

Abstract
Human cytomegalovirus (HCMV) virions are structurally complex, and the mechanisms by which they are assembled are poorly understood, especially with respect to the cytoplasmic phase of assembly, during which the majority of the tegument is acquired and final envelopment occurs. These processes occur at a unique cytoplasmic structure called the assembly complex, which is formed through a reorganization of the cellular secretory apparatus. The HCMV tegument protein UL99 (pp28) is essential for viral replication at the stage of secondary envelopment. We previously demonstrated that UL99 interacts with the essential tegument protein UL94 in infected cells as well as in the absence of other viral proteins. Here we show that UL94 and UL99 alter each other's localization and that UL99 stabilizes UL94 in a binding-dependent manner. We have mapped the interaction between UL94 and UL99 to identify the amino acids of each protein that are required for their interaction. Mutation of these amino acids in the context of the viral genome demonstrates that HCMV is completely defective for replication in the absence of the interaction between UL94 and UL99. Further, we demonstrate that in the absence of their interaction, both UL94 and UL99 exhibit aberrant localization and do not accumulate at the assembly complex during infection. Taken together, our data suggest that the interaction between UL94 and UL99 is essential for the proper localization of each protein to the assembly complex and thus for the production of infectious virus.
INTRODUCTION
Human cytomegalovirus (HCMV) is a ubiquitous member of the betaherpesvirus family. HCMV infection is predominantly asymptomatic in healthy individuals but is a significant cause of morbidity and mortality in those with compromised immune function. Further, congenital HCMV infection can lead to severe and permanent disability in infected children and is the leading infectious cause of congenital birth defects in the United States (14).
HCMV encodes at least 25 tegument proteins that are incorporated into the virus particle during virion assembly (4, 11, 28). These tegument proteins are released into the host cell upon infection and thus are available to function immediately, prior to the onset of viral gene expression. However, many tegument proteins function in the late phase of infection and are essential for proper virion assembly and morphogenesis (1, 16, 20, 24, 25). Despite a growing number of reports identifying viral proteins that are essential for these processes, the molecular mechanisms by which HCMV particles are assembled remain poorly understood.
Many HCMV tegument and glycoproteins localize late in infection to a juxtanuclear structure unique to HCMV infection that is referred to as the assembly complex (AC). The AC is formed through a dramatic relocalization of various components of the cellular secretory apparatus and is thought to be the site of cytoplasmic tegumentation and particle envelopment (6, 9, 13, 18). While the formation of the AC is a well-documented phenomenon, the events that occur in the AC and that result in the formation of mature virus particles remain elusive. Although the mechanisms of virion assembly are not well understood, these processes are thought to be mediated largely by protein-protein interactions.
We and others have reported an interaction between the tegument proteins UL94 and UL99 (12, 15, 27). This interaction occurs during infection as well as in the absence of other viral proteins. UL94 and UL99 are herpesvirus core genes that are conserved among all members of the herpesvirus family. In addition to their conservation at the genetic level, the interaction between UL94 and UL99 homologs also appears to be conserved (7, 29). This high level of evolutionary conservation suggests that the interaction between these tegument proteins plays an important role in viral replication. However, it should be noted that some UL94 and UL99 homologs are not essential for the replication of other herpesviruses in cell culture (2, 3, 17), whereas deletion of UL94 or UL99 results in a complete block in HCMV replication at the stage of secondary envelopment (16, 24). Therefore, it is likely that the function(s) of UL94 and UL99 during HCMV replication is different from or nonredundant with that of their homologs in other herpesviruses.
UL99 is essential for final envelopment of virions in the cytoplasm. In the absence of UL99, early events of virus replication, including viral gene expression and DNA replication, proceed normally. However, late in infection, partially tegumented, nonenveloped, DNA-containing nucleocapsids accumulate in the cytoplasm (24). Thus, a UL99-deletion mutant is incapable of producing infectious extracellular virus. We recently showed that the phenotype of a UL94-null virus is virtually identical to that of previously characterized UL99-deletion mutants (16). In addition to the absence of enveloped virions in the cytoplasm, we also demonstrated that UL99 exhibits aberrant localization in the absence of UL94 expression. This observation is consistent with the results of a previous study showing that the proper localization of UL99 to the AC during infection requires the function of another viral late gene (23). Based on these data, we hypothesize that the interaction between UL94 and UL99 is essential for the proper localization of UL99 to the AC and thus for HCMV replication.
To test this hypothesis, the amino acid residues of both UL94 and UL99 that are involved in their interaction were identified through mutagenesis of each protein. Mutations in either UL94 or UL99 that were found to abolish their interaction were then incorporated into the viral genome. The resulting mutant viruses were completely defective for replication. Further phenotypic analysis of these mutants indicates that both UL94 and UL99 exhibit aberrant localization and do not accumulate at the AC in the absence of their interaction. Taken together, these data suggest that the interaction between UL94 and UL99 is essential for the proper localization of UL94 and UL99 to the AC and for HCMV replication.
MATERIALS AND METHODS
Cell culture and virus infections.
Human foreskin fibroblast (HFF) cells were cultured in Dulbecco's modified Eagle's medium supplemented with 10% (vol/vol) fetal calf serum (HyClone), 100 units/ml penicillin, and 100 μg/ml streptomycin in an atmosphere of 5% CO2 at 37°C. For protein stability experiments, medium containing cycloheximide at a concentration of 50 μg/ml was added to 293T cells at 48 h posttransfection with hemagglutinin (HA)-tagged UL94 (HAUL94) or FLAG-tagged UL99 (FLAGUL99) expression plasmids. MG132 was used at a final concentration of 10 μM. For HCMV infections, HFF cells were infected for 2 h at 37°C. Following 2 h incubation, the inoculum was removed and replaced with fresh complete medium. All infections were carried out with HCMV strain AD169 derived from either the ADCRE (33) or ADCREGFP (5) bacterial artificial chromosome (BAC).
Cloning and site-directed mutagenesis.
Plasmids containing single amino acid substitutions were generated using a QuikChange Lightning site-directed mutagenesis kit (Stratagene) according to the manufacturer's protocol. Briefly, UL94pENTR/D-TOPO (15) was used as the template for PCR mutagenesis using complementary mutagenic primers containing the desired nucleotide changes. Residual template was digested using DpnI enzyme, and mutagenized plasmids were transformed into electrocompetent Escherichia coli cells. Plasmids were isolated, and direct sequencing was employed to verify the presence of the desired mutations. UL99 internal deletion mutant plasmids were generated by PCR amplification of FLAGUL99-dest-pcDNA3.1 (15) to generate linear PCR products missing the desired nucleotide sequences. Linear amplification products were gel purified, phosphorylated with T4-PNK (NEB), and ligated with T4 DNA ligase (NEB). Resulting plasmids were transformed into electrocompetent E. coli cells. Plasmids were isolated, and direct sequencing was employed to verify the presence of the desired mutations.
Yeast two-hybrid screen.
The yeast two-hybrid screen was performed using the ProQuest two-hybrid system (Invitrogen) as previously described (15). Briefly, bait and prey plasmids were transformed into Saccharomyces cerevisiae strain MaV203 using a Frozen-EZ Yeast Transformation II kit (Zymo Research) according to the manufacturer's protocol. Transformants were selected on minimal synthetic agar medium containing dropout supplements lacking leucine and tryptophan (Leu-Trp−; Clontech). Interactions were identified through activation of the HIS3 reporter gene. To assay for activation of the HIS3 reporter gene, multiple colonies from each Leu-Trp− plate were resuspended in 0.9% NaCl and spotted onto synthetic complete agar medium containing dropout supplements lacking leucine, tryptophan, and histidine (Leu-Trp-His−; Clontech). Plates were incubated for 3 days at 30°C and assessed for growth.
Western blotting.
Total protein was harvested by trypsinization, centrifugation, and lysis in radioimmunoprecipitation assay (RIPA) lysis buffer (50 mM Tris, pH 7.4, 150 mM NaCl, 0.4 mM EDTA, 1% IPEGAL detergent, 0.1% SDS, 0.5% deoxycholate) containing protease inhibitor cocktail (Roche). Protein concentration was determined by the method of Bradford. Lysates were boiled in 2× SDS sample buffer (62.5 mM Tris-HCl, pH 6.8, 2.5% SDS, 20% glycerol, 1% β-mercaptoethanol). Proteins were separated by SDS-PAGE, and Western blotting was performed. Proteins were separated by electrophoresis on 10% polyacrylamide gels and transferred to nitrocellulose membranes (Optitran; Whatman). Membranes were blocked in 5% nonfat dry milk and probed with primary and secondary antibodies. Immunoreactive proteins were detected by use of an enhanced chemiluminescence system (Thermo).
Coimmunoprecipitation.
At 24 h prior to transfection, 1 × 106 293T cells were plated onto 6-cm dishes. On the next day, medium was changed and the cells were transfected with 2 μg each of FLAG- and HA-tagged expression constructs. Transfections were performed using a ProFection mammalian transfection system (Promega) according to the manufacturer's protocol. Total protein was harvested at 48 h posttransfection by trypsinization, centrifugation, and lysis in NP-40 lysis buffer (50 mM Tris, pH 7.4, 150 mM NaCl, 0.5% NP-40, 0.75% IPEGAL) containing protease inhibitor cocktail (Roche). Immunoprecipitations were performed with 200 μg of total protein incubated with 3 μl rabbit anti-FLAG antibody for 2 h at 4°C. Protein complexes were precipitated with protein A/G plus agarose (Santa Cruz Biotechnology) for 2 h at 4°C. Immunoprecipitates were washed 3 times with NP-40 lysis buffer and boiled in 2× SDS sample buffer (62.5 mM Tris-HCl, pH 6.8, 2.5% SDS, 20% glycerol, 1% β-mercaptoethanol). Proteins were separated by SDS-PAGE, and Western blotting was performed as described above.
Antibodies.
The following antibodies used for Western blot analysis were obtained from commercial sources: mouse anti-HA (16B12; Covance), rat anti-HA (3F10; Roche), mouse anti-UL99 (1207; Rumbaugh-Goodwin Institute), mouse anti-FLAG (M2; Sigma-Aldrich), rabbit polyclonal anti-FLAG (F-7245; Sigma-Aldrich), and mouse anti-tubulin (TU-02; Santa Cruz Biotechnology). The following horseradish peroxidase-conjugated secondary antibodies were used for detection: goat anti-mouse IgG (Molecular Probes), goat anti-mouse IgM (Chemicon), and goat anti-rabbit IgG (Zymax). Alexa 546-conjugated Fab2 goat anti-mouse (Molecular Probes) and Alexa 488-conjugated goat anti-rat (Molecular Probes) antibodies were used for immunofluorescence analysis.
Recombinant virus generation.
Recombinant BAC DNAs were generated using a two-step Red recombination protocol in E. coli GS1783 as previously described (26). Briefly, GS1783 cells containing parental ADCRE (33) or ADCREGFP (5) BAC were grown to mid-log phase and induced to express Red recombination proteins at 42°C for 15 min. Induced cells were made competent and transformed with 100 ng purified PCR product composed of the kanamycin/I-SceI cassette flanked by complementary viral DNA sequence containing the desired mutation. PCR products were generated using the appropriate oligonucleotide primers and pEP-Kan-S2 plasmid template (26). Transformed cells were plated on LB agar plates containing chloramphenicol and kanamycin. Resulting colonies were screened for proper insertion of the Kan/I-SceI cassette by PCR. GS1783 cells containing the desired Kan/I-SceI BAC were induced to express I-SceI in medium containing 1% l-arabinose and plated on LB agar plates containing 1% arabinose and chloramphenicol. Resulting colonies were replica plated to identify clones that were kanamycin sensitive, indicating removal of the Kan/I-SceI cassette. Recombinant BACs were screened by restriction digestion, PCR, and DNA sequencing to confirm the presence of the desired mutations. Recombinant viruses were generated by transfecting ∼1 μg BAC DNA and 5 μg pp71-pCGN expression plasmid into 5 × 106 HFF cells via electroporation (950 μF, 260 V). Cells were seeded into dishes, and infectious virus was harvested when 100% cytopathic effect was observed. Infectious titers for all viruses were determined by plaque assay on HFF cells.
Complementation of UL94 mutant virus.
The UL94C250A mutant virus was propagated in cells stably transduced with LEX lentivirus (Open Biosystems) expressing HAUL94 (HAUL94LEX lentivirus) as previously described (16). Cells transduced with the HAUL94LEX lentivirus were selected for by culture in medium containing 1.5 μg/ml puromycin.
Immunofluorescence analysis.
HFF cells on glass coverslips were washed in phosphate-buffered saline (PBS) and fixed in 4% paraformaldehyde in PBS at room temperature for 10 min. Fixed cells were washed and permeabilized in PBS-T (PBS, 0.05% Tween 20, 0.1% Triton X-100). Cells were blocked with PBS-T, 0.5% bovine serum albumin, and 1% goat serum for 30 min, followed by sequential incubation with primary and secondary antibodies in a humidified chamber at 37°C. Cells were incubated with Hoechst to stain nuclei and mounted in 90% glycerol. Fluorescence was visualized on a Zeiss Axiovert 40 CFL microscope. Images were taken using a Jenoptik ProgRes C10 plus camera equipped with ProgRes CapturePro (version 2.8.0) software.
RESULTS
UL94 and UL99 alter each other's localization.
We and others have shown that both UL94 and UL99 exhibit a different pattern of localization in infected cells compared to the localization observed when each protein is expressed alone (16, 19). We sought to determine whether UL94 and UL99 influence each other's subcellular localization in the absence of other viral proteins. HFF cells were transfected with plasmids expressing HA-tagged UL94, UL99, or both. Cells were fixed and stained for immunofluorescence analysis at 48 h posttransfection. As shown in Fig. 1, when UL94 was expressed alone we observed a diffuse pattern of localization throughout both the nucleus and cytoplasm of the transfected cell, as previously described (Fig. 1, top row) (16). When UL99 was transfected alone, UL99 protein was observed throughout the cytoplasm with an irregular punctate appearance, likely due to its association with cytoplasmic membranes of the endoplasmic reticulum-Golgi intermediate compartment (ERGIC), as previously described, (Fig. 1, middle row) (23). In contrast, when UL94 and UL99 were expressed together, the subcellular localization of UL94 and UL99 was dramatically altered, in that UL94 protein was largely excluded from the nucleus and both proteins displayed an identical perinuclear pattern of localization (Fig. 1, bottom row). These data demonstrate that UL94 and UL99 alter each other's localization in the absence of other viral proteins.
Fig 1.
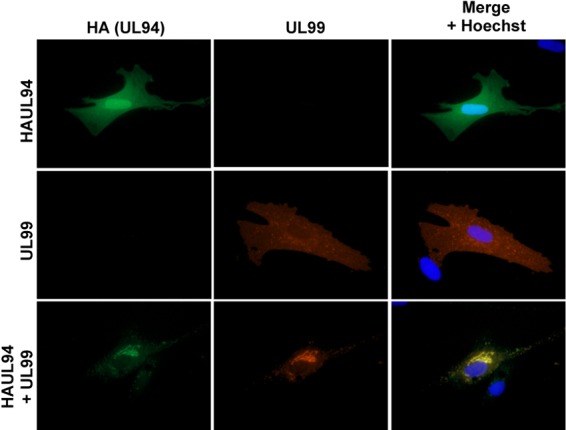
UL94 and UL99 alter each other's localization. HFF cells were transfected with plasmid DNA expressing HAUL94, UL99, or both. Cells were fixed and stained for immunofluorescence analysis at 48 h posttransfection. UL94 was detected with anti-HA antibody (green). UL99 was detected with anti-UL99 antibody (red). Nuclei are stained with Hoechst (blue). Images are representative of results obtained from five independent transfections.
UL94 protein is stabilized in the presence of UL99.
We and others have shown that UL94 and UL99 interact in the absence of other viral proteins (Fig. 2A) (12, 15, 27). In cells cotransfected with UL94 and UL99, we routinely observed a modest but reproducible increase in the levels of UL94 protein compared to the levels of UL94 observed when transfected alone (Fig. 2A, cell lysates; compare lanes 2 and 3). In contrast, UL99 is present in equal amounts regardless of the presence of UL94 (data not shown). This led us to hypothesize that UL99 stabilizes UL94.
Fig 2.
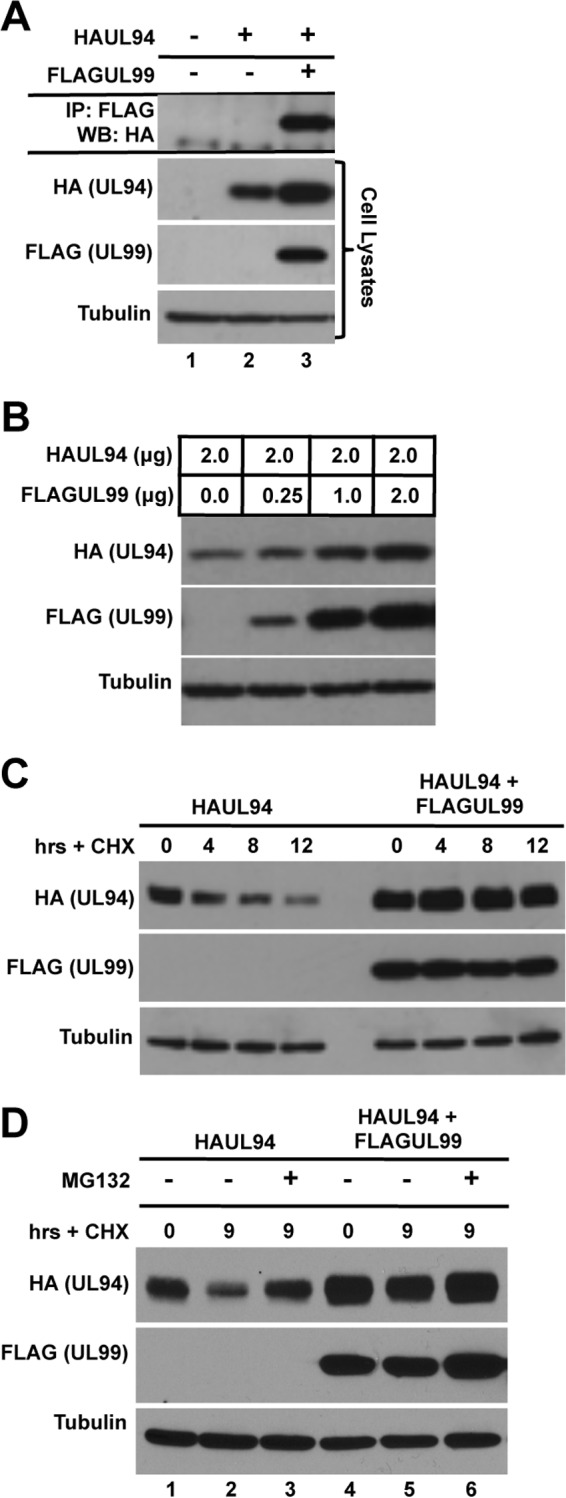
UL99 interacts with and stabilizes UL94 protein. (A) 293T cells were transfected with HAUL94 alone or with FLAGUL99. Total protein was harvested at 48 h posttransfection, and immunoprecipitation (IP) was performed with anti-FLAG antibody. Immune complexes were separated by SDS-PAGE, and Western blotting (WB) was performed with anti-HA antibody. (B) 293T cells were cotransfected with the indicated amounts of plasmid DNA expressing HAUL94 and FLAGUL99. Total protein was harvested at 48 h posttransfection, and levels of UL94 and UL99 protein were analyzed by Western blotting. (C) 293T cells were transfected with plasmid DNA expressing HAUL94 alone or cotransfected with FLAGUL99, as indicated. Cells were treated with cycloheximide (CHX) at 48 h posttransfection. Total protein was harvested at the indicated times after cycloheximide treatment, and levels of UL94 and UL99 were analyzed by Western blotting. (D) 293T cells were transfected with plasmid DNA expressing HAUL94 alone or cotransfected with FLAGUL99, as indicated. Cells were treated with cycloheximide and MG132 at 48 h posttransfection, as indicated. Total protein was harvested at the indicated times after treatment, and levels of UL94 and UL99 were analyzed by Western blotting.
To test this hypothesis, we first sought to determine whether UL99 expression leads to an increase in UL94 protein levels in a dose-dependent manner. Cells were cotransfected with a static amount of HAUL94 plasmid and increasing amounts of FLAGUL99 plasmid, as indicated. Total protein was harvested at 48 h posttransfection for Western blot analysis. As shown in Fig. 2B, we observed an increase in the levels of UL94 protein that positively correlated with the increasing amount of UL99 transfected. Tubulin was used as a loading control to ensure that equal amounts of cell lysates were analyzed.
This increase in the levels of UL94 observed upon cotransfection with increasing amounts of UL99 led us to predict that the half-life of UL94 protein may be extended in the presence of UL99. To assess whether UL99 influences the half-life of UL94 protein, cells were transfected with either HAUL94 alone or cotransfected with HAUL94 and FLAGUL99 expression plasmids. Cycloheximide was added to the culture medium at 48 h posttransfection to inhibit new protein synthesis. Total protein was harvested at the indicated times after cycloheximide treatment for Western blot analysis. We observed that in the absence of UL99, levels of UL94 protein were rapidly reduced upon the inhibition of new protein synthesis (Fig. 2C, left). In contrast, in the presence of UL99, UL94 levels remained constant and were significantly higher than those observed when UL94 was expressed alone (Fig. 2C, right).
Finally, to elucidate the mechanism by which UL94 degradation occurs in the absence of UL99, we investigated whether UL94 levels could be restored in the presence of cycloheximide by proteasome inhibition. Cells were transfected with HAUL94 plasmid alone or cotransfected with FLAGUL99 as indicated and treated with cycloheximide with or without the proteasome inhibitor MG132. Total protein was harvested at 9 h posttreatment and analyzed by Western blotting. Inhibition of proteasome activity resulted in a restoration of UL94 levels to those observed in untreated cells (Fig. 2D; compare lanes 1 and 3). Taken together, the data shown in Fig. 2 suggest that UL94 protein is stabilized by UL99 in a proteasome-dependent manner.
Conserved cysteine residues of UL94 are involved in binding UL99.
To investigate the functional significance of the interaction between UL94 and UL99, we first sought to identify the amino acids of both UL94 and UL99 that are required for their binding. We started by constructing a panel of UL94 truncation mutants through stepwise deletions from both the N and C termini of the 345-amino-acid UL94 protein. These truncations were then tested for their ability to bind UL99 in a yeast two-hybrid assay. We found that all truncations of UL94 protein completely abolished the ability of UL94 to bind UL99 in the yeast two-hybrid assay (data not shown). Next, we analyzed the expression of the UL94 truncation mutants in transfected cells to assess the expression levels of truncated forms of UL94 protein. We were unable to detect appreciable levels of any of the truncated forms of UL94 by Western blot analysis (data not shown). These results suggest that UL94 is sensitive to mutation, in that the full length of the UL94 protein may be required for the proper expression and stability of the protein, and are consistent with those from other studies attempting mutagenesis of UL94 or its herpes simplex virus 1 (HSV-1) homolog UL16 (8, 12, 32).
As a herpesvirus core gene, UL94 is conserved among all members of the Herpesviridae family, and alignment of UL94 with its homologs reveals the presence of several conserved amino acid residues in the C terminus of the protein (31). We predicted that due to the conservation of the interaction between UL94 and UL99 homologs in other herpesviruses, the interaction between UL94 and UL99 may involve these conserved residues. Therefore, we mutated several cysteine residues that are conserved among UL94 homologs and have been shown to be important for binding of the HSV-1 homologs UL16 and UL11 (32). The amino acid sequence of UL94 that encompasses the conserved cysteine residues is shown in alignment with the sequence of selected UL94 homologs in Fig. 3A. The cysteines mutated for our mapping studies are highlighted. Each of the conserved cysteine residues was mutated to alanine using site-directed mutagenesis. As a control mutation, we also mutated the cysteine residue at amino acid position 204, as it is not conserved among the UL94 homologs. We then tested these mutants for their ability to bind UL99 in the yeast two-hybrid assay (Fig. 3B). We found that all of the cysteine mutants retained their ability to bind UL99, with the exception of the C250A mutant. We also observed a moderate but reproducible decrease in the growth of yeast cells expressing the C252A mutation when plated on selective medium, suggesting that this mutation may cause a decrease in UL99 binding and thus a reduction in the activation of the HIS3 reporter gene necessary for growth on plates lacking histidine.
Fig 3.

Mapping UL94 interaction domains. (A) Amino acid sequence alignment of residues 200 to 256 of HCMV UL94 with its homologs in varicella-zoster virus (VZV), herpes simplex virus 1 (HSV1), Epstein-Barr virus (EBV), and human herpesvirus 6 (HHV6). Cysteine residues that were mutated to alanine for mapping studies are indicated with an asterisk. (B) Yeast cells were cotransformed with bait plasmid expressing wild-type (WT) UL99 and prey plasmid expressing wild-type UL94 or the indicated UL94 mutant. Interactions were assessed by growth of cotransformed cells on medium lacking histidine. (C) 293T cells were transfected with plasmid DNA expressing the indicated HAUL94 alone (left) or with FLAGUL99 (right). Total protein was harvested at 48 h posttransfection, and levels of UL94 and UL99 were analyzed by Western blotting. (D) 293T cells were cotransfected with plasmid DNA expressing the indicated HAUL94 and FLAGUL99. Total protein was harvested at 48 h posttransfection, and immunoprecipitation was performed with anti-FLAG antibody. Immune complexes were separated by SDS-PAGE, and Western blotting was performed with anti-HA antibody. (E) 293T cells were cotransfected with plasmid DNA expressing the indicated HAUL94 and FLAGUL99 and treated with MG132 for 9 h prior to protein harvest, as indicated. Total protein was harvested at 48 h posttransfection, and immunoprecipitation was performed with anti-FLAG antibody. Immune complexes were separated by SDS-PAGE, and Western blotting was performed with anti-HA antibody.
To verify the results of the yeast two-hybrid assay, we analyzed the ability of the UL94 cysteine mutants to bind UL99 by coimmunoprecipitation from transfected cells. Due to the apparent sensitivity of UL94 to mutation, we first sought to determine whether the cysteine mutations affected the levels of UL94 expression. Cells were transfected with plasmids expressing wild-type or mutant HAUL94 as indicated. Total protein was harvested at 48 h posttransfection for Western blot analysis. As shown in Fig. 3C, mutation of the cysteines at positions 200 and 204 had little effect on the levels of UL94 expression compared to those for wild-type UL94 (Fig. 3C, left, lanes 1 to 3). In contrast, the remaining cysteine mutations at amino acid positions 227, 250, 252, and 256 each resulted in a decrease in the levels of UL94 expression compared to that for wild type (Fig. 3C, left, lanes 4 to 7). We next asked whether the level of mutant forms of UL94 could be increased by cotransfection of UL99, as was observed with wild-type UL94. As shown in Fig. 3C, we observed a significant increase in the levels of the UL94 C227A and C256A mutants in the presence of UL99 (Fig. 3C, lanes 4 and 7; compare the left and right panels). In contrast, we observed only a modest increase in the levels of UL94 in cells transfected with the UL94 C250A and C252A mutants in the presence of UL99 (Fig. 3C, lanes 5 and 6; compare the left and right panels). Interestingly, the C227A and C256A mutants that appeared to be stabilized by UL99 were those that also retained their ability to bind UL99 in the yeast two-hybrid assay. Conversely, the UL94 C250A and C252A mutants that exhibited little stabilization by UL99 also exhibited no or significantly reduced binding to UL99 in the yeast two-hybrid assay (Fig. 3B). These results suggest that stabilization of UL94 by UL99 requires their interaction and that mutant forms of UL94 that are unable to bind UL99 also show a defect in expression levels.
To confirm the results of the yeast two-hybrid assay, we assayed each of the UL94 cysteine mutants for their ability to bind UL99 by coimmunoprecipitation. As shown in Fig. 3D (top), we observed significant levels of UL94 immunoprecipitated with UL99 in cells transfected with wild-type, C200A, C204A, C227A, and C256A UL94. We were also able to detect UL94 in the immunoprecipitate of cells transfected with the C252A mutant; however, the level was dramatically reduced compared to that of wild-type UL94 (Fig. 3D, lane 7). We did not observe detectable levels of the UL94 C250A mutant in the immunoprecipitate (Fig. 3D, lane 6); however, the levels of the UL94 C250A mutant present in the cell lysate were also reduced compared to those for wild-type UL94.
To determine whether the absence of an interaction between UL94 C250A and UL99 resulted from the low level of UL94 expression, we treated transfected cells with MG132 to increase the levels of the UL94 C250A protein in the cell lysate. Coimmunoprecipitation was performed as described above, and the levels of UL94 C250A bound to UL99 were determined by Western blot analysis. We found that while UL94 C250A is stabilized under conditions of proteasome inhibition (Fig. 3E, cell lysates, lane 4), we were still unable to detect appreciable amounts of UL94 C250A in the immunoprecipitate.
Taken together, these results are consistent with the results of the yeast two-hybrid analysis and demonstrate that the cysteine residues at amino acid positions 250 and 252 of UL94 are likely involved in binding UL99. These results are also consistent with mutational analysis of HSV-1 UL16, which showed that the same cysteines are required for binding UL11 (32). Based on the combination of the yeast two-hybrid assay and coimmunoprecipitation results, we chose the UL94 C250A mutant for analysis of the interaction between UL94 and UL99 in the context of infection.
UL99 amino acids 37 to 39 are required for binding UL94.
In addition to identifying the amino acids in UL94 that are required for the interaction with UL99, we also sought to map the interaction with respect to the amino acid residues of UL99 that are required for binding UL94. UL99 is a 190-amino-acid myristoylated tegument protein. Several mutational analyses have been performed to identify the domains of UL99 that are necessary and sufficient for its essential function. Generation of C-terminal UL99 truncations in the context of the viral genome demonstrated that the N-terminal 57 amino acids of UL99 are necessary and sufficient for both wild-type virus replication and incorporation of UL99 into virions (10, 23). Later studies confirmed these results and further defined an essential region of UL99 between amino acids 34 and 43. Mutant viruses lacking these essential regions are completely defective for replication and exhibit defects in the trafficking of UL99 to the AC and in its retention there. These defects ultimately result in the absence of virion envelopment and thus the production of infectious progeny (21–23).
Based on these previous studies, we began our mutational analysis of UL99 by assaying a panel of UL99 C-terminal truncation mutants for their ability to bind UL94 in the yeast two-hybrid assay. We generated constructs expressing the first 123, 90, 61, and 33 amino acids of UL99, as these mutants have been previously tested for their ability to support virus replication (23). In addition, we tested two internal deletion mutants lacking amino acids 26 to 33 or 26 to 43 of UL99 (UL99 Δ26-33 and UL99 Δ26-43, respectively). As shown in Fig. 4A, the first 61 amino acids of UL99 are sufficient for binding UL94, whereas a truncation containing only the first 33 amino acids of UL99 (UL99 1-33) does not bind UL94. In addition, while deletion of amino acids 26 to 33 does not affect the ability of UL99 to bind UL94, extending the deletion to encompass amino acids 26 to 43 completely abolishes binding to UL94. The results obtained from the yeast two-hybrid analysis were confirmed by coimmunoprecipitation analysis from transfected cells (data not shown). Taken together these results suggest that the region of UL99 required for binding UL94 is located between amino acids 34 and 43 and support the hypothesis that the interaction between UL94 and UL99 is essential for virus replication, as mutant forms of UL99 that are incapable of supporting virus replication (UL99 1-33 and UL99 Δ26-43) are also defective for binding UL94.
Fig 4.

Mapping UL99 interaction domains. (A) Yeast cells were cotransformed with bait plasmid expressing wild-type UL99 or the indicated UL99 mutant and prey plasmid expressing UL94. Interactions were assessed by growth of cotransformed cells on medium lacking histidine. (B) 293T cells were cotransfected with plasmid DNA expressing HAUL94 and the indicated FLAGUL99. Total protein was harvested at 48 h posttransfection, and immunoprecipitation was performed with anti-FLAG antibody. Immune complexes were separated by SDS-PAGE, and Western blotting was performed with anti-HA antibody.
We next wanted to confirm the yeast two-hybrid assay results as well as further define the amino acids of UL99 that are critical for binding UL94. We therefore constructed three additional internal deletions spanning the region from amino acids 34 to 43. We then analyzed the ability of these UL99 internal deletion mutants to bind UL94 by coimmunoprecipitation from transfected cells. As shown in Fig. 4B (top), we were able to detect binding of UL99 Δ26-33 to UL94 at levels similar to the levels observed with wild-type UL99 (Fig. 4B, lanes 2 and 3). In contrast, we observed no binding between UL99 Δ26-43 and UL94 (Fig. 4B, lane 4), confirming the results of the yeast two-hybrid assay. We also demonstrated that deletion of amino acids 37 to 39 completely abolished binding of UL99 to UL94, whereas deletion of the amino acids directly adjacent to these residues had no effect on binding (Fig, 4B, lanes 5 to 7). Analysis of the levels of UL94 in the lysates of cells cotransfected with these UL99 mutants further demonstrated that in the absence of the interaction between UL94 and UL99, there was a decrease in the level of UL94 expression (Fig. 4B, UL94 cell lysates, lanes 4 and 6). In contrast, UL99 Δ26 to 43 and UL99 Δ37-39 were expressed at levels equal to or greater than those of wild-type UL99, suggesting that UL94 does not stabilize UL99.
These results demonstrate that amino acids 37 to 39 of UL99 are essential for the interaction between UL99 and UL94. We therefore chose the UL99 Δ37-39 mutation to further investigate the functional significance of the interaction between UL94 and UL99 in the context of infection.
Mutations that abolish the interaction between UL94 and UL99 also prevent virus replication.
The ultimate goal of our mapping studies was to identify mutations that abolish the interaction between UL94 and UL99 to then investigate the functional relevance of those mutations in the context of viral infection. To this end, we generated recombinant bacterial artificial chromosome (BAC) constructs incorporating the UL94 C250A mutation and the UL99 Δ37-39 mutation into the HCMV genome. In addition, we generated control constructs carrying the UL94 C204A and UL99 Δ40-43 mutations, neither of which has any effect on the interaction between UL94 and UL99. As additional controls, the UL94 C250A and UL99 Δ37-39 mutations were repaired to confirm the absence of secondary mutations elsewhere in the genome. The BAC constructs generated and the relative locations of the mutations inserted are depicted in Fig. 5A and D. Recombinant BACs were generated using the UL94HA ADCREGFP parental BAC (15), as described in Materials and Methods. Generation of the desired recombinant BAC was verified by restriction digestion, PCR, and direct sequencing of BAC DNA (data not shown). To assess the production of replication-competent virus, HFF cells were cotransfected with BAC DNA and pp71 expression plasmid. Generation of infectious virus was monitored by cytopathic effect and green fluorescent protein (GFP) expression from the viral genome.
Fig 5.

Generation of recombinant viruses. (A) Schematic diagram of UL94 mutations incorporated into the viral genome. Nucleotide changes incorporated to change cysteine residues to alanine are indicated. AA, amino acid. (B) UL94 mutant virus stocks were generated from HAUL94-complementing HFF cells. Viruses were then used to infect noncomplementing HFF cells at a multiplicity of 0.01 PFU/cell, and total virus was harvested at 21 days postinfection. Titers of progeny virus were determined on HAUL94-complementing HFF cells. (C) HFF cells were infected at a multiplicity of 0.05 PFU/cell, and total protein was harvested at 120 h postinfection. Levels of viral proteins were analyzed by Western blotting. IE1/2, immediate-early proteins 1 and 2. (D) UL99 mutant BACs were transfected into HFF cells, and virus replication was monitored by cytopathic effect and GFP expression from the viral genome. Recovery of infectious virus from BAC-transfected cells for each mutant is indicated. Rep, repair.
Visual analysis of GFP expression in cells transfected with UL94 C250A BAC DNA showed the presence of single GFP-positive cells representing those that were initially transfected with BAC DNA; however, we did not observe any subsequent spread of GFP expression to other cells in the culture. This result suggests that the UL94 C250A mutant is completely defective for viral growth. HFF cells transfected with the UL94 C204A control mutant and the UL94 C250A repair BAC exhibited significant cytopathic effect as well as spread of GFP expression indistinguishable from that observed in cells transfected with the UL94HA parental BAC (data not shown), suggesting that these constructs are competent for virus replication. Because the UL94 C250A mutation results in a block in virus replication, we used a previously described UL94 complementation system to generate stocks of this mutant for further analysis (16). UL94 C250A BAC DNA was transfected into HAUL94-complementing HFF cells. Virus was harvested when cells exhibited 100% cytopathic effect, and then the titer on complementing cells was determined. The resulting UL94 C250A mutant virus grew slowly and to low titers (∼104 PFU/ml), similar to the previously described UL94-null virus (16).
UL94HA (16), UL94 C204A, UL94 C250A, and UL94 C250A repair viruses were then used to infect HFF cells at a multiplicity of 0.01 PFU/cell to measure the levels of replication for each mutant. Total progeny virus from both cells and supernatant were harvested from infected cells at 21 days postinfection, and titers on HAUL94-complementing cells were determined. Figure 5B shows that while UL94HA, UL94 C204A, and UL94 C250A repair viruses replicated to similar titers by 21 days postinfection, the UL94 C250A mutant did not produce any infectious progeny virus. These results demonstrate that the cysteine residue at amino acid position 250 of UL94 is essential for virus replication.
Because the C250A mutation results in a decrease in UL94 expression levels in transfected cells, we also sought to determine the levels of UL94 expression in cells infected with the UL94 C250A mutant virus. HFF cells were infected at a multiplicity of 0.05 PFU/cell with either the UL94 C250A or UL94 C250A repair virus, and total protein was harvested at 96 h postinfection for Western blot analysis. As shown in Fig. 5C, we observed comparable levels of both immediate-early genes and UL99 in cells infected with the UL94 C250A mutant or the UL94 C250A repair virus, suggesting that the C250A mutation does not affect the initiation of infection or the expression of viral late genes. Consistent with the results of our transfection studies, there was a decrease in the levels of UL94 C250A compared to those of wild-type UL94, indicating that this mutation also affects the stability of UL94 in the context of infection. However, we were able to detect appreciable amounts of UL94 C250A protein, suggesting that the absolute growth defect observed with the UL94 C250A mutant virus is not likely due to a complete absence of UL94 expression.
We also generated several recombinant BAC constructs carrying mutations in the UL99 open reading frame to investigate the significance of the interaction between UL94 and UL99 during infection (Fig. 5D). Transfection of HFF cells with the UL99 Δ37-39 mutant indicated that this mutation causes a complete inhibition of viral growth, as evidenced by the appearance of single GFP-positive cells with the absence of subsequent spread of infection. In contrast, cells transfected with the UL99 Δ40-43 or the UL99 Δ37-39 repair BAC produced infectious virus to levels similar to those produced by the parental UL94HA BAC (data not shown). Our ability to recover infectious virus from the UL99 mutant BAC-transfected cells is summarized in Fig. 5D. Despite repeated attempts to complement the growth defect of the UL99 Δ37-39 mutant using HFF cells expressing wild-type UL99 from a retroviral vector, we were unable to generate any infectious virus containing the UL99 Δ37-39 mutation for further analysis. Nonetheless, the results of our BAC transfections strongly suggest that amino acids 37 to 39 of UL99 are required for virus replication as well as for the interaction between UL94 and UL99.
Proper localization of UL94 and UL99 during infection requires their interaction.
We previously demonstrated that UL99 exhibits aberrant subcellular localization during infection in the absence of UL94 (16). Based on those results and the observation that UL94 and UL99 are capable of altering each other's localization in transfected cells, we hypothesized that the proper localization of both proteins during the course of infection is dependent on their ability to interact. To test this hypothesis, we sought to determine the subcellular localization of UL94 and UL99 in cells infected with viruses containing mutations that abolish their interaction.
Because we have a complementation system for propagating UL94 mutant viruses, we were able to assess the localization of UL94 and UL99 in the context of infection by directly infecting HFF cells with the virus generated from HAUL94-complementing cells. For immunofluorescence analysis of infected cells, HFF cells were infected with the UL94 C250A or control viruses at a multiplicity of 0.01 PFU/cell. These viruses were produced from the parental BAC ADCRE UL94HA, which does not express GFP, allowing for the simultaneous immunofluorescent staining of both UL94 and UL99. Cells were fixed at 96 h postinfection and stained for UL94 and UL99 using anti-HA and anti-UL99 antibodies, respectively. As shown in Fig. 6A, during infection with the UL94HA virus, UL94 (green) and UL99 (red) both accumulated at the juxtanuclear AC, as previously described (Fig. 6A, top row) (16). Similar results were observed in cells infected with the UL94 C204A control mutant virus (Fig. 6A, second row). In contrast, during infection with the UL94 C250A virus, both UL94 and UL99 exhibited largely diffuse localization throughout the cytoplasm and did not appear to accumulate to appreciable levels at the AC (Fig. 6A, third row). When the UL94 C250A mutation was repaired, we observed a restoration of juxtanuclear localization for both UL94 and UL99 identical to that observed during infection with the UL94HA virus (Fig. 6A, bottom row). These results demonstrate that the cysteine residue at amino acid position 250 of UL94 is required for UL99 binding, virus replication, and the proper localization of both UL94 and UL99 to the AC during infection.
Fig 6.
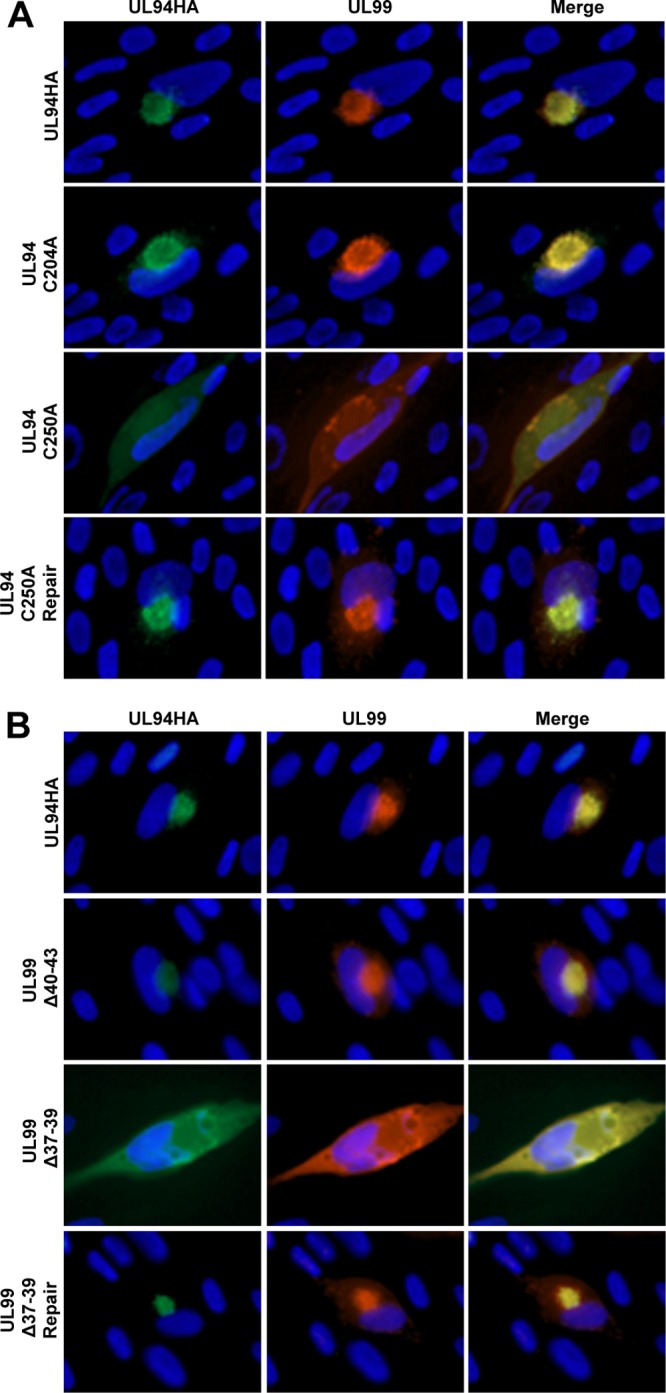
Localization of UL94 and UL99 during infection. (A) HFF cells were infected with the indicated virus at a multiplicity of 0.01 PFU/cell. Cells were fixed at 96 h postinfection, and immunofluorescence analysis was performed with the indicated antibodies to visualize viral proteins. (B) HFF cells were transfected with the indicated BAC DNA. Cells were fixed at 8 days posttransfection, and immunofluorescence analysis was performed with the indicated antibodies to visualize viral proteins. UL94 was detected with anti-HA antibody (green). UL99 was detected with anti-UL99 antibody (red). Nuclei are stained with Hoechst (blue). Images are representative of results obtained from three independent infections (A) or BAC transfections (B).
As mentioned previously, we were unable to complement the growth defect of the UL99 Δ37-39 mutant by providing UL99 in trans. We therefore assessed the localization of UL94 and UL99 in BAC-transfected cells, a method that has previously been used to analyze the phenotype of mutants in the absence of sufficient complementation to generate virus stocks (1, 25, 30). UL99 mutations were inserted into the parental ADCRE UL94HA BAC that does not express GFP, allowing for the simultaneous staining of UL94 and UL99, as described above. HFF cells were transfected with BAC DNA and fixed for immunofluorescence analysis at 8 days posttransfection to allow sufficient expression of viral late genes (1).
In cells transfected with the UL94HA parental BAC, we observed the expected juxtanuclear localization of both UL94 and UL99 at the AC (Fig. 6B, top row). A similar pattern of localization was observed in cells transfected with the UL99 Δ40-43 BAC (Fig. 6B, second row), demonstrating that a mutation that does not affect the interaction between UL94 and UL99 also does not prevent their localization to the AC. In contrast, in cells transfected with the UL99 Δ37-39 BAC, both UL94 and UL99 exhibited diffuse cytoplasmic localization (Fig. 6B, third row), suggesting that neither protein is properly trafficking to the AC. Repairing the UL99 Δ37-39 mutation resulted in a restoration of the wild-type pattern of localization for both UL94 and UL99 (Fig. 6B, bottom row), demonstrating that the aberrant localization observed in cells transfected with the UL99 Δ37-39 BAC results specifically from the UL99 Δ37-39 mutation. These data suggest that amino acids 37 to 39 of UL99 are required for the interaction between UL94 and UL99, virus replication, and the proper localization of both UL94 and UL99 to the AC in the context of infection.
DISCUSSION
The mechanisms that facilitate the assembly of mature HCMV particles in the cytoplasm of infected cells are poorly understood. While the events that result in virion maturation remain largely undefined, many reports have demonstrated a crucial role for tegument proteins in virion assembly and morphogenesis. UL99 (pp28) is essential for secondary envelopment of virions in the cytoplasm. In the absence of UL99, partially tegumented but nonenveloped capsids accumulate in the cytoplasm of infected cells (24). Our previous characterization of a UL94-null mutant demonstrated that the growth phenotype of HCMV in the absence of UL94 expression is virtually identical to that of a UL99-deletion mutant (16).
UL94 and UL99 physically interact in the absence of other viral proteins (12, 15, 27). We also demonstrated that this interaction occurs during late times in infection, when progeny virus is being assembled and released from infected cells (15). Further, the proper localization of UL99 to the AC in infected cells requires the expression of viral late genes (23). Based on these observations and the fact that deletion of either UL94 or UL99 results in an absolute defect in secondary envelopment, we hypothesized that the interaction between UL94 and UL99 is essential for HCMV replication at the stage of virion assembly.
Both UL94 and UL99 exhibit different patterns of localization in transfected cells compared to the localization observed during infection (16, 19). Therefore, we first investigated whether UL94 and UL99 are capable of altering each other's localization in the absence of other infected-cell proteins. Figure 1 demonstrates that the localization of each protein is altered compared to that observed when both proteins are expressed together, demonstrating that UL94 and UL99 are able to redirect each other's localization in the absence of infection. These results are consistent with the observation that UL99 exhibits aberrant localization in infected cells in the absence of UL94 (16).
We repeatedly observed during the course of our immunoprecipitation analyses that the levels of UL94 detected by Western blotting were reproducibly lower in the absence of UL99 expression (Fig. 2A). We further demonstrated that UL99 is capable of effecting an increase in UL94 protein levels in a dose-dependent manner (Fig. 2B), that the half-life of the UL94 protein is extended in the presence of UL99 (Fig. 2C), and that the degradation of UL94 in the absence of UL99 occurs in a proteasome-dependent manner (Fig. 2D). In contrast, levels of UL99 remain unaffected by cotransfection with increasing amounts of UL94 (data not shown). Further, analysis of UL94 protein levels in cells transfected with mutant forms of either UL94 or UL99 that abolish their ability to interact demonstrates that this stabilization effect is dependent on their interaction (Fig. 3C and 4B). These results suggest that any mutation that disrupts the interaction between UL94 and UL99 will also result in a decrease in UL94 protein levels.
To assess the functional significance of the interaction between UL94 and UL99 during HCMV infection, we sought to identify the amino acids of each protein that are involved in their binding. Site-directed mutagenesis of conserved cysteine residues of UL94 demonstrated that the cysteine residue at amino acid position 250 is involved in UL99 binding (Fig. 3B and D). Incorporation of this mutation into the viral genome revealed that C250 of UL94 is essential for virus replication, as indicated by the complete inability of this mutant to generate infectious progeny (Fig. 5B). Further, we observed that in cells infected with the UL94 C250A mutant virus, neither UL94 nor UL99 appeared to localize to the AC and instead remained largely diffuse throughout the cytoplasm (Fig. 6A). Taken together, these results suggest that amino acid C250 of UL94 is required for the interaction between UL94 and UL99, for the proper localization of each protein to the AC, and for virus replication. In addition to disrupting the interaction between UL94 and UL99, mutation of UL94 C250 also resulted in a decrease in UL94 expression that could not be restored by coexpression of UL99 (Fig. 3C and D). Therefore, caution must be taken when interpreting the effect of the UL94 C250A mutation on virus replication. While our results with the UL94 C250A mutant are consistent with the hypothesis that the interaction between UL94 and UL99 is essential for virus replication, we cannot rule out the possibility that the growth defect observed is due to decreased levels of UL94 expression in cells infected with the UL94 C250A mutant. However, we were able to detect UL94 during infection with the UL94 C250A mutant virus by immunofluorescence and Western blot analysis (Fig. 5C and 6A). Further, the observation that UL94 is stabilized in the presence of UL99 and that this stabilization requires their interaction, along with the results of the UL99 mapping studies, strongly suggests that the interaction between UL94 and UL99 is required for virus replication.
Our mapping studies also led to the identification of amino acids 37 to 39 of UL99 as the region involved in binding UL94 (Fig. 4B). These amino acids are part of a region that has previously been demonstrated to be essential for UL99 function and thus for virus replication (23). Deletion of UL99 amino acids 37 to 39 in the context of the viral genome resulted in a complete inhibition of the production of infectious virus (Fig. 5D). Further, immunofluorescence analysis of UL94 and UL99 in cells transfected with the UL99 Δ37-39 mutant genome demonstrated that both proteins are aberrantly localized compared to the pattern of localization observed in cells transfected with the UL94HA or control BAC (Fig. 6B).
A previous report demonstrated that UL99 multimerizes in both transfected and infected cells and is detected in a predominantly dimeric form by nondenaturing gel electrophoresis and sedimentation analysis (22). Mutagenesis carried out to map the amino acids required for the UL99 self-interaction showed that amino acids 34 to 43 of UL99 are important but not essential for its dimerization. Fluorescence resonance energy transfer (FRET) analysis was used to demonstrate that UL99 multimerization occurs only after its localization to the AC (22). Given the possibility of temporal differences in the association of UL99 with UL94 and with itself, essential roles for UL99 multimerization and for the interaction between UL94 and UL99 during infection are not mutually exclusive. For example, it is possible that the interaction between UL94 and UL99 occurs first and is responsible for trafficking UL99 to the AC. Once there, UL94 may dissociate from the region of UL99 encompassing amino acids 37 to 39, allowing UL99 multimerization. Further studies would be necessary to elucidate the individual roles of the UL94-UL99 interaction and the multimerization of UL99 in the proper assembly of virions.
Based on our results, it appears that the interaction between UL94 and UL99 is required for each protein to traffic to the AC during infection. Analysis of the sequence requirements for trafficking of UL99 to the AC demonstrated that at steady state late in infection (>6 days postinfection), only those forms of UL99 containing at least the first 50 amino acids were detected at the AC (23). Further, the localization of UL99 to the AC required a late viral gene product, as evidenced by a defect in UL99 trafficking in the presence of a viral DNA synthesis inhibitor (23). Phenotypic analysis of UL99 mutant viruses demonstrated that the localization of UL99 to the AC is required for secondary envelopment of virions (21). Therefore, we propose a model in which the interaction between UL94 and UL99 results in the localization of UL99 to the AC and in its retention there, likely through additional interactions of the UL94-UL99 complex with other yet to be identified proteins. Further studies are under way to identify additional binding partners of UL94 and UL99 and to elucidate the spatial and temporal relationships between viral and cellular protein interactions that are involved in the assembly and envelopment of infectious HCMV virions.
ACKNOWLEDGMENTS
We are grateful to Greg Smith (Northwestern University) for kindly providing the E. coli GS1783 cells and Klaus Osterrieder (Cornell University) for providing the pEP-Kan-S2 plasmid for the BAC recombinations. We are also grateful to William Britt (University of Alabama at Birmingham) for kindly providing plasmid vectors for the UL99 Δ26-33 and Δ26-43 internal deletions. We also thank Steve Rice and Peter Southern for critically reading the manuscript.
This work was supported in part by NIH grant AI059340 (to W.A.B.) and by grant number T32DE007288 from the National Institute of Dental and Craniofacial Research.
The content is solely our responsibility and does not necessarily represent the official views of the National Institute of Dental and Craniofacial Research or the National Institutes of Health.
Footnotes
Published ahead of print 3 July 2012
REFERENCES
- 1. AuCoin DP, Smith GB, Meiering CD, Mocarski ES. 2006. Betaherpesvirus-conserved cytomegalovirus tegument protein ppUL32 (pp150) controls cytoplasmic events during virion maturation. J. Virol. 80:8199–8210 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2. Baines JD, Roizman B. 1991. The open reading frames UL3, UL4, UL10, and UL16 are dispensable for the replication of herpes simplex virus 1 in cell culture. J. Virol. 65:938–944 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3. Baines JD, Roizman B. 1992. The UL11 gene of herpes simplex virus 1 encodes a function that facilitates nucleocapsid envelopment and egress from cells. J. Virol. 66:5168–5174 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4. Baldick CJ, Shenk T. 1996. Proteins associated with purified human cytomegalovirus particles. J. Virol. 70:6097–6105 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5. Cantrell SR, Bresnahan WA. 2005. Interaction between the human cytomegalovirus UL82 gene product (pp71) and hDaxx regulates immediate-early gene expression and viral replication. J. Virol. 79:7792–7802 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6. Das S, Vasanji A, Pellettl PE. 2007. Three-dimensional structure of the human cytomegalovirus cytoplasmic virion assembly complex includes a reoriented secretory apparatus. J. Virol. 81:11861–11869 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7. Fossum E, et al. 2009. Evolutionarily conserved herpesviral protein interaction networks. PLoS Pathog. 5:e1000570 doi:10.1371/journal.ppat.1000570 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8. Harper AL, et al. 2010. Interaction domains of the UL16 and UL21 tegument proteins of herpes simplex virus. J. Virol. 84:2963–2971 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9. Homman-Loudiyi M, Hultenby K, Britt W, Soderberg-Naucler C. 2003. Envelopment of human cytomegalovirus occurs by budding into Golgi-derived vacuole compartments positive for gB, rab 3, trans-Golgi network 46, and mannosidase II (vol, 2003). J. Virol. 77:3191–3203 (Erratum, 77:8179.) [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10. Jones TR, Lee SW. 2004. An acidic cluster of human cytomegalovirus UL99 tegument protein is required for trafficking and function. J. Virol. 78:1488–1502 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11. Kalejta RF. 2008. Tegument proteins of human cytomegalovirus. Microbiol. Mol. Biol. Rev. 72:249–265 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12. Liu YL, et al. 2009. The tegument protein UL94 of human cytomegalovirus as a binding partner for tegument protein pp28 identified by intracellular imaging. Virology 388:68–77 [DOI] [PubMed] [Google Scholar]
- 13. Moorman NJ, Sharon-Friling R, Shenk T, Cristea IM. 2010. A targeted spatial-temporal proteomics approach implicates multiple cellular trafficking pathways in human cytomegalovirus virion maturation. Mol. Cell. Proteomics 9:851–860 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14. Pass RF. 2001. Cytomegalovirus, p 2675–2706 In Knipe DM, Howley PM. (ed), Fields virology, 4th ed, vol 2 Lippincott, Williams & Williams, Philadelphia, PA [Google Scholar]
- 15. Phillips SL, Bresnahan WA. 2011. Identification of binary interactions between human cytomegalovirus virion proteins. J. Virol. 85:440–447 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16. Phillips SL, Bresnahan WA. 2012. The human cytomegalovirus (HCMV) tegument protein UL94 is essential for secondary envelopment of HCMV virions. J. Virol. 86:2523–2532 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17. Sadaoka T, Yoshii H, Imazawa T, Yamanishi K, Mori Y. 2007. Deletion in open reading frame 49 of varicella-zoster virus reduces virus growth in human malignant melanoma cells but not in human embryonic fibroblasts. J. Virol. 81:12654–12665 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18. Sanchez V, Greis KD, Sztul E, Britt WJ. 2000. Accumulation of virion tegument and envelope proteins in a stable cytoplasmic compartment during human cytomegalovirus replication: characterization of a potential site of virus assembly. J. Virol. 74:975–986 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19. Sanchez V, Sztul E, Britt WJ. 2000. Human cytomegalovirus pp28 (UL99) localizes to a cytoplasmic compartment which overlaps the endoplasmic reticulum-Golgi-intermediate compartment. J. Virol. 74:3842–3851 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20. Schauflinger M, et al. 2011. The tegument protein UL71 of human cytomegalovirus is involved in late envelopment and affects multivesicular bodies. J. Virol. 85:3821–3832 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21. Seo JY, Britt WJ. 2007. Cytoplasmic envelopment of human cytomegalovirus requires the postlocalization function of tegument protein pp28 within the assembly compartment. J. Virol. 81:6536–6547 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22. Seo JY, Britt WJ. 2008. Multimerization of tegument protein pp28 within the assembly compartment is required for cytoplasmic envelopment of human cytomegalovirus. J. Virol. 82:6272–6287 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23. Seo JY, Britt WJ. 2006. Sequence requirements for localization of human cytomegalovirus tegument protein pp28 to the virus assembly compartment and for assembly of infectious virus. J. Virol. 80:5611–5626 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24. Silva MC, Yu QC, Enquist L, Shenk T. 2003. Human cytomegalovirus UL99-encoded pp28 is required for the cytoplasmic envelopment of tegument-associated capsids. J. Virol. 77:10594–10605 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25. Tandon R, Mocarski ES. 2008. Control of cytoplasmic maturation events by cytomegalovirus tegument protein pp150. J. Virol. 82:9433–9444 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26. Tischer BK, von Einem J, Kaufer B, Osterrieder N. 2006. Two-step Red-mediated recombination for versatile high-efficiency markerless DNA manipulation in Escherichia coli. Biotechniques 40:191–197 [DOI] [PubMed] [Google Scholar]
- 27. To A, et al. 2011. Yeast two hybrid analyses reveal novel binary interactions between human cytomegalovirus-encoded virion proteins. PLoS One 6:e17796 doi:10.1371/journal.pone.0017796 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28. Varnum SM, et al. 2004. Identification of proteins in human cytomegalovirus (HCMV) particles: the HCMV proteome. 78:10960–10966 (Erratum, 78:13395.) [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29. Vittone V, et al. 2005. Determination of interactions between tegument proteins of herpes simplex virus type 1. J. Virol. 79:9566–9571 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30. White EA, Clark CL, Sanchez V, Spector DH. 2004. Small internal deletions in the human cytomegalovirus IE2 gene result in nonviable recombinant viruses with differential defects in viral gene expression. J. Virol. 78:1817–1830 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31. Wing BA, Lee GCY, Huang ES. 1996. The human cytomegalovirus UL94 open reading frame encodes a conserved herpesvirus capsid/tegument-associated virion protein that is expressed with true late kinetics. J. Virol. 70:3339–3345 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32. Yeh PC, Meckes DG, Wills JW. 2008. Analysis of the interaction between the UL11 and UL16 tegument proteins of herpes simplex virus. J. Virol. 82:10693–10700 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33. Yu D, Smith GA, Enquist LW, Shenk T. 2002. Construction of a self-excisable bacterial artificial chromosome containing the human cytomegalovirus genome and mutagenesis of the diploid TRL/IRL13 gene. J. Virol. 76:2316–2328 [DOI] [PMC free article] [PubMed] [Google Scholar]