH (glyceraldehyde-3-phosphate dehydrogenase) mRNA was also analyzed to serve as an internal control of RT-PCR. (B) Effects of anti-IFN-α/β antibodies and a PHx on HBV. Mice were injected with 200 μg of the control IgG or the anti-IFN-α/β antibodies as indicated, followed by a PHx. Mice were sacrificed 3 days after the PHx for the isolation of the liver for HBV RI DNA, RNA, and core protein analyses. Note that the order of samples in lanes 9 to 12 is slightly different from that in lanes 1 to 8.
Effects of HBV on the expression of HNF3γ, Stat3, and SOCS3 in the mouse liver after a PHx.
Our recent studies indicated that IFN-α/β can induce the expression of HNF3γ and activate Stat3 in the liver of HBV transgenic mice that produced a low level of HBV (17). These two transcription factors are both required to mediate the effects of IFN-α/β to activate HBV enhancer I for the stimulation of HBV gene expression and viral replication (17). To test whether a PHx and the ensuing liver regeneration could also induce the expression of HNF3γ and activate Stat3, we performed a Western blot analysis on these two protein factors 1 day after a PHx. As shown in Fig. 4A, although the PHx had no effect on the protein levels of HNF3α, HNF3β, and lamin-β in the liver of all four HBV transgenic mouse lines, it increased the expression level of HNF3γ in only Tg08 and Tg31 mice, which produced low levels of HBV. When Stat3 was analyzed, the PHx was not found to affect the overall expression level of this protein. However, it was found to activate Stat3, as evidenced by the significant increase of the phospho-Stat3 level, in all four HBV transgenic mouse lines (Fig. 4B). The PHx had only a marginal effect on the activation of Stat3 in the control, nontransgenic mice.
Fig 4.

Analysis of the effects of a PHx on HNF3 and Stat3. HBV transgenic mice were subjected to a PHx and sacrificed the next day (D1) for the isolation of the liver. The resected liver was used as the day 0 (D0) control. (A) Western blot analysis for the expression of the three HNF3 isoforms HNF3α, HNF3β, and HNF3γ. Lamin-β was also analyzed to serve as a loading control. (B) Western blot analysis of Stat3 and phosphor-Stat3 (Y705) in the liver of naïve control mice and HBV transgenic mice. The asterisk denotes a nonspecific band which was not always observed in our experiments. The α-actin protein was also analyzed to serve as the loading control.
To understand why Stat3 was significantly activated in HBV transgenic mice after a PHx, we analyzed the expression of SOCS3, a negative regulator of Stat3 (2, 12). We first analyzed the expression level of SOCS3 in the liver of naïve mice. As shown in Fig. 5A, in agreement with the previous report (2), the expression of SOCS3 was slightly elevated 1 day after the PHx in naïve mice. In contrast, the expression of SOCS3 in the liver of all four HBV transgenic mouse lines was reduced to an almost-undetectable level 1 day after the PHx (Fig. 5A). As SOCS3 is the negative regulator of Stat3, its loss of expression in the transgenic mouse liver after the PHx provided an explanation for the activation of Stat3 in the liver of these HBV transgenic mice.
Fig 5.
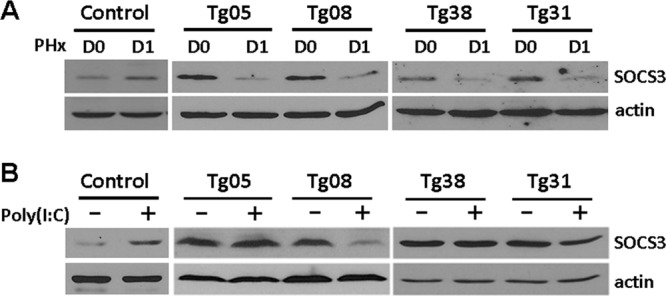
Analysis of the effect of a PHx on the expression of SOCS3. (A) Western blot analysis of SOCS3 in the liver of naïve control mice and HBV transgenic mice 1 day after the PHx. (B) Western blot analysis of SOCS3 in the liver of naïve control mice and HBV transgenic mice after poly(I·C) injection. For poly(I·C) injection, HBV mice with matched HBeAg levels were injected with saline (−) or poly(I·C) (+) and sacrificed a day later for the isolation of the liver for a Western blot analysis.
As our previous studies indicated that poly(I·C), which can induce the interferon response, activated Stat3 in the liver of Tg08 and Tg31 mice, which produced low levels of HBV, but not in the liver of Tg05 and Tg38 mice, which produced high and moderate levels of HBV, respectively (17), we also determined the possible effect of poly(I·C) on SOCS3 expression in the liver of these HBV transgenic mouse lines. Again, we first analyzed the effect of poly(I·C) on the expression of SOCS3 in control naïve mice. As shown in Fig. 5B, poly(I·C) increased the SOCS3 expression level in the control mouse liver. However, it had no effect on SOCS3 in Tg05 and Tg38 mice and reduced its expression level in Tg08 and Tg31 mice. These results indicated that HBV can inhibit the SOCS3 expression induced by poly(I·C) in mice that produced low levels of HBV. The inverse relationship between the expression levels of SOCS3 and the levels of activated Stat3 after the poly(I·C) treatment is again consistent with a negative role of SOCS3 in the activation of Stat3 in HBV transgenic mice.
No apparent effect of HBV on liver regeneration and hepatocellular proliferation.
Stat3 is transiently activated after a PHx for the entry of quiescent hepatocytes into cell cycles for liver regeneration. Its activation peaked at 2 h after the PHx and became almost undetectable after 8 h (2, 12). The ablation of SOCS3 expression in hepatocytes has been show to prolong the activation of Stat3 and accelerate liver regeneration (2, 12). The observation that HBV can suppress the expression of SOCS3 and prolong the activation of Stat3 prompted us to investigate whether HBV can affect liver regeneration after a PHx. HBV transgenic mice and control naïve mice with the same genetic background were subjected to a 70% PHx and sacrificed at different time points after surgery to measure the liver weight. As shown in Fig. 6A, the mouse liver underwent rapid regeneration and reached approximately 100% of the original liver weight 10 days after the surgery. There was no statistically significant difference (P > 0.05) in the liver regeneration rates at any time point after the PHx between control mice and any of the HBV transgenic mouse lines, whether they carried the wild-type HBV genome or the X-null HBV genome. To further compare the hepatocellular proliferation rates among different mouse groups, liver tissue sections were stained for PCNA, a nuclear protein associated with the cell proliferation, at different time points after the PHx. The representative staining results of one of the HBV transgenic mouse lines are shown in Fig. 6B, and the percentages of PCNA-positive cells of different mouse groups at different time points after the PHx are shown in Fig. 6C. The statistical analysis of PCNA-positive cells at different time points after the PHx revealed no significant difference among different mouse groups: there were few PCNA-positive hepatocytes on day 0, more than 80% PCNA-positive hepatocytes on day 3, and approximately 60% and 30% PCNA-positive hepatocytes on day 7 and day 10, respectively. These results indicated that in spite of its ability to inhibit SOCS3 expression and prolong Stat3 activation, HBV did not affect liver regeneration and hepatocellular proliferation in mice, whether or not the HBx protein was expressed.
Fig 6.

(A) Liver regeneration rates of control and HBV transgenic mice. Age- and sex-matched HBV transgenic mice and control, nontransgenic mice were subjected to a 70% PHx and sacrificed at the indicated time points for liver weight measurements. Three mice were analyzed per time point, and the graph shows the means. The average liver weight prior to PHx was defined as 100%. (B) PCNA staining of the regenerating mouse liver. Only the staining result of the HBV transgenic mouse line Tg05 is shown. However, other HBV transgenic mouse lines and the control mice showed similar results. (C) Percentage of PCNA-positive cells. No statistical difference was observed between control mice and any of the HBV transgenic mouse lines.
DISCUSSION
Guidotti et al. previously showed that liver injury and regeneration induced by a PHx can inhibit HBV replication in transgenic mice (6). By conducting a similar study using four different HBV transgenic mouse lines, we now demonstrate that the effect of liver injury and regeneration on HBV replication is actually dependent on mouse lines. A PHx suppressed HBV replication in mice that produced a high level of HBV, marginally suppressed HBV replication in mice that produced a moderate level of HBV, and enhanced HBV replication in mice that produced a low level of HBV (Fig. 1). In their previous studies, Guidotti et al. (6) used transgenic mice that produced a high level of HBV (>108 genome copies/ml serum), and that was probably the reason why they observed only the suppression of HBV replication by a PHx. The viral-load-dependent effect of a PHx on HBV replication was confirmed by our hydrodynamic injection experiments, in which we found that a PHx enhanced the replication of HBV in mice injected with 14 μg or less of the HBV genomic DNA and inhibited the replication of HBV in mice injected with 20 μg or more of the HBV DNA (Fig. 2). Our studies further indicated that the effects of a PHx on HBV were mediated by IFN-α/β, as these effects were able to be abolished by the antibodies directed against IFN-α/β (Fig. 3). These findings were consistent with those of our previous studies, which demonstrated that IFN-α/β, and IFN-β in particular, could positively or negatively regulate HBV replication in a viral-load-dependent manner (17).
Guidotti et al. showed that a PHx suppressed HBV DNA replication at a posttranscriptional step, possibly by inhibiting the formation or facilitating the degradation of viral core particles (6). Our studies with the Tg05 mouse line, which produced a high level of HBV, revealed a similar result (Fig. 1A and B). We found that a PHx and liver regeneration did not affect HBV RNA levels. However, it reduced the core protein level as well as the HBV DNA level in both the liver and serum. This result is consistent with a posttranscriptional suppressive mechanism on HBV replication. Previously, we found that the injection of poly(I·C) to induce the interferon response reduced the HBV RNA level in Tg05 mice (17). As the HBV RNA level was not affected by a PHx, additional factors, such as other cytokines induced by a PHx, might be involved in the regulation of HBV RNA transcription and/or stability. Note that a slight reduction of HBV RNA levels by a PHx was observed in naïve mice injected with 20, 24, or 32 μg HBV genomic DNA (Fig. 2). This reduction might be due to the induction of immune responses and the production of inflammatory cytokines, as, unlike HBV transgenic mice, naïve mice are not immunotolerant to HBV introduced by the hydrodynamic injection.
In contrast to HBV transgenic mouse lines that produced a high or moderate level of HBV, a PHx enhanced HBV replication in mouse lines that produced a low level of HBV, apparently by stimulating viral RNA transcription, since the increase of viral DNA replication was accompanied by a concomitant increase of viral RNA and core protein levels (Fig. 1C and D and 2). These results are consistent with our previous finding that IFN-α/β stimulated HBV replication when viral load was low by activating enhancer I of the HBV genome to enhance HBV gene expression (17). IFN-α/β accomplished this task by inducing the expression of HNF3γ and activating Stat3, both of which then bind cooperatively to enhancer I in the HBV genome (17). Similarly, a PHx induced the expression of HNF3γ in mice that produced a low level of HBV but not in mice that produced a high or moderate level of HBV (Fig. 4A). This induction of HNF3γ by a PHx in mice that produced a low level of HBV may explain why the PHx increased the HBV RNA levels and enhanced HBV replication only in these low-HBV-producing mouse lines.
Unlike interferons, which activated Stat3 only in the presence of a low level of HBV (17), a PHx activated Stat3 in all four of our HBV transgenic mouse lines (Fig. 4B). Interestingly, the activation of Stat3 was associated with the loss of SOCS3 expression in HBV transgenic mice after the PHx (Fig. 5B). SOCS3 is a gene that is activated by Stat3. It inhibits the activation of Stat3 by disrupting its upstream signaling pathway and provides a negative feedback regulation for Stat3 (2, 12). Thus, its loss of expression may explain the prolonged activation of Stat3 in HBV transgenic mice after the PHx. It is unclear how HBV inhibited the expression of SOCS3 after the PHx. This inhibition does not require HBx, since Tg31 and Tg38 mice carried the X-null HBV genome. We had examined the mechanism of this inhibition by injecting HBV transgenic mice with poly(I·C). We found that poly(I·C) increased the liver expression level of SOCS3 in control mice, but it was not able to do so in Tg05 and Tg38 mice and, on the contrary, reduced the SOCS3 expression in Tg08 and Tg31 mice (Fig. 5B). Thus, the inhibition of SOCS3 expression in HBV transgenic mice that produced a low level of HBV (i.e., Tg08 and Tg31 mice) after a PHx might be due to the effect of IFN-α/β. The reason why a PHx could also inhibit the expression of SOCS3 in Tg05 and Tg38 mice, which produced high and moderate levels of HBV, respectively, but poly(I·C) could not is unclear. It might be because in mice that produced a high or moderate level of HBV, HBV gene products, such as the polymerase, which has been shown to interfere with the interferon signaling pathway (19), interfered with the Jak-Stat signaling pathway and prevented the activation of Stat3 and the induction of SOCS3. This interference, however, is overcome by a PHx, which induced the expression of additional cytokines to activate other signaling pathways. Clearly, there is an interesting interplay between HBV and the Stat3-SOCS3 regulatory circuit, and further research in this area will likely generate very interesting results.
Stat3 is transiently activated after liver injury and is important for the entry of quiescent hepatocytes into cell cycles (2, 12). The level of activated Stat3 peaked at 2 h after the PHx and reduced to the background level by 8 h after the PHx (2). Ablating the expression of SOCS3 in mouse hepatocytes has been shown to prolong the activation of Stat3 and accelerate hepatocellular proliferation and liver regeneration (12). Curiously, in spite of the ability of HBV to inhibit the expression of SOCS3 and prolong the activation of Stat3 after the PHx, HBV had no apparent effect on the hepatocellular proliferation rate and liver regeneration (Fig. 6). Nevertheless, as Stat3 has been shown to play an important role in promoting hepatocarcinogenesis (8), it is conceivable that its prolonged activation during chronic liver injury caused by HBV infection may contribute to the development of hepatocellular carcinoma in HBV patients.
In conclusion, by using HBV transgenic mice as a model and PHx to induce liver injury, we demonstrated that liver injury and regeneration could have positive and negative effects on HBV replication depending on the viral load. In addition, we demonstrated that HBV can perturb the Stat3-SOCS3 regulatory circuit, which may have important consequences in HBV pathogenesis and carcinogenesis during chronic HBV infection in patients.
ACKNOWLEDGMENTS
We thank Michelle MacVeigh-Aloni and the Histology subcore of the USC Research Center for Liver Diseases for help with the PCNA staining.
This work was supported by the Public Health Service grant P01CA123328 from the National Cancer Institute.
Footnotes
Published ahead of print 20 June 2012
REFERENCES
- 1. Akerman P, et al. 1992. Antibodies to tumor necrosis factor-alpha inhibit liver regeneration after partial hepatectomy. Am. J. Physiol. 263:G579–G585 [DOI] [PubMed] [Google Scholar]
- 2. Campbell JS, et al. 2001. Expression of suppressors of cytokine signaling during liver regeneration. J. Clin. Invest. 107:1285–1292 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3. Fausto N, Campbell JS, Riehle KJ. 2006. Liver regeneration. Hepatology 43:S45–S53 [DOI] [PubMed] [Google Scholar]
- 4. Fujiyoshi M, Ozaki M. 2011. Molecular mechanisms of liver regeneration and protection for treatment of liver dysfunction and diseases. J. Hepatobiliary Pancreat. Sci. 18:13–22 [DOI] [PubMed] [Google Scholar]
- 5. Greene AK, Puder M. 2003. Partial hepatectomy in the mouse: technique and perioperative management. J. Investig. Surg. 16:99–102 [PubMed] [Google Scholar]
- 6. Guidotti LG, Matzke B, Chisari FV. 1997. Hepatitis B virus replication is cell cycle independent during liver regeneration in transgenic mice. J. Virol. 71:4804–4808 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7. Guidotti LG, Matzke B, Schaller H, Chisari FV. 1995. High-level hepatitis B virus replication in transgenic mice. J. Virol. 69:6158–6169 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8. He G, Karin M. 2011. NF-κB and STAT3—key players in liver inflammation and cancer. Cell Res. 21:159–168 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9. Iwai M, Cui TX, Kitamura H, Saito M, Shimazu T. 2001. Increased secretion of tumour necrosis factor and interleukin 6 from isolated, perfused liver of rats after partial hepatectomy. Cytokine 13:60–64 [DOI] [PubMed] [Google Scholar]
- 10. Michalopoulos GK, DeFrances MC. 1997. Liver regeneration. Science 276:60–66 [DOI] [PubMed] [Google Scholar]
- 11. Pahlavan PS, Feldmann RE, Jr, Zavos C, Kountouras J. 2006. Prometheus' challenge: molecular, cellular and systemic aspects of liver regeneration. J. Surg. Res. 134:238–251 [DOI] [PubMed] [Google Scholar]
- 12. Riehle KJ, et al. 2008. Regulation of liver regeneration and hepatocarcinogenesis by suppressor of cytokine signaling 3. J. Exp. Med. 205:91–103 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13. Riehle KJ, Dan YY, Campbell JS, Fausto N. 2011. New concepts in liver regeneration. J. Gastroenterol. Hepatol. 26(Suppl 1):203–212 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14. Schindler C, Plumlee C. 2008. Inteferons pen the JAK-STAT pathway. Semin. Cell Dev. Biol. 19:311–318 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15. Seeger C, Zoulim F, Mason WS. 2007. Hepadnaviruses, p 2977–3029 In Knipe DM, Howley PM. (ed), Fields virology, 5th ed Lippincott Williams & Wilkins, Philadelphia, PA [Google Scholar]
- 16. Sir D, et al. 2010. The early autophagic pathway is activated by hepatitis B virus and required for viral DNA replication. Proc. Natl. Acad. Sci. U. S. A. 107:4383–4388 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17. Tian Y, Chen WL, Ou JH. 2011. Effects of interferon-alpha/beta on HBV replication determined by viral load. PLoS Pathog. 7:e1002159 doi:10.1371/journal.ppat.1002159 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18. Trautwein C, Rakemann T, Niehof M, Rose-John S, Manns MP. 1996. Acute-phase response factor, increased binding, and target gene transcription during liver regeneration. Gastroenterology 110:1854–1862 [DOI] [PubMed] [Google Scholar]
- 19. Wang H, Ryu WS. 2010. Hepatitis B virus polymerase blocks pattern recognition receptor signaling via interaction with DDX3: implications for immune evasion. PLoS Pathog. 6:e1000986 doi:10.1371/journal.ppat.1000986 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20. Xu Z, et al. 2002. Enhancement of hepatitis B virus replication by its X protein in transgenic mice. J. Virol. 76:2579–2584 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21. Yang PL, Althage A, Chung J, Chisari FV. 2002. Hydrodynamic injection of viral DNA: a mouse model of acute hepatitis B virus infection. Proc. Natl. Acad. Sci. U. S. A. 99:13825–13830 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22. Zheng Y, Li J, Johnson DL, Ou JH. 2003. Regulation of hepatitis B virus replication by the ras-mitogen-activated protein kinase signaling pathway. J. Virol. 77:7707–7712 [DOI] [PMC free article] [PubMed] [Google Scholar]