

While most of the enzymes responsible for tRNA modifications in Escherichia coli were known, the enzyme that introduces the m2A modification at purine 37 of some tRNAs was not. The authors of this study show that the missing enzyme is the methyltransferase RlmN responsible for introducing the m2A modification at position 2503 of the 23S rRNA, thus demonstrating that RlmN is a dual-specificity enzyme. The authors go on to use a tRNA chimera that should also be useful for the study of other tRNA modifications, to identify the specificity determinants. They also report that strains lacking rlmN have an error-prone phenotype (expected from loss of m2A2503 modification of rRNA) rather than a hyperaccurate phenotype (expected from loss of m2A37 modification of tRNA).
Keywords: RNA modification, anticodon region, peptidyl transferase center, yfgB, radical SAM enzymes, translational misreading
Abstract
Modifying RNA enzymes are highly specific for substrate—rRNA or tRNA—and the target position. In Escherichia coli, there are very few multisite acting enzymes, and only one rRNA/tRNA dual-specificity enzyme, pseudouridine synthase RluA, has been identified to date. Among the tRNA-modifying enzymes, the methyltransferase responsible for the m2A synthesis at purine 37 in a tRNA set still remains unknown. m2A is also present at position 2503 in the peptidyl transferase center of 23S RNA, where it is introduced by RlmN, a radical S-adenosyl-L-methionine (SAM) enzyme. Here, we show that E. coli RlmN is a dual-specificity enzyme that catalyzes methylation of both rRNA and tRNA. The ΔrlmN mutant lacks m2A in both RNA types, whereas the expression of recombinant RlmN from a plasmid introduced into this mutant restores tRNA modification. Moreover, RlmN performs m2A37 synthesis in vitro using a tRNA chimera as a substrate. This chimera has also proved useful to characterize some tRNA identity determinants for RlmN and other tRNA modification enzymes. Our data suggest that RlmN works in a late step during tRNA maturation by recognizing a precise 3D structure of tRNA. RlmN inactivation increases the misreading of a UAG stop codon. Since loss of m2A37 from tRNA is expected to produce a hyperaccurate phenotype, we believe that the error-prone phenotype exhibited by the ΔrlmN mutant is due to loss of m2A from 23S rRNA and, accordingly, that the m2A2503 modification plays a crucial role in the proofreading step occurring at the peptidyl transferase center.
INTRODUCTION
Ribosomal and transfer RNAs (rRNAs and tRNAs, respectively) are post-transcriptionally modified in all living organisms. Modification enzymes specifically recognize the target RNA molecule and incorporate the chemical modifications at the precise position. The resulting modified nucleosides can alter RNA chemistry and structure and generally contribute to fine-tune protein synthesis (Grosjean 2005).
tRNA exhibits a larger number and wider variety of modifications than rRNA. In Escherichia coli tRNAs, 31 distinct types of modified nucleosides have been identified (Björk and Hagervall 2005), and the set of enzymes responsible for their synthesis has almost been fully characterized (for brief review, see Benítez-Páez et al. 2010).
The modifications found in the tRNA anticodon loop collectively optimize codon-anticodon recognition, maintain the translational reading frame, and shape the anticodon architecture (Agris 2004, 2008; Björk and Hagervall 2005; Agris et al. 2007). Loss of anticodon modifications, particularly at the wobble position (position 34) and conserved purine 37, frequently impairs gene expression and affects a range of bacterial phenotypic traits, including virulence, pathogenicity, biological fitness, and response to stress conditions (Petrullo et al. 1983; Elseviers et al. 1984; Gray et al. 1992; Li and Bjork 1995; Bjork et al. 2001; Urbonavicius et al. 2001; Gong et al. 2004; Sha et al. 2004; Cho and Caparon 2008; Golovina et al. 2009; Benítez-Páez et al. 2010). Purine 37 modifications, 3′-adjacent to the anticodon, negate intra-loop base-pairing and, thereby, ensure correct anticodon loop width (Dao et al. 1994). Moreover, they contribute to order the structure of the loop by improving the stacking of its 3′ side and to stabilize the first base pair of the codon-anticodon interaction (Björk and Hagervall 2005; Agris et al. 2007; Agris 2008). In E. coli tRNAs, G37 is always modified to m1G37, whereas A37 may or may not be modified. A weak A-U interaction at the first position of the codon generally requires a complex hydrophobic or charged modification (i6A- or t6A-type) at position 37, whereas a more stable G-C interaction admits a simpler, hydrophobic modification (m1G, m2A, m6A) or an unmodified A37 (Agris 1996; Björk and Hagervall 2005).
Among the tRNA-modifying enzymes acting at position 37 of the E. coli tRNAs, the methyltransferase responsible for m2A synthesis is still unknown. C2-methyladenosine is present at position 37 of tRNAArgICG, tRNAAspQUC, tRNAGlncmnm5s2UUG, tRNAGlnCUG, tRNAGlumnm5s2UUC, and tRNAHisQUG from E. coli (Czerwoniec et al. 2009). Identification of the gene encoding this tRNA modifying enzyme could shed light on the role of m2A37 in translation and bacterial physiology.
The m2A nucleoside is also present at A2503 in 23S rRNA, where it is introduced by RlmN. m2A2503 is located at the entrance of the nascent peptide exit tunnel and has been involved in the ribosomal mechanism that promotes translation arrest in response to specific peptide sequences (Vazquez-Laslop et al. 2010; Ramu et al. 2011). Lack of methylation of A2503 has been reported to produce a subtle yet significant effect on cell fitness and to increase linezolid resistance (Toh et al. 2008; Gao et al. 2010; Lamarre et al. 2011). RlmN and its evolutionarily related resistance enzyme Cfr belong to the radical S-adenosyl-L-methionine (SAM) enzyme superfamily, and both use protein-free rRNA as a substrate (Atta et al. 2010; Kaminska et al. 2010; Yan et al. 2010; Grove et al. 2011; Yan and Fujimori 2011).
Modifying RNA enzymes are highly specific for substrate (RNA type) and the target position. In E. coli, there are very few multisite acting enzymes (RluD, RsmA, RlmKL, TruA, and DusABC), and only one enzyme, the pseudouridine synthase RluA, has been shown to have dual specificity, recognizing both rRNA and tRNA (Fig. 1). In this work, we demonstrate that E. coli RlmN catalyzes the incorporation of m2A at position 37 of tRNAs. Thus, RlmN is the first example of a dual-specificity RNA methylase which catalyzes m2A synthesis in both rRNA and tRNA. Moreover, we show that inactivation of the rlmN gene results in an error-prone phenotype, which highlights a role for nucleoside m2A in translational accuracy.
FIGURE 1.

RlmN is the first example of a methyltransferase working on both rRNA and tRNA. The figure shows the distribution of the Escherichia coli modified nucleotides by RNA type. The position of each modification (in parentheses) and the enzyme(s) responsible for its synthesis are indicated. Only two enzymes, RluA and the herein studied RlmN, have dual substrate specificity, recognizing both rRNA and tRNA. (*) Modifications of 16S rRNA; (unk) unknown enzyme; (‡) modification h5C2501 is partial and dependent on the growth phase (Andersen et al. 2004; Havelund et al. 2011).
RESULTS AND DISCUSSION
RlmN catalyzes m2A formation in tRNA
Synthesis of m2A involves the addition of a methyl group to an amidine carbon of the adenine, which is a highly chemically challenging reaction because of the low nucleophilicity of the target carbon atom. RlmN and its closely related homolog Cfr catalyze this reaction on 23S rRNA (Toh et al. 2008; Giessing et al. 2009; Yan et al. 2010; Grove et al. 2011; Yan and Fujimori 2011). Therefore, as a first attempt to identify the enzyme responsible for synthesizing m2A in tRNAs, we performed a search for RlmN/Cfr paralogs in databases. However, BLAST searches of the E. coli and other bacterial genomes using conserved amino acid blocks of the RlmN family as queries revealed no paralog that might function in tRNA modification, which is in agreement with previous studies (Toh et al. 2008; Kaminska et al. 2010).
Then, we tested the hypothesis that RlmN is also responsible for the modification of tRNAs. This hypothesis was based on the observation that m2A-carrying tRNAs share certain sequence similarity with 23S rRNA in the region including the target nucleoside (Fig. 2A). Moreover, the global L-shaped structure of tRNAs resembles the local structure of 23S rRNA required for recognition by RlmN (helices 89 and 90–92) (Fig. 2B). Fractions of tRNA and rRNA (23S rRNA-enriched) purified from ΔrlmN and wild-type strains were analyzed by high-performance liquid chromatography (HPLC) after digestion with nuclease P1 and alkaline phosphatase. Nucleoside m2A, which elutes at ∼54.5 min, was found to be absent in the hydrolysates of rRNA and tRNA purified from the ΔrlmN mutant (Fig. 3A). To test whether the presence of m2A in tRNA is truly dependent on RlmN activity, the sequence encoding the full-length RlmN protein with an N-terminal histidine tag was cloned into pET15b, and the resulting plasmid was used to transform the ΔrlmN mutant. The recombinant plasmid was able to restore the presence of m2A in bulk tRNA (Fig. 3B). Note that the in vivo modification reaction proceeded efficiently in a host strain lacking the T7 RNA polymerase gene. Low-level synthesis of the His-RlmN recombinant protein could be due to transcription mediated by bacterial RNA polymerase from the T7 promoter (nonspecific transcription) and/or other plasmid promoters. Under such conditions, the His-RlmN expression was so low that it could not be detected by Western blotting with an anti-His antibody (data not shown). In spite of this, incorporation of m2A into tRNA was indicative of the RlmN expression from the plasmid-borne rlmN gene.
FIGURE 2.

Sequence and structural comparisons of the RlmN substrates. (A) Sequence and secondary structure of the peptidyl transferase region of the E. coli 23S rRNA. Modification m2A at position 2503 is highlighted in black. The remaining post-transcriptional modifications in the region are highlighted in gray. Note location of helices 90–92, which are crucial for recognition by RlmN. In the bottom right box, a short multiple sequence alignment (seven positions in length) of 23S rRNA and the tRNAs carrying m2A37 is used to search for the potential identity determinants recognized by the m2A-synthesizing enzyme. Positions at the consensus pattern are represented according to the IUPAC nucleotide code. Positions 35 and 36 of substrate tRNAs showing a significant conservation pattern were chosen herein for subsequent studies. (B) The three-dimensional structure corresponding to 2455–2580 nucleotides of the Escherichia coli 23S rRNA sequence was extracted from the 3OAS structure stored in the PDB database and shown in ribbon representation (left-hand side) using UCSF Chimera viewer (Pettersen et al. 2004). Helices required for optimal RlmN action (Yan et al. 2010) are depicted in color, and the target nucleoside for the RlmN-mediated methylation is highlighted in red. The three-dimensional, global structure of the Escherichia coli tRNAGlnCUG (extracted from the 2RE8 PDB record) is shown in ribbon representation on the right-hand side. The D, anticodon, and T stem–loops are depicted in color, resembling the secondary structures shown on the left-hand side. The target nucleoside for RlmN-mediated modification is also highlighted in red.
FIGURE 3.

RlmN modifies both rRNA and tRNA. (A) rRNA/tRNA HPLC-nucleoside profile of the Escherichia coli ΔrlmN mutant. After isolation and P1 digestion, bulk rRNA (left) and tRNA (right) from the wild-type and ΔrlmN strains were separately analyzed by HPLC. Nucleoside m2A was identified by its elution time and UV spectra. Note that nucleoside t6A (present at position 37 of some tRNAs that are not RlmN substrates) is unaffected in the ΔrlmN cells. (B) In vivo complementation of the ΔrlmN mutant. Recombinant protein His-RlmN, expressed from pET15b-his-rlmN, restores the m2A synthesis in ΔrlmN cells (right panel). Wild-type and ΔrlmN cells containing pET15b (left and middle panels, respectively) were used as controls. Absorbance was monitored at 260 nm. (mAU) Absorbance units ×10−3.
Construction and characterization of tRNAChimeraUUG
To further investigate the effect of RlmN activity on tRNA, we planned to express tRNAGlncmnm5s2UUG from a tRNA scaffold system (Ponchon and Dardel 2007; Ponchon et al. 2009), which could facilitate the production, purification, and mutational analysis of the recombinant tRNA. This approach has been successfully used by our group in a previous study involving the tRNALeuCmAA isoacceptor (Benítez-Píez et al. 2010). However, no accumulation of several chimeric constructs involving tRNAGlncmnm5s2UUG and other tRNA substrates was herein detected, probably because they are unstable and undergo degradation by endogenous RNases (see Materials and Methods). In contrast, we observed the overexpression of tRNAChimeraUUG, which was constructed by replacing the anticodon stem–loop (ASL) of the tRNA scaffold with the ASL of tRNAGlncmnm5s2UUG (Fig. 4). The HPLC profile of tRNAChimeraUUG indicated the presence of the normally occurring modifications in the tRNA scaffold (such as s4U8, m7G46, T54, D16/20, and Ψ55) (see Fig. 4A) when overexpressed in E. coli (Ponchon and Dardel 2007). Additionally, we identified the five modifications typically present in the tRNAGlncmnm5s2UUG ASL (Czerwoniec et al. 2009), with Um at position 32, s2U and cmnm5U at position 34, m2A at position 37, and Ψ at position 38 (whose presence was inferred from the increase of the Ψ peak area in the chromatogram) (Fig. 4B). These data suggest that tRNAChimeraUUG may be an appropriate model to investigate RlmN activity. In fact, an A37C replacement in the chimeric tRNA, and the overproduction of the wild-type tRNAChimeraUUG in ΔrlmN cells allowed us to demonstrate that the m2A modification is confined to position 37 and that it is dependent on RlmN activity, respectively (Fig. 4C).
FIGURE 4.

RlmN methylates tRNAChimeraUUG both in vivo and in vitro. (A) The sequence and secondary structure of the scaffold tRNA is shown together with a representative section of its HPLC chromatogram (D and Ψ modifications elute at early times and are not included in the figure). (B) The tRNAChimeraUUG sequence (red nucleotides indicate the ASL from tRNAGlncmnm5s2UUG) is shown together with a section of the corresponding chromatogram. (C) Synthesis in vivo of m2A on tRNAChimeraUUG (left panel) is prevented by mutation A37C (middle panel) or by the expression of the wild-type tRNAChimeraUUG in ΔrlmN cells (right panel). (D) In vitro methylation of tRNAChimeraUUG purified from ΔrlmN cells is carried out by in vitro-reconstituted RlmN in a SAM-dependent manner (red lines). Reconstituted RlmN does not work on in vitro synthesized tRNAGlnUUG (black lines). Solid and discontinuous lines indicate that the modification reactions were performed in the presence and absence of SAM, respectively. Absorbance was monitored at 260 nm. (mAU) Absorbance units ×10−3.
In vitro methylation of tRNA by RlmN
Purification of the enzymatically competent RlmN protein demands strict anaerobic conditions to allow the proper assembly of its Fe-S cluster (Yan et al. 2010). An in vitro reconstitution of an Fe-S-bound RlmN protein was herein performed by the degassing and N2 purging of buffers, reagents, and solutions as an alternative method to using a glovebox device (Yan et al. 2010). N2 purging efficiently removes O2 from aqueous solutions, leaving oxygen levels at 0.2–0.4 ppm (Butler et al. 1994). The recombinant His-RlmN protein purified by this procedure was able to catalyze the synthesis of m2A in a SAM-dependent manner on tRNAChimeraUUG purified from ΔrlmN cells (Fig. 4D). In vitro accumulation of m2A was calculated to be ∼10%–20% of that observed in vivo by comparing the peak areas of m2A (normalized to the cytidine area) from the HPLC profiles of the in vitro (Fig. 4D) and in vivo (Fig. 4C) modified tRNA. Interestingly, when we attempted to assay RlmN activity using in vitro-transcribed tRNAGlnUUG or tRNAArgACG, no trace of m2A was observed in the HPLC analysis of the reaction products (Fig. 4D; data not shown). These results suggest that unmodified tRNA is not a suitable substrate for RlmN, as also observed for other tRNA modifying enzymes (Benítez-Páez et al. 2010).
tRNA determinants for RlmN and other tRNA-modifying enzymes
The ability of RlmN to modify rRNA and tRNA supports our initial hypothesis that both RNAs have common identity determinants for the enzyme (see Fig. 2). At the sequence level, positions 35 and 36 in the tRNAs recognized by RlmN show a clear pattern of nucleotide conservation (G or C at position 36 and a pyrimidine at position 35), as also observed at the counterpart positions in 23S rRNA (Fig. 2A). Therefore, we performed site-directed mutagenesis on the plasmid containing tRNAChimeraUUG to produce the U35A, U35G, G36U, and G36A changes and to analyze their effect on m2A synthesis. All the tRNA mutants were overexpressed at similar or slightly lower levels than tRNAChimeraUUG (Supplemental Fig. S1). The results of HPLC analysis are summarized in Table 1. We found that the accumulation of m2A in the mutants was ∼20%–50% lower than in the wild-type tRNAChimeraUUG, which suggests that U35 and G36 work as identity determinants for RlmN.
TABLE 1.
Mutations affecting the modification status of tRNAChimeraUUG

Post-transcriptional modifications at position 37 may change to match a mutational change in the nucleotide at position 36 (Yarus 1982; Raftery and Yarus 1987). However, the decrease in m2A produced by the mutations at G36 in tRNAChimeraUUG was not accompanied by an accumulation of alternative adenosine nucleosides at position 37. Specifically, we observed neither t6A accumulation in mutant G36U (whose anticodon 5′-UUU is characteristic of tRNALysmnm5s2UUU) nor (ms2)i6A accumulation in mutant G36A (whose anticodon 5′-UUA may recognize the stop codons 5′-UAA and 5′-UAG). The G36A mutant is a putative substrate for MiaA, the enzyme that introduces i6A37 into tRNA reading codons starting with U, as a first step in the synthesis of the final modification ms2i6A37. MiaA predominantly recognizes a conserved consensus sequence A36A37A38 in the anticodon loop of tRNA (Chimnaronk et al. 2009). However, some tRNA chimeras carrying a sequence A36A37Ψ38 have been shown to carry both ms2i6A37 and m2A37 (Yarus et al. 1986). Apparently, this is not the case for our mutant chimera G36A which, even though containing a sequence A36A37Ψ38, is not recognized by MiaA. Modification t6A37, found in ANN decoding tRNAs (such as tRNALysmnm5s2UUU), is dependent on activity of RimN (previously named YrdC), although the identity elements recognized by this enzyme have not yet been determined (El Yacoubi et al. 2009). Lack of t6A37 in mutant G36U indicates that the generation of an anticodon that properly matches an ANN codon does not suffice for recognition by RimN.
Additionally, we observed that mutation of U35 and G36 affects modifications at positions of tRNAChimeraUUG other than position 37. Thus, mutations G36A and G36U diminish the accumulation of Ψ and s4U to a certain extent, while mutation G36A also lowers the s2U level to ∼50% (Table 1). MnmA catalyzes s2U34 formation in tRNA species with anticodons 5′-UUG (glutamine), 5′-UUU (lysine), and 5′-UUC (glutamate) by recognizing U34 and U35 in a base-specific manner (Numata et al. 2006). Our results suggest that the presence of an A at position 36 (instead of G, U, or C) hinders the MnmA function. As expected, mutations U35A and U35G drastically impaired the synthesis of s2U34 by MnmA in the chimeric tRNA and, consequently, accumulation of the cmnm5s2U nucleoside (Table 1). Strikingly, both mutations induced the incorporation of the cmo5 group at the wobble uridine. This modification is present in tRNAs that read codons in family boxes specific for Leu, Ser, Pro, Val, Thr, and Ala (Nasvall et al. 2004; Björk and Hagervall 2005; Nasvall et al. 2007). Formation of cmo5U34 in mutants U35A (with anticodon 5′-UAG) and U35G (with anticodon 5′-UGG) is consistent with the modification status at the wobble uridine in tRNALeucmo5UAG and tRNAProcmo5UGG. Therefore, an A or G nucleotide at position 35 of the composite tRNA appears as a critical identity determinant for its recognition by the cmo5U34 synthesizing enzymes.
A further comparison of the 23S rRNA and tRNAChimeraUUG sequences revealed some additional similarities which could be used for recognition by RlmN. A Ψ residue is located 3′-adjacent to the RlmN-target nucleotide in both RNAs, and a 2′-O-methylation is present in the pyrimidine separated by four nucleotides toward the 5′ end from the RlmN target (Figs. 2A, 4B). Thus, we decided to explore the effect of inactivating genes truA and trmJ (responsible for pseudouridilation at position 38 and 2′-O-methylation at U32, respectively) on m2A accumulation in tRNAChimeraUUG. No significant differences were found between the wild-type strain and mutants ΔtruA and ΔtrmJ (Table 1).
In order to investigate the role of the tRNA structure in the recognition by RlmN, we attempted to clone truncated versions of the tRNAChimeraUUG lacking either the D stem–loop or the T stem–loop. Unfortunately, no overexpression of any truncated mutant was obtained.
Inactivation of rlmN decreases translational accuracy
Nucleoside m2A is located in functionally important regions of tRNA and rRNA. However, there are no data available on a role of m2A in translational accuracy. To test the fidelity phenotype of the ΔrlmN mutant, we used a nonsense readthrough assay in which decoding of a lacZ premature stop codon by near-cognate tRNAs is required to produce β-galactosidase activity. This assay has previously proved useful to characterize the effect of several tRNA modification enzymes (Petrullo et al. 1983; Elseviers et al. 1984; Yim et al. 2003). As shown in Table 2, mutations ΔmiaA (causing the loss of ms2i6A37 in a tRNA set), and ΔmnmE and ΔmnmA (both impairing wobble uridine modifications in certain tRNAs), used here as controls, produce an error-restrictive phenotype, i.e., they lower the readthrough level in relation to the wild-type strain. These results suggest that tRNAs that are substrates for MiaA, MnmE, and MnmA misread the lacZ UAG codon, recognizing it as a near-cognate codon and contributing to the natural low-level readthrough detected in wild-type cells. Presumably, loss of the MiaA-, MnmE-, and MnmA-dependent modifications destabilizes the weak pairing of tRNAs with the stop codon at the decoding center, thus reducing the readthrough level.
TABLE 2.
rlmN inactivation confers an error-prone phenotype
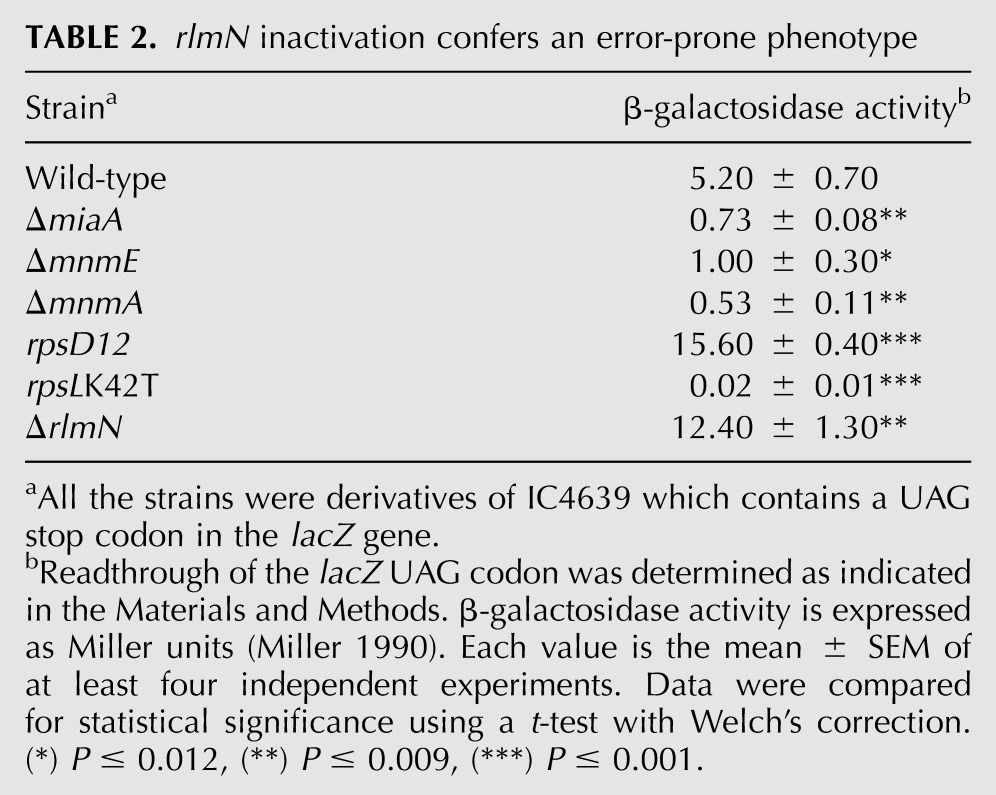
The readthrough assay has also proved useful to assess the effects of changes in ribosomal components on translational accuracy (Petrullo et al. 1983). In our hands (see Table 2), the assay successfully characterized the well-known error-prone and error-restrictive phenotype conferred by mutations rpsD12 (in ribosomal protein S4) and rpsLK42T (in ribosomal protein S12), respectively (Petrullo et al. 1983; Kurland et al. 1996). Mutations in proteins S4 and S12 affect the conformational changes occurring at the decoding center during the tRNA selection step (Vallabhaneni and Farabaugh 2009), thus stimulating the selection or rejection of near-cognate tRNAs.
Interestingly, rlmN inactivation produces a clear error-prone phenotype since it increases the UAG readthrough levels in relation to the wild-type strain (Table 2). This effect is opposite to that observed in ΔmiaA, where loss of modification at position 37 decreases the UAG readthrough. All the tRNAs that are substrates for RlmN carry G or C at position 36, which involves a noncanonical interaction with the first base of the UAG stop codon. Considering that most purine 37 modifications are required for stabilization of the crucial first base pair of the anticodon–codon interaction, it appears unlikely that the error-prone phenotype of the ΔrlmN mutant is due to loss of m2A from tRNAs because this would mean that m2A-lacking tRNAs form more stable interactions with the lacZ stop codon than fully modified tRNAs.
Alternatively, we propose that the error-prone phenotype exhibited by the ΔrlmN mutant is due to loss of m2A from ribosomal 23S. Nucleotide m2A2503 of 23S RNA resides in the peptidyl transferase center (PTC). Besides its crucial role in peptide-bond formation, this center is involved in the discrimination of near-cognate tRNAs during the proofreading step (Jenner et al. 2010). The location of A2503 suggests that loss of m2A may alter conformational changes in the PTC, allowing the accommodation of an incorrect A-site tRNA and the subsequent peptidyl-transfer catalysis, thus increasing the readthrough levels. If this were the case, it would indicate that the rRNA modification catalyzed by RlmN serves to optimize ribosomal fidelity.
Concluding remarks
We show herein that RlmN is the methyltransferase responsible for m2A synthesis at position 37 in a group of E. coli tRNAs. RlmN activity on tRNA was demonstrated in vivo by the absence of m2A in total tRNA purified from a ΔrlmN mutant (Fig. 3A) and by restoration of tRNA methylation upon the expression of a plasmid-encoded rlmN gene in the same strain (Fig. 3B). In vivo RlmN activity on tRNA was confirmed by the presence of m2A in a chimeric tRNA (tRNAChimeraUUG) purified from the wild-type strain and by the absence of the modification when this tRNA was purified from the ΔrlmN mutant (Fig. 4C, left and right panels). Moreover, the A37C replacement in tRNAChimeraUUG prevented the RlmN-dependent formation of m2A, indicating that this nucleoside is confined to position 37 (Fig. 4C, left and middle panels). We also demonstrate that RlmN, after in vitro reconstitution of its iron-sulfur cluster, carries out the synthesis in vitro of m2A on tRNAChimeraUUG purified from ΔrlmN cells (Fig. 4D). Therefore, our data evidence that RlmN is a dual-specificity enzyme capable of catalyzing m2A formation in both rRNA and tRNA. This is the first example of a methyltransferase that works on both types of RNAs (Fig. 1).
The tRNA chimera herein constructed also proves useful to study the tRNA identity determinants for RlmN (Table 1). By using site-directed mutagenesis on the plasmid containing tRNAChimeraUUG, we produced the U35A, U35G, G36U, and G36A variants. We found that change G36U, as well as the other variants albeit to a lesser extent, reduce m2A accumulation, suggesting that nucleosides at positions 35 and 36 of the anticodon help the recognition of tRNA by RlmN.
RlmN in vitro modifies tRNAChimeraUUG purified from ΔrlmN cells but not two unmodified substrates (tRNAGlnUUG and tRNAArgACG) obtained by in vitro transcription (Fig. 4D; data not shown). This suggests that RlmN works in a late step during tRNA maturation. No modification in the ASL region of tRNAChimeraUUG seems crucial for m2A synthesis (Table 1), but modifications in other tRNA regions contributing to stabilizing the L-shaped tertiary structure may be required for RlmN action. RlmN activity on 23S rRNA is dependent on the presence of helices 90–92 (Yan et al. 2010). Likewise, RlmN may require a precise 3D structure of tRNA to perform m2A synthesis at position 37. Unfortunately, we were unable to obtain stable tRNAChimeraUUG derivatives lacking the D or the T stem–loop. Construction of new, stable derivatives of tRNA substrates for RlmN or the determination of crystal structures of RlmN in the apo- and tRNA-bound forms might help characterize the tRNA identity elements for RlmN and discover what they have in common with those in 23S rRNA.
The overexpression in a wild-type strain of tRNAChimeraUUG and its mutants also provides valuable information about the identity determinants used by certain tRNA modifying enzymes, such as RimN and CmoA/CmoB, whose tRNA recognition mechanisms remain to be investigated (Table 1).
RlmN inactivation produces an error-prone phenotype since it increases the misreading of a UAG stop codon (Table 2). This phenotype is opposite to the error-restrictive phenotype expected for the mutations removing modifications at position 37 and exemplified by ΔmiaA (Table 2). Therefore, we suspect that the misreading effect of ΔrlmN is due to the lack of m2A in the rRNA, which would facilitate the accommodation of an incorrect tRNA in the PTC. In the readthrough assay herein used, the error-prone effect produced by loss of m2A from the PTC could compensate for the error-restrictive effect due to the absence of m2A in tRNAs. Certainly, the simultaneous location of m2A in tRNA and rRNA complicates the study of its precise role in translational accuracy. The construction of in vivo and in vitro systems allowing a combination of fully modified tRNAs with m2A-lacking ribosomes, or vice versa, would facilitate the analysis of the individual effect of m2A on rRNA or tRNA functioning, respectively.
Modifications in rRNAs cluster in ribosomal regions of functional importance and are thought to collectively contribute to the fine-tuning of the translation process (Decatur and Fournier 2002; Chow et al. 2007). However, there is so far no known role for these modifications in ribosomal accuracy, except for dimethylation introduced by KsgA (RsmA) into A1518 and A1519 of 16S rRNA, and the N4 and 2′-O-methylations introduced by RsmH and RsmI proteins at position 1402 of 16S rRNA (van Buul et al. 1984; Kimura and Suzuki 2010). If, as we believe, the error-prone phenotype of the ΔrlmN mutant is due to the lack of m2A in 23S rRNA, then our data would represent the first example of how a single 23S RNA modification at the PTC plays a crucial role in ribosomal accuracy.
MATERIALS AND METHODS
Sequence analysis and candidate selection
Using the RlmN sequence from E. coli (Uniprot id P36979) as a query, a Blastp search against the nonredundant Reference Sequences Database at NCBI (Pruitt et al. 2012) was conducted with default parameters (http://blast.ncbi.nlm.nih.gov/Blast.cgi) (Altschul et al. 1997). We manually explored the phylogenetic distribution of RlmN homologs in major Eubacteria phyla (i.e., Acidobacteria, Actinobacteria, Bacteroidetes, Chloroflexi, Chlamydiae, Cyanobacteria, Deinococci, Firmicutes, Fusobacteria, Proteobacteria, Spirochaetes, Tenericutes, and Thermotogae). Based on pairwise comparisons with alignment coverage >75% and alignment score >60 bits, we retrieved more than 130 different sequences that are representative of diversity for each bacterial phylum. The full set of RlmN homologs was aligned using the Probcons software, v1.12, with 1000 passes of iterative refinement (Do et al. 2005) followed by a filtering for gaps. An RlmN amino acid profile was constructed using the HMMER v2.3.2 algorithm and its default parameters (Eddy 1998). Then, search against nonredundant nucleotide and protein databases from Escherichia coli K-12 was carried out with the TBLASTN and Blastp algorithms, respectively, using the different conserved blocks of amino acids obtained from HMMER as queries. Afterward, no paralogs of RlmN were found. We extended this search to other model bacterial genomes and obtained similar results.
Bacterial strains, plasmids, oligonucleotides, and DNA manipulations
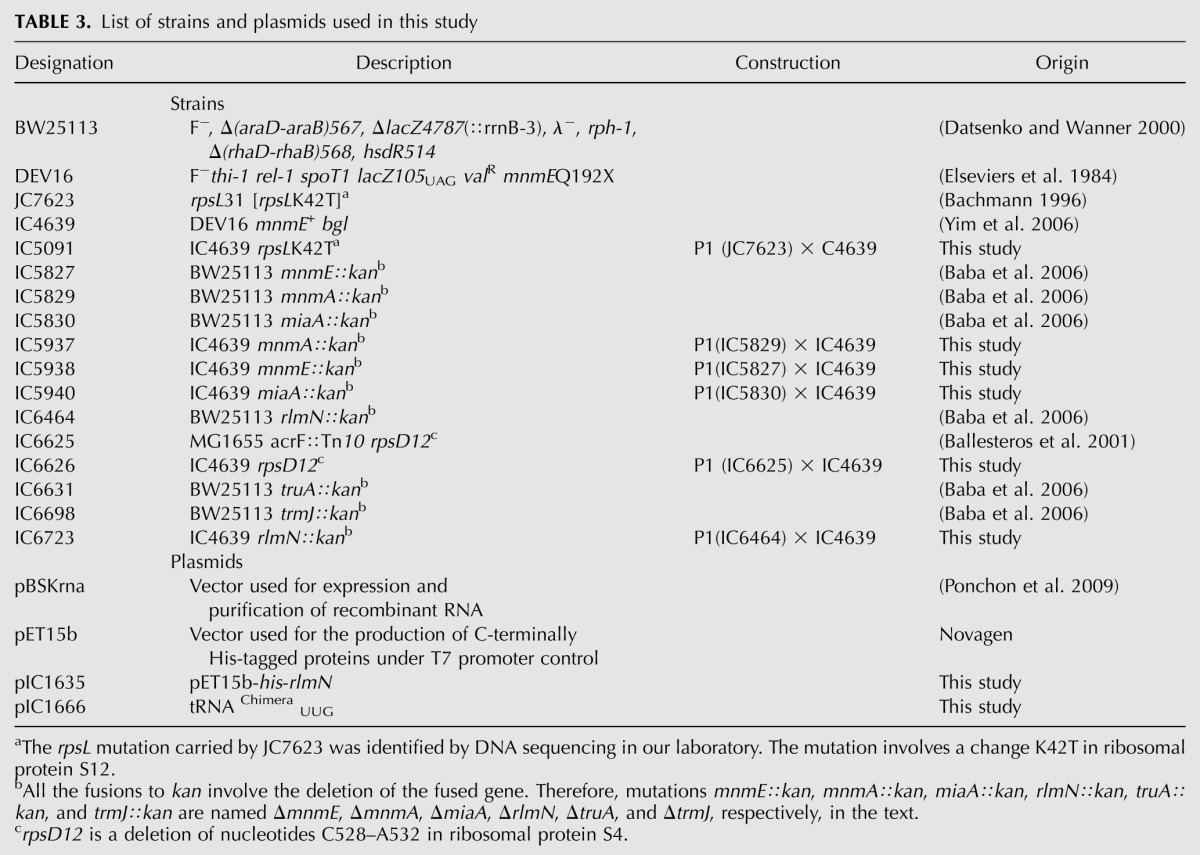
E. coli strains and plasmids used in this study are listed in Table 3. Oligonucleotides and primers are described in Supplemental Table S1. P1 transductions were performed following standard procedures (Miller 1990). Kanamycin-resistant transductants were verified by PCR using a primer flanking the gene replaced with the kan cassette and an internal primer for kan, as recommended (Datsenko and Wanner 2000). The transductants carrying the rpsLK42T or rpsD12 allele were verified by PCR amplification and sequencing. DNA manipulations were carried out according to standard protocols. Point mutations were obtained by site-directed mutagenesis with appropriate PCR primers. The resulting constructs were verified by DNA sequencing.
TABLE 3.
List of strains and plasmids used in this study
Isolation of bulk rRNA/tRNA and analysis of the RNA modification status by HPLC
Bacterial strains were grown overnight in LB medium, 1/100 diluted in 100 mL of the same medium and then grown to an OD600 of 0.7–0.8. Cells were harvested by centrifugation and resuspended in 0.4 mL of buffer-A (25 mM Tris pH 7.4, 60 mM KCl, 10 mM MgCl2). Lysozyme (2 mg, Sigma) was added, and the suspension was incubated at 37°C for 15–20 min. The cell suspension was lysed by three freeze-thaw cycles using liquid nitrogen; then 0.6 mL of buffer-B (buffer-A supplied with 0.6% Brij35, 0.2% Na-deoxycholate, 0.02% SDS) and 0.1 mL of phenol (equilibrated to pH 4.3 with citrate) were added and mixed. The suspension was incubated on ice for 15 min, and the aqueous phase was extracted twice with 1 vol of phenol. RNA was precipitated with 2.5 vol of cold ethanol containing 1% (w/v) potassium acetate. The pellet was washed with 70% ethanol and dissolved in 2 mL of buffer R200 (100 mM Tris-H3PO4, pH 6.3, 15% ethanol, 200 mM KCl) prior to running over a Nucleobond AX500 column (Macherey-Nagel), pre-equilibrated with 10 mL of the same buffer. The column was washed once with 6 mL of R200 and once with 2 mL of R650 (R200 with 650 mM KCl). tRNA was eluted with 6 mL of R650 buffer and precipitated with 0.7 vol of isopropanol, washed with 70% ethanol and redissolved in water. After tRNA elution, columns were washed with 6 mL of R1200 (R200 with 1.2 M KCl), which led to elution of 16S-enriched rRNA, and subsequently with 6 mL R1500 (R200 with 1.5 M KCl), which permitted us to obtain a 23S-enriched rRNA fraction. Given that the m2A modification resides only in 23S rRNA, this 23S-enriched rRNA fraction was considered appropriate for the HPLC analysis.
For the HPLC analysis of nucleosides, 35 μg of bulk tRNA and 70 μg of 23S-enriched rRNA were hydrolyzed with nuclease P1 (Sigma) by overnight incubation in water with 1 mM ZnSO4, followed by treatment with E. coli alkaline phosphatase (Sigma) at pH 8.3 for 2 h. The hydrolysate was analyzed by HPLC using a Develosil 5-μm C30 RP-Aqueous column (Phenomenex) with gradient elution to obtain optimal separations of nucleosides. Buffer A contained 2.5% methanol, 10 mM NH4H2PO4, pH 5.1, while buffer B contained 25% methanol, 10 mM NH4H2PO4, pH 5.3. The time for gradient elution was extended to 70 min. Nucleoside modifications were evaluated at a 260-nm wavelength using the EZchrom Elite software.
In vivo complementation
The rlmN open reading frame from Escherichia coli was amplified using the oligonucleotides: 5′-CGCCCATGGGTCATCATCACCATCACCATTCTGAACAATTAGTCACACCTGAA and 3′-GCCGGATCCTTATCAGACCGCTTTAATGTCGATG encoding the NcoI and BamHI restriction sites (bold) and the N-terminal histidine tag (italics). The PCR product was digested and inserted into an NcoI/BamHI linearized pET15b plasmid by incubation with T4 ligase at 16°C overnight. The pET15b-his-rlmN construct was used to transform the BW25113 ΔrlmN strain; the empty pET15b plasmid was used to transform BW25113 wild-type and BW25113 ΔrlmN cells as controls. Bulk tRNA isolation from these plasmid-bearing strains and HPLC-nucleoside analysis were carried out as above.
Construction, purification, and in vivo analysis of tRNAChimeraUUG and its derivatives
Initially, the gene for tRNAGlncmnm5s2UUG was inserted into the scaffold tRNA (at the EcoRV site) carried by pBSKrna (Ponchon et al. 2009). When we attempted to overexpress this construct in DH5α cells (as well as in MC4100, BW25113, or MG1655), following the recommended protocol (Ponchon et al. 2009), we were unable to observe accumulation of the construct from bulk tRNA preparations analyzed by agarose gel electrophoresis (data not shown) or to recover any fraction by the Chaplet Column Chromatography method (Suzuki and Suzuki 2007). This also occurred with other RlmN tRNA substrates such as tRNAArgICG, tRNAAspQUC, tRNAGlumnm5s2UUC, and tRNAHisQUG. Therefore, it seems that some chimera constructs are unstable and that they undergo degradation by native RNases. We finally constructed a tRNAChimeraUUG by replacing the ASL present in the tRNA scaffold with the ASL of tRNAGlncmnm5s2UUG (see Fig. 4). This procedure was assisted by designing chimera constructs in the ssDNA version, followed by PCR amplification and cloning into an EcoRI/PstI linearized pBSKrna plasmid. The resulting tRNAChimeraUUG was stably overproduced and could be purified by the standard method. Subsequently, several directed mutagenesis assays were done to introduce the following changes: A37C, U35A, U35G, G36U, and G36A. Using a similar procedure, we constructed truncated versions of tRNAChimeraUUG by deleting the D or the T stem–loops. However, these constructs were unstable since no accumulation was observed. All stable tRNAChimeraUUG versions were purified by the Chaplet Column Chromatography method (Suzuki and Suzuki 2007) with the DNA probe biotin5′-TGGCGCCCGAACAGGGACTTGAACCC, complementary to the scaffold human cytosolic tRNALys3 moiety of the tRNAChimeraUUG, and immobilized in a HiTrap Streptavidin HP column (GE Healthcare). Then, 25 μg of purified tRNAChimeraUUG (or its mutant derivatives) was P1 hydrolyzed and analyzed by HPLC. Modifications s4U, D, m7G, T, and Ψ, reported to be present in the tRNA scaffold after in vivo overproduction in E. coli cells (Ponchon and Dardel 2007), were observed according to their UV spectra (spectra at 260 nm-wavelength range) and elution time (Gehrke and Kuo 1989). Nucleoside quantitation was obtained from the peak areas in the HPLC chromatograms with the EZchrom Elite software. Each nucleoside was normalized to the cytidine area, and the normalized data from mutants were referred to normalized data from the wild-type control.
Quantitation of tRNAChimeraUUG and derivatives
Quantitative Real-Time PCR (qRT-PCR) analysis was performed to measure the expression of tRNAChimeraUUG and derivatives in wild-type cells. Total RNA was isolated from cultures under identical growth conditions to those used for chimeric tRNA purification. Amplification of chimeric tRNAs and native tRNALysmnm5s2UUU was carried out using specific primers (Supplemental Table S1). qRT-PCR was performed in a one-step format using the SYBR Green PCR Master Mix (Applied Biosystems), 150 ng of total RNA, 5 units of MultiScribe Reverse Transcriptase (Applied Biosystems), and a StepOne Real Time PCR instrument (Applied Biosystems). Relative quantitation of chimeric tRNA expression was calculated with StepOne Software v2.0 (Applied Biosystems) using the expression of native tRNALysmnm5s2U as the reference sample.
Determining RlmN in vitro activity on tRNA
Radical SAM proteins containing Fe-S clusters, pivotal for their activity, require further processing to allow a proper assembly of this chemical cluster in vitro (Pierrel et al. 2002; Yan et al. 2010). Recombinant RlmN enzyme has been previously overproduced to carry out in vitro methylations of 23S rRNA (Yan et al. 2010). To obtain an active RlmN protein, we overexpressed recombinant His-RlmN from pET15b-his-rlmN in ArticExpress cells (Agilent Technologies) at 13°C in LB medium supplemented with 0.2 mM FeCl3 and 0.5 mM IPTG for 24 h. His-RlmN was purified by affinity chromatography (TALON, Clontech) using 100 mM Tris-HCl pH 8.0, 250 mM NaCl buffer, which was previously degassed and extensively purged with N2 to reduce O2 levels (to 0.2–0.4 ppm) in the solution as described previously (Butler et al. 1994). The in vitro reconstitution of the His-RlmN Fe-S cluster was performed by also employing degassed and N2 purged stock solutions. Purified His-RlmN was diluted in 1.5 mL of 100 mM Tris-HCl pH 7.5, 2 mM DTT, and incubated at 25°C with gentle shaking for 20 min. Then, 0.015 mL of FeCl3 solution (2 mM DTT, 200 mM FeCl3, and 10 mM cysteine) were added, and incubation with gentle shaking was allowed for a further 30-min period. Finally, the Fe-S cluster assembly was initiated by the addition of 0.2 mM Na2S, and incubation was extended to 3–4 h with gentle shaking at 25°C. The protein solution was centrifuged for 5 min at 10,000g to remove precipitated Fe. Then, the supernatant was recovered, and His-RlmN was washed and concentrated in an Amicon device equipped with a 30-kDa MW cut off membrane (Millipore). Proper Fe-S assembly was noted by obtaining a brownish fraction of His-RlmN at the end of this process. To assay the RlmN methyltransferase activity in vitro, the reaction mix (degassed and N2 purged) contained 100 mM Tris-HCl, pH 7.5, 250 mM NaCl, 5 mM KCl, 5 mM MgCl2, 2 mM sodium dithionite, 1 mM SAM, 15 μg of in vivo-transcribed tRNAChimeraUUG (obtained from ΔrlmN cells), and 40 μM His-RlmN. After 2 h at 37°C with gentle shaking, the methylation reaction was stopped by the addition of 1 vol of phenol pH 4.3, and tRNA contained in the aqueous phase was ethanol-precipitated, P1-digested, and HPLC-analyzed.
Readthrough assay
Strain IC4639 and derivatives contain a lacZ gene with an in-frame UAG stop codon. Misreading of this codon was quantitated by measuring β-galactosidase activity as previously described (Elseviers et al. 1984; Miller 1990) with minor modifications. Cultures were supplemented with 1.0 mM IPTG in order to enhance β-galactosidase activity. Samples for the assays were taken from exponentially growing cultures (OD600 ∼ 0.6) after at least 10 generations of steady-state growth. Data were compared for statistical significance using a t-test with Welch's correction.
SUPPLEMENTAL MATERIAL
Supplemental material is available for this article.
ACKNOWLEDGMENTS
We thank Dr. Luc Ponchon for the gift of the pBSKrna plasmid. We also thank the National BioResource Project (NIG, Japan) and the E. coli genetic Stock Center (CGSC) for providing E. coli strains used in this study. This work has been supported by the Spanish Ministry of Science and Innovation (BFU2007-66509 and BFU2010-19737 grants) and the Generalitat Valenciana (APE-010/11 and ACOMP/2012/065) to M.E.-A., and a PhD fellowship from the Prince Felipe Research Centre to A.B.-P.
Footnotes
Article published online ahead of print. Article and publication date are at http://www.rnajournal.org/cgi/doi/10.1261/rna.033266.112.
REFERENCES
- Agris PF 1996. The importance of being modified: Roles of modified nucleosides and Mg2+ in RNA structure and function. Prog Nucleic Acid Res Mol Biol 53: 79–129 [DOI] [PubMed] [Google Scholar]
- Agris PF 2004. Decoding the genome: A modified view. Nucleic Acids Res 32: 223–238 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Agris PF 2008. Bringing order to translation: The contributions of transfer RNA anticodon-domain modifications. EMBO Rep 9: 629–635 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Agris PF, Vendeix FA, Graham WD 2007. tRNA's wobble decoding of the genome: 40 years of modification. J Mol Biol 366: 1–13 [DOI] [PubMed] [Google Scholar]
- Altschul SF, Madden TL, Schaffer AA, Zhang J, Zhang Z, Miller W, Lipman DJ 1997. Gapped BLAST and PSI-BLAST: A new generation of protein database search programs. Nucleic Acids Res 25: 3389–3402 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Andersen TE, Porse BT, Kirpekar F 2004. A novel partial modification at C2501 in Escherichia coli 23S ribosomal RNA. RNA 10: 907–913 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Atta M, Mulliez E, Arragain S, Forouhar F, Hunt JF, Fontecave M 2010. S-adenosylmethionine-dependent radical-based modification of biological macromolecules. Curr Opin Struct Biol 20: 684–692 [DOI] [PubMed] [Google Scholar]
- Baba T, Ara T, Hasegawa M, Takai Y, Okumura Y, Baba M, Datsenko KA, Tomita M, Wanner BL, Mori H 2006. Construction of Escherichia coli K-12 in-frame, single-gene knockout mutants: The Keio collection. Mol Syst Biol 2: 2006.0008 doi: 10.1038/msb4100050 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bachmann D. 1996. Derivations and genotypes of some mutant derivatives of Escherichia coli K-12. In Escherichia coli and Salmonella: Cellular and molecular biology (ed. F Neidhardt, R Curtis), pp. 2460–2488. American Society for Microbiology, Washington, DC. [Google Scholar]
- Ballesteros M, Fredriksson A, Henriksson J, Nystrom T 2001. Bacterial senescence: Protein oxidation in non-proliferating cells is dictated by the accuracy of the ribosomes. EMBO J 20: 5280–5289 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Benítez-Páez A, Villarroya M, Douthwaite S, Gabaldon T, Armengod ME 2010. YibK is the 2′-O-methyltransferase TrmL that modifies the wobble nucleotide in Escherichia coli tRNALeu isoacceptors. RNA 16: 2131–2143 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Björk GR, Hagervall TG 2005. Transfer RNA modification. In EcoSal—Escherichia coli and Salmonella: Cellular and molecular biology (ed. RCI Böck et al.). ASM Press, Washington, DC [Google Scholar]
- Bjork GR, Jacobsson K, Nilsson K, Johansson MJ, Bystrom AS, Persson OP 2001. A primordial tRNA modification required for the evolution of life? EMBO J 20: 231–239 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Butler IB, Schoonen MA, Rickard DT 1994. Removal of dissolved oxygen from water: A comparison of four common techniques. Talanta 41: 211–215 [DOI] [PubMed] [Google Scholar]
- Chimnaronk S, Forouhar F, Sakai J, Yao M, Tron CM, Atta M, Fontecave M, Hunt JF, Tanaka I 2009. Snapshots of dynamics in synthesizing N6-isopentenyladenosine at tRNA anticodon. Biochemistry 48: 5057–5065 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Cho KH, Caparon MG 2008. tRNA modification by GidA/MnmE is necessary for Streptococcus pyogenes virulence: A new strategy to make live attenuated strains. Infect Immun 76: 3176–3186 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Chow CS, Lamichhane TN, Mahto SK 2007. Expanding the nucleotide repertoire of the ribosome with post-transcriptional modifications. ACS Chem Biol 2: 610–619 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Czerwoniec A, Dunin-Horkawicz S, Purta E, Kaminska KH, Kasprzak JM, Bujnicki JM, Grosjean H, Rother K 2009. MODOMICS: A database of RNA modification pathways. 2008 update. Nucleic Acids Res 37: D118–D121 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Dao V, Guenther R, Malkiewicz A, Nawrot B, Sochacka E, Kraszewski A, Jankowska J, Everett K, Agris PF 1994. Ribosome binding of DNA analogs of tRNA requires base modifications and supports the “extended anticodon”. Proc Natl Acad Sci 91: 2125–2129 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Datsenko KA, Wanner BL 2000. One-step inactivation of chromosomal genes in Escherichia coli K-12 using PCR products. Proc Natl Acad Sci 97: 6640–6645 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Decatur WA, Fournier MJ 2002. rRNA modifications and ribosome function. Trends Biochem Sci 27: 344–351 [DOI] [PubMed] [Google Scholar]
- Do CB, Mahabhashyam MS, Brudno M, Batzoglou S 2005. ProbCons: Probabilistic consistency-based multiple sequence alignment. Genome Res 15: 330–340 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Eddy SR 1998. Profile hidden Markov models. Bioinformatics 14: 755–763 [DOI] [PubMed] [Google Scholar]
- El Yacoubi B, Lyons B, Cruz Y, Reddy R, Nordin B, Agnelli F, Williamson JR, Schimmel P, Swairjo MA, de Crecy-Lagard V 2009. The universal YrdC/Sua5 family is required for the formation of threonylcarbamoyladenosine in tRNA. Nucleic Acids Res 37: 2894–2909 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Elseviers D, Petrullo LA, Gallagher PJ 1984. Novel E. coli mutants deficient in biosynthesis of 5-methylaminomethyl-2-thiouridine. Nucleic Acids Res 12: 3521–3534 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Gao W, Chua K, Davies JK, Newton HJ, Seemann T, Harrison PF, Holmes NE, Rhee HW, Hong JI, Hartland EL, et al. 2010. Two novel point mutations in clinical Staphylococcus aureus reduce linezolid susceptibility and switch on the stringent response to promote persistent infection. PLoS Pathog 6: e1000944 doi: 10.1371/journal.ppat.1000944 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Gehrke CW, Kuo KC 1989. Ribonucleoside analysis by reversed-phase high-performance liquid chromatography. J Chromatogr 471: 3–36 [DOI] [PubMed] [Google Scholar]
- Giessing AM, Jensen SS, Rasmussen A, Hansen LH, Gondela A, Long K, Vester B, Kirpekar F 2009. Identification of 8-methyladenosine as the modification catalyzed by the radical SAM methyltransferase Cfr that confers antibiotic resistance in bacteria. RNA 15: 327–336 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Golovina AY, Sergiev PV, Golovin AV, Serebryakova MV, Demina I, Govorun VM, Dontsova OA 2009. The yfiC gene of E. coli encodes an adenine-N6 methyltransferase that specifically modifies A37 of tRNA1Val(cmo5UAC). RNA 15: 1134–1141 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Gong S, Ma Z, Foster JW 2004. The Era-like GTPase TrmE conditionally activates gadE and glutamate-dependent acid resistance in Escherichia coli. Mol Microbiol 54: 948–961 [DOI] [PubMed] [Google Scholar]
- Gray J, Wang J, Gelvin SB 1992. Mutation of the miaA gene of Agrobacterium tumefaciens results in reduced vir gene expression. J Bacteriol 174: 1086–1098 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Grosjean H. 2005. Fine tuning of RNA functions by modification and editing. In Topics in current genetics (ed. S Hohmann). Springer Verlag, New York, NY. [Google Scholar]
- Grove TL, Benner JS, Radle MI, Ahlum JH, Landgraf BJ, Krebs C, Booker SJ 2011. A radically different mechanism for S-adenosylmethionine-dependent methyltransferases. Science 332: 604–607 [DOI] [PubMed] [Google Scholar]
- Havelund JF, Giessing AMB, Hansen T, Rasmussen A, Scott LG, Kirpekar F 2011. Identification of 5-hydroxycytidine at position 2501 concludes characterization of modified nucleosides in E. coli 23S rRNA. J Mol Biol 411: 529–536 [DOI] [PubMed] [Google Scholar]
- Jenner L, Demeshkina N, Yusupova G, Yusupov M 2010. Structural rearrangements of the ribosome at the tRNA proofreading step. Nat Struct Mol Biol 17: 1072–1078 [DOI] [PubMed] [Google Scholar]
- Kaminska KH, Purta E, Hansen LH, Bujnicki JM, Vester B, Long KS 2010. Insights into the structure, function, and evolution of the radical-SAM 23S rRNA methyltransferase Cfr that confers antibiotic resistance in bacteria. Nucleic Acids Res 38: 1652–1663 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kimura S, Suzuki T 2010. Fine-tuning of the ribosomal decoding center by conserved methyl-modifications in the Escherichia coli 16S rRNA. Nucleic Acids Res 38: 1341–1352 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kurland C, Hughes D, Ehrenberg M 1996. Limitations of translational accuracy. In Escherichia coli and Salmonella: Cellular and molecular biology (ed. F Neidhardt et al.), pp. 979–1004. ASM Press, Washington, DC [Google Scholar]
- Lamarre JM, Howden BP, Mankin AS 2011. Inactivation of the indigenous methyltransferase RlmN in Staphylococcus aureus increases linezolid resistance. Antimicrob Agents Chemother 55: 2989–2991 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Li JN, Bjork GR 1995. 1-Methylguanosine deficiency of tRNA influences cognate codon interaction and metabolism in Salmonella typhimurium. J Bacteriol 177: 6593–6600 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Miller JH. 1990. A short course in bacterial genetics: A laboratory manual and handbook for Escherichia coli and related bacteria. Cold Spring Harbor Laboratory Press, Cold Spring Harbor, NY. [Google Scholar]
- Nasvall SJ, Chen P, Bjork GR 2004. The modified wobble nucleoside uridine-5-oxyacetic acid in tRNAPro(cmo5UGG) promotes reading of all four proline codons in vivo. RNA 10: 1662–1673 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Nasvall SJ, Chen P, Bjork GR 2007. The wobble hypothesis revisited: Uridine-5-oxyacetic acid is critical for reading of G-ending codons. RNA 13: 2151–2164 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Numata T, Ikeuchi Y, Fukai S, Suzuki T, Nureki O 2006. Snapshots of tRNA sulphuration via an adenylated intermediate. Nature 442: 419–424 [DOI] [PubMed] [Google Scholar]
- Petrullo LA, Gallagher PJ, Elseviers D 1983. The role of 2-methylthio-N6-isopentenyladenosine in readthrough and suppression of nonsense codons in Escherichia coli. Mol Gen Genet 190: 289–294 [DOI] [PubMed] [Google Scholar]
- Pettersen EF, Goddard TD, Huang CC, Couch GS, Greenblatt DM, Meng EC, Ferrin TE 2004. UCSF Chimera—a visualization system for exploratory research and analysis. J Comput Chem 25: 1605–1612 [DOI] [PubMed] [Google Scholar]
- Pierrel F, Bjork GR, Fontecave M, Atta M 2002. Enzymatic modification of tRNAs: MiaB is an iron-sulfur protein. J Biol Chem 277: 13367–13370 [DOI] [PubMed] [Google Scholar]
- Ponchon L, Dardel F 2007. Recombinant RNA technology: The tRNA scaffold. Nat Methods 4: 571–576 [DOI] [PubMed] [Google Scholar]
- Ponchon L, Beauvais G, Nonin-Lecomte S, Dardel F 2009. A generic protocol for the expression and purification of recombinant RNA in Escherichia coli using a tRNA scaffold. Nat Protoc 4: 947–959 [DOI] [PubMed] [Google Scholar]
- Pruitt KD, Tatusova T, Brown GR, Maglott DR 2012. NCBI Reference Sequences (RefSeq): Current status, new features and genome annotation policy. Nucleic Acids Res 40: D130–D135 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Raftery LA, Yarus M 1987. Systematic alterations in the anticodon arm make tRNAGlu-Suoc a more efficient suppressor. EMBO J 6: 1499–1506 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ramu H, Vazquez-Laslop N, Klepacki D, Dai Q, Piccirilli J, Micura R, Mankin AS 2011. Nascent peptide in the ribosome exit tunnel affects functional properties of the A-site of the peptidyl transferase center. Mol Cell 41: 321–330 [DOI] [PubMed] [Google Scholar]
- Sha J, Kozlova EV, Fadl AA, Olano JP, Houston CW, Peterson JW, Chopra AK 2004. Molecular characterization of a glucose-inhibited division gene, gidA, that regulates cytotoxic enterotoxin of Aeromonas hydrophila. Infect Immun 72: 1084–1095 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Suzuki T, Suzuki T 2007. Chaplet column chromatography: Isolation of a large set of individual RNAs in a single step. Methods Enzymol 425: 231–239 [DOI] [PubMed] [Google Scholar]
- Toh SM, Xiong L, Bae T, Mankin AS 2008. The methyltransferase YfgB/RlmN is responsible for modification of adenosine 2503 in 23S rRNA. RNA 14: 98–106 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Urbonavicius J, Qian Q, Durand JM, Hagervall TG, Bjork GR 2001. Improvement of reading frame maintenance is a common function for several tRNA modifications. EMBO J 20: 4863–4873 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Vallabhaneni H, Farabaugh PJ 2009. Accuracy modulating mutations of the ribosomal protein S4-S5 interface do not necessarily destabilize the rps4-rps5 protein–protein interaction. RNA 15: 1100–1109 [DOI] [PMC free article] [PubMed] [Google Scholar]
- van Buul CP, Visser W, van Knippenberg PH 1984. Increased translational fidelity caused by the antibiotic kasugamycin and ribosomal ambiguity in mutants harbouring the ksgA gene. FEBS Lett 177: 119–124 [DOI] [PubMed] [Google Scholar]
- Vazquez-Laslop N, Ramu H, Klepacki D, Kannan K, Mankin AS 2010. The key function of a conserved and modified rRNA residue in the ribosomal response to the nascent peptide. EMBO J 29: 3108–3117 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Yan F, Fujimori DG 2011. RNA methylation by radical SAM enzymes RlmN and Cfr proceeds via methylene transfer and hydride shift. Proc Natl Acad Sci 108: 3930–3934 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Yan F, LaMarre JM, Rohrich R, Wiesner J, Jomaa H, Mankin AS, Fujimori DG 2010. RlmN and Cfr are radical SAM enzymes involved in methylation of ribosomal RNA. J Am Chem Soc 132: 3953–3964 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Yarus M 1982. Translational efficiency of transfer RNA's: Uses of an extended anticodon. Science 218: 646–652 [DOI] [PubMed] [Google Scholar]
- Yarus M, Cline S, Raftery L, Wier P, Bradley D 1986. The translational efficiency of tRNA is a property of the anticodon arm. J Biol Chem 261: 10496–10505 [PubMed] [Google Scholar]
- Yim L, Martínez-Vicente M, Villarroya M, Aguado C, Knecht E, Armengod ME 2003. The GTPase activity and C-terminal cysteine of the Escherichia coli MnmE protein are essential for its tRNA modifying function. J Biol Chem 278: 28378–28387 [DOI] [PubMed] [Google Scholar]
- Yim L, Moukadiri I, Bjork GR, Armengod ME 2006. Further insights into the tRNA modification process controlled by proteins MnmE and GidA of Escherichia coli. Nucleic Acids Res 34: 5892–5905 [DOI] [PMC free article] [PubMed] [Google Scholar]