

Abstract
Kaposi's sarcoma-associated herpesvirus (KSHV) establishes sustained latent persistence in susceptible cells. This is dependent on the latency-associated nuclear antigen (LANA). Understanding how LANA transcription is regulated thus aids our fundamental understanding of KSHV biology. Two hundred ninety-four base pairs are sufficient to regulate LANA transcription in response to the viral RTA protein and RBPjκ. The same region controls K14/viral G-protein-coupled receptor (vGPCR) transcription in the opposite direction. We used a quantitative analysis in conjunction with specific nucleotide substitutions and defined gain-of-function and loss-of-function RTA mutants to dissect this region. We used a bidirectional reporter driving red and green luciferase to study the LANApi and K14p promoters simultaneously. This established that LANApi/K14p functions as a canonical bidirectional promoter. Both were TATA dependent. K14p was favored by ∼50-fold in this context. Eliminating the distal LANApi TATA box increased maximal output and lowered the induction threshold (T) of K14p even further. Two RBPjκ binding sites were independently required; however, at high concentrations of RTA, direct interactions with an RTA-responsive element (RRE) could complement the loss of one RBPjκ binding site. Intracellular Notch (ICN) was no longer able to activate RBPjκ in the viral context. This suggests a model whereby KSHV alters ICN-RBPjκ gene regulation. When the architecture of this pair of head-to-head RBPjκ binding sites is changed, the sites now respond exclusively to the viral transactivator RTA and no longer to the host mediator ICN.
INTRODUCTION
Kaposi's sarcoma-associated herpesvirus (KSHV) is a human oncogenic gammaherpesvirus. The KSHV genome is 137,000 bp long and encodes more than 70 open reading frames (ORFs). KSHV is associated with Kaposi's sarcoma (KS), primary effusion lymphoma (PEL), and multicentric Castleman's disease (MCD). Viral transcription is tightly regulated and can be divided into two well-defined states (22, 31). (i) During the lytic phase, the genome replicates and every viral promoter is active. (ii) During latency, the viral genome persists within the nucleus as a circular plasmid (episome) and is subject to the same regulation as human chromosomes (27, 66, 79). As a result, this minichromosome is transcriptionally silent, with the exception of some key genes: the KSHV latency locus and a few genes that respond to cell-type-specific and environmental stimuli. The KSHV latency locus encodes vital viral genes, which drive latent episome persistence: for instance, the latency-associated nuclear antigen (LANA) gene, as well as all viral microRNAs (8, 21, 24, 37, 67, 78, 82). Latent genes are central to KSHV tumorigenesis, since abrogation of LANA protein expression by small interfering RNA (siRNA) results in a loss of the KSHV plasmid and induction of apoptosis (26). Conversely, LANA expression can drive B cell hyperplasia in vivo (23, 72).
LANA, vCyc, vFLIP, the viral microRNAs (miRNAs), and kaposin are transcribed via alternative splicing from a single promoter. Other promoters can regulate kaposin, vCyc/vFLIP, and the microRNAs separately from LANA (4, 5, 8, 21, 48, 67, 71, 78). The LANA promoter ensures the coordinated expression of this KSHV latent gene cluster, including all viral microRNA. Elucidating the molecular details of this regulation can be expected to contribute significantly to our understanding of KSHV persistence and the AIDS-defining malignancies, KS and PEL. A contiguous ∼1,200-bp fragment contains all cis regulatory elements to ensure constitutive LANA promoter (LANApc) activity (21, 32–34). The LANA promoter is never methylated and is free of repressive histone marks (16, 27, 79). Thus, this locus provides the opportunity to investigate general principles of promoter structure and function in a defined genomic context; however, the situation is more complicated.
During latency, LANApc, a largely constitutive promoter, drives transcription of the LANA mRNA (Fig. 1A). It initiates transcription at position 127880. It is B cell specific in transgenic mice (33) and constitutively active in a large number of tissue culture cell lines. During reactivation, additional promoters are used. These are termed the LANApi, which is a promoter that can drive LANA transcription in response to the viral immediate-early transactivator RTA, and K14p, which is a promoter that drives a large K14/viral G-protein-coupled receptor (vGPCR) mRNA. Transcripts initiating from these two promoters have thus far been detected only in lytically reactivating cells. Nested within the LANApc untranslated region (UTR) is the bidirectional LANApi/K14p promoter (Fig. 1A), which is the subject of this study. The LANApi/K14 promoter is small (297 bp), and both transcription start sites (TSSs) are inactive during latency despite being located within an open chromatin environment. The LANApi/K14p region is part of a large class of bidirectional regulatory regions. Approximately 10% of human open reading frames (ORFs) are regulated via bidirectional promoters (50, 80, 81). Bidirectional promoters regulate two ORFs positioned 5′ to 5′ (“head to head”) on opposite DNA strands. The TSSs for the majority of these bidirectional promoters are separated by less than 400 bp. Members of bidirectional gene pairs tend to utilize shared cis regulatory elements. Another human gammaherpesvirus, Epstein-Barr virus (EBV), also exploits this mode of regulation (35).
Fig 1.

KSHV genomic organization of the LANApi and K14p TSSs. (A) Schematic depiction of the nucleotide positions (according to reference 70) spanning the LANA and K14 coding regions within the KSHV latency locus (vCyclin, vFLIP, and vGPCR are omitted for simplicity); SD, splice donor site; SA, splice acceptor site. LANApi, LANApc, and K14p TSSs as previously identified are shown. Also shown are CTCF binding sites as previously identified (76). (B) Sequence of the cloned genomic fragment used in analyses. Regulatory elements are indicated by text and boxed. Mutated nucleotides described in this report are shown in lowercase text, and nucleotide positions are indicated in the key below. Horizontal triangles indicate relative RBPjκ directionality; REV, location of the internal reversion mutant.
A number of studies have looked at either K14p or LANApi by itself (46, 52, 57, 75) but thus far not at both in the bidirectional context. The LANApi TSS is positioned 313 bp upstream of the LANA protein translation initiation site (57, 75), and the K14p TSS initiates transcription 35 bp upstream of the K14 translation initiation site on the opposite strand (18, 41, 60). The LANApi TSS utilizes a canonical TATA element (57, 75). A K14p TATA element has been predicted but not yet confirmed by functional studies. In sum, features of the LANApi/K14p pair resemble the architectural (spacing and strand identity) and functional (coexpression and coregulation) features of bidirectional promoters.
The LANApi and K14p TSSs are induced by the KSHV RTA transactivator. In fact, K14p is the most highly RTA-induced TSS in the entire KSHV genome (20). The RTA-mediated transactivation of LANApi and K14p displays a strict reliance upon two binding sites (Fig. 1B) for the human transcriptional adaptor RBPjκ (52, 57). The RTA-RBPjκ complex binds to these sites and activates the TSS. RTA functions as a multimer (6, 65). The situation is more complex, however, since RTA can also bind DNA directly through a loosely defined RTA-responsive element (RRE) (17, 73, 74) and thus can activate other viral promoters independent of RBPjκ. Further, RBPjκ can be activated by its host partner intracellular Notch (ICN) independent of any viral proteins (reviewed in reference 58). The LANApi/K14p region contains two RBPjκ elements, as well as a centrally located RRE, which could function to regulate K14p, LANApi, or both.
To further understand this regulation, we applied a quantitative model combined with single nucleotide and single amino acid mutant alleles of the cis recognition sequence elements and the RTA DNA binding domain. Transactivators (trans inputs) and their cognate sequence elements (cis inputs) coalesce to regulate mRNA production in the form of a transcriptional regulatory circuit (40). These regulatory circuits can be described in quantitative terms using the Hill function (see equation 1) (39, 40, 44, 69). This equation relates promoter output to transactivator concentration. Traditional, enzymatic studies use purified proteins and enzyme activity relates to the enzyme protein concentration. In our transfection studies, we did not know the intracellular RTA concentration, but we established that within the boundaries of our transfection series, doubling the input of a transfected expression plasmid resulted in a linear increase in the RTA protein. Hence, we can use this equation to characterize promoter behavior.
Three parameters describe the Hill function: the maximal output, measured herein as RLU s−1max; T, the induction threshold; and n, the Hill coefficient, which is a measure of cooperativity. By determining these parameters, we can make inferences about regulation and the biochemical mechanism of action.
| (1) |
This mathematical framework is well known and has also been applied to study promoter activity in transfected cells (39, 40, 44, 69). For instance, the induction threshold T determines at which activator concentration the promoter is 50% active. A promoter with a lower T will be more active at lower transactivator concentrations. The Hill coefficient, n, indicates the cooperativity of the response. A Hill coefficient of >1 indicates a high degree of cooperativity and a step-like, “all-or-nothing” response curve; a Hill coefficient of ≤1 indicates a more gradual response. The maximal output provides a measure of promoter strength. A weak promoter will produce fewer transcripts (specifically, initiation events per unit time) than a strong promoter. One way to understand maximal output is as follows: at the limit, the promoter initiates as many new transcripts per time unit as possible. Adding more specific transactivator no longer increases this rate, which is determined by how fast the general transcription factor complex can assemble and “reset” at the TATA element.
Using this framework, we investigated the LANApi/K14p response to RTA and report two new findings. (i) Our studies revealed a competitive relationship between the two TSSs, i.e., we found that one function of the LANApi TSS is to dampen the K14p response to RTA. (ii) Since LANApi/K14p contains two RBPjκ sites, we expected RTA-dependent transactivation to be highly cooperative. This was not the case. We found an unconventional utilization of the head-to-head RBPjκ element pair reminiscent of sequence-paired site (SPS) Notch signaling (1, 10, 63) but evolved to respond solely to the viral transactivator and no longer to ICN.
MATERIALS AND METHODS
Plasmids.
All plasmids were sequence verified and match nucleotide positions 127583 to 128879 of the BC-1 KSHV genome (70). Site-directed mutations are shown in Fig. 1B. LANApi and K14p single reporter constructs (pDD2002 and pDD2005, respectively) were generated by cloning the genomic portion indicated above from pDD919 (nucleotides [nt] 127583 to 127879 in pBluescript II(KS+) [Stratagene]) into pCBG68 Basic and pCBR Basic (Promega Corp.) via SmaI/HindIII and KpnI/SmaI restriction sites, respectively. The bidirectional reporter construct (pDD2000) was generated by cloning the green luciferase isoform (pCBG68 Basic) into pDD919 via HindIII/SalI restriction sites, with a secondary subcloning step to incorporate the red luciferase isoform (pCBR Basic) via ligation of blunt-ended (End-It DNA repair kit; Epicentre), SmaI/BamHI-digested pCBR Basic, with the SmaI-digested, green-only vector intermediate. The LANApi single reporter, K14p single reporter, and bidirectional reporter variants, LANApiTBP (pDD2026, -2027, and -2029_3-1), RBP A (pDD2024, -2028, and -2031), RBP B (pDD2022, -2023, and -2030), and K14pTBP mutants (pDD2013, -2015, and -2016), respectively, were generated using the GeneTailor site-directed mutagenesis system (Invitrogen) and the GeneAmp high-fidelity PCR system (Applied Biosystems). The bidirectional mutant variants, RRE (pDD2038) and 611-843 REV (pDD2034), were designed as synthetic oligonucleotides (Blue Heron Biotechnology Inc.) with flanking HindIII sites for subcloning into the HindIII self-ligated bidirectional empty vector (pDD2045). Regulatory elements were identified using the software program Alibaba 2.1 and a literature review. The wild-type (WT) ORF50 expression vector was a kind gift from J. Choe (28). 4X-RBPjκ Luc (4X-CBF Luc) was generously provided by S. D. Hayward (30). The ORF50 mutant expression vectors (ORF50 KK/EE and ORF50 R161A) were generous gifts from G. Miller (13, 15). The Flag-tagged human intracellular Notch- and Myc-tagged RTA (61)-encoding constructs were kindly provided by J. Jung. Myc-tagged ORF50 KK/EE (pDD2032) and ORF50 R161A (pDD2033) were generated using the same methods as in reference 34.
Tissue culture and transfection.
SLK cells (29) were cultured in Dulbecco's modified Eagle's medium (DMEM) supplemented with 5% fetal bovine serum, penicillin (0.05 μg/ml), and streptomycin (5 U/ml) (Invitrogen Inc.) at 37°C under 5% CO2. SLK cells were seeded at a density of 1.0 × 104 cells per well in 96-well plates (Sarstedt). The next day, transfection mixes were prepared using the RoboGo liquid handling system (Aviso) (or by hand in the case of Fig. 8 only) and then mixed with incomplete medium (DMEM without serum or antibiotic) and Superfect (Qiagen) as per the manufacturer's instructions. Total DNA was normalized with pBluescript II(KS+) (pDD223) such that a total 0.5 μg of total input DNA was transfected per well in all experiments. Similar to previous observations (75), coincident expression of ORF50 consistently led to deviations in the expression of β-galactosidase, rendering this value unreliable for normalization purposes. We therefore relied on extensive biological replicates. Transfections utilized the MWG RoboGo liquid handling system and were performed in triplicate, at least three different times. Those transfections that were performed by hand were performed in duplicate, at least two different times.
Fig 8.

Directionality of RTA-RBPjκ transactivation. (A) Depiction of the REV mutant (sequence reversal of −2 bp relative to TATA elements) in the bidirectional reporter. RRE refers to the predicted RTA direct DNA binding element. (B) LANApi output in the WT (black bars) and REV (white bars) bidirectional promoter variants in response to KKEE or WT RTA. (C) K14p output in the WT (black bars) and REV (white bars) bidirectional promoter variants in response to KKEE or WT RTA.). **, P ≤ 0.01; P values were determined by Student's t test.
Luciferase data acquisition.
Cells were lysed with 100 μl 1× cell Culture lysis reagent (Promega), undergoing gentle orbital rotation for 10 min at room temperature. Lysate was then mixed with the Chromaglo luciferase assay system (Promega) substrate or with luciferase assay system (Promega) substrate as per the manufacturer's instructions. Luciferase activity was measured using a FLUOstar Optima 96-well luminometer (BMG Labtech). Red and green signal outputs were separated as per the manufacturer's instructions using a 590-nm long-pass and 510/60-nm filter (Chroma Corp.), respectively. Luciferase activity was measured from each well for 10 s at 1-s intervals, with the final values derived by the luminometer software as the average of all interval readings (n = 10), such that the output therein was expressed as relative light units observed per second (RLU s−1obs). Filter correction was achieved using the Chroma-Luc technology calculator (Promega).
Data fitting and analysis.
Raw luciferase data in the form of RLU s−1obs was generated via titration of WT RTA, KKEE RTA, or R161A RTA, with input values of RTA-encoding expression vector ranging from 0 to 86.5, 0 to 76.9, and 0 to 76.9 nM, respectively. Because equal molecular weights were input to maintain the optimal 0.5-μg/well transfection conditions outlined above in all experiments, the different molecular amounts between the input RTA expression vectors are reflective of the differences in double-stranded molecular weights. The raw output curves were fit to the Hill function (see equation 1) via nonlinear least-squares regression (38), with three freely varying parameters: RLU s−1max, T (expressed in nM), and n (the Hill coefficient). Independent regression analyses were performed on each individual titration curve to generate independent values of RLU s−1max, T, and n for each individual trial (i.e., n ≥ 9 data points per titration curve). Global values were subsequently calculated by fitting the averaged outputs across all runs from each condition to generate a global fit, with standard error derived from the variance among individual runs. Initial analyses revealed adherence to first-order Hill kinetics (see Fig. S1 in the supplemental material); as such, we fixed the Hill coefficient to n = 1 for subsequent calculations (see equation 2) (19, 39, 64). The apparent kcat and efficiency T were calculated as described above (see equations 3 and 4). The initial concentration (Et) used to calculate the apparent kcat was defined as the concentration (in nM) of measurable input reporter construct (held constant throughout each titration curve). This definition expresses the observed output as a function of total detectable molecular quantities, and other definitions (such as potential binding sites, etc.) arbitrarily dilute this relationship. Due to the differences in molecular weight, the single reporter alone, bidirectional reporter, and single reporter in trans (1:1) thus had corresponding values of Et of 138 nM, 99 nM, and 69 nM, respectively.
Immunoblotting.
SLK cells were transfected with Myc-tagged WT RTA, Myc-tagged KKEE RTA, Myc-tagged R161A RTA, or Flag-tagged ICN expression vectors as described above. After 4°C 1× phosphate-buffered saline (PBS) rinsing, two of three wells were harvested, rinsed again, and pooled, with subsequent lysis via RIPA buffer (50 mM Tris-HCl [pH 8.0], 150 mM NaCl, 1% NP-40, 0.5% deoxycholate [DOC], 0.1% SDS, 1 mM NaVO3, 1 mM dithiothreitol [DTT], 1× protease inhibitor cocktail [Sigma], and 1 mM phenylmethylsulfonyl fluoride [PMSF]). Total protein from lysates was normalized via a bovine serum albumin (BSA) assay (Pierce) and separated on 10% SDS-PAGE followed by transfer to PVDF membrane (GE Healthcare). Membranes were blocked in 5% nonfat milk in PBST (0.1% Tween 20). Antibodies were diluted in blocking buffer at 1:1,000 for anti-Myc (Cell Signaling), 1:5,000 for anti-Flag (Sigma), 1:5,000 for anti-β-actin (Sigma), and 1:5,000 for anti-mouse horseradish peroxidase (HRP) (Vector Labs). Luciferase analysis was carried out on the remaining wells to ensure that epitope-tagged RTA variants responded similarly to responses of untagged variants (data not shown).
RESULTS
LANApi and K14p form a bidirectional promoter.
To understand LANApi and K14p, we cloned the KSHV genomic region encompassing both TSSs (nt 127583 to 127879 [70]) into a dual reporter context (Fig. 2A). The LANApi TSS drove a green-emitting luciferase isoform and the K14p TSS drove a red-emitting luciferase isoform on the same plasmid. Previous experiments by us and others (46, 52, 57, 75) always used either one or the other promoter but never investigated both TSSs within the same construct.
Fig 2.

LANApi and K14p are bidirectional promoters activation by RTA. (A) Single and bidirectional reporter constructs. LANApi directs expression of a green luciferase isoform, and K14p directs expression of an isogenic red luciferase isoform. (B) The response to RTA for LANApi in single (triangles) and bidirectional (circles) reporter input contexts. (C) The K14p response to RTA in single (triangles) and bidirectional (circles) reporter input contexts. (D) RTA expression over the range of input expression vector as assayed by Western blotting. (E) Induction threshold ± SE for each TSS.
We transfected the bidirectional-reporter vector and single-reporter controls with increasing amounts of an RTA transactivator expression construct into SLK cells. SLK cells are derived from a KS lesion but do not carry KSHV (29). As a control, we switched the isoforms in some experiments and obtained the same response curves (data not shown). We used 12 different amounts of RTA expression plasmid to obtain high-resolution response curves. Western blotting confirmed expression of RTA upon transfection (Fig. 2D).
| (2) |
To test the hypothesis that RTA cooperatively transactivates either TSS, the response (RLU s−1obs) to increasing amounts of input RTA-encoding expression vector was fit to the Hill function. We found no evidence of cooperativity (i.e., Hill coefficient ≤ 1; see Fig. S1 in the supplemental material). This was surprising for a transactivator like RTA, which functions as a dimer or tetramer. The RTA response curves exhibited first-order kinetics. These can be modeled by the simpler equation 2 (19, 64). It suggests that the RTA transactivation complex does not assemble from individual monomers cooperatively at the DNA interface but that the slowest and thus rate-determining step is RTA dimerization/tetramerization. Our data suggest that in intact cells, RTA dimerization/tetramerization happens prior to DNA binding.
Bidirectional promoter is dominated by K14p.
Both TSSs were responsive to RTA when transfected as single reporters (Fig. 2B and C, triangles) or when positioned in the bidirectional orientation (Fig. 2B and C, circles). Regardless of context, however, the K14p TSS had 10- to 100-fold-higher maximal output than the LANApi TSS. K14p was a much stronger promoter.
Since these experiments were conducted in transfected cells rather than with purified components in vitro, they take into account all molecular interactions that lead to luciferase output. Thus, rather than absolute numbers, the relative comparison of LANApi and K14p is important. We assume that the only variable in our experiments is the amount of RTA, and we conducted a series of validation experiments to support our data (see Fig. S2 in the supplemental material). Each data point is the result of three technical replicates. Each titration experiment was conducted in at least three biological replicates on different days. We switched the colored luciferase isoforms to verify that the response curves were defined by the cis elements rather than the reporter. We conducted real-time quantitative PCR (qPCR) for each luciferase reporter mRNA to show that the level of luciferase activity was linearly correlated with the level of mRNA and that the amount of transfected reporter DNA was the same among all replicates (see Fig. S2 in the supplemental material). These experiments validated that the luciferase readings are an accurate reflection of TSS activity.
Because the RTA response could be described by a first-order kinetic fit, we were able to calculate the parameters which characterize such a response. The first is kcat, as shown in equation 3 (25):
| (3) |
kcat describes the promoter behavior at saturating levels of a specific transactivator. A higher kcat value indicates more transcription initiation events per unit time. In kinetic studies, it relates the maximal output to the enzyme concentration (Et). Here, calculating an apparent kcat allowed us to directly compare experiments that used the bidirectional promoter and experiments that used the single reporter (as well as mutants) and also to normalize across biological replicates that transfect different amounts of luciferase reporter constructs.
K14p had a kcat value of 713 ± 64 RLU s−1, and LANApi had a kcat value of 175 ± 32 RLU s−1 (Fig. 3A), verifying the differential promoter strength we observed when analyzing the raw data in each individual experiment.
Fig 3.

RTA transactivation based on fit to noncooperative response model (Hill constant = 1). (A) The maximal output is shown for each TSS when assayed as single reporters alone (black bars) or in the bidirectional reporter (white bars). (B) The efficiency of RTA-mediated induction is shown for each TSS as single reporters (black bars) or in the bidirectional reporter (white bars). “*” indicates P ≤ 0.05; “**” indicates P ≤ 0.01. P values were determined by Student's t test.
LANApi TATA element limits K14p activity.
We observed a consistent increase in the induction threshold T (Fig. 2E) for both TSSs when assayed in the bidirectional context compared to results in the single-reporter context. For K14p, the induction threshold T shifted from 4.2 nM input vector DNA for the single reporter to 13.1 nM in the bidirectional-reporter context. For LANApi, the induction threshold T shifted from 7.4 nM for the single-reporter to 33.7 nM in the bidirectional-reporter context.
A second, very informative and perhaps more relevant parameter (2, 25) is the ratio of kcat to T (see equation 4). This ratio is the promoter efficiency. It can be thought of as promoter activity at physiological levels, i.e., at and around the induction threshold T (the analog to T in enzyme biochemistry is the Km, though that strictly applies only to purified proteins). It is the most biologically relevant comparator among promoters. A promoter can be very strong (high kcat), but if the induction threshold T is high, it will yield little output at low concentrations of transactivator. Conversely, a promoter can be very sensitive, i.e., become active at very low concentrations of transactivator, and still not yield much output if kcat is low.
| (4) |
For each LANApi and K14p, the efficiency was much lower in the bidirectional context than in the single-reporter context (Fig. 3B, compare filled bars to empty bars). This reduction in efficiency when assayed in the bidirectional promoter construct compared to the single-reporter context was statistically significant, with a P value of ≤0.005 for LANApi and a P value of ≤0.05 for K14p using Student's t test. Similar differences were observed regardless of the luciferase isoform assayed (data not shown). These results provided the first indication that in the bidirectional context, more RTA is required for induction than in the single-reporter context. One model to explain this difference proposes that both TSSs compete for a central common RTA binding site. In the single-reporter context, the distal TSS is not functional. Even though a preinitiation complex may assemble, there is no ORF to transcribe and no poly(A) site to support efficient transcription. In the bidirectional reporter, preinitiation complexes assemble around both TATA elements and now can compete for the arriving RTA complex. This model predicts that the distal TATA element negatively regulates promoter activity, i.e., the LANApi TATA would inhibit K14p and vice versa (Fig. 4A).
Fig 4.

The LANApi-proximal TATA box binding protein (LANApi TATA) element limits K14p output. (A) Schematic illustrating the positions of the LANApi and K14p-proximal TATA elements (LANApi TATA [triangle] and K14pTATA [diamond], respectively) that were mutated in the bidirectional reporter construct. (B) LANApi response to RTA in the presence (WT; cross) or absence of TSS-proximal (LANApi TATA; triangles) or TSS-distal (K14p TATA; diamonds) elements. (C) K14p response to WT RTA in the presence (WT, X's) or absence of TSS-proximal (K14p TATA; diamonds) or TSS-distal (LANApi TATA; triangles) elements. Note the 10-fold change in scale between panel B and panel C.
To test this hypothesis, we inactivated the predicted TATA elements through site-directed mutagenesis. LANApi activity was dependent on a proximal TATA element. If we mutated the proximal LANApi TATA element, activity was abolished (Fig. 4B, arrow and red triangles). K14p activity also was dependent upon the proximal TATA element (Fig. 4C, gray diamonds). Note that panels B and C have different scales to account for the different overall activities. These experiments confirm the LANApi TATA element and define the K14p TATA element, which had been predicted but was not previously experimentally verified.
Deletion of the distal, (LANApi) TATA element dramatically increased the activity of K14p (Fig. 4C, red triangles and arrow). This observation is consistent with the aforementioned model in which the distal LANApi element inhibits K14p in the context of the bidirectional promoter. By comparison, deletion of the distal (K14p) TATA element only marginally affected LANApi activity (Fig. 4B, gray diamonds). This observation introduces asymmetry into the model. The LANApi TATA element inhibits the K14p TSS, but the K14p TATA element has no impact on the LANApi TSS. In the absence of the LANApi TATA element, the K14p promoter became hyperresponsive to RTA. This prompted us to hypothesize that a second purpose of LANApi was the inhibition of K14p, more than driving the expression of the LANA protein.
Direct DNA binding by RTA augments promoter efficiency.
Binding of RTA to the bidirectional LANApi/K14p region is mediated by RBPjκ, which is the essential downstream mediator of Notch signaling (reviewed in reference 42). Two RBPjκ elements, RBP A and RBP B, mediate the response to RTA (Fig. 5A) (52, 57). To verify these observations, we mutated each in the context of the bidirectional reporter construct. As predicted, mutation of either RBPjκ element abolished promoter activity (Fig. 5B and C; please note the 10-fold difference in scale between the two panels). This demonstrates that both RBP elements were necessary.
Fig 5.

Both RBPjκ elements are essential. (A) Schematic depicting the position of the LANApi- and K14p-proximal RBPjκ elements (RBP A [circle] and RBP B [square], respectively) mutated for analysis in the bidirectional reporter construct. (B) LANApi response to RTA in the presence (WT; crosses) or absence of TSS-proximal (RBP A; circles) or TSS-distal (RBP B; squares) RBPjκ elements. (C) K14p response to WT RTA in the presence (WT; Xs) or absence of TSS-proximal (RBP B; squares) or TSS-distal (RBP A; circles) elements. Note the change in scale between panels 5B and C.
In addition to RBPjκ-mediated DNA recognition, RTA can also bind DNA directly (9, 15, 51, 65, 68). To test the hypothesis that direct RTA-DNA interactions were important, we used an RTA mutant with enhanced DNA binding capacity, called KKEE (13, 15). KKEE RTA increased the activity for both LANApi and K14p TSSs in the context of the bidirectional reporter. The maximal output increased from the WT level of ∼17,000 to ∼130,000 RLU s−1 for the LANApi promoter and from the WT RTA level of ∼70,000 to ∼880,000 RLU s−1 for the K14p. This translated into an increase in kcat of 7.3-fold for LANApi and 12.4-fold for K14p. In contrast, another mutant, RTA R161A, which is capable of interaction with RBPjκ (12) but no longer binds DNA directly, decreased the efficiency for each TSS compared to results with wild-type RTA (Fig. 6A and D). This demonstrated that in addition to binding via RBPjκ, RTA binds directly to a binding site within K14p/LANApi and that this RTA-DNA interaction modulates the activation efficiency of either TSS.
Fig 6.

Enhanced DNA binding by RTA augments promoter activity. (A) Log LANApi efficiency in response to increased RTA DNA binding (KKEE RTA) or loss of RTA DNA binding (R161A RTA). (B and C) Efficiency of the LANApi response to KKEE, WT, or R161A RTA in the absence of proximal LANApi TATA (B) or distal K14p TATA (C). Note that in the absence of the proximal TATA element (B), we did not see significant reporter activity. (D) K14p efficiency in response to increased RTA DNA binding (KKEE RTA) or loss of RTA DNA binding (R161A RTA). (E and F) Efficiency of the K14p response to KKEE, WT, or R161A RTA in the absence of distal LANApi TATA (E) or proximal K14p TATA (F). Note that in the absence of the proximal TATA element (F), we did not see significant reporter activity. *, P ≤ 0.05; ** P ≤ 0.01; ****, P ≤ 0.001; P values were determined by Student's t test, and dotted lines indicate mean WT efficiency.
To test the hypothesis that the KKEE mutant phenotype was dependent on an intact promoter, we analyzed TSS efficiency for the TATA box mutants. Enhanced DNA binding by RTA could not compensate for a mutated proximal TATA box, presumably because there was no RNA polymerase II (Pol II) complex to activate in the first place. We recorded only a minimal signal (Fig. 6B and F). The DNA binding mutant RTA R161A was worse than the wild type under all conditions tested. As before, we observed an increase in K14p efficiency if the distal TATA box was compromised (Fig. 6D and E). This was not the case for LANApi (Fig. 6A and C). More subtle differences were also evident. In the wild-type situation, the KKEE mutant enhanced efficiency (Fig. 6A and D); however, in the context of a mutated distal TATA element (Fig. 6E and C), this was no longer the case. In sum, the hypermorphic phenotype of the KKEE mutant can be attributed solely to increased DNA binding. The dependency on the general transcription complex remains.
To test the hypothesis that at saturating concentrations RTA no longer depends on its own DNA binding activity, we used the R161A mutant, which is deficient in binding to the RTA-responsive element (RRE) but still binds to RBPjκ. First, we analyzed RTA R161A in the context of the wild-type promoter. There was no significant difference between the WT and the R161A mutant (Fig. 7A, and D, yellow bars). As expected, the KKEE mutant had increased activity (Fig. 7A and D, blue bars). The maximal output was 26,458 ± 3,993 RLU s−1 for the R161 mutant, compared to 17,275 ± 3,181 RLU s−1 for the WT RTA for LANApi and 80,527 ± 9,735 RLU s−1 for the R161 mutant for K14p, compared to 70,439 ± 6,347 RLU s−1 for the WT RTA for K14p. Thus, direct RTA-DNA interactions are not required at saturating concentrations of RTA. All contacts are established through RBPJκ.
Fig 7.

At saturating levels, RTA DNA binding becomes dispensable. (A) Normalized LANApi maximal output (analogous to kcat for enzymes) in response to increased RTA DNA binding (KKEE RTA) or loss of RTA DNA binding (R161A RTA) mutant. (B and C) LANApi response in the absence of proximal RBP A (B) or distal RBP B (C). (D) Normalized K14p maximal output in response to increased RTA DNA binding (KKEE RTA) or loss of RTA DNA binding (R161A RTA). (E and F) K14p response to KKEE, WT, or R161A RTA in the absence of distal RBP A (E) or proximal RBP B (F). The red star indicates a significant (P ≤ 0.05) difference between WT and R161A RTA only in K14p with a mutated RBP A element (E). *, P ≤ 0.05; **, P ≤ 0.01; ***, P ≤ 0.005; ****, P ≤ 0.001. P values were determined by Student's t test; dotted lines indicate the mean WT promoter response to WT RTA.
What would happen in the absence of an RBPjκ binding element? Inactivation of either RBP A or RBP B severely reduces the activation by RTA. This was the main message of the results shown in Fig. 5. However, the power of our quantitative analysis and the large number of replicates and dose steps allows us to compare accurate parameters, such as maximal output, shown in Fig. 7, among promoters of vastly different activities. At saturating concentrations, the DNA binding mutant R161 was no worse than the wild type on the LANApi promoter. (Fig. 7B and C, yellow). The situation was different for K14p (Fig. 7E and F, yellow). In the absence of a functional RBP A site, R161A RTA had a lower maximal output than the WT on K14p (Fig. 7E, red star). In the absence of a functional RBP B site, which is proximal to K14p, the R161A DNA binding mutant was identical to the wild type. This suggests that the RRE contributes to the activity of K14p. Note that loss of any one RBP site severely cripples the promoter. Hence, this difference should not be overinterpreted.
In each case the KKEE mutant could substitute for the loss of an RBP site (Fig. 7, blue bars). In the context of a wild-type promoter, KKEE RTA was better than WT RTA, as previously observed. In the context of an RBP-deficient promoter, KKEE RTA was able to complement the cis defect and restore kcat to wild-type levels. These data demonstrate that improved interactions between RTA and an RRE DNA element in the case of KKEE RTA can complement for a loss of interaction between RTA-RBPjκ and a single RBP site.
The absence of functional DNA binding by RTA increased the activation threshold for both TSSs. The induction threshold was 126.4 ± 2.7 nM for the R161 expression plasmid, compared to 33.7 ± 8.3 nM for the WT for the LANApi TSS, and 80 ± 14.2 nM for the R161 mutant, compared to 13.1 ± 5.5 nM for the WT for the K14p TSS. Of note, R161 also makes normal RBPjκ interactions. This verifies two aspects of our model: (i) direct RTA-DNA contacts increase the responsiveness of these TSSs at low concentrations of RTA, and (ii) at saturating concentrations, the direct RTA-DNA contacts can complement RBPjκ element defects.
The direct RTA-DNA contact is mediated by a regulatory element, termed the RTA Response Element (RRE), which is present between the two RBP elements (Fig. 8A). Previously this RRE element was shown to have less of an impact upon transactivation than the RBPjκ elements (52, 57). We confirmed these observations. Loss of the RRE site led to equivalent reductions in output for both TSSs when assayed with any RTA variant (data not shown). This suggests that three cis elements, RBP A, RBP B, and RRE, together define the response behavior of the K14/LANApi bidirectional promoter.
Sequence architecture between TBP elements provided directionality for RBPjκ transactivation.
Since RTA binding sites function in tandem, transactivation could in principle act toward either side. In isolation, RTA-RBPjκ can bind to and activate promoters independent of orientation (51). In contrast, we hypothesized that spacing of the RBPjκ elements imparted directionality to the bidirectional KSHV promoter. To test this hypothesis, we switched the orientation of the intervening sequence between the two TATA elements (Fig. 8A). We then cotransfected either the WT or KEEE RTA at a 1:1 molar ratio. Reversing the sequence of binding sites relative to LANApi increased the output of LANApi (Fig. 8B). This was true for either mutant. KKEE RTA in this experiment served as a better RTA. The reverse was not true. Reversing the sequence of binding sites relative to K14p did not increase the output of K14p (Fig. 8C). This demonstrates that the specific orientation and distance of the two RBPjκ binding sites and the RRE element confer preferential activation of the K14p TSS.
RBPjκ binding sites are RTA specific and insensitive to Notch.
Given the stringent reliance of this viral promoter upon the effector of Notch signaling, RBPjκ, we hypothesized that overexpression of the host ligand might also activate these TSSs. The host ligand for RBPjκ is the transcriptionally active, cleaved form of the human Notch protein: intracellular Notch (ICN). Unlike RTA, ICN does not have RBPjκ-independent DNA binding activity and thus cannot engage the RRE. We transfected cells with LANApi, K14p, or a positive control consisting of 4 RBPjκ sites. The positive control containing four RBPjκ sites (31) responded as expected (Fig. 9A); however, neither K14p nor LANApi responded to activation by ICN. Western blotting confirmed ICN expression (Fig. 9B). Thus, even though the RBPjκ elements are essential with regard to RTA-mediated induction of either TSS, their orientation within the viral promoter can no longer use ICN.
Fig 9.

Response to intracellular Notch (ICN). (A) Fold change in relative luciferase activity in response to ICN for each TSS in the bidirectional reporter (LANApi, circles; K14p, triangles) or the TSS of positive control 4X-RBPjκ Luc (diamonds). (B) Expression of ICN, detected by Western blotting. (C) Illustration of the unequal RBPjκ head-to-head spacing within the LANApi/K14p bidirectional promoter. Shown are the distances in bp between the two RBP elements (top) and between each RBP element and the central RRE. Also shown are the two essential TATA elements for each promoter.
We had observed a similar phenotype when we profiled KSHV gene expression in response to ICN and RTA (11). One possibility to explain this phenotype comes from recent observations that revealed a requirement for ICN target loci to have a head-to-head, paired RBP element orientation as well as strict spacing requirements (26–28). In human promoters the canonical architecture is spatially restricted to a maximum distance of ∼15 to 19 bp between adjacent RBPjκ sites. In contrast, 79 base pairs bridge the two head-to-head RBPjκ sites in the KSHV promoter (Fig. 9C). By extending the sequence spacing, the RBPjκ pair can no longer be bridged by ICN but requires multimers of a different transactivator: RTA.
DISCUSSION
The KSHV latency control region plays multiple roles during the viral life cycle. During de novo infection, the immediate-early transactivator RTA is brought in with the virion (3, 46). This results in an initial burst of lytic and latent transcription (43). During this phase, RTA drives LANA and K14/vGPCR protein expression from the lytic LANApi and K14/vGPCR promoters (54). Eventually the constitutive LANA promoter takes over. It is active independent of RTA or other viral proteins (33, 34). As LANA protein accumulates, it also inhibits RTA (36, 45). This leads to a stable autoregulatory loop during which LANA activates its own promoter (32, 34). Reexpression of RTA is necessary and sufficient for KSHV reactivation (55, 56, 77, 84). Yet LANA is dispensable for KSHV reactivation and replication in KSHV and related gammaherpesviruses (7, 49, 53, 59, 83). Perhaps LANApi evolved not to regulate LANA protein expression during lytic replication. Perhaps the unique architecture of two divergent RTA-dependent TSSs in such close proximity reveals a novel molecular mechanism of RTA/RBPjκ/ICN regulation. To investigate these conjectures, we studied the regulation of LANApi and of K14p in quantitative terms.
This report demonstrates that the LANApi/K14p locus constitutes a bona fide KSHV bidirectional promoter. This is the first functional verification of a bidirectional promoter within the KSHV genome; others are predicted (I. B. Hilton, D. Wang, and D. P. Dittmer, unpublished). Bidirectional transcription has been identified in Epstein-Barr-Virus (EBV) (35, 47). It was studied in yeast and other eukaryotic model systems (62). By applying the same quantitative framework to KSHV, we have generated a detailed understanding of the functional roles that cis (regulatory elements) and trans (DNA binding) factors play in the transactivation of this genetic circuit. Each TSS (LANApi, K14p) is responsive to RTA when positioned on the same reporter. At least three shared RTA-responsive elements (RBP A, RBP B, and RRE) modulate this response. There exists a selective advantage for the K14p TSS. K14p is 10× to 100× more active than LANApi.
This report demonstrates a competitive relationship between the two TATA elements. This is reflected in a decreased efficiency for each TSS in the bidirectional reporter relative to the single-reporter-only input condition. The distal LANApi TATA element negatively regulates and limits K14p. A similar phenotype has been seen for some bidirectional promoters in yeast (62). Here too, mutation of one TATA element resulted in upregulation of the other. In the majority of those cases, however, the second distal TATA box initiates a noncoding or “cryptic” transcript. In the case of KSHV LANApi, the resulting transcript has the potential to encode the entire LANA ORF. We speculate that perhaps the LANApi TATA box evolved for two purposes: first, to drive transcription of the LANA protein during lytic replication, but more importantly to fine-tune the expression of K14 and vGPCR.
The interaction between RTA and RBPjκ is well established (12, 46, 51, 52, 54, 57, 65, 68, 75). Biochemical evidence suggests that RTA functions as a tetramer. This higher-order RTA complex then stabilizes RBPjκ on the DNA and changes conformation such that the transactivation event takes place (6, 9, 14, 65). Here we have shown that while both RBP elements are critical, their importance is differential. The RBP B element is located 71 bp from the K14p TATA element (Fig. 10). The RBP A element is located only 7 bp from the LANApi TATA element. This introduces asymmetry. The asymmetry is also reflected in the spacing between the RRE and the RBPjκ elements. The RRE is spaced 17 bp away from the K14p-proximal RBP B element. This is the same spacing as for ICN-dependent RBPjκ pairs. In contrast, the RRE is spaced 50 bp away from the LANApi-proximal RBP A element. The net result is that K14p is 10 to 15 times more active at physiological, low concentrations of RTA. Last, we show that ICN no longer activates these paired RBP sites, because of extended spacing, and that now RTA is required as the bridging factor.
Fig 10.
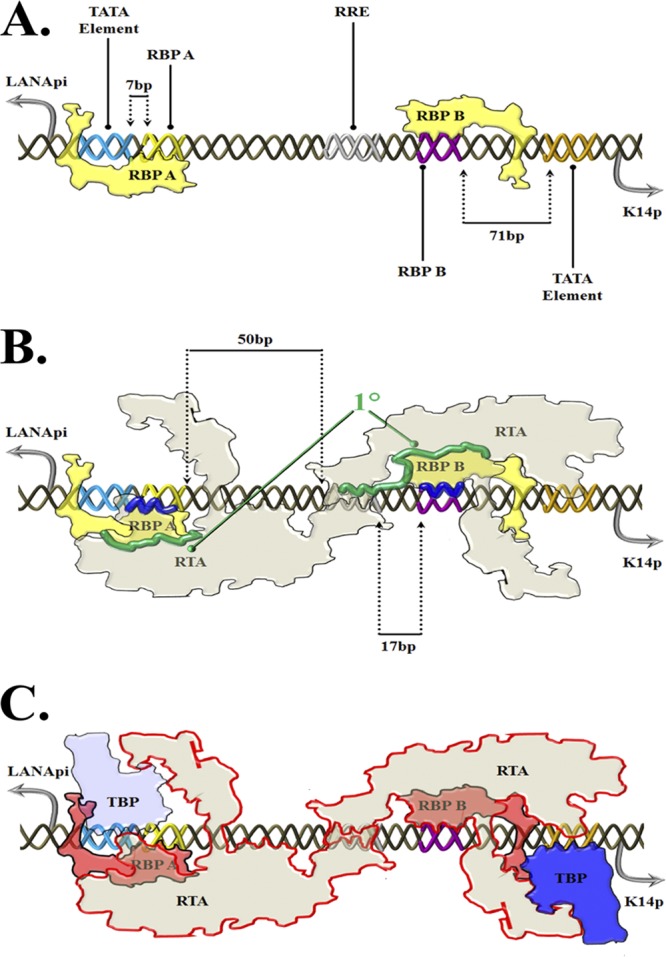
Summary and putative model. Shown is an artist's rendition of a general model of RTA/RBPjκ binding to bidirectional LANApi/K14p. (A) “Ground state.” RBPjκ elements are dynamically bound. RBPjκ complex bound to the RBP A cis site represses LANApi via shielding of the LANApi TATA cis element due to immediate (7 bp) proximity. RBPjκ complex bound to the RBP B cis site does not repress K14p because of distance (71 bp). (B) “Recognition event.” The interaction between the DNA surface and RTA is guided by interaction with both RBPjκ elements (1°; green) and the RTA protein complexes (presumably tetramer or higher [65]). Note that the RRE cis element also is recognized by the RTA protein complex. The distance between the RRE cis element and the RBP A cis element is 50 bp; that between the RRE and RBP B is only 17 bp. (C) “Activation state.” We envision a third state accompanied by overall conformational changes upon recruitment of the TBP complex. This is mediated by RTA DNA binding-dependent selective stabilization of proximal RBPjκ elements and is conveyed via low (LANApi)- or high (K14p)-activity TATA elements. Structural and cis regulatory discrepancies result in a suboptimal (LANApi) and optimal (K14p) asymmetric transactivation exclusively via RTA. The abbreviations are as follows: TATA, DNA consensus sequence element; TBP, TATA binding complex consisting of TATA binding protein and TAFs; RRE, DNA sequence element, RBPA/B RBPjκ complex consisting of one or more RBPjκ proteins and associated, undetermined cellular factors.
Taken together, we suggest the following model (Fig. 10). Based on prior work on RBPjκ (58), we assume that in the ground state RBPjκ exists as a transcriptional repressor of both the LANApi and K14p TSSs (Fig. 10A). RBPjκ bound to RBP A effectively occludes other molecules from binding the LANApi TATA element due to extreme proximity (7 bp). In contrast, 71 base pairs separate the RBP B element from the K14p TATA element. Thus, the repression mediated by RBP B is less potent, resulting in a preassembly of the basal machinery at the K14p TSS. The RTA complex (likely tetrameric) then interfaces with the RBPjκ-bound DNA using the RRE to position the higher-order complex in favor of RBP B (Fig. 10B). Only 17 bp separates RRE from RBP B, compared to 50 bp which separates RRE from RBP A. This asymmetric assembly leads to an approximately 10-fold advantage for the K14p TSS over LANApi (Fig. 10C).
Since both LANA and vGPCR mediate important phenotypes in KSHV persistence, transmission, and tumorigenesis, the detailed dissection of this regulatory unit may contribute to our overall understanding of KSHV biology and gene regulation.
Supplementary Material
ACKNOWLEDGMENTS
We thank Nat Moorman, Blossom Damania, and Tim Elston for insightful discussions. We thank Mindy Feng for assistance with robotic hardware.
Generous donations of reagents were provided by George Miller, Diane Hayward, and Jae Jung. This work was supported by Public Health Service grant CA109232 to D.P.D. I.B.H. was supported by virology training grant T32AI007419.
Footnotes
Published ahead of print 27 June 2012
Supplemental material for this article may be found at http://jvi.asm.org/.
REFERENCES
- 1. Arnett KL, et al. 2010. Structural and mechanistic insights into cooperative assembly of dimeric Notch transcription complexes. Nat. Struct. Mol. Biol. 17:1312–1317 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2. Bauer C, Osman AM, Cercignani G, Gialluca N, Paolini M. 2001. A unified theory of enzyme kinetics based upon the systematic analysis of the variations of k(cat), K(M), and k(cat)/K(M) and the relevant DeltaG(0 not equal) values—possible implications in chemotherapy and biotechnology. Biochem. Pharmacol. 61:1049–1055 [DOI] [PubMed] [Google Scholar]
- 3. Bechtel JT, Winant RC, Ganem D. 2005. Host and viral proteins in the virion of Kaposi's sarcoma-associated herpesvirus. J. Virol. 79:4952–4964 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4. Bieleski L, Hindley C, Talbot SJ. 2004. A polypyrimidine tract facilitates the expression of Kaposi's sarcoma-associated herpesvirus vFLIP through an internal ribosome entry site. J. Gen. Virol. 85:615–620 [DOI] [PubMed] [Google Scholar]
- 5. Bieleski L, Talbot SJ. 2001. Kaposi's sarcoma-associated herpesvirus vCyclin open reading frame contains an internal ribosome entry site. J. Virol. 75:1864–1869 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6. Bu W, Carroll KD, Palmeri D, Lukac DM. 2007. Kaposi's sarcoma-associated herpesvirus/human herpesvirus 8 ORF50/Rta lytic switch protein functions as a tetramer. J. Virol. 81:5788–5806 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7. Budt M, Hristozova T, Hille G, Berger K, Brune W. 2011. Construction of a lytically replicating Kaposi's sarcoma-associated herpesvirus. J. Virol. 85:10415–10420 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8. Cai X, Cullen BR. 2006. Transcriptional origin of Kaposi's sarcoma-associated herpesvirus microRNAs. J. Virol. 80:2234–2242 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9. Carroll KD, et al. 2006. Kaposi's sarcoma-associated herpesvirus lytic switch protein stimulates DNA binding of RBP-Jk/CSL to activate the Notch pathway. J. Virol. 80:9697–9709 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10. Cave JW, Loh F, Surpris JW, Xia L, Caudy MA. 2005. A DNA transcription code for cell-specific gene activation by notch signaling. Curr. Biol. 15:94–104 [DOI] [PubMed] [Google Scholar]
- 11. Chang H, Dittmer DP, Shin YC, Hong Y, Jung JU. 2005. Role of Notch signal transduction in Kaposi's sarcoma-associated herpesvirus gene expression. J. Virol. 79:14371–14382 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12. Chang PJ, Boonsiri J, Wang SS, Chen LY, Miller G. 2010. Binding of RBP-Jkappa (CSL) protein to the promoter of the Kaposi's sarcoma-associated herpesvirus ORF47 (gL) gene is a critical but not sufficient determinant of transactivation by ORF50 protein. Virology 398:38–48 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13. Chang PJ, Miller G. 2004. Autoregulation of DNA binding and protein stability of Kaposi's sarcoma-associated herpesvirus ORF50 protein. J. Virol. 78:10657–10673 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14. Chang PJ, Shedd D, Miller G. 2008. A mobile functional region of Kaposi's sarcoma-associated herpesvirus ORF50 protein independently regulates DNA binding and protein abundance. J. Virol. 82:9700–9716 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15. Chang PJ, Shedd D, Miller G. 2005. Two subclasses of Kaposi's sarcoma-associated herpesvirus lytic cycle promoters distinguished by open reading frame 50 mutant proteins that are deficient in binding to DNA. J. Virol. 79:8750–8763 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16. Chen J, et al. 2001. Activation of latent Kaposi's sarcoma-associated herpesvirus by demethylation of the promoter of the lytic transactivator. Proc. Natl. Acad. Sci. U. S. A. 98:4119–4124 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17. Chen J, Ye F, Xie J, Kuhne K, Gao SJ. 2009. Genome-wide identification of binding sites for Kaposi's sarcoma-associated herpesvirus lytic switch protein, RTA. Virology 386:290–302 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18. Chiou CJ, et al. 2002. Patterns of gene expression and a transactivation function exhibited by the vGCR (ORF74) chemokine receptor protein of Kaposi's sarcoma-associated herpesvirus. J. Virol. 76:3421–3439 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19. Chow CC, Ong KM, Dougherty EJ, Simons SS., Jr 2011. Inferring mechanisms from dose-response curves. Methods Enzymol. 487:465–483 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20. Damania B, et al. 2004. Comparison of the Rta/Orf50 transactivator proteins of gamma-2-herpesviruses. J. Virol. 78:5491–5499 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21. Dittmer D, et al. 1998. A cluster of latently expressed genes in Kaposi's sarcoma-associated herpesvirus. J. Virol. 72:8309–8315 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22. Dittmer DP. 2003. Transcription profile of Kaposi's sarcoma-associated herpesvirus in primary Kaposi's sarcoma lesions as determined by real-time PCR arrays. Cancer Res. 63:2010–2015 [PubMed] [Google Scholar]
- 23. Fakhari FD, Jeong JH, Kanan Y, Dittmer DP. 2006. The latency-associated nuclear antigen of Kaposi sarcoma-associated herpesvirus induces B cell hyperplasia and lymphoma. J. Clin. Invest. 116:735–742 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24. Ganem D. 2010. KSHV and the pathogenesis of Kaposi sarcoma: listening to human biology and medicine. J. Clin. Invest. 120:939–949 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25. Garrett R, Grisham CM. 2010. Biochemistry, 4th ed Brooks/Cole, Cengage Learning, Belmont, CA [Google Scholar]
- 26. Godfrey A, Anderson J, Papanastasiou A, Takeuchi Y, Boshoff C. 2005. Inhibiting primary effusion lymphoma by lentiviral vectors encoding short hairpin RNA. Blood 105:2510–2518 [DOI] [PubMed] [Google Scholar]
- 27. Gunther T, Grundhoff A. 2010. The epigenetic landscape of latent Kaposi sarcoma-associated herpesvirus genomes. PLoS Pathog. 6:e1000935 doi:10.1371/journal.ppat.1000935 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28. Gwack Y, Byun H, Hwang S, Lim C, Choe J. 2001. CREB-binding protein and histone deacetylase regulate the transcriptional activity of Kaposi's sarcoma-associated herpesvirus open reading frame 50. J. Virol. 75:1909–1917 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29. Herndier BG, et al. 1994. Characterization of a human Kaposi's sarcoma cell line that induces angiogenic tumors in animals. AIDS 8:575–581 [DOI] [PubMed] [Google Scholar]
- 30. Hsieh JJ, et al. 1996. Truncated mammalian Notch1 activates CBF1/RBPJk-repressed genes by a mechanism resembling that of Epstein-Barr virus EBNA2. Mol. Cell. Biol. 16:952–959 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31. Jenner RG, Alba MM, Boshoff C, Kellam P. 2001. Kaposi's sarcoma-associated herpesvirus latent and lytic gene expression as revealed by DNA arrays. J. Virol. 75:891–902 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32. Jeong J, Papin J, Dittmer D. 2001. Differential regulation of the overlapping Kaposi's sarcoma-associated herpesvirus vGCR (orf74) and LANA (orf73) promoters. J. Virol. 75:1798–1807 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33. Jeong JH, Hines-Boykin R, Ash JD, Dittmer DP. 2002. Tissue specificity of the Kaposi's sarcoma-associated herpesvirus latent nuclear antigen (LANA/orf73) promoter in transgenic mice. J. Virol. 76:11024–11032 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34. Jeong JH, et al. 2004. Regulation and autoregulation of the promoter for the latency-associated nuclear antigen of Kaposi's sarcoma-associated herpesvirus. J. Biol. Chem. 279:16822–16831 [DOI] [PubMed] [Google Scholar]
- 35. Jimenez-Ramirez C, et al. 2006. Epstein-Barr virus EBNA-3C is targeted to and regulates expression from the bidirectional LMP-1/2B promoter. J. Virol. 80:11200–11208 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36. Jin Y, et al. 2012. Carboxyl-terminal amino acids 1052 to 1082 of the latency-associated nuclear antigen (LANA) interact with RBP-Jkappa and are responsible for LANA-mediated RTA repression. J. Virol. 86:4956–4969 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37. Kang H, Lieberman PM. 2009. Cell cycle control of Kaposi's sarcoma-associated herpesvirus latency transcription by CTCF-cohesin interactions. J. Virol. 83:6199–6210 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38. Kemmer G, Keller S. 2010. Nonlinear least-squares data fitting in Excel spreadsheets. Nat. Protoc. 5:267–281 [DOI] [PubMed] [Google Scholar]
- 39. Kim HD, O'Shea EK. 2008. A quantitative model of transcription factor-activated gene expression. Nat. Struct. Mol. Biol. 15:1192–1198 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40. Kim HD, Shay T, O'Shea EK, Regev A. 2009. Transcriptional regulatory circuits: predicting numbers from alphabets. Science 325:429–432 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41. Kirshner JR, Staskus K, Haase A, Lagunoff M, Ganem D. 1999. Expression of the open reading frame 74 (G-protein-coupled receptor) gene of Kaposi's sarcoma (KS)-associated herpesvirus: implications for KS pathogenesis. J. Virol. 73:6006–6014 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42. Kovall RA, Blacklow SC. 2010. Mechanistic insights into Notch receptor signaling from structural and biochemical studies. Curr. Top. Dev. Biol. 92:31–71 [DOI] [PubMed] [Google Scholar]
- 43. Krishnan HH, et al. 2004. Concurrent expression of latent and a limited number of lytic genes with immune modulation and antiapoptotic function by Kaposi's sarcoma-associated herpesvirus early during infection of primary endothelial and fibroblast cells and subsequent decline of lytic gene expression. J. Virol. 78:3601–3620 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44. Kuhlman T, Zhang Z, Saier MH, Jr, Hwa T. 2007. Combinatorial transcriptional control of the lactose operon of Escherichia coli. Proc. Natl. Acad. Sci. U. S. A. 104:6043–6048 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 45. Lan K, Kuppers DA, Verma SC, Robertson ES. 2004. Kaposi's sarcoma-associated herpesvirus-encoded latency-associated nuclear antigen inhibits lytic replication by targeting Rta: a potential mechanism for virus-mediated control of latency. J. Virol. 78:6585–6594 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 46. Lan K, et al. 2005. Induction of Kaposi's sarcoma-associated herpesvirus latency-associated nuclear antigen by the lytic transactivator RTA: a novel mechanism for establishment of latency. J. Virol. 79:7453–7465 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 47. Laux G, Economou A, Farrell PJ. 1989. The terminal protein gene 2 of Epstein-Barr virus is transcribed from a bidirectional latent promoter region. J. Gen. Virol. 70:3079–3084 [DOI] [PubMed] [Google Scholar]
- 48. Li H, Komatsu T, Dezube BJ, Kaye KM. 2002. The Kaposi's sarcoma-associated herpesvirus K12 transcript from a primary effusion lymphoma contains complex repeat elements, is spliced, and initiates from a novel promoter. J. Virol. 76:11880–11888 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 49. Li Q, Zhou F, Ye F, Gao SJ. 2008. Genetic disruption of KSHV major latent nuclear antigen LANA enhances viral lytic transcriptional program. Virology 379:234–244 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 50. Li YY, et al. 2006. Systematic analysis of head-to-head gene organization: evolutionary conservation and potential biological relevance. PLoS Comput. Biol. 2:e74 doi:10.1371/journal.pcbi.0020074 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 51. Liang Y, Chang J, Lynch SJ, Lukac DM, Ganem D. 2002. The lytic switch protein of KSHV activates gene expression via functional interaction with RBP-Jkappa (CSL), the target of the Notch signaling pathway. Genes Dev. 16:1977–1989 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 52. Liang Y, Ganem D. 2004. RBP-J. (CSL) is essential for activation of the K14/vGPCR promoter of Kaposi's sarcoma-associated herpesvirus by the lytic switch protein RTA. J. Virol. 78:6818–6826 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 53. Lu F, Day L, Gao SJ, Lieberman PM. 2006. Acetylation of the latency-associated nuclear antigen regulates repression of Kaposi's sarcoma-associated herpesvirus lytic transcription. J. Virol. 80:5273–5282 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 54. Lu J, Verma SC, Cai Q, Robertson ES. 2011. The single RBP-Jkappa site within the LANA promoter is crucial for establishing Kaposi's sarcoma-associated herpesvirus latency during primary infection. J. Virol. 85:6148–6161 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 55. Lukac DM, Kirshner JR, Ganem D. 1999. Transcriptional activation by the product of open reading frame 50 of Kaposi's sarcoma-associated herpesvirus is required for lytic viral reactivation in B cells. J. Virol. 73:9348–9361 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 56. Lukac DM, Renne R, Kirshner JR, Ganem D. 1998. Reactivation of Kaposi's sarcoma-associated herpesvirus infection from latency by expression of the ORF 50 transactivator, a homolog of the EBV R protein. Virology 252:304–312 [DOI] [PubMed] [Google Scholar]
- 57. Matsumura S, Fujita Y, Gomez E, Tanese N, Wilson AC. 2005. Activation of the Kaposi's sarcoma-associated herpesvirus major latency locus by the lytic switch protein RTA (ORF50). J. Virol. 79:8493–8505 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 58. Miele L. 2011. Transcription factor RBPJ/CSL: a genome-wide look at transcriptional regulation. Proc. Natl. Acad. Sci. U. S. A. 108:14715–14716 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 59. Moorman NJ, Willer DO, Speck SH. 2003. The gammaherpesvirus 68 latency-associated nuclear antigen homolog is critical for the establishment of splenic latency. J. Virol. 77:10295–10303 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 60. Nador RG, et al. 2001. Expression of Kaposi's sarcoma-associated herpesvirus G protein-coupled receptor monocistronic and bicistronic transcripts in primary effusion lymphomas. Virology 287:62–70 [DOI] [PubMed] [Google Scholar]
- 61. Nakamura H, et al. 2003. Global changes in Kaposi's sarcoma-associated virus gene expression patterns following expression of a tetracycline-inducible Rta transactivator. J. Virol. 77:4205–4220 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 62. Neil H, et al. 2009. Widespread bidirectional promoters are the major source of cryptic transcripts in yeast. Nature 457:1038–1042 [DOI] [PubMed] [Google Scholar]
- 63. Ong CT, et al. 2006. Target selectivity of vertebrate notch proteins. Collaboration between discrete domains and CSL-binding site architecture determines activation probability. J. Biol. Chem. 281:5106–5119 [DOI] [PubMed] [Google Scholar]
- 64. Ong KM, Blackford JA, Jr, Kagan BL, Simons SS, Jr, Chow CC. 2010. A theoretical framework for gene induction and experimental comparisons. Proc. Natl. Acad. Sci. U. S. A. 107:7107–7112 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 65. Palmeri D, Carroll KD, Gonzalez-Lopez O, Lukac DM. 2011. Kaposi's sarcoma-associated herpesvirus Rta tetramers make high-affinity interactions with repetitive DNA elements in the Mta promoter to stimulate DNA binding of RBP-Jk/CSL. J. Virol. 85:11901–11915 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 66. Pantry SN, Medveczky PG. 2009. Epigenetic regulation of Kaposi's sarcoma-associated herpesvirus replication. Semin. Cancer Biol. 19:153–157 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 67. Pearce M, Matsumura S, Wilson AC. 2005. Transcripts encoding K12, v-FLIP, v-cyclin, and the microRNA cluster of Kaposi's sarcoma-associated herpesvirus originate from a common promoter. J. Virol. 79:14457–14464 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 68. Persson LM, Wilson AC. 2010. Wide-scale use of Notch signaling factor CSL/RBP-Jkappa in RTA-mediated activation of Kaposi's sarcoma-associated herpesvirus lytic genes. J. Virol. 84:1334–1347 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 69. Rosenfeld N, Young JW, Alon U, Swain PS, Elowitz MB. 2005. Gene regulation at the single-cell level. Science 307:1962–1965 [DOI] [PubMed] [Google Scholar]
- 70. Russo JJ, et al. 1996. Nucleotide sequence of the Kaposi sarcoma-associated herpesvirus (HHV8). Proc. Natl. Acad. Sci. U. S. A. 93:14862–14867 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 71. Sarid R, Flore O, Bohenzky RA, Chang Y, Moore PS. 1998. Transcription mapping of the Kaposi's sarcoma-associated herpesvirus (human herpesvirus 8) genome in a body cavity-based lymphoma cell line (BC-1). J. Virol. 72:1005–1012 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 72. Sin SH, Fakhari FD, Dittmer DP. 2010. The viral latency-associated nuclear antigen augments the B-cell response to antigen in vivo. J. Virol. 84:10653–10660 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 73. Song MJ, Deng H, Sun R. 2003. Comparative study of regulation of RTA-responsive genes in Kaposi's sarcoma-associated herpesvirus/human herpesvirus 8. J. Virol. 77:9451–9462 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 74. Song MJ, Li X, Brown HJ, Sun R. 2002. Characterization of interactions between RTA and the promoter of polyadenylated nuclear RNA in Kaposi's sarcoma-associated herpesvirus/human herpesvirus 8. J. Virol. 76:5000–5013 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 75. Staudt MR, Dittmer DP. 2006. Promoter switching allows simultaneous transcription of LANA and K14/vGPCR of Kaposi's sarcoma-associated herpesvirus. Virology 350:192–205 [DOI] [PubMed] [Google Scholar]
- 76. Stedman W, et al. 2008. Cohesins localize with CTCF at the KSHV latency control region and at cellular c-myc and H19/Igf2 insulators. EMBO J. 27:654–666 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 77. Sun R, et al. 1998. A viral gene that activates lytic cycle expression of Kaposi's sarcoma-associated herpesvirus. Proc. Natl. Acad. Sci. U. S. A. 95:10866–10871 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 78. Talbot SJ, Weiss RA, Kellam P, Boshoff C. 1999. Transcriptional analysis of human herpesvirus-8 open reading frames 71, 72, 73, K14, and 74 in a primary effusion lymphoma cell line. Virology 257:84–94 [DOI] [PubMed] [Google Scholar]
- 79. Toth Z, et al. 2010. Epigenetic analysis of KSHV latent and lytic genomes. PLoS Pathog. 6:e1001013 doi:10.1371/journal.ppat.1001013 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 80. Trinklein ND, et al. 2004. An abundance of bidirectional promoters in the human genome. Genome Res. 14:62–66 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 81. Wei W, Pelechano V, Jarvelin AI, Steinmetz LM. 2011. Functional consequences of bidirectional promoters. Trends Genet. 27:267–276 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 82. Wen KW, Damania B. 2010. Kaposi sarcoma-associated herpesvirus (KSHV): molecular biology and oncogenesis. Cancer Lett. 289:140–150 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 83. Wen KW, Dittmer DP, Damania B. 2009. Disruption of LANA in rhesus rhadinovirus generates a highly lytic recombinant virus. J. Virol. 83:9786–9802 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 84. Xu Y, et al. 2005. A Kaposi's sarcoma-associated herpesvirus/human herpesvirus 8 ORF50 deletion mutant is defective for reactivation of latent virus and DNA replication. J. Virol. 79:3479–3487 [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.