

Abstract
We have used electron paramagnetic resonance (EPR) to probe the homo- and heterooligomeric interactions of reconstituted sarcoplasmic reticulum Ca-ATPase (SERCA) and its regulator phospholamban (PLB). SERCA is responsible for restoring calcium to the sarcoplasmic reticulum to allow muscle relaxation, whereas PLB inhibits cardiac SERCA unless phosphorylated at Ser16. To determine whether changes in protein association play essential roles in regulation, we detected the microsecond rotational diffusion of both proteins using saturation transfer EPR. Peptide synthesis was used to create a fully functional and monomeric PLB mutant with a spin label rigidly coupled to the backbone of the transmembrane helix, while SERCA was reacted with a Cys-specific spin label. Saturation transfer EPR revealed that sufficiently high lipid/protein ratios minimized self-association for both proteins. Under these dilute conditions, labeled PLB was substantially immobilized after co-reconstitution with unlabeled SERCA, reflecting their association to form the regulatory complex. Ser16 phosphorylation slightly increased this immobilization. Complementary measurements with labeled SERCA showed no change in mobility after co-reconstitution with unlabeled PLB, regardless of its phosphorylation state. We conclude that phosphorylating monomeric PLB can relieve SERCA inhibition without changes in the oligomeric states of these proteins, indicating a structural rearrangement within the heterodimeric regulatory complex.
Introduction
Muscle relaxation is induced by the active transport of the divalent calcium ion (Ca2+) from the cytoplasm into the sarcoplasmic reticulum (SR). The SR Ca2+-ATPase (SERCA) is a 994-residue enzyme that transports two Ca2+ into the SR lumen per ATP hydrolyzed (Fig. 1, left) (1), resulting in muscle relaxation. In cardiac and slow-twitch muscle, SERCA activity is allosterically regulated by phospholamban (PLB) (2), a 52-residue membrane protein consisting of a 30-residue transmembrane helix and a 16-residue cytosolic helix connected by a short, flexible loop (3) (Fig. 1, right). PLB inhibits SERCA by decreasing its apparent Ca2+ affinity (increasing KCa) (4). SERCA inhibition is relieved physiologically either by micromolar [Ca2+] or through PLB phosphorylation at Ser16 by protein kinase A (5), and can also be relieved by PLB point mutations (6–8).
Figure 1.
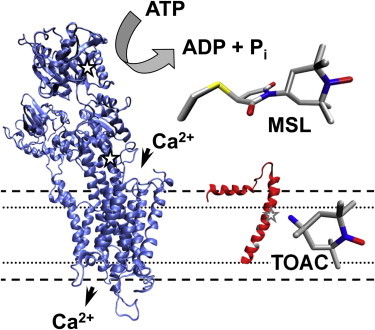
Structural models of SERCA (left, crystal structure 1IWO (76)) and monomeric PLB (right, hybrid NMR structure 2KB7 (77)). All experiments in this study were conducted with the monomeric form of PLB. The amino-acid TOAC (bottom right), containing a nitroxide spin label rigidly coupled to the α-carbon, is incorporated at position 36 (shaded star) on PLB; PLB residues 41 and 46 (shaded) are mutated to Phe and Ala. For unlabeled PLB, position 36 is Ala. For spin-labeled SERCA, MSL (top right) is attached at Cys344/Cys364 (solid star) (78).
The regulatory mechanism of the SERCA-PLB complex has considerable therapeutic relevance, because inadequate calcium transport has been strongly linked to heart failure (9). Emerging treatments aim to improve cardiac performance by increasing SERCA activity through increased SERCA expression (10), allosteric activation of SERCA by small molecules (11), or reduced PLB inhibitory potency (12,13). Elucidating this mechanism would provide a roadmap for the rational design of PLB mutants and drugs designed to activate SERCA.
However, this regulatory mechanism remains controversial. Cross-linking (14) and immunoprecipitation (15) studies suggest that dissociation of the SERCA-PLB complex is required (Fig. 2, Dissociation Model). However, spectroscopic studies suggest that PLB remains bound to SERCA after activation by Ser16 phosphorylation (16–18), Ca2+ addition (19), or PLB loss-of-function mutations (13,18), indicating that a structural rearrangement within the complex can relieve inhibition (Fig. 2, Subunit Model). Other studies have also identified multiple PLB conformations within the regulatory complex that correlate with different inhibitory states (17,20).
Figure 2.
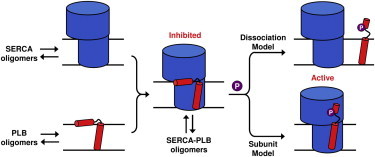
Oligomeric interactions of SERCA and PLB. Both proteins have homooligomeric interactions (left), and bind to form an inhibited SERCA-PLB complex (center). Phosphorylation of PLB relieves inhibition (right), which may occur through dissociation of the complex (Dissociation Model), or a structural rearrangement within the complex (Subunit Model).
Oligomeric interactions
Distinguishing these regulatory mechanisms is made more difficult by the variety of protein-protein interactions in this system. Although the fundamental inhibitory complex is proposed to be a SERCA-PLB heterodimer (2,21,22) (Fig. 2, center), both proteins form homooligomers that can affect SERCA function (Fig. 2). SERCA may form functioning dimer and trimer states (23,24), while larger aggregates lead to inhibition (25,26). Numerous studies have examined the oligomeric state of SERCA in native (27–29) and reconstituted (25) membranes, and several have investigated the role of the lipid/protein molar ratio (L/P) in modulating SERCA aggregation (26,30,31), all based on the proportionality between oligomeric size and rotational correlation time (32).
In cardiac SR, SERCA inhibition and aggregation are both relieved by micromolar Ca2+ or PLB phosphorylation (33). Native PLB equilibrates between monomers and homopentamers, and the latter can be destabilized by mutation (typically leading to increased SERCA inhibition) or stabilized by phosphorylation (leading to decreased SERCA inhibition). Thus, inhibition depends, at least in part, on the concentration of PLB monomer (21). To simplify analysis and focus on SERCA-PLB interactions, we have used 1), a monomeric PLB mutant, C36A/C41F/C46A-phospholamban (AFA-PLB, referred to below as PLB) (34) in which oligomerization is eliminated by replacing Cys residues 36, 41, and 46 with Ala, Phe, and Ala, respectively, and 2), sufficiently high L/Ps to minimize self-association of both SERCA and PLB. Thus, we are able to focus solely on the heterodimeric interactions between these two proteins.
In this study, we have characterized the heterooligomerization of PLB and SERCA using electron paramagnetic resonance (EPR), while minimizing the homooligomerization of both proteins. Rotational motions detected by EPR can be sensitive to oligomeric interactions, because the rotational correlation time (τR) of the spin-labeled protein is proportional to the protein’s transmembrane volume (32). However, conventional EPR is sensitive only to correlation times in the nanosecond range, which is appropriate for rotation of aqueous proteins, but not membrane-embedded proteins, where the viscosity (and thus τR) is orders-of-magnitude greater. Even in fluid lipid bilayers, τR values for most integral membrane proteins are in the microsecond range.
These slow rotational motions are best measured by saturation transfer EPR (STEPR) (35,36), which requires the use of spin probes that are immobilized on the labeled protein. In this study, PLB rotational motion was detected by incorporating the spin label 2,2,6,6-tetramethylpiperidine-1-oxyl-4-amino-4-carboxylic acid (TOAC) into the peptide backbone at position 36 in the transmembrane helix (Fig. 1), while SERCA rotational dynamics was detected after covalent reaction with a maleimide spin label, 4-maleimido-TEMPO (MSL). Both labels have been shown to be rigidly attached to their respective proteins, making them sensitive to rigid-body rotational diffusion that directly reflects the size of the oligomeric complex (27,37). We used STEPR to first find conditions where homooligomerization was minimized, and then to detect the formation of SERCA-PLB heterooligomers so as to discriminate between the Dissociation Model and the Subunit Model (see Fig. 2).
Methods
Peptide synthesis
C36TOAC, C41F, C46A, PLB (36-TOAC-AFA-PLB) and C36A, C41F, C46A PLB (AFA-PLB) were synthesized as described previously (37–39). Fmoc-TOAC was purchased from Toronto Research Chemicals (North York, Ontario, Canada) and Fmoc-protected amino acids were purchased from EMD Chemicals (Philadelphia, PA). Peptides were purified by reverse-phase HPLC on a preparative C8 column (Grace Vydac, Hesperia, CA) using H2O + 0.1% trifluoroacetic acid and isopropanol as mobile phases. Peptides were characterized by mass spectrometry and sodium dodecyl-sulfate polyacrylamide gel electrophoresis (SDS-PAGE). Protein concentrations were quantified using a bicinchoninic-acid assay (Thermo Fisher Scientific, Rockford IL).
SERCA purification and spin labeling
SR vesicles were prepared from rabbit fast-twitch muscle (40), then resuspended to 25 mg/mL total protein in SR buffer (30 mM 3-(n-morpholino)propanesulfonic acid (MOPS), 0.3 M sucrose, pH 7.0). SERCA was purified by reactive red chromatography (41), omitting reducing agents. For spin labeling, SERCA was diluted to 10 mg/mL with SR buffer, to which 300 μM MSL was added from a 50 mM stock in dimethylformamide. After stirring 1 h at 25°C, labeled vesicles were diluted 10-fold in SR buffer and centrifuged 45 min at 30,000 × g (4°C) to remove unreacted MSL. The resulting pellet was resuspended in SR buffer and purified as above. Protein concentrations were quantified using a bicinchoninic-acid assay.
Reconstitution of SERCA and PLB
Functional reconstitution of SERCA and PLB was carried out as described previously, using 4:1 1,2-dioleoyl-sn-glycero-3-phosphocholine (DOPC)/1,2-dioleoyl-sn-glycero-3-phosphoethanolamine (DOPE) (mol/mol) to yield unilamellar vesicles that are optimal for reproducing physiological SERCA-PLB regulatory function (42,43). These vesicles have an average diameter of 100 nm (43) and thus rotate too slowly to affect our saturation transfer EPR spectra (27,45). Final buffer composition of EPR samples was 50 mM MOPS, 50 mM KCl, 5 mM MgCl2, 0.5 mM ethylene glycol-bis(2-aminoethylether)-n,n,n′,n′-tetraacetic acid (EGTA), 210 μM CaCl2, pH 7.0, resulting in pCa 6.5 to give optimal regulation of SERCA by PLB (19). Identical buffer conditions were used in functional assays, except that [CaCl2] was varied. An enzyme-coupled ATPase assay was used to measure the Ca-dependence of SERCA activity as described previously in Lockamy et al. (13) and Reddy et al. (42).
As in previous publications from our laboratory, EPR experiments were performed at 4°C to maintain protein stability during lengthy STEPR experiments, while functional measurements were performed at 25°C to enhance SERCA activity and permit comparison to published work. PLB inhibitory potency is only weakly dependent on temperature (46). For phosphorylation, the PLB/lipid/detergent mixture (containing 20 μM PLB, before SERCA addition) was incubated 2 h at 25°C with 200 IU/mL bovine protein kinase A catalytic subunit (Sigma, St. Louis, MO) and 1 mM MgATP. Complete PLB phosphorylation (>92%) was confirmed by quantitative Western blotting (47) using 10–20% tris-tricine gels (Bio-Rad, Hercules, CA).
EPR spectroscopy and data analysis
Before EPR experiments, samples were centrifuged 1 h at 200,000 × g (4°C) and the pellets resuspended in minimal buffer; 40 μL samples were loaded into 22-gauge Teflon tubing (Amazon Supply, Miami Lakes, FL) threaded through a glass capillary (Wiretrol; Drummond Scientific Company, Broomall, PA) flame-sealed at one end. The Teflon tubing extended 1 cm from the end of the capillary and was plugged with Critoseal (Krackeler Scientific, Albany, NY). Spectra were acquired at X-band (9.5 GHz) with an EleXsys E500 spectrometer equipped with the ER 4122 SHQ cavity (Bruker BioSpin, Billerica, MA). Temperature was maintained at 4 ± 0.2°C with a quartz Dewar insert and a nitrogen-gas-flow temperature controller. Spectra were acquired after sample deoxygenation under nitrogen gas (∼1 h) to eliminate the dependence of spectra on oxygen accessibility (48). Microwave field amplitude H1 (G) was calibrated with a solution of 0.9 mM PADS, 50 mM K2CO3 (48).
Spectral acquisition parameters were set according to Squier and Thomas (48). Conventional (V1, first harmonic, in-phase) EPR spectra were acquired with H1 = 0.14 G microwave field amplitude, 100 kHz modulation frequency, 2 G modulation amplitude, and 120 G sweep width. V1 spectra used for normalization were acquired as above except that H1 was set to 0.032 G. STEPR (V2′, second harmonic, out-of-phase) spectra were acquired with 50 kHz modulation frequency, 5 G modulation amplitude, and 120 G sweep width. After null phase determination by minimizing the signal at nonsaturating microwave intensity (H1 = 0.032 G), H1 was increased to 0.25 G and the V2′ spectrum recorded. All EPR spectra were baseline-corrected and then normalized, either to ∫∫V1 (conventional EPR) or to ∫∫V1 / H1 for H1 = 0.032 G (STEPR). V1 EPR spectra near the slow-motion limit were analyzed by evaluating lineshape features ΔL′ (the outer half-width of the low-field line, which increases with spin-spin interaction and rotational motion) and 2T||′ (the splitting between outer extrema, which decreases with rotational motion but not with spin-spin interaction) (49,50).
For V2′ spectra, effective rotational correlation times were calculated from an integrated intensity parameter (IST), using a correlation plot created from MSL-hemoglobin samples in glycerol (48). IST reports an effective rotational correlation time that is minimally sensitive to modulation phase errors and thus provides greater precision than provided by lineshape parameters (48).
Data plots were produced with Origin 8.1 (OriginLab, Northampton, MA). Error bars are mean ± SE (n = 3–5). When no error bar is visible, this indicates that SE is smaller than the plotted symbol.
Results
Functional assays
To confirm that 36-TOAC-PLB retained regulatory function, we reconstituted it with SERCA into 4:1 (mol/mol) 1,2-dioleoyl-sn-glycero-3-phosphocholine/1,2-dioleoyl-sn-glycero-3-phosphoethanolamine (DOPC/DOPE) vesicles at 700 lipids/SERCA (mol/mol) and measured Ca-dependent ATPase activity using a coupled-enzyme assay (42). Results were fit by the Hill equation,
| (1) |
where V is the ATPase rate, Vmax is the ATPase rate at saturating [Ca2+], n is the Hill coefficient, and pKCa is the pCa (−log10[Ca2+]) where activity is half-maximal. PLB inhibitory potency, defined as the decrease in pKCa, was indistinguishable between our spin-labeled construct and unlabeled PLB controls (Fig. 3). At 10 PLB/SERCA, where inhibition is maximal, pKCa shifted from 6.48 ± 0.01 to 6.00 ± 0.01 (inhibitory potency = 0.48 ± 0.02). When PLB was completely phosphorylated (pPLB, >92%), inhibition was partially reversed, increasing pKCa to 6.30 ± 0.01 (inhibitory potency = 0.18 ± 0.02) and restoring SERCA Ca2+ sensitivity to the same extent as phosphorylating unlabeled PLB (Fig. 3). At 0.5 PLB/SERCA, matching the conditions of some experiments below, the inhibitory potency of 36-TOAC-PLB was 0.28 ± 0.03 (unphosphorylated) and 0.12 ± 0.04 (phosphorylated). These results, including the partial reversal of inhibition by phosphorylation, are similar to those reported previously for other PLB derivatives (17,37) and demonstrate that 36-TOAC-PLB regulates SERCA normally. We also used functional assays to determine whether reacting SERCA with MSL affected its interaction with PLB. Although MSL decreased Vmax by ∼30% as reported previously (29), pKCa regulation by PLB was unaltered, indicating that the label does not perturb regulatory interactions between the two proteins.
Figure 3.
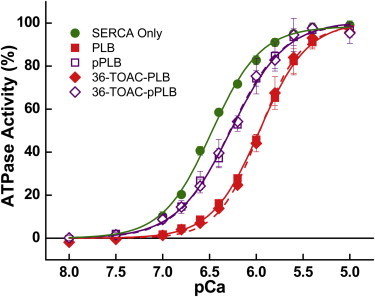
Ca-dependence of ATPase activity. Samples were reconstituted at 700 lipids/SERCA and 10 PLB/SERCA (mol/mol). ATP turnover was measured at 25°C, fit by Eq. 1, and normalized to Vmax. SERCA-only controls (circles) were compared to samples containing PLB (solid squares), 36-TOAC-PLB (solid diamonds), and their pSer16 counterparts (open squares and diamonds). pKCa values obtained from the fits were used to quantify SERCA inhibition, and are summarized in Table S1 in the Supporting Material.
Monomeric mutant AFA-PLB does not aggregate
Our goal was to find conditions in which self-association is minimized for both PLB and SERCA. We used conventional and saturation transfer EPR to examine spin-labeled PLB or SERCA reconstituted at several L/P. Conventional EPR of 36-TOAC-PLB detected only a marginal decrease in outer splitting (2T||′, Fig. 4 a) with increasing L/P, indicating little or no change in nanosecond rotational dynamics. However, lipid dilution did lead to a substantial decrease in the linewidth (ΔL′) (Fig. 4 b), which could arise from decreased nanosecond rotational mobility (49,50), but this is inconsistent with the lack of increased splitting (Fig. 4 b). Thus, the linewidth decrease is due to decreased dipolar broadening from spin-spin interactions, but the observed broadening is precisely that expected for a random two-dimensional arrangement of monomers in the membrane (51), and would be much greater if PLB oligomers were present, as demonstrated by Fig. S1 and Fig. S2 in the Supporting Material. STEPR spectra support this interpretation (see Fig. S3). Thus, this PLB construct remains predominantly monomeric at all L/P tested here.
Figure 4.
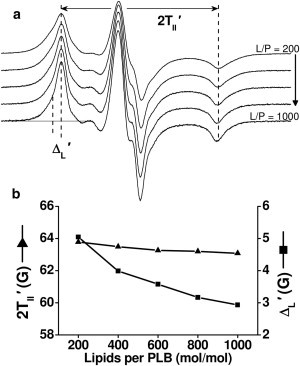
Dependence of conventional EPR spectra of 36-TOAC-PLB on lipid/protein ratio. (a) Spectra of 36-TOAC-PLB reconstituted at 200–1000 L/P. (b) Outer splitting (2T||′, triangles) and low-field outer linewidth (ΔL′, squares) as a function of L/P. The vertical scales were set so that a change in rotational motion would yield changes in ΔL′ and 2T||′ of similar magnitude (but opposite sign) (49,50). STEPR spectra are given in Fig. S3.
SERCA does not aggregate above 600 L/P
We conducted similar lipid-dilution experiments with MSL-SERCA. Conventional EPR spectra (see Fig. S4) have no significant dependence on L/P, confirming the previous finding that this spin label binds rigidly to SERCA and undergoes no nanosecond rotational dynamics (27). Thus, any changes in STEPR spectra (Fig. 5 a) are due to changes in microsecond rotational motion of SERCA about an axis perpendicular to the membrane (27). These STEPR spectra show that the effective rotational correlation time τR decreases substantially from L/P = 200–600, to a limiting value of ∼12 μs for L/P > 600. This τR is consistent with the rotational diffusion of monomeric or dimeric SERCA (28) based on the Saffman-Delbrück equation (32), which predicts that τR is proportional to protein transmembrane volume and lipid viscosity. Assuming a membrane height of 4 nm (52) and membrane viscosity of 2–5 P (53), the Saffman-Delbrück model yields correlation times of 2–4 μs and 8–19 μs for SERCA monomers and dimers, respectively. We conclude that SERCA undergoes concentration-dependent self-association, but that this dependence is negligible for L/P > 600, suggesting the presence of stable SERCA monomers or small oligomers. Therefore, further experiments were performed at 700 lipids per SERCA to eliminate the formation of homooligomers, allowing us to focus on the SERCA-PLB interaction.
Figure 5.
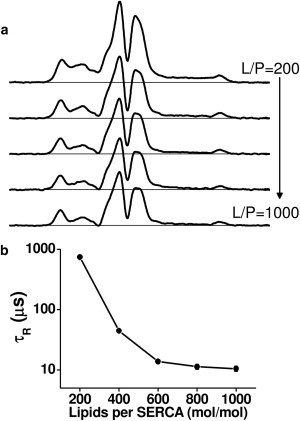
STEPR of MSL-SERCA as a function of lipid/protein molar ratio (L/P). (a) Representative STEPR (V2′) spectra of reconstituted MSL-SERCA. (b) Effective rotational correlation times (τR). Conventional EPR (V1) spectra are given in Fig. S4.
Effects of phosphorylation and SERCA binding on rotational dynamics of spin-labeled PLB
We co-reconstituted 36-TOAC-PLB with SERCA to investigate PLB rotational dynamics within the regulatory complex, using 700 lipids/SERCA and 0.5 PLB/SERCA to minimize both homooligomers and unbound PLB. Conventional EPR spectra of co-reconstituted samples (PLB + SERCA, pPLB + SERCA) and their PLB-only controls (PLB, pPLB) are all near the rigid limit and are virtually identical (Fig. 6, a and c), indicating no change in nanosecond dynamics or self-association of PLB upon phosphorylation and/or SERCA binding. The V1 EPR spectra in Fig. 6 a contain subtle shoulders on the low- and high-field peaks. These could arise from spin-spin interactions (Fig. 5, and see Fig. S1) or the presence of slow restricted-amplitude rotation (54). However, the V1 spectra are not affected by phosphorylation and/or SERCA addition, so the effects on V2′ spectra (Fig. 6 b) must arise from changes in microsecond rotational dynamics.
Figure 6.
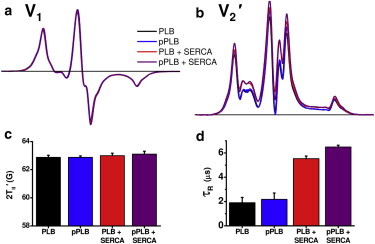
Effects of Ser16 phosphorylation and SERCA on EPR of 36-TOAC-PLB. (a) Conventional (V1) EPR spectra and (b) STEPR (V2′) spectra of 36-TOAC-PLB as affected by phosphorylation (pPLB) and/or SERCA (0.5 PLB/SERCA). (c) Outer splitting 2T||′ measured from conventional EPR spectra in panel a. (d) Effective rotational correlation times from STEPR spectra in panel b. Typical normalized spectra are shown in panels a and b. (Bar graphs in panels c and d show mean ± SE for n = 3–5 trials.)
In contrast to the invariant conventional (V1) spectra (Fig. 6, a and c), STEPR (V2′) revealed significant changes in microsecond dynamics (Fig. 6, b and d). SERCA decreased PLB mobility substantially, with τR increasing from 1.9 ± 0.4 μs (PLB) to 5.6 ± 0.2 μs (PLB + SERCA), consistent with the large increase in SERCA binding. Ser16 phosphorylation had no significant effect on PLB dynamics in the absence of SERCA (Fig. 6, b and d), but increased τR slightly in the presence of SERCA, from 5.6 ± 0.2 μs to 6.5 ± 0.1 μs (Fig. 6 d). This larger τR is probably not due to increased binding to SERCA (because that would increase inhibition) or to increased PLB aggregation (because that is inconsistent with the V1 spectra in Fig. 6 a), so it must be due to some structural change of the SERCA-PLB complex that alters either the tilt of the PLB TM domain or the frictional interaction of the SERCA-PLB complex with lipid (55–57). In any case, it is clear that PLB does not dissociate from SERCA after phosphorylation, because this would cause a substantial decrease in the rotational correlation time, making the blue (pPLB) and purple (pPLB + SERCA) spectra identical (Fig. 6, c and d). Indeed, a slight increase in τR was observed due to phosphorylation (Fig. 6 d), and the blue and purple spectra are quite different (Fig. 6 c), so Fig. 6 clearly contradicts the Dissociation Model and strongly supports the Subunit Model (Fig. 2).
Rotational dynamics of spin-labeled SERCA reconstituted with PLB
To determine whether the results of Fig. 6 were affected by changes in SERCA aggregation, we performed identical experiments except using MSL-SERCA and unlabeled PLB. Both V1 (see Fig. S5) and V2′ (Fig. 7) detect no change in SERCA rotational dynamics due to PLB or pPLB. The addition of 0.5 PLB per SERCA should increase the transmembrane volume (and thus the rotational correlation time) by only ∼5% (32), within the experimental error of our measurements (∼10%, Fig. 6 d). This indicates that the aggregation state of SERCA is not significantly perturbed by PLB binding or phosphorylation under these dilute conditions (700 lipids/SERCA and 0.5 PLB/SERCA), despite substantial functional effects. These findings indicate that regulatory effects of PLB do not arise solely from modulating SERCA aggregation, although such phosphorylation-dependent aggregation does contribute to functional regulation in cardiac SR (33).
Figure 7.
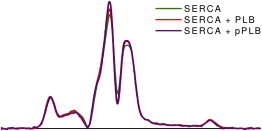
STEPR (V2′) spectra of MSL-SERCA. Experimental conditions are as in Fig. 6, using MSL-SERCA and unlabeled PLB. Rotational correlation times did not vary significantly among these samples and were consistent with the data in Fig. 5 at 700 lipids/SERCA (13 ± 4 μs). Conventional EPR spectra also showed no significant changes (see Fig. S5).
Rotational correlation times determined from STEPR spectra are effective, and depend on several parameters that are not easily controlled or evaluated, such as probe orientation relative to the diffusion axis (58) and spin label environment (48). In addition, it is likely that the TM domain of PLB has residual small-amplitude rotational flexibility relative to SERCA. These factors probably contribute to the significant differences between the V1 spectra of the SERCA-PLB complex with labeled PLB (Fig. 6 a) or labeled SERCA (see Fig. S5), and to the shorter τR values determined from V2′ spectra of the complex with labeled PLB (Fig. 6 b), compared to labeled SERCA (Fig. 7). Therefore, our key conclusions come from comparing the V2′ spectra of a particular spin-labeled protein (36-TOAC-PLB or MSL-SERCA), as perturbed by factors (protein-protein interactions, protein concentration, and phosphorylation) that do not affect the V1 spectra.
Discussion
We have used EPR to investigate the tendencies of SERCA and the monomeric AFA-PLB to form homo- and heterooligomers in reconstituted membranes. By systematically varying the lipid/protein molar ratio (L/P), we found that SERCA self-association occurs at low L/P but is eliminated for L/P > 600 (Fig. 5), even in the presence of 0.5 PLB/SERCA (Fig. 7). We also showed that TOAC-labeled PLB does not self-associate under the conditions of this study (Fig. 4). Thus, the data in Fig. 6 reflect directly the rotational dynamics of PLB alone or in complex with SERCA. Because phosphorylation and SERCA binding caused no significant effects on PLB nanosecond dynamics as detected by conventional EPR (Fig. 6, left), the changes in STEPR (Fig. 6, right) are due to microsecond rigid-body dynamics of PLB. Thus, SERCA does immobilize PLB by binding it, but phosphorylation does not dissociate PLB from SERCA, supporting the Subunit Model (Fig. 2, bottom right).
AFA-PLB is monomeric in lipid bilayers at high L/P, even after phosphorylation
Although the null-Cys PLB mutant AAA-PLB, similar to AFA-PLB, was found previously to be monomeric by SDS-PAGE (38,59), solid-state NMR studies suggested that AAA-PLB is substantially aggregated in reconstituted membranes at the extremely low L/P value of 20 (60). 36-TOAC-AFA-PLB shows substantial broadening of the V1 spectrum at 20 L/P, consistent with aggregation (see Fig. S1), along with dramatic effects on the V2′ spectrum (see Fig. S2), showing that PLB aggregation is detectable if present. In contrast, our EPR experiments performed between 200 and 1000 L/P did not reveal significant PLB aggregation (Fig. 4). This is the most direct demonstration to date that AFA-PLB behaves as a monomer in reconstituted bilayers, whether phosphorylated or not. In previous studies, it was shown that the pentameric propensity of wild-type PLB increases with phosphorylation (21), and that SERCA preferentially binds to the PLB monomer (61). Thus, it remains likely that the decrease in PLB monomer concentration due to phosphorylation plays a significant role in relieving SERCA inhibition (21,62). However, our results show that some relief of inhibition by phosphorylation occurs even under conditions where PLB oligomers do not form.
SERCA self-association occurs only at low L/P and is not induced by PLB at high L/P
The oligomeric state of SERCA for optimal function has been postulated to range from monomeric to tetrameric (24,28,63–65), whereas larger aggregates have been shown to decrease SERCA activity, whether induced by cross-linking (66), cationic peptides (67,68), cationic local anesthetics (69), mismatch in lipid bilayer thickness (70), small-molecule SERCA inhibitors (23,71), or PLB (33,72). Most importantly, inhibition relief in cardiac SR by either Ca2+ or phosphorylation correlates with decreased SERCA aggregation (33). This study does not contradict these findings; SERCA aggregation probably plays a significant physiological role in both skeletal and cardiac SR. However, our results show that at low protein concentrations in the membrane (at high L/P), significant changes in SERCA inhibition by PLB occur without significant changes in SERCA aggregation (Fig. 7).
Binding of TOAC-spin-labeled PLB to SERCA is clearly detected by STEPR
STEPR shows that reconstitution with SERCA decreases the microsecond mobility of 36-TOAC-AFA-PLB substantially, consistent with binding (Fig. 6, b and d), whereas conventional EPR detects no change in nanosecond motion or PLB aggregation. This complex formation is consistent with FRET from SERCA to PLB in a similar reconstituted system (20).
Phosphorylation causes a structural change in the SERCA-PLB complex, not dissociation
Phosphorylation of SERCA-bound PLB at Ser16 does not increase its rotational mobility, and thus does not dissociate it from SERCA under conditions of our study (Fig. 6). In fact, an increase in the effective rotational correlation time is observed, indicating a structural change in the SERCA-PLB complex that decreases mobility or changes the tilt of the PLB TM domain. This result supports the Subunit Model (Fig. 2, bottom right), though future studies are needed to characterize the structural change. Because the concentrations of SERCA and PLB, as well as the PLB/SERCA ratio, are much higher in cardiac SR than in the samples considered here, it is unlikely that phosphorylation of PLB causes significant dissociation under physiological conditions. Whereas previous studies have found that PLB is in dynamic equilibrium between free and SERCA-bound states (13,73), our data indicate that PLB phosphorylation does not perturb this binding equilibrium significantly.
This study used the SERCA1a isoform from skeletal SR. There is no known difference between the functional or physical interaction of PLB with SERCA1a and SERCA2a (62,74,75), but future studies with the SERCA2a isoform will be needed to rule this out. Previous studies using spin or fluorescent probes of phosphorylated PLB interacting with SERCA are consistent with the conclusions of this study (16–18). However, to our knowledge, this is the first study of the SERCA-PLB interaction using a probe rigidly coupled to the transmembrane domain of PLB, thus reporting reliably the rotational mobility of PLB and showing that Ser16 phosphorylation does not dissociate the inhibitory transmembrane domain (38) of PLB from SERCA.
Conclusions
We have used STEPR to detect the microsecond rotational mobilities of SERCA and AFA-PLB in reconstituted membranes, providing direct insight into their oligomeric interactions, as perturbed by phosphorylation. At L/P ≥ 600 and PLB/SERCA = 0.5, SERCA does not change its state of self-association due to PLB or pPLB, both of which are strongly immobilized by SERCA binding. Under these conditions, relief of SERCA inhibition must be due to a structural change within the SERCA-PLB complex, not to dissociation of the complex.
Acknowledgments
EPR experiments were performed at the Biophysical Spectroscopy Center. Computational resources were provided by the Minnesota Supercomputing Institute. We thank Edmund Howard, Ryan Mello, and Yuri Nesmelov for EPR assistance and helpful discussions, Elizabeth Lockamy for advice and training on functional assays, and Octavian Cornea for assistance in preparing the manuscript.
This work was supported in part by National Institutes of Health grants No. GM27906 and No. AR057220 (to D.D.T.). Z.M.J. was supported by National Institutes of Health grants No. AR007612 and No. GM008700, J.E.M. was supported by National Institutes of Health Training grant No. AR007612, and K.D.T. was supported by a Predoctoral Fellowship from the American Heart Association (grant No. 0615710Z). J.E.M. was enrolled in the graduate program in Physics and Astronomy.
Supporting Material
References
- 1.Møller J.V., Olesen C., Nissen P. The sarcoplasmic Ca2+-ATPase: design of a perfect chemi-osmotic pump. Q. Rev. Biophys. 2010;43:501–566. doi: 10.1017/S003358351000017X. [DOI] [PubMed] [Google Scholar]
- 2.MacLennan D.H., Kranias E.G. Phospholamban: a crucial regulator of cardiac contractility. Nat. Rev. Mol. Cell Biol. 2003;4:566–577. doi: 10.1038/nrm1151. [DOI] [PubMed] [Google Scholar]
- 3.Zamoon J., Mascioni A., Veglia G. NMR solution structure and topological orientation of monomeric phospholamban in dodecylphosphocholine micelles. Biophys. J. 2003;85:2589–2598. doi: 10.1016/s0006-3495(03)74681-5. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Cantilina T., Sagara Y., Jones L.R. Comparative studies of cardiac and skeletal sarcoplasmic reticulum ATPases. Effect of a phospholamban antibody on enzyme activation by Ca2+ J. Biol. Chem. 1993;268:17018–17025. [PubMed] [Google Scholar]
- 5.Tada M., Kadoma M. Regulation of the Ca2+ pump ATPase by cAMP-dependent phosphorylation of phospholamban. Bioessays. 1989;10:157–163. doi: 10.1002/bies.950100505. [DOI] [PubMed] [Google Scholar]
- 6.Simmerman H.K., Jones L.R. Phospholamban: protein structure, mechanism of action, and role in cardiac function. Physiol. Rev. 1998;78:921–947. doi: 10.1152/physrev.1998.78.4.921. [DOI] [PubMed] [Google Scholar]
- 7.MacLennan D.H., Kimura Y., Toyofuku T. Sites of regulatory interaction between calcium ATPases and phospholamban. Ann. N. Y. Acad. Sci. 1998;853:31–42. doi: 10.1111/j.1749-6632.1998.tb08254.x. [DOI] [PubMed] [Google Scholar]
- 8.Ha K.N., Traaseth N.J., Veglia G. Controlling the inhibition of the sarcoplasmic Ca2+-ATPase by tuning phospholamban structural dynamics. J. Biol. Chem. 2007;282:37205–37214. doi: 10.1074/jbc.M704056200. [DOI] [PubMed] [Google Scholar]
- 9.Hasenfuss G., Pieske B. Calcium cycling in congestive heart failure. J. Mol. Cell. Cardiol. 2002;34:951–969. doi: 10.1006/jmcc.2002.2037. [DOI] [PubMed] [Google Scholar]
- 10.Lipskaia L., Chemaly E.R., Hajjar R.J. Sarcoplasmic reticulum Ca2+ ATPase as a therapeutic target for heart failure. Expert Opin. Biol. Ther. 2010;10:29–41. doi: 10.1517/14712590903321462. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Cornea R.L., Gruber S.J., Thomas D.D. High-throughput FRET assay yields allosteric SERCA activators. J. Biomol. Screen. 2012 doi: 10.1177/1087057112456878. Accepted July 8, 2012. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Kaye D.M., Preovolos A., Power J.M. Percutaneous cardiac recirculation-mediated gene transfer of an inhibitory phospholamban peptide reverses advanced heart failure in large animals. J. Am. Coll. Cardiol. 2007;50:253–260. doi: 10.1016/j.jacc.2007.03.047. [DOI] [PubMed] [Google Scholar]
- 13.Lockamy E.L., Cornea R.L., Thomas D.D. Functional and physical competition between phospholamban and its mutants provides insight into the molecular mechanism of gene therapy for heart failure. Biochem. Biophys. Res. Commun. 2011;408:388–392. doi: 10.1016/j.bbrc.2011.04.023. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Chen Z., Akin B.L., Jones L.R. Mechanism of reversal of phospholamban inhibition of the cardiac Ca2+-ATPase by protein kinase A and by anti-phospholamban monoclonal antibody 2D12. J. Biol. Chem. 2007;282:20968–20976. doi: 10.1074/jbc.M703516200. [DOI] [PubMed] [Google Scholar]
- 15.Toyoshima C., Asahi M., MacLennan D.H. Modeling of the inhibitory interaction of phospholamban with the Ca2+ ATPase. Proc. Natl. Acad. Sci. USA. 2003;100:467–472. doi: 10.1073/pnas.0237326100. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Negash S., Yao Q., Squier T.C. Phospholamban remains associated with the Ca2+- and Mg2+-dependent ATPase following phosphorylation by cAMP-dependent protein kinase. Biochem. J. 2000;351:195–205. doi: 10.1042/0264-6021:3510195. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Karim C.B., Zhang Z., Thomas D.D. Phosphorylation-dependent conformational switch in spin-labeled phospholamban bound to SERCA. J. Mol. Biol. 2006;358:1032–1040. doi: 10.1016/j.jmb.2006.02.051. [DOI] [PubMed] [Google Scholar]
- 18.Gruber S.J., Haydon S., Thomas D.D. Phospholamban mutants compete with wild type for SERCA binding in HEK cells. Biochem. Biophys. Res. Commun. 2012;420:236–240. doi: 10.1016/j.bbrc.2012.02.125. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Mueller B., Karim C.B., Thomas D.D. Direct detection of phospholamban and sarcoplasmic reticulum Ca-ATPase interaction in membranes using fluorescence resonance energy transfer. Biochemistry. 2004;43:8754–8765. doi: 10.1021/bi049732k. [DOI] [PubMed] [Google Scholar]
- 20.Li J., James Z.M., Thomas D.D. Structural and functional dynamics of an integral membrane protein complex modulated by lipid headgroup charge. J. Mol. Biol. 2012;418:379–389. doi: 10.1016/j.jmb.2012.02.011. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Cornea R.L., Jones L.R., Thomas D.D. Mutation and phosphorylation change the oligomeric structure of phospholamban in lipid bilayers. Biochemistry. 1997;36:2960–2967. doi: 10.1021/bi961955q. [DOI] [PubMed] [Google Scholar]
- 22.MacLennan D.H., Toyofuku T., Kimura Y. Sites of regulatory interaction between calcium ATPases and phospholamban. Basic Res. Cardiol. 1997;92(Suppl 1):11–15. doi: 10.1007/BF00794063. [DOI] [PubMed] [Google Scholar]
- 23.Mersol J.V., Kutchai H., Thomas D.D. Self-association accompanies inhibition of Ca-ATPase by thapsigargin. Biophys. J. 1995;68:208–215. doi: 10.1016/S0006-3495(95)80176-1. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Mahaney J.E., Thomas D.D., Froehlich J.P. Intermolecular interactions in the mechanism of skeletal muscle sarcoplasmic reticulum Ca2+-ATPase (SERCA1): evidence for a triprotomer. Biochemistry. 2008;47:13711–13725. doi: 10.1021/bi801024a. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Hidalgo C., Thomas D.D., Ikemoto N. Effect of the lipid environment on protein motion and enzymatic activity of sarcoplasmic reticulum calcium ATPase. J. Biol. Chem. 1978;253:6879–6887. [PubMed] [Google Scholar]
- 26.Squier T.C., Thomas D.D. Relationship between protein rotational dynamics and phosphoenzyme decomposition in the sarcoplasmic reticulum Ca-ATPase. J. Biol. Chem. 1988;263:9171–9177. [PubMed] [Google Scholar]
- 27.Thomas D.D., Hidalgo C. Rotational motion of the sarcoplasmic reticulum Ca2+-ATPase. Proc. Natl. Acad. Sci. USA. 1978;75:5488–5492. doi: 10.1073/pnas.75.11.5488. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Birmachu W., Thomas D.D. Rotational dynamics of the Ca-ATPase in sarcoplasmic reticulum studied by time-resolved phosphorescence anisotropy. Biochemistry. 1990;29:3904–3914. doi: 10.1021/bi00468a017. [DOI] [PubMed] [Google Scholar]
- 29.Horváth L.I., Dux L., Marsh D. Saturation transfer electron spin resonance of Ca2+-ATPase covalently spin-labeled with β-substituted vinyl ketone- and maleimide-nitroxide derivatives. Effects of segmental motion and labeling levels. Biophys. J. 1990;58:231–241. doi: 10.1016/S0006-3495(90)82368-7. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Thomas D.D., Bigelow D.J., Hidalgo C. Rotational dynamics of protein and boundary lipid in sarcoplasmic reticulum membrane. Biophys. J. 1982;37:217–225. doi: 10.1016/S0006-3495(82)84671-7. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Napier R.M., East J.M., Lee A.G. State of aggregation of the (Ca2+ + Mg2+)-ATPase studied using saturation-transfer electron spin resonance. Biochim. Biophys. Acta. 1987;903:365–373. doi: 10.1016/0005-2736(87)90227-6. [DOI] [PubMed] [Google Scholar]
- 32.Saffman P.G., Delbrück M. Brownian motion in biological membranes. Proc. Natl. Acad. Sci. USA. 1975;72:3111–3113. doi: 10.1073/pnas.72.8.3111. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.Voss J., Jones L.R., Thomas D.D. The physical mechanism of calcium pump regulation in the heart. Biophys. J. 1994;67:190–196. doi: 10.1016/S0006-3495(94)80469-2. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.Mascioni A., Karim C., Veglia G. Solid-state NMR and rigid body molecular dynamics to determine domain orientations of monomeric phospholamban. J. Am. Chem. Soc. 2002;124:9392–9393. doi: 10.1021/ja026507m. [DOI] [PubMed] [Google Scholar]
- 35.Thomas D.D., Dalton L.R., Hyde J.S. Rotational diffusion studied by passage saturation transfer electron paramagnetic resonance. J. Chem. Phys. 1976;65:3006–3024. [Google Scholar]
- 36.Thomas D.D. Rotational diffusion of membrane proteins. In: Ragan C.I., Cherry R.J., editors. Techniques for the Analysis of Membrane Proteins. Chapman and Hall; London, UK: 1986. [Google Scholar]
- 37.Karim C.B., Kirby T.L., Thomas D.D. Phospholamban structural dynamics in lipid bilayers probed by a spin label rigidly coupled to the peptide backbone. Proc. Natl. Acad. Sci. USA. 2004;101:14437–14442. doi: 10.1073/pnas.0402801101. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38.Karim C.B., Marquardt C.G., Thomas D.D. Synthetic null-cysteine phospholamban analogue and the corresponding transmembrane domain inhibit the Ca-ATPase. Biochemistry. 2000;39:10892–10897. doi: 10.1021/bi0003543. [DOI] [PubMed] [Google Scholar]
- 39.Karim C.B., Zhang Z., Thomas D.D. Synthesis of TOAC spin-labeled proteins and reconstitution in lipid membranes. Nat. Protoc. 2007;2:42–49. doi: 10.1038/nprot.2007.2. [DOI] [PubMed] [Google Scholar]
- 40.Eletr S., Inesi G. Phospholipid orientation in sarcoplasmic membranes: spin-label ESR and proton MNR studies. Biochim. Biophys. Acta. 1972;282:174–179. doi: 10.1016/0005-2736(72)90321-5. [DOI] [PubMed] [Google Scholar]
- 41.Coll R.J., Murphy A.J. Purification of the CaATPase of sarcoplasmic reticulum by affinity chromatography. J. Biol. Chem. 1984;259:14249–14254. [PubMed] [Google Scholar]
- 42.Reddy L.G., Cornea R.L., Thomas D.D. Defining the molecular components of calcium transport regulation in a reconstituted membrane system. Biochemistry. 2003;42:4585–4592. doi: 10.1021/bi026995a. [DOI] [PubMed] [Google Scholar]
- 43.Lévy D., Gulik A., Rigaud J.L. Reconstitution of the sarcoplasmic reticulum Ca2+-ATPase: mechanisms of membrane protein insertion into liposomes during reconstitution procedures involving the use of detergents. Biochim. Biophys. Acta. 1992;1107:283–298. doi: 10.1016/0005-2736(92)90415-i. [DOI] [PubMed] [Google Scholar]
- 44.Reference deleted in proof.
- 45.Baroin A., Bienvenue A., Devaux P.F. Spin-label studies of protein-protein interactions in retinal rod outer segment membranes. Saturation transfer electron paramagnetic resonance spectroscopy. Biochemistry. 1979;18:1151–1155. doi: 10.1021/bi00574a005. [DOI] [PubMed] [Google Scholar]
- 46.Karim C.B., Paterlini M.G., Thomas D.D. Role of cysteine residues in structural stability and function of a transmembrane helix bundle. J. Biol. Chem. 2001;276:38814–38819. doi: 10.1074/jbc.M104006200. [DOI] [PubMed] [Google Scholar]
- 47.Ablorh N.A., Miller T., Thomas D.D. Accurate quantitation of phospholamban phosphorylation by immunoblot. Anal. Biochem. 2012;425:68–75. doi: 10.1016/j.ab.2012.01.028. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 48.Squier T.C., Thomas D.D. Methodology for increased precision in saturation transfer electron paramagnetic resonance studies of rotational dynamics. Biophys. J. 1986;49:921–935. doi: 10.1016/S0006-3495(86)83720-1. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 49.Goldman S.A., Bruno G.V., Freed J.H. Estimating slow-motional rotational correlation times for nitroxides by electron spin resonance. J. Phys. Chem. 1972;76:1858–1860. [Google Scholar]
- 50.Mason R.P., Freed J.H. Estimating microsecond rotational correlation times from lifetime broadening of nitroxide electron spin resonance spectra near the rigid limit. J. Phys. Chem. 1974;78:1321–1323. [Google Scholar]
- 51.Scarpelli F., Drescher M., Huber M. Aggregation of transmembrane peptides studied by spin-label EPR. J. Phys. Chem. B. 2009;113:12257–12264. doi: 10.1021/jp901371h. [DOI] [PubMed] [Google Scholar]
- 52.Uhríková D., Balgavý P., Kuklin A. Small-angle neutron scattering study of the n-decane effect on the bilayer thickness in extruded unilamellar dioleoylphosphatidylcholine liposomes. Biophys. Chem. 2000;88:165–170. doi: 10.1016/s0301-4622(00)00211-8. [DOI] [PubMed] [Google Scholar]
- 53.Cherry R.J., Godfrey R.E. Anisotropic rotation of bacteriorhodopsin in lipid membranes. Comparison of theory with experiment. Biophys. J. 1981;36:257–276. doi: 10.1016/S0006-3495(81)84727-3. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 54.Howard E.C., Lindahl K.M., Thomas D.D. Simulation of saturation transfer electron paramagnetic resonance spectra for rotational motion with restricted angular amplitude. Biophys. J. 1993;64:581–593. doi: 10.1016/S0006-3495(93)81417-6. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 55.Beth A.H., Hustedt E.J. Saturation Transfer EPR: Rotational Dynamics of Membrane Proteins. In: Eaton S.S., Eaton G.R., Berliner L.J., editors. Springer; New York: 2005. pp. 369–408. [Google Scholar]
- 56.Marsh D., Jost M., Toniolo C. TOAC spin labels in the backbone of alamethicin: EPR studies in lipid membranes. Biophys. J. 2007;92:473–481. doi: 10.1529/biophysj.106.092775. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 57.Marsh D. Saturation transfer EPR studies of slow rotational motion in membranes. Appl. Magn. Reson. 2007;31:387–410. [Google Scholar]
- 58.Hustedt E.J., Beth A.H. The sensitivity of saturation transfer electron paramagnetic resonance spectra to restricted amplitude uniaxial rotational diffusion. Biophys. J. 2001;81:3156–3165. doi: 10.1016/S0006-3495(01)75952-8. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 59.Fujii J., Maruyama K., MacLennan D.H. Expression and site-specific mutagenesis of phospholamban. Studies of residues involved in phosphorylation and pentamer formation. J. Biol. Chem. 1989;264:12950–12955. [PubMed] [Google Scholar]
- 60.Hughes E., Clayton J.C., Middleton D.A. Probing the oligomeric state of phospholamban variants in phospholipid bilayers from solid-state NMR measurements of rotational diffusion rates. Biochemistry. 2005;44:4055–4066. doi: 10.1021/bi0482351. [DOI] [PubMed] [Google Scholar]
- 61.Reddy L.G., Jones L.R., Thomas D.D. Depolymerization of phospholamban in the presence of calcium pump: a fluorescence energy transfer study. Biochemistry. 1999;38:3954–3962. doi: 10.1021/bi981795d. [DOI] [PubMed] [Google Scholar]
- 62.Yao Q., Chen L.T., Bigelow D.J. Oligomeric interactions between phospholamban molecules regulate Ca-ATPase activity in functionally reconstituted membranes. Biochemistry. 2001;40:6406–6413. doi: 10.1021/bi002891t. [DOI] [PubMed] [Google Scholar]
- 63.Mahaney J.E., Froehlich J.P., Thomas D.D. Conformational transitions of the sarcoplasmic reticulum Ca-ATPase studied by time-resolved EPR and quenched-flow kinetics. Biochemistry. 1995;34:4864–4879. doi: 10.1021/bi00014a044. [DOI] [PubMed] [Google Scholar]
- 64.Mahaney J.E., Albers R.W., Froehlich J.P. Phospholamban inhibits Ca2+ pump oligomerization and intersubunit free energy exchange leading to activation of cardiac muscle SERCA2a. Ann. N. Y. Acad. Sci. 2003;986:338–340. doi: 10.1111/j.1749-6632.2003.tb07208.x. [DOI] [PubMed] [Google Scholar]
- 65.Froehlich J.P., Taniguchi K., Albers R.W. Complex kinetic behavior in the Na,K- and Ca-ATPases. Evidence for subunit-subunit interactions and energy conservation during catalysis. Ann. N. Y. Acad. Sci. 1997;834:280–296. doi: 10.1111/j.1749-6632.1997.tb52259.x. [DOI] [PubMed] [Google Scholar]
- 66.Squier T.C., Hughes S.E., Thomas D.D. Rotational dynamics and protein-protein interactions in the Ca-ATPase mechanism. J. Biol. Chem. 1988;263:9162–9170. [PubMed] [Google Scholar]
- 67.Voss J., Birmachu W., Thomas D.D. Effects of melittin on molecular dynamics and Ca-ATPase activity in sarcoplasmic reticulum membranes: time-resolved optical anisotropy. Biochemistry. 1991;30:7498–7506. doi: 10.1021/bi00244a019. [DOI] [PubMed] [Google Scholar]
- 68.Mahaney J.E., Kleinschmidt J., Thomas D.D. Effects of melittin on lipid-protein interactions in sarcoplasmic reticulum membranes. Biophys. J. 1992;63:1513–1522. doi: 10.1016/S0006-3495(92)81736-8. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 69.Karon B.S., Geddis L.M., Thomas D.D. Anesthetics alter the physical and functional properties of the Ca-ATPase in cardiac sarcoplasmic reticulum. Biophys. J. 1995;68:936–945. doi: 10.1016/S0006-3495(95)80269-9. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 70.Cornea R.L., Thomas D.D. Effects of membrane thickness on the molecular dynamics and enzymatic activity of reconstituted Ca-ATPase. Biochemistry. 1994;33:2912–2920. doi: 10.1021/bi00176a022. [DOI] [PubMed] [Google Scholar]
- 71.Karon B.S., Mahaney J.E., Thomas D.D. Halothane and cyclopiazonic acid modulate Ca-ATPase oligomeric state and function in sarcoplasmic reticulum. Biochemistry. 1994;33:13928–13937. doi: 10.1021/bi00250a048. [DOI] [PubMed] [Google Scholar]
- 72.Birmachu W., Voss J.C., Thomas D.D. Protein and lipid rotational dynamics in cardiac and skeletal sarcoplasmic reticulum detected by EPR and phosphorescence anisotropy. Biochemistry. 1993;32:9445–9453. doi: 10.1021/bi00087a024. [DOI] [PubMed] [Google Scholar]
- 73.Robia S.L., Campbell K.S., Thomas D.D. Förster transfer recovery reveals that phospholamban exchanges slowly from pentamers but rapidly from the SERCA regulatory complex. Circ. Res. 2007;101:1123–1129. doi: 10.1161/CIRCRESAHA.107.159947. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 74.Fujii J., Maruyama K., MacLennan D.H. Co-expression of slow-twitch/cardiac muscle Ca2+-ATPase (SERCA2) and phospholamban. FEBS Lett. 1990;273:232–234. doi: 10.1016/0014-5793(90)81092-3. [DOI] [PubMed] [Google Scholar]
- 75.Toyofuku T., Kurzydlowski K., MacLennan D.H. Identification of regions in the Ca2+-ATPase of sarcoplasmic reticulum that affect functional association with phospholamban. J. Biol. Chem. 1993;268:2809–2815. [PubMed] [Google Scholar]
- 76.Toyoshima C., Nomura H. Structural changes in the calcium pump accompanying the dissociation of calcium. Nature. 2002;418:605–611. doi: 10.1038/nature00944. [DOI] [PubMed] [Google Scholar]
- 77.Traaseth N.J., Shi L., Veglia G. Structure and topology of monomeric phospholamban in lipid membranes determined by a hybrid solution and solid-state NMR approach. Proc. Natl. Acad. Sci. USA. 2009;106:10165–10170. doi: 10.1073/pnas.0904290106. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 78.Bigelow D.J., Inesi G. Frequency-domain fluorescence spectroscopy resolves the location of maleimide-directed spectroscopic probes within the tertiary structure of the Ca-ATPase of sarcoplasmic reticulum. Biochemistry. 1991;30:2113–2125. doi: 10.1021/bi00222a016. [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.