

Abstract
Enveloped viruses can incorporate host cell membrane proteins during the budding process. Here we demonstrate that mumps virus (MuV) and vesicular stomatitis virus (VSV) assemble to include CD46 and CD55, two host cell regulators which inhibit propagation of complement pathways through distinct mechanisms. Using viruses which incorporated CD46 alone, CD55 alone, or both CD46 and CD55, we have tested the relative contribution of these regulators in resistance to complement-mediated neutralization. Virion-associated CD46 and CD55 were biologically active, with VSV showing higher levels of activity of both cofactors, which promoted factor I-mediated cleavage of C3b into iC3b as well as decay-accelerating factor (DAF) activity against the C3 convertase, than MuV. Time courses of in vitro neutralization with normal human serum (NHS) showed that both regulators could delay neutralization, but viruses containing CD46 alone were neutralized faster and more completely than viruses containing CD55 alone. A dominant inhibitory role for CD55 was most evident for VSV, where virus containing CD55 alone was not substantially different in neutralization kinetics from virus harboring both regulators. Electron microscopy showed that VSV neutralization proceeded through virion aggregation followed by lysis, with virion-associated CD55 providing a delay in both aggregation and lysis more substantial than that conferred by CD46. Our results demonstrate the functional significance of incorporation of host cell factors during virion envelope assembly. They also define pathways of virus complement-mediated neutralization and suggest the design of more effective viral vectors.
INTRODUCTION
The complement system constitutes a complex group of both soluble and cell-associated proteins that together form an integral part of host defense against pathogens (reviewed in references 5, 8, and 18). Although complement is considered part of the innate immunity system involved in recognition of viruses and direct neutralization of infectivity, it can also have profound effects on adaptive immunity, through recruitment and stimulation of leukocytes, antigen presentation to immune cells, and activation of T and B cell responses (5, 8, 18). The ability of viruses to activate complement as well as counteract complement pathways can play important roles in viral pathogenesis (e.g., see references 10, 29, and 41). In addition, it is increasingly clear that a greater understanding of complement interactions with viruses will be needed for the design of more effective viral vaccines and therapeutic vectors (31, 39). The overall goal of the work described here was to understand the mechanisms by which the negative-strand RNA viruses mumps virus (MuV) and vesicular stomatitis virus (VSV) limit complement-mediated neutralization.
The complement cascade can be initiated through three main pathways: the classical pathway, lectin pathway, or alternative pathway (8, 24, 36). These three pathways converge on a central component, C3, which is activated by cleavage into C3a and C3b. C3a serves as a potent anaphylatoxin to promote inflammation. C3b can bind covalently to viral components to aid in opsonization and phagocytosis. The association of C3b with components further downstream, such as C5 through C9, can lead to formation of the membrane attack complex (MAC), which is capable of lysing virus particles or infected cells (reviewed in references 8 and 41). Progression of the complement cascade depends on assembly of C3b with other cleavage products from C4, C2, and factor B to form the C3 convertase (19), a protein complex which functions to amplify the signal by further cleavage of C3 molecules in a feedback loop. The alternative pathway C3 convertase complex consists of C3b together with a factor B cleavage product to make C3bBb. The classical pathway C3 convertase consists of C4 and C2 cleavage products to make C4bC2a, which is essential for propagation along the pathway leading to MAC formation.
Nearly half of the complement components serve to regulate activity (22), a finding that reflects the need to control complement to avoid inappropriate activation and potential damage to normal cells and healthy tissues (e.g., see reference 2). Self-regulation of complement pathways involves the actions of a family of soluble and cell-associated proteins called regulators of complement activation (RCA). Some of these regulators limit complement pathways by targeting C3 or C4 cleavage products through two major mechanisms: (i) by acting as a cofactor to promote proteolytic cleavage of C3b or C4b by the complement protease factor I or (ii) by accelerating the disassociation of alternative or classical pathway convertases. Here, we have tested the relative contribution of these two regulatory mechanisms in resistance of MuV and VSV to complement-mediated neutralization.
The cofactor CD46 is a glycosylated integral membrane RCA protein expressed on a wide range of tissues and cell types as multiple isoforms due to differential splicing (21, 35, 37). CD46 combines with factor I to mediate inactivation of C3b into iC3b, rendering it incapable of integrating with convertases and thereby arresting propagation of the pathway. CD46 can also serve as a cofactor for factor I-mediated cleavage of C4b into C4c and C4d along the classical pathway or lectin pathway, but this cofactor activity is much less efficient than that seen for C3b cleavage (reviewed in references 21 and 22).
The decay-accelerating factor (DAF) CD55 is a heavily glycosylated RCA that is membrane associated through a glycosylphosphatidylinositol (GPI) linkage (23). The mechanism by which CD55 blocks propagation of the complement pathway is distinct from that of CD46, since CD55 lacks cofactor activity (26, 32, 34). Instead, CD55 functions to disrupt complement pathways at the level of the convertases, being able to either inhibit convertase formation or rapidly promote the decay of a previously formed convertase.
Viruses have evolved mechanisms to inhibit or to delay the neutralizing effects of complement (5, 41). For example, a number of enveloped viruses have been shown to recruit cell-associated RCA proteins into budding particles (9). Examples of this include influenza virus, vaccinia virus, and human immunodeficiency virus type 1 (HIV 1), which have been shown to incorporate CD55, CD59, and/or CD46 into progeny virions (38, 40, 42, 44). However, in many cases the relative contribution of these individual molecules to neutralization remains unclear.
It has been known for many years that complement is an important factor in neutralization of a range of nonsegmented negative-strand RNA viruses, including the paramyxoviruses and rhabdoviruses (11, 14, 25, 45, 46, 47). For example, the closely related paramyxoviruses parainfluenza virus type 5 (PIV5) and MuV both preferentially activate the alternative pathway, and this can contribute to the efficiency of neutralization by human serum (14, 25). In contrast, VSV activates and is neutralized by the classical pathways, through an antibody-dependent or -independent mechanism (3, 27). These findings raised the question of whether these negative-strand RNA viruses have mechanisms to limit activation and/or amplification of the complement cascade.
We have previously shown that PIV5 and MuV incorporate CD46 into progeny virions (17) and virus containing CD46 gained partial resistance to neutralization by normal human serum (NHS). Here we demonstrate that both CD46 and CD55 are associated with the paramyxovirus MuV and rhabdovirus VSV. Our results support a model whereby CD46 can delay neutralization of MuV and VSV, but the system's impact is much less that that seen with virus harboring CD55. These results demonstrate the functional significance of incorporation of host cell factors during virion assembly, pathways of virus neutralization, and the design of more effective viral vectors.
MATERIALS AND METHODS
Complement proteins and reagents.
The complement proteins C3, C3b, C4, C1, C2, factor B, factor D, factor I, and factor H and guinea pig serum were purchased from Complement Technologies (Tyler, TX). Recombinant CD55 (rCD55) and CD46 were procured from R&D Systems (Minneapolis, MN) and Sino Biologicals (China), respectively. Gelatin Veronal buffered saline (GVBO) and GVB with calcium and magnesium (GVB++) were from Complement Technologies, while GVB with EDTA (GVBE) was from Sigma (St. Louis, MO). Dextrose GVB (DGVB++) contained half-ionic-strength GVB with 2.5% dextrose (pH 7.4). Phosphate-buffered saline (PBS) contained 10 mM sodium phosphate and 145 mM sodium chloride (pH 7.4). Sheep and rabbit red blood cells (RBCs) were from Lampire Biologicals (Pipersville, PA).
Cells and viruses.
Chinese hamster ovary (CHO) cells that overexpress the CYT2 isoform of human CD46 (CHO-CD46) and the control drug-resistant CHO-K1 cells were kindly provided by Denis Gerlier (University of Lyon) (13) and were maintained as described previously (17). CHO cells stably expressing CD55 (CHO-CD55) were derived by transfecting CHO or CHO-CD46 cells with a plasmid encoding CD55 (kindly provided by Shuji Miyagawa [28, 33]), followed by selection for resistance to 100 μg/ml G418 or both G418 and hygromycin (for CHO-CD46/55 cells). The Enders strain of MuV (ATCC VR-1379) and Orsay strain of VSV were grown in CHO cells supplemented with 2% heat-inactivated (HI) serum. Viruses were purified by sucrose gradient ultracentrifugation as described previously (16). Time-dependent virus neutralization by NHS was carried out as previously described (16), with remaining infectivity determined by plaque assay on CV-1 cells. Results are the average of six reactions, with the significance of data points calculated using Student's t test.
Fluorescence-activated cell sorter (FACS) analysis.
To determine cell surface expression of CD46 and CD55, cells were harvested at 24 h postplating, washed twice with PBS, and centrifuged. The resulting pellet was suspended in 100 μl of PBS and incubated with mouse anti-CD46 (R&D, MN) and/or rabbit anti-CD55 (Santa Cruz Biotechnology, Santa Cruz, CA). Following the washes, the cell pellets were suspended in PBS and incubated with anti-mouse Alexa Fluor 488 and/or anti-rabbit Alexa Fluor 633. Analysis was carried out on a FACSCalibur machine (BD Biosciences, San Diego, CA).
EM.
Electron microscopy (EM) analysis of sucrose gradient-purified MuV or VSV was carried as previously described (16). Briefly, the viruses were placed on carbon-coated 200-mesh gold grids (EM Sciences, PA) and incubated in a humidified chamber for 5 min, before blocking with a solution of 1% bovine serum albumin in PBS. Samples were probed with a mouse monoclonal anti-CD46 antibody (1 μg/10 μl; R&D), mouse monoclonal anti-CD55 antibody (1 μg/10 μl; Millipore, CA), or rabbit anti-CD55 polyclonal antibody (1:5 dilution; Santa Cruz Biotechnology). Samples were then incubated with 10 μl of a 1:10 dilution of anti-mouse 6-nm gold-labeled antibody (for anti-CD46) and/or anti-mouse or anti-rabbit 12-nm gold-labeled antibody for (anti-CD55; Jackson ImmunoResearch, CA). Samples were negatively stained with 2% phosphotungstic acid and analyzed with a Technai transmission electron microscope. To study the effect of NHS on virion morphology, 10 μl of VSV (1 × 105 PFU) was incubated with a 1:5 dilution of NHS for the time periods indicated in the appropriate figure, followed by staining and imaging as described above.
Western blotting.
Cell lysates or gradient-purified virions were analyzed for levels of CD46, CD55, P protein, or M protein by SDS-PAGE, followed by Western blot analysis. The blots were probed with antibodies against either CD46 or CD55 (Santa Cruz Biotechnology, Santa Cruz, CA) at a dilution of 1:500. Polyclonal rabbit serum specific for the PIV5 P protein was used to detect MuV P protein, due to the extensive cross-reactivity. Blots were visualized by horseradish peroxidase-conjugated antibodies and enhanced chemiluminescence (Pierce Chemicals).
To quantitate levels of virion-associated regulators, dilutions of purified CD46 or CD55 were run on SDS-gels along with 10 μg of purified virus. Levels of virion-associated regulators were estimated visually by comparison to the known concentration of standards. In the case of virus preparations with nonspecific cleavage of regulators, all bands were included in the estimation.
Factor I cofactor activity assay.
MuV and VSV particles generated from CHO cells were purified by sucrose gradient ultracentrifugation, and protein concentrations were determined by bicinchoninic acid assay before use in the cofactor assays. The factor I-mediated cofactor activity on C3b was analyzed as described earlier (17). Briefly, 5 μg of purified VSV or 10 μg of purified MuV was incubated with 3 μg of C3b and 100 ng of factor I for different time periods at 37°C in a total volume of 20 μl of PBS. Samples were analyzed on a 9% SDS-polyacrylamide gel, after the reactions were stopped by adding sample buffer containing SDS and boiling. After gel electrophoresis, protein bands were visualized with GelCode blue stain reagent (Thermo Scientific, IL).
Alternative pathway and classical pathway decay-acceleration activity assay.
Alternative pathway C3 convertase activity associated with purified virions was measured by forming the enzyme C3bBb on rabbit RBCs as described earlier (30). C3b deposition to form rabbit RBCs-C3b was done by incubating rabbit RBCs with C3, factor B, and factor D in the presence of NiCl2. Finally, C3bBb was assembled on rabbit RBCs-C3b by mixing 1 × 109 cells in 70 μl with 21 μg of factor B, 0.36 μg of factor D, and 7.5 μl of GVB containing 3.5 mM NiCl2. After 5 min incubation at 37°C, further assembly was blocked by addition of EDTA to a final concentration of 10 mM. The convertase assay was carried out in a total volume of 25 μl by mixing various concentrations of viruses or rCD55 with 10 μl of rabbit RBCs-C3bBb and incubating at 37°C for 10 min. Afterward, 25 μl of NHS (1:5) in GVB containing 20 mM EDTA was added and samples were incubated for 20 min at 37°C. After incubation, 100 μl of GVBE was added to the samples and the remaining cells were pelleted. Percent rabbit RBC lysis was calculated by determination of the absorbance at 405 nm and normalized to the results with 100% lysis in the absence of inhibitor.
To measure classical pathway C3 convertase activity, EAC142 was assembled on sheep erythrocytes. Sheep erythrocytes were sensitized by adding anti-sheep erythrocyte antibody (MP Biomedicals, Solon, OH) at a dilution of 1:80 and washed. C1 (0.5 μg) diluted in 100 μl of DGVB++ was added to 4 × 109 anti-sheep erythrocytes, and the mixture was incubated for 20 min at 30°C. The cells were then washed with DGVB++, followed by the addition of 1.5 μg of C4, and the mixture was incubated for a further 20 min at 30°C. Finally, EAC142 was formed by addition of 0.5 μg of C2 and incubation of the mixture for 5 min at 30°C. In the convertase assay, various concentrations of virions or rCD55 were incubated with 10 μl of EAC142 at 22°C for 5 min in a total final volume 25 μl. Afterward, 100 μl guinea pig serum (1:100 in DGVB++ with 40 mM EDTA) was added as a source of complement factors, and samples were incubated at 37°C for 30 min. After centrifugation, percent lysis was calculated by determination of the absorbance at 405 nm.
RESULTS
MuV containing both CD46 and CD55 is delayed in neutralization by NHS.
To determine the effect of cellular regulators on virion neutralization, purified MuV was produced in CHO cells, which do not express complement regulators, or in HeLa cells, which express both CD46 and CD55. Western blotting showed high levels of CD46 and CD55 associated with purified MuV from HeLa cells but not from CHO cells (Fig. 1A). To determine the effect of virion-associated regulators on complement-mediated neutralization, NHS was incubated with 100 PFU of HeLa- or CHO-derived MuV, and at various times after addition of NHS, remaining infectivity was determined by plaque assay. As shown in Fig. 1B, CHO-derived virus was rapidly neutralized, with no infectivity remaining at 15 min posttreatment. In contrast, neutralization of HeLa-derived MuV was delayed. No neutralization was seen with HI NHS (data not shown, but see below). While these results indicate that regulators can delay neutralization, they do not distinguish between a contribution from CD46 versus a contribution from CD55.
Fig 1.
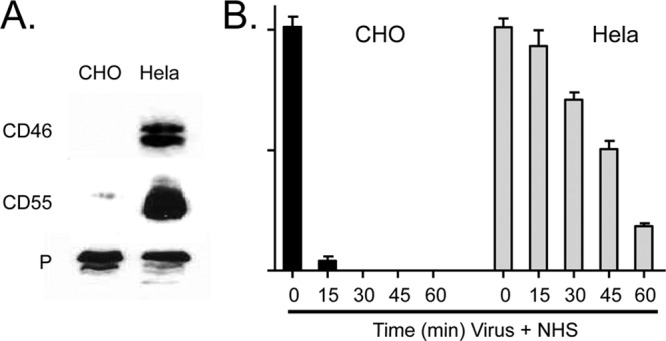
NHS-mediated neutralization of HeLa-grown MuV is delayed compared to that of CHO-grown MuV. (A) MuV was grown in HeLa or CHO cells and purified by gradient centrifugation. Virions were analyzed for levels of CD55, CD46, and P protein by Western blotting. (B) One hundred PFU of virus was incubated with NHS for the indicated times, and the remaining infectivity was determined by plaque assay. No loss of infectivity was seen using HI NHS.
Generation of MuV and VSV particles containing CD46, CD55, or both regulators.
CHO cells were engineered to express CD46 alone, CD55 alone, or a combination of both CD46 and CD55 by transfection of cDNA plasmids and selection of cells for drug resistance. Analysis of CD46 and CD55 expression in cell lysates by Western blotting (Fig. 2A) showed that CD46 was expressed at approximately the same level in CD46 and CD46/55 cells, whereas CD55 was expressed in CD55 cells to a higher level than in the double-positive CD46/55 cells. These results were confirmed by analysis of cell surface expression by immunostaining and FACS analysis (Fig. 2B). The levels of cell surface expression of CD55 and CD46 were similar to the endogenous levels of expression on a range of human cell lines. For example, the mean fluorescence intensities (MFIs) of CD55 on HeLa, A549, and MCF-7 cells were 264, 134, and 113, respectively (means of two experiments). Likewise, the MFIs of CD46 on HeLa, A549, and MCF-7 cells were 413, 418, and 394, respectively.
Fig 2.
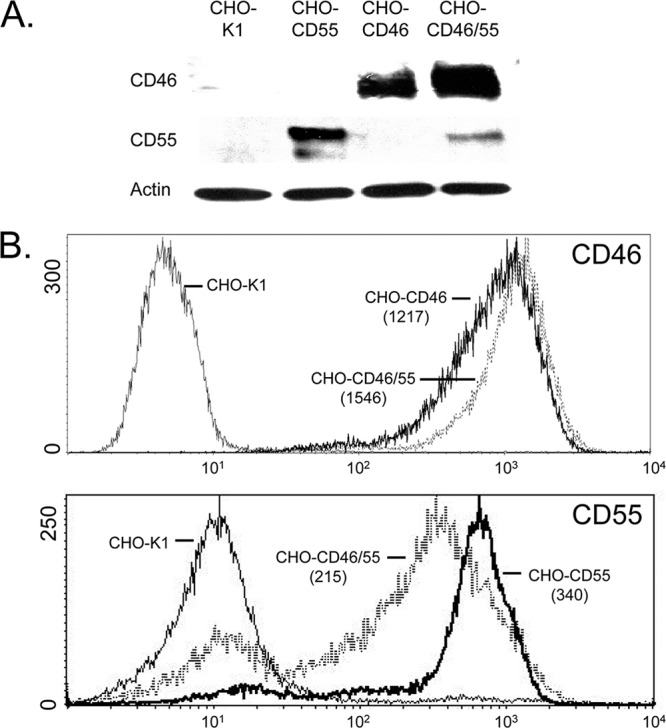
CD55 and CD46 expression in CHO cell lines. (A) Western blotting. Lysates from control CHO-K1, CHO-CD55, CHO-CD46, or double-positive CHO-CD46/55 cells were analyzed by Western blotting for expression of CD46, CD55, and actin. (B) Cell surface expression of CD46 and CD55. CHO cells expressing CD46, CD55, or both regulators were analyzed by flow cytometry for cell surface expression of CD46 and CD55. MFIs (arbitrary units on y axis; number of cells on x axis) are shown in parentheses.
To determine the extent to which CD46 and CD55 were associated with MuV particles, purified virus from control CHO-K1 cells or CD55- and CD46-expressing CHO cells was analyzed by Western blotting. As shown in Fig. 3A, virus derived from CHO-CD46 cells contained slightly more CD46 than that found for virus from the CHO-CD46/55 cells. Likewise, virus derived from CHO-CD55 cells contained slightly more CD55 than that found for virus derived from CHO-CD46/55 cells. Western blotting of multiple virus preparations indicated that, on average, ∼20 and ∼10 ng of CD46 were associated with 1 μg of purified MuV from CD46 and CD46/55 cells, respectively (Table 1). Likewise, ∼25 and ∼10 ng of CD55 were associated with 1 μg of purified MuV from CD55 and CD46/55 cells, respectively. To confirm the association of these regulators with virions, MuV particles grown in the different CHO cell lines were probed with antibodies to CD46 or CD55 and gold-labeled secondary antibody before visualization by EM. As shown in Fig. 3B, virus particles showed clear immunostaining for CD55 or CD46 that was clustered along one face of the virus particle (open and white arrows) as described previously for CD46 association with PIV5 (17). In the case of virus grown in the CD46/CD55 cells, staining for these two regulators clustered on opposite faces of the virion particle (Fig. 3B, right, open and white arrows).
Fig 3.

CD46 and CD55 are associated with purified MuV virions. (A) Western blotting. MuV derived from the indicated cell lines was purified by gradient centrifugation and analyzed by Western blotting for the presence of CD46 or CD55. Levels of viral P protein were analyzed as a loading control. (B) EM analysis. Purified virus derived from CHO cells expressing CD46 (left), CD55 (middle), or both CD46 and CD55 (right) was treated with the indicated antibodies, followed by 6-nm (CD46) or 12-nm (CD55) colloidal gold-labeled goat anti-mouse antibody (top row). Control samples were treated with secondary antibody alone (bottom row). Samples were analyzed by EM at a magnification of ×55,000. Bars, 0.1 μm. White and open arrows, locations of staining for CD46 and CD55, respectively; 1° and 2°, primary and secondary antibodies, respectively.
Table 1.
Levels of CD55 and CD46 associated with purified MuV and VSV
| Virusa | Levelb (ng) |
|
|---|---|---|
| CD55 | CD46 | |
| MuV CHO-CD46 | 20 | |
| MuV CHO-CD55 | 25 | |
| MuV CHO-CD46/55 | 10 | 10 |
| VSV CHO-CD46 | 25 | |
| VSV CHO-CD55 | 50 | |
| VSV CHO-CD46/55 | 25 | 25 |
Viruses were grown in CHO cells expressing CD46, CD55, or CD46/55 and purified by gradient centrifugation.
Levels of CD55 and CD46 were determined by Western blotting and compared to standards of purified protein.
Purified VSV that was derived from the CD46- and CD55-expressing cells was also analyzed for the levels of these two regulators. As shown in Fig. 4A and Table 1, ∼25 ng of CD46 was associated with 1 μg of purified VSV from CD46 and CD46/55 cells, and ∼50 and ∼25 ng of CD55 were associated with 1 μg of purified VSV from CD55 and CD46/55 cells, respectively. Similar to what we found for MuV, immunostaining of purified virions showed distinct clustering of CD46 (Fig. 4B, white arrow) and CD55 (open arrows) on opposite faces of the VSV particles.
Fig 4.

CD46 and CD55 are associated with purified VSV virions. (A) Western blotting. Purified VSV derived from the indicated cell lines was analyzed by Western blotting for the presence of CD46 or CD55. Levels of viral M protein were analyzed as a loading control. (B) EM analysis. Purified virus derived from CHO cells expressing CD46 (left), CD55 (middle), or both CD46 and CD55 (right) was treated with the indicated antibodies, followed by 6-nm (CD46) or 12-nm (CD55) colloidal gold-labeled goat anti-mouse antibody (top row). Control samples were treated with secondary antibody alone (bottom row). Samples were analyzed by EM at a magnification of ×55,000. Bars, 0.1 μm. White and open arrows, locations of staining for CD46 and CD55, respectively.
MuV- and VSV-associated CD46 displays cofactor activity that promotes factor I-mediated cleavage of C3b.
A key level of complement regulation involves inactivation of C3b into iC3b, a highly specific cleavage event catalyzed by protease factor I along with a cofactor, CD46 or factor H. To directly test if virion-associated CD46 was functional in cofactor activity, factor I-mediated cleavage of C3b into iC3b was reconstituted in vitro using purified virions and purified commercially available components as described previously (4). The appearance of iC3b cleavage products was monitored by SDS-PAGE and Coomassie blue staining. As shown in Fig. 5 (left), the C3b-alpha′ chain remained intact in the presence of purified MuV (lane 1) but in the positive-control sample (lane 2) was cleaved into 67-kDa and 43-kDa fragments when incubated with purified factor I and factor H (compare lanes 1 and 2). Incubation of MuV derived from CHO-CD46 cells with factor I and C3b resulted in a time-dependent disappearance of the C3b alpha′ chain and the corresponding appearance of both the 43- and 67-kDa fragments (Fig. 5, left; compare lanes 3 and 10). Using 25 μg of MuV virions, approximately half of the C3b alpha′ chain was cleaved by 45 min (compare lane 1 with lane 5 [asterisked]). Similar results were found with MuV derived from CHO-CD46/55 cells (Fig. 5, bottom left). As reported previously (17), MuV derived from CHO-K1 cells completely lacked cofactor activity (data not shown).
Fig 5.

Cofactor activity associated with MuV and VSV grown in CD46 and CD46/55 CHO cells. Twenty-five micrograms or 5 μg of purified MuV and VSV derived from CHO-CD46 or CHO-CD46/55 cells was incubated for the indicated times with purified C3b and factor I as detailed in Materials and Methods. The cleavage of the alpha′ chain of C3b into the 67- and 43-kDa products was monitored by SDS-PAGE and Coomassie blue staining. Lanes 1 and 2, samples showing negative-control starting products and positive-control products generated by cleavage of C3b-alpha′ by factor I and factor H. The positions of factor H, alpha′, and beta chains of C3b and the 67- and 43-kDa cleavage products are indicated. *, approximate time point when half of C3b-alpha′ was cleaved.
VSV derived from CHO-CD46 and CHO-CD46/55 cells also showed strong cofactor activity for factor I-mediated cleavage of C3b (Fig. 5, right). By comparison to the results presented above using 25 μg of MuV, the rate of cleavage catalyzed by only 5 μg VSV-associated CD46 was much faster. As shown in Fig. 5, approximately half of the C3b alpha′ chain was cleaved using VSV particles by 30 to 45 min (compare lane 1 with starting material with lane 7 [asterisked] in Fig. 5, right). There was no substantial difference in cofactor activity between VSV derived from CHO-CD46 and CHO-CD46/55 cells, consistent with the similar levels of CD46 associated with these virion preparations (Table 1). VSV derived from CHO-K1 cells completely lacked cofactor activity (data not shown). These data provide direct evidence that purified MuV and VSV particles contain a functional cofactor activity that promotes C3b cleavage.
MuV- and VSV-associated CD55 displays DAF activity that inhibits C3 convertase activity.
Progression of the alternative pathway is dependent on the formation of the C3 convertase C3bBb, a complex that can be disrupted by CD55 DAF activity. To determine the DAF activity associated with virions derived from CD55-expressing cells, rabbit RBCs were used to assemble the C3bBb convertase from purified C3, factor B, and factor D, as described in Materials and Methods. When exposed to complement, these convertase-containing cells are lysed due to formation of the MAC (30). Virion-associated DAF activity was determined as a function of the ability of CD55 to inhibit convertase activity and thus prevent cell lysis, which was set at 100% for control samples lacking CD55.
As shown in Fig. 6A and B, purified recombinant rCD55 had potent dose-dependent DAF activity, with approximately half of the C3 convertase activity inhibited using 5 ng of recombinant protein, MuV and VSV derived from control CHO-K1 cells showed little DAF activity, and the convertase-containing RBCs were efficiently lysed by complement (100% convertase activity). MuV and VSV derived from either the CHO-CD55 cells or the CHO-CD46/55 cells showed a significant dose-dependent inhibition of C3 convertase activity, but this was less potent than that seen with rCD55. For example, 1 μg of MuV and VSV from CHO-CD55 cells was estimated to contain 25 and 50 ng of CD55 (Table 1), but the DAF activity was lower than that seen with 10 ng of rCD55 protein. For VSV, there was little difference in DAF activity associated with virus from the two CHO cell lines, consistent with only an ∼2-fold difference in CD55 levels (Table 1). For MuV, virus derived from the CHO-CD46/55 or CHO-CD55 cells displayed DAF activity that was significantly higher than that of virus derived from control cells. These data indicate that DAF activity against the alternative pathway convertase is very similar for MuV and VSV derived from either CHO-CD55 or CHO-CD46/55 cells, despite differences in the levels of CD55 associated with virions.
Fig 6.

Alternative and classical pathway DAF activity associated with MuV and VSV particles. Purified MuV (A and C) or VSV (B and D) that was derived from the indicated cells was assayed for decay-accelerating activity against the alternative pathway (AP) (A and B) or classical pathway (CP) (C and D) C3 convertase as detailed in Materials and Methods. C3-mediated hemolytic activity is expressed as a percentage of the lysed control sample set at 100%. Data are from triplicate samples, and error bars represent standard deviations. (A and B) #, P < 0.001 when comparing results for 0.25 to 1 μg of MuV-CD55 and MuV-CD46/55 with those for 0.25 to 10 μg from control virus derived from CHO-K1 cells or 0.25 to 1 μg of VSV-CD55 and VSV-CD46/55 with those for 1 to 5 μg from control virus derived from CHO-K1 cells; (C and D) #, P < 0.001 when comparing results for 0.5 to 2.5 μg VSV-CD55 with those for 1 to 10 μg control virus and comparing results for 2.5 to 10 μg MuV-CD55 and MuV-CD46/55 with those for the corresponding amount (μg) of control virus; *, P < 0.01 when comparing results for 0.25 to 10 μg VSV-CD46/55 with those for 1 to 10 μg control virus derived from CHO-K1 cells.
Virus particles were also tested for DAF activity against the classical pathway C3 convertase; (Fig. 6C and D). Here, the C4bC2a complex was assembled on antibody-sensitized sheep RBCs by sequential addition of purified C1, C2, and C4, and then these RBCs were used in assays with purified MuV and VSV particles. Inhibition of C3 convertase activity was assayed by decreases in hemolysis, As shown in Fig. 6C and D, the levels of rCD55 required to achieve 50% inhibition of the classical pathway convertase (10 to 50 ng) were higher than those required for inhibition of alternative pathway convertase (1 to 10 ng), consistent with the known specificity of CD55 (20). For viruses from CHO-CD55 cells, more MuV (2.5 μg) than VSV (∼1 μg) was required to inhibit 50% of convertase activity. These data indicate that virion-associated CD55 activity against the classical pathway convertase is very similar between MuV and VSV, with the exception of VSV that contains CD55 only.
Virion-associated CD46 and CD55 show different effects on complement-mediated neutralization of MuV and VSV.
A time course of complement-mediated neutralization of viruses containing CD46 and/or CD55 was carried out as described above for Fig. 1. As shown in Fig. 7A for NHS from two different donors, MuV from CHO-K1 cells was neutralized very rapidly, with almost no infectivity remaining by 30 to 45 min. MuV containing CD46 alone or CD55 alone was more resistant to neutralization than control virus, with CD55 having a more potent effect on delaying neutralization (e.g., at the 30-min time point). MuV containing both CD46 and CD55 showed the lowest rate of neutralization and displayed resistance that was clearly distinct from that seen with MuV containing only CD55. These results were seen with several dilutions of sera (Fig. 8A), and there was no neutralization of any of the CHO-derived viruses by HI sera (Fig. 8B). These data show that while CD55 alone has a more potent inhibitory effect on neutralization kinetics than CD46 alone, there was clear additional resistance conferred by having both CD46 and CD55.
Fig 7.

Rate of in vitro neutralization of CD46- and CD55-containing MuV and VSV. One hundred PFU of MuV (A) or VSV (B) derived from the indicated cell lines was incubated with NHS from two different donors for the indicated times, and then remaining infectivity was determined by plaque assay. As a control, virus was incubated with PBS alone for 1 h. Data are from six independent reactions, and error bars represent standard deviations. ^, P < 0.0001 when comparing virus with CD46 only to virus from control CHO-K1 cells; *, P < 0.0001 when comparing virus containing CD55 only to virus containing CD46 only.
Fig 8.

Effect of NHS dilution and HI NHS on neutralization of various CHO-grown viruses. (A) Effect of NHS dilution. One hundred PFU of the indicated viruses was incubated for 1 h with the indicated dilutions of NHS, and remaining infectivity was determined by plaque assay. (B) Effect of HI NHS. One hundred PFU of the indicated viruses was incubated for 1 h with PBS, NHS, or HI NHS, and remaining infectivity was determined by plaque assay.
Virion-associated CD55 had a much stronger influence on neutralization of VSV than on that of MuV. This is evident in Fig. 7B, where the neutralization of VSV containing CD46 alone was much faster and more complete than that seen with virus containing CD55 alone or CD55 in combination with CD46. The dominant effect of CD55 was reinforced by the finding that the kinetics of neutralization of VSV containing CD55 only was nearly the same as that of VSV containing both CD55 and CD46. These data indicate that while both regulators can delay VSV neutralization, CD55 has a much greater impact than CD46.
As an alternative approach to determine the effect of virion-associated regulators on VSV neutralization, virus derived from the four CHO cell lines was treated with NHS and visualized at various time points by electron microscopy. As shown in Fig. 9, VSV from control CHO-K1 cells showed rapid aggregation within 5 min of treatment. Although the micrographs were not quantitated, many virions appeared to be lysed, as evidenced by the loss of the bullet-shape morphology and release of nucleocapsid-like structures (arrows). Previous data indicate that this lysis does not occur with C3-deficient serum (16). There were very few intact virions evident by 30 min of treatment. VSV containing CD46 also displayed rapid aggregation, and there were few intact virions detected by 30 min of treatment (Fig. 9). In contrast, CD55-containing VSV derived from either CHO-CD55 or CHO-CD46/55 cells did not show aggregation or lysis until very late after treatment. For example, as shown in Fig. 10 for samples taken at 30 min after treatment, VSV containing CD55 was largely intact with only a few lysed particles. By 60 min of treatment, more aggregation and lysis were detected. Thus, by comparison of data from 30-min time points for each of the CHO-derived viruses, the degree of protection from lysis is much greater when CD55 is present. In addition, these data suggest that VSV neutralization by NHS proceeds through a mechanism involving virion aggregation followed by virion lysis.
Fig 9.

Electron micrographs of in vitro neutralization of VSV derived from CHO-K1 and CHO-CD46 cells. Purified VSV particles derived from the indicated CHO cells were incubated for the indicated times at 37°C with NHS and then analyzed by negative staining and EM. Arrowheads, apparently intact virions; arrows, clearly lysed particles or nucleocapsid-like structures. Bars, 0.1 μm.
Fig 10.

Electron micrographs of in vitro neutralization of VSV derived from CHO-CD55 and CHO-CD46/55 cells. Purified VSV particles derived from the indicated CHO cells were incubated for the indicated times at 37°C with NHS and then analyzed by negative staining and EM. Arrowheads, apparently intact virions, arrows, clearly lysed particles or nucleocapsid-like structures. Bars, 0.1 μm.
DISCUSSION
Here we demonstrate that MuV and VSV assemble their lipid envelopes to include both CD55 and CD46, two important host cell membrane proteins that inhibit complement by different mechanisms and can have profound effects on the ability of these viruses to delay complement-mediated neutralization. The overall goal of the work presented here was to determine the relative importance of CD46 and CD55 for MuV and VSV neutralization. Our results support a model whereby virion-associated CD55 is a potent inhibitor that contributes to a delay in VSV and MuV neutralization. While CD46 is also an important regulator, the extent of its relative impact on the kinetics and extent of neutralization is less than that of CD55.
It has long been known that enveloped viruses can incorporate or exclude host cell membrane proteins during the budding process (48), raising the important question of the functional significance of assembly of a host membrane protein in a virion envelope. We have shown that the integral transmembrane protein CD46 and the GPI-linked protein CD55 are both abundantly incorporated into VSV and MuV particles. One of our most striking findings from immunogold labeling and EM was that CD55 and CD46 appear to cluster on opposing faces of both VSV and MuV particles. Analysis of more than 20 independent EM grids showed that CD46 and CD55 cluster to opposite faces in >80% of virions and in the case of VSV are almost always located on the sides of the particle rather than the tip or base. While CD55 and CD46 also clustered on opposing faces of MuV particles, the pleomorphic structure made the separation less clear. For VSV, assembly of membrane proteins into budding virions likely involves clustering into distinct microdomains, some of which can differ in size and in protein content (7). The GPI-linked CD55 is associated with lipid rafts, defined by insolubility in cold nonionic detergents (43), whereas CD46 is considered a conventional nonraft transmembrane protein. Thus, while these two proteins differ in how they are associated with the plasma membrane, their presence in distinct membrane microdomains may be the mechanism behind different locations in the virion envelope. Incorporation could also be influenced by protein-protein interactions through cytoplasmic tails. While CD46 exists as isoforms with different cytoplasmic tails (21), our CHO transfectants express only the CYT2 form of CD46, and it remains to be determined if other isoforms can be incorporated as well. Finally, while virus derived from cells expressing more CD55 had more virion-associated CD55 (e.g., CHO-CD55 versus CHO-CD46/55), VSV always incorporated the same level or more of these two regulators than did MuV. Taken together, the signals that direct a particular RCA into virions could be a combination of location in particular membrane microdomains, the nature of the association with the lipid bilayer, potential protein-protein interactions during budding, and overall concentration at the site of budding.
While both CD55 and CD46 were incorporated into VSV and MuV particles, available evidence suggests that there may be some specificity to which RCAs are associated with a particular virion envelope. For example, Shaw and coworkers (40) showed that influenza virus can incorporate the inhibitory protein CD59, which acts to inhibit MAC assembly, but CD46 was not found in purified virions. This raises the interesting possibility that enveloped RNA viruses may have evolved to selectively incorporate host inhibitors depending on the particular complement pathway activated by an individual virus type. Consistent with this hypothesis, influenza virus can activate the lectin or classical pathways (15) and does not incorporate CD46 (40), which is more potent in inhibition of the alternative pathway (21, 22). Conversely, PIV5 activates the alternative pathways, incorporates abundant levels of CD46, and has low cofactor activity against the classical and lectin pathway factor C4b (17). There is evidence for differential expression of complement factors and regulators in cells from distinct organ systems, such as the brain or respiratory tract (6, 46). This raises the hypothesis that the incorporation of a particular RCA may influence the tissue or cell tropism for a particular virus and could be an important factor in the extent of dissemination.
A strength of our data is the quantitation of the biological activity of virion-associated regulators, a factor more relevant than the level of protein alone. CD46 has been shown to be much more active against the alternative pathway than the classical pathway (22). Since MuV activates the alternative pathway and VSV activates the classical pathway (3, 14, 25, 27), we predicted that CD46 would have a higher impact on neutralization of MuV than that of VSV. Cofactor activity for VSV containing CD46 was much greater than that for MuV, consistent with the finding of more CD46 associated with VSV than that seen with MuV (Table 1). However, the impact that CD46 played on resistance to neutralization was approximately the same for both viruses. Thus, while CD46 delayed neutralization compared to its neutralization of virus from control CHO-K1 cells, the effect was less than that seen with CD55 alone. The added effect of CD46 over that which is seen for CD55 is evident for MuV, a finding consistent with previous work showing that CD55 and CD46 can act synergistically on cell surfaces (22).
CD55 alone had a more potent effect than CD46 alone on the delay in virus neutralization, and this effect was most evident in the case of VSV. Here, the kinetics and extent of neutralization of VSV containing CD55 alone or containing both CD55 plus CD46 were very similar, despite a nearly 2-fold difference in the levels found in particles. This may indicate that virion-associated DAF activity reaches a threshold and cannot be increased with more CD55. This higher resistance of VSV-CD55 than VSV-CD46 to neutralization may reflect the fact that CD55 inhibits both the classical pathway convertase, which is activated by VSV, and the alternative pathway convertase, which is essential for amplifying the initial input into the complement cascade. Thus, VSV neutralization depends on both classical pathway activation and alternative pathway amplification, and these dual targets of CD55 action can enhance its potency for VSV over that for MuV, which activates only the alternative pathway.
We have previously shown that treatment of PIV5 and MuV with NHS results in either particle aggregation or virion lysis, depending on the cell type from which the virus was derived (16). This raised the hypothesis that virion-associated RCAs could influence the mechanism of neutralization. Using the easily recognized VSV bullet shape in EM analyses, a time course of neutralization of VSV lacking regulators with NHS showed a rapid aggregation of particles, followed by a slightly slower lysis which resulted in release of nucleocapsid-like structures. VSV containing only CD46 was slightly delayed in these steps. In contrast, CD55-containing VSV showed a substantial delay in both aggregation and lysis, with no overt difference between VSV containing CD55 alone or CD55 plus CD46. While similar results were found with MuV, the pleomorphic virion structure made the analysis less clear with MuV than with VSV. Thus, for VSV, the main impact of CD55 is on the rate and extent of aggregation/lysis, but the viruses with different regulators all proceed down a similar mechanism of inactivation. This finding does not rule out the possibility that the mechanism of neutralization may be influenced by other RCAs, such as CD59, which is involved in inhibition of MAC formation, or by the concentration of complement factors in microenvironments other than serum.
VSV is currently being developed as a therapeutic vector (1), and PIV5 and MuV show promise as viral therapeutic vectors (e.g., see reference 12). Our results show that incorporation of either CD55 or CD46 can profoundly affect the survival of viral vectors in the face of complement activation. Our work has implications for the development of more effective vectors which could be designed to control the incorporation of cellular RCAs such as CD55 and CD46. It would be important to achieve a balance between survival of a vector in a host long enough to have a therapeutic effect and safety concerns that arise from enhancing resistance to neutralization.
ACKNOWLEDGMENTS
We are grateful to Ellen Young, Aaron Graff, and Margie McKenzie for excellent technical help, Denis Gerlier (University of Lyon) for the CHO-K1 and CHO-CD46 cell lines, and Shuji Miyagawa (Osaka University) for the CD55-expressing plasmid.
This work was supported by NIH grant AI083253 (to G.D.P.) and AI015892 (to D.S.L.).
Footnotes
Published ahead of print 3 July 2012
REFERENCES
- 1. Ahmed M, Puckett S, Lyles DS. 2010. Susceptibility of breast cancer cells to an oncolytic matrix (M) protein mutant of vesicular stomatitis virus. Cancer Gene Ther. 17:883–892 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2. Atkinson JP, Liszewski MK, Richards A, Kavanagh D, Moulton EA. 2005. Hemolytic uremic syndrome: an example of insufficient complement regulation on self tissue. Ann. N. Y. Acad. Sci. 1056:144–152 [DOI] [PubMed] [Google Scholar]
- 3. Beebe DP, Cooper NR. 1981. Neutralization of vesicular stomatitis virus (VSV) by human complement requires a natural IgM antibody present in human serum. J. Immunol. 126:1562–1568 [PubMed] [Google Scholar]
- 4. Bernet J, Mullick J, Panse Y, Parab PB, Sahu A. 2004. Kinetic analysis of the interactions between vaccinia virus complement control protein and human complement proteins C3b and C4b. J. Virol. 78:9446–9457 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5. Blue CE, Spiller OB, Blackbourn DJ. 2004. The relevance of complement to virus biology. Virology 319:176–184 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6. Bolger MS, et al. 2007. Complement levels and activity in the normal and LPS-injured lung. Am. J. Physiol. Lung Cell. Mol. Physiol. 292:L748–L759 [DOI] [PubMed] [Google Scholar]
- 7. Brown EL, Lyles DS. 2005. Pseudotypes of vesicular stomatitis virus with CD4 formed by clustering of membrane microdomains during budding. J. Virol. 79:7077–7086 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8. Carroll MC. 2004. The complement system in regulation of adaptive immunity. Nat. Immunol. 5:981–986 [DOI] [PubMed] [Google Scholar]
- 9. Cummings KL, Waggoner SN, Tacke R, Hahn YS. 2007. Role of complement in immune regulation and its exploitation by virus. Viral Immunol. 20:505. [DOI] [PubMed] [Google Scholar]
- 10. Delgado MF, Polack FP. 2004. Involvement of antibody, complement and cellular immunity in the pathogenesis of enhanced respiratory syncytial virus disease. Expert Rev. Vaccines 3:693–700 [DOI] [PubMed] [Google Scholar]
- 11. Devaux P, Christiansen D, Plumet S, Gerlier D. 2004. Cell surface activation of the alternative complement pathway by the fusion protein of measles virus. J. Gen. Virol. 85:1665–1673 [DOI] [PubMed] [Google Scholar]
- 12. Gainey MD, Manuse MJ, Parks GD. 2008. Hyperfusogenic F protein enhances oncolytic potency of an SV5 P/V mutant without compromising sensitivity to type I interferon. J. Virol. 82:9369–9380 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13. Gerlier D, et al. 1994. Measles virus receptor properties are shared by several CD46 isoforms differing in extracellular regions and cytoplasmic tails. J. Gen. Virol. 75:2163–2171 [DOI] [PubMed] [Google Scholar]
- 14. Hirsch RL, Wolinsky JS, Winkelstein JA. 1986. Activation of the alternative complement pathway by mumps infected cells: relationship to viral neuraminidase activity. Arch. Virol. 87:181–190 [DOI] [PubMed] [Google Scholar]
- 15. Jayasekara JP, Moseman EA, Carroll MC. 2007. Natural antibody and complement mediate neutralization of influenza virus in the absence of prior immunity. J. Virol. 81:3487–3494 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16. Johnson JB, Capraro GA, Parks GD. 2008. Differential mechanisms of complement mediated neutralization of the closely related paramyxoviruses simian virus 5 and mumps virus. Virology 376:112–123 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17. Johnson JB, Grant K, Parks GD. 2009. The paramyxoviruses simian virus 5 and mumps virus recruit host cell CD46 to evade complement-mediated neutralization. J. Virol. 83:7602–7611 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18. Kemper C, Atkinson JP. 2007. T-cell regulation: with complements from innate immunity. Nat. Rev. Immunol. 7:9–18 [DOI] [PubMed] [Google Scholar]
- 19. Kerr MA. 1980. The human complement system: assembly of the classical pathway C3 convertase. Biochem. J. 189:173–181 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20. Kim DD, Song WC. 2006. Membrane complement regulatory proteins. Clin. Immunol. 118:127–136 [DOI] [PubMed] [Google Scholar]
- 21. Liszewski MK, Post TW, Atkinson JP. 1991. Membrane cofactor protein (MCP or CD46): newest member of the regulators of complement activation gene cluster. Annu. Rev. Immunol. 9:431–455 [DOI] [PubMed] [Google Scholar]
- 22. Liszewski MK, Fang CJ, Atkinson JP. 2008. Inhibiting complement activation on cells at the step of C3 cleavage. Vaccine 26(Suppl 8):I22–I27 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23. Lublin DM, Atkinson JP. 1989. Decay-accelerating factor: biochemistry, molecular biology and function. Annu. Rev. Immunol. 7:35–58 [DOI] [PubMed] [Google Scholar]
- 24. Markiewski MM, Lambris JD. 2007. The role of complement in inflammatory diseases from behind the scenes into the spotlight. Am. J. Pathol. 171:715–727 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25. McSharry JJ, Pickering RJ, Caliguiri LA. 1981. Activation of the alternative complement pathway by enveloped viruses containing limited amounts of sialic acid. Virology 114:507–515 [DOI] [PubMed] [Google Scholar]
- 26. Medof ME, Kinoshita T, Nussenzweig V. 1984. Inhibition of complement activation on the surface of cells after incorporation of decay-accelerating factor (DAF) into their membranes. J. Exp. Med. 160:1558–1578 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27. Mills BJ, Beebe DP, Cooper NR. 1979. Antibody-independent neutralization of vesicular stomatitis virus by human complement. II. Formation of VSV-lipoprotein complexes in human serum and complement-dependent viral lysis. J. Immunol. 123:2518–2524 [PubMed] [Google Scholar]
- 28. Miyagawa S, et al. 2004. Delta-short consensus repeat 4-decay accelerating factor (DAF; CD55) inhibits complement-mediated cytolysis but not NK cell-mediated cytolysis. J. Immunol. 173:3945–3952 [DOI] [PubMed] [Google Scholar]
- 29. Morrison TE, Fraser RJ, Smith PN, Mahalingam S, Heise MT. 2007. Complement contributes to inflammatory tissue destruction in a mouse model of Ross River virus-induced disease. J. Virol. 81:5132–5143 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30. Mullick J, et al. 2005. Identification of complement regulatory domains in vaccinia virus complement control protein. J. Virol. 79:12382–12393 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31. Nayak S, Herzog RW. 2010. Progress and prospects: immune responses to viral vectors. Gene Ther. 17:295–304 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32. Nicholson-Weller A, Burge J, Fearon DT, Weller PF, Austen KF. 1982. Isolation of a human erythrocyte membrane glycoprotein with decay-accelerating activity for C3 convertases of the complement system. J. Immunol. 129:184–189 [PubMed] [Google Scholar]
- 33. Okura E, et al. 2008. Differential human serum-mediated neutralization of PERV released from pig cells transfected with variants of hDAF. Xenotransplantation 15:365–373 [DOI] [PubMed] [Google Scholar]
- 34. Pangburn MK, Schreiber RD, Muller-Eberhard HJ. 1983. Deficiency of an erythrocyte membrane protein with complement regulatory activity in paroxysmal nocturnal hemoglobinuria. Proc. Natl. Acad. Sci. U. S. A. 80:5430–5434 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35. Post TW, Liszewski MK, Adams EM, Tedja I, Miller EA. 1991. Membrane cofactor protein of the complement system: alternative splicing of serine/threonine/proline-rich exons and cytoplasmic tails produces multiple isoforms that correlate with protein phenotype. J. Exp. Med. 174:93–102 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36. Roozendaal R, Carroll MC. 2006. Emerging patterns in complement-mediated pathogen recognition. Cell 125:29–32 [DOI] [PubMed] [Google Scholar]
- 37. Russell SR, Sparrow RL, McKenzie IFC, Purcell DFJ. 1992. Tissue-specific and allelic expression of the complement regulator CD46 is controlled by alternative splicing. Eur. J. Immunol. 22:1513–1518 [DOI] [PubMed] [Google Scholar]
- 38. Saifuddin M, et al. 1997. Human immunodeficiency virus type 1 incorporates both glycosyl phosphatidylinositol-anchored CD55 and CD59 and integral membrane CD46 at levels that protect from complement-mediated destruction. J. Gen. Virol. 78:1907–1911 [DOI] [PubMed] [Google Scholar]
- 39. Schauber-Plewa C, Simmons A, Tuerk M, Pacheco C, Veres G. 2005. Regulatory proteins are incorporated into lentiviral vectors and protect particles against complement inactivation. Gene Ther. 12:238–245 [DOI] [PubMed] [Google Scholar]
- 40. Shaw ML, Stone KL, Colangelo CM, Gulcicek EE, Palese P. 2008. Cellular proteins in influenza virus particles. PLoS Pathog. 4:e1000085 doi:10.1371/journal.ppat.1000085 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41. Stoermer KA, Morrison TE. 2011. Complement and viral pathogenesis. Virology 411:362–373 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42. Stoiber H, Pinter C, Siccardi AG, Clivio A, Dierich MP. 1996. Efficient destruction of human immunodeficiency virus in human serum by inhibiting the protective action of complement factor H and decay accelerating factor (DAF, CD55). J. Exp. Med. 183:307–310 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43. Stuart AD, Eustace HE, McKee TA, Brown TD. 2002. A novel cell entry pathway for a DAF-using human enterovirus is dependent on lipid rafts. J. Virol. 76:9307–9322 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44. Vanderplasschen A, Mathew E, Hollinshead M, Sim RB, Smith GL. 1998. Extracellular enveloped vaccinia virus is resistant to complement because of incorporation of host complement control proteins into its envelope. Proc. Natl. Acad. Sci. U. S. A. 95:7544–7549 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 45. Vasantha S, et al. 1988. Interaction of a nonneutralizing IgM antibody and complement in parainfluenza virus neutralization. Virology 167:433–441 [PubMed] [Google Scholar]
- 46. Veerhuis R, Nielsen HM, Tenner AJ. 2011. Complement in the brain. Mol. Immunol. 48:1592–1603 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 47. Welsh RM., Jr 1977. Host cell modification of lymphocytic choriomeningitis virus and Newcastle disease virus altering viral inactivation by human complement. J. Immunol. 118:348–354 [PubMed] [Google Scholar]
- 48. Zavada J. 1982. The pseudotype paradox. J. Gen. Virol. 63:15–24 [DOI] [PubMed] [Google Scholar]