

The transcriptome of kinetoplastid mitochondria undergoes extensive RNA editing that inserts and deletes uridine residues (U's) to produce mature mRNAs. The editosome is a multiprotein complex that provides endonuclease, TUTase, exonuclease, and ligase activities required for RNA editing. The editosome's KREPB4 and KREPB5 proteins are essential for editosome integrity and parasite viability and contain semi-conserved motifs corresponding to zinc finger, RNase III, and PUF domains, but to date no functional analysis of these domains has been reported. The authors show here that various point mutations to KREPB4 and KREPB5 identify essential domains and suggest that these proteins do not themselves perform RNase III catalysis. The data presented are consistent with the hypothesis that KREPB4 and KREPB5 form intermolecular heterodimers with the catalytically active editing endonucleases, which is unprecedented among known RNase III proteins.
Keywords: RNA editing, editosome, RNase III, Trypanosoma brucei, endonuclease, endoribonuclease
Abstract
The transcriptome of kinetoplastid mitochondria undergoes extensive RNA editing that inserts and deletes uridine residues (U's) to produce mature mRNAs. The editosome is a multiprotein complex that provides endonuclease, TUTase, exonuclease, and ligase activities required for RNA editing. The editosome's KREPB4 and KREPB5 proteins are essential for editosome integrity and parasite viability and contain semi-conserved motifs corresponding to zinc finger, RNase III, and PUF domains, but to date no functional analysis of these domains has been reported. We show here that various point mutations to KREPB4 and KREPB5 identify essential domains, and suggest that these proteins do not themselves perform RNase III catalysis. The zinc finger of KREPB4 but not KREPB5 is essential for editosome integrity and parasite viability, and mutation of the RNase III signature motif in KREPB5 prevents integration into editosomes, which is lethal. Isolated TAP-tagged KREPB4 and KREPB5 complexes preferentially associate with components of the deletion subcomplex, providing additional insights into editosome architecture. A new alignment of editosome RNase III sequences from several kinetoplastid species implies that KREPB4 and KREPB5 lack catalytic activity and reveals that the PUF motif is present in the editing endonucleases KREN1, KREN2, and KREN3. The data presented here are consistent with the hypothesis that KREPB4 and KREPB5 form intermolecular heterodimers with the catalytically active editing endonucleases, which is unprecedented among known RNase III proteins.
INTRODUCTION
The dramatic remodeling of the mitochondrial transcripts in Trypanosoma brucei mitochondria by RNA editing involves the insertion of thousands and deletion of hundreds of uridylylates (U's) to generate mature mRNAs (Stuart et al. 2005; Hajduk and Ochsenreiter 2010; Aphasizhev and Aphasizheva 2011). Template guide RNAs (gRNAs) specify editing sites and provide the information to recode these RNAs by forming an expanding double-stranded (ds) RNA duplex with their target mRNAs. Each gRNA typically contains information for multiple editing sites, and most mRNAs require several gRNAs during the course of editing. Multiprotein complexes called editosomes catalyze RNA editing steps of cleavage by site-specific endonuclease, U addition by 3′ terminal uridylyl-transferase (TUTase), U removal by 3′ U-specific exoribonuclease (exoUase), and RNA rejoining by ligase.
More than 1000 different editing sites are present in the mitochondrial transcriptome, representing a vast diversity of substrates that editosomes modify. The mechanism by which editosomes recognize various editing sites is incompletely understood, although experiments have identified three kinetoplastid RNA editing endonucleases (KRENs)—KREN1, KREN2, and KREN3—whose activities are dependent on substrate recognition (Carnes et al. 2005, 2008; Trotter et al. 2005). The editing endonucleases act on distinct substrates, with current data indicating that KREN1 cleaves deletion sites, KREN2 cleaves most insertion editing sites, and KREN3 cleaves COII insertion editing sites. Due to the complexity of recognizing distinct bona fide editing sites (both insertion and deletion) among many potential substrates, understanding endonucleolytic cleavage is of particular importance.
The three editing endonucleases are required for viability, as are the conserved catalytic residues in the single conserved RNase III domain they possess (Carnes et al. 2005, 2008; Trotter et al. 2005; Macrae and Doudna 2007). Because all characterized RNase III endonucleases function as dimers that typically cleave both strands of a dsRNA duplex and because recent experiments have indicated that the editing endonucleases are present as a single copy per editosome, we have hypothesized that they form a dimeric RNase III domain with either KREPB4 or KREPB5 (Macrae and Doudna 2007; Carnes et al. 2008, 2011). Degeneracy in the RNase III motifs of both KREPB4 and KREPB5 makes it unclear whether they retain catalytic capacity, as they lack the amino acids that are universally conserved in the active site of all known RNase III enzymes (Worthey et al. 2003). If KREN1, KREN2, and KREN3 form intermolecular heterodimers with KREPB4 or KREPB5, the catalytic activity could result in only mRNA being cleaved, allowing gRNA to be recycled.
KREPB4 and KREPB5 are also essential, and loss of either protein results in the loss of intact editosomes and editosome proteins (Wang et al. 2003; Babbarwal et al. 2007). An initial bioinformatic analysis of editosome proteins used a combination of approaches to create alignments and identify putative motifs in KREN1, KREN2, KREN3, KREPB4, and KREPB5 (Worthey et al. 2003). In addition to the RNase III motifs identified in KREN1, KREN2, KREN3, KREPB4, and KREPB5, this analysis found they had a U1-like zinc finger and either a dsRNA binding motif (dsRBM; in KREN1, KREN2, and KREN3) or PUF domain (KREPB4 and KREPB5). Curiously, this analysis generated overlapping RNase III and PUF domains in which amino acids E284 of KREPB4 and E236 of KREPB5 represented residues conserved in both the RNase III and PUF domains.
Each of the site-specific endonucleases, KREN1, KREN2, and KREN3 (Panigrahi et al. 2006; Carnes et al. 2008, 2011), is found in a compositionally distinct ∼20S editosome. These ∼20S editosomes contain a common set of 12 proteins and a mutually exclusive endonuclease and partner protein(s): KREN1 editosomes exclusively contain KREPB8 and exoUase KREX1; KREN2 editosomes exclusively contain KREPB7; KREN3 editosomes exclusively contain KREPB6. The common set of editosome proteins includes the heterotrimeric (Schnaufer et al. 2003) insertion subcomplex (KREPA1, KRET2, and KREL2), the heterotrimeric deletion subcomplex (KREPA2, KREX2, and KREL1), as well as KREPA3, KREPA4, KREPA5, KREPA6, KREPB4, and KREPB5. By use of a combination of yeast two-hybrid analysis and subcomplex reconstitution with recombinant proteins, several direct protein–protein interactions among editosome proteins were identified for members of the common set of editosome proteins (Schnaufer et al. 2010); however, no interactions were identified for any of the endonucleases or their partner proteins. No interactions for KREPB4 were identified, but KREPB5 was shown to interact with KREPA3. Thus, although KREPB4 and KREPB5 are essential for editosome structural integrity, very little is known about how they interact with other editosome proteins.
We show here that point mutations in the zinc finger domain of KREPB4 and the degenerate RNase III motif of KREPB5 prevent normal function of the editosome and are lethal when exclusively expressed. Isolation of editosome complexes via tagged wild-type and mutant KREPB4 or KREPB5 reveals a preferential association with components of the deletion subcomplex. Mutation of the zinc finger domain of KREPB4, but not KREPB5, results in predominant isolation of an editosome subcomplex and prevents cleavage activity. A single point mutation to the RNase III motif of KREPB5 prevents isolation of intact editosomes, which is consistent with the lethal phenotype observed when it is exclusively expressed. Other mutations to KREPB4 and KREPB5 produce no observed defect in function and suggest that these proteins are noncatalytic. This conclusion is further supported by a new alignment of editosome RNase III sequences from several kinetoplastid species that suggests that both KREPB4 and KREPB5 lack amino acids required for catalytic activity. This alignment also reveals that the PUF motif, previously only identified in KREPB4 and KREPB5, is present in the editing endonucleases as well. Our results are consistent with the hypothesis that KREPB4 and KREPB5 can form the noncatalytic half of an RNase III heterodimer with the characterized editing endonucleases.
RESULTS
Sequence alignment of putative functional domains in KREPB4 and KREPB5
Sequence analysis predicts domains potentially important for the function of KREPB4 and KREPB5. Because several additional editosome protein sequences have become available since the original sequence alignments of KREN1, KREN2, KREN3, KREPB4, and KREPB5 were published, we performed a new alignment of these proteins using MUSCLE (Edgar 2004) with 63 sequences from various kinetoplastid species (Supplemental Table 1). Identified domains of interest in the new alignment are represented by sequences from T. brucei and Trypanosoma cruzi in Figure 1 (all aligned sequences for RNase III C-terminal motif and PUF domain in Supplemental Fig. 1). The alignment of the zinc finger motif and RNase III signature motif (Fig. 1A,B) was largely similar to the previously published alignment (Worthey et al. 2003). However, the sequence originally identified as being a part of both the RNase III C-terminal motif and a PUF domain in KREPB4 and KREPB5 is significantly shifted in the new alignment. This shift in alignment was also observed using ClustalW (Larkin et al. 2007) and ClustalO (Sievers et al. 2011; data not shown). In particular, the previous alignment places E284 of KREPB4 and E236 of KREPB5 at the position of the conserved catalytic glutamate of the RNase III motif and also has these amino acids in an overlapping PUF domain. In the new alignment, E284 of KREPB4 and E236 of KREPB5 are shifted further C-terminally and demonstrate noticeable sequence similarity with amino acids in KREN1, KREN2, and KREN3. A motif search of these protein sequences using Motif Scan (Pagni et al. 2004) identified the PUF domain in KREPB4 from several Trypanosoma species (e.g., T. brucei had an expectation value of 0.00063 by pfam_fs), but the dsRBM domain was not detected in any of the tested sequences. The 63 aligned kinetoplastid sequences show significant similarity with the identified KREPB4 PUF domain, and have universal conservation of the histidine that would be predicted to form stacking interactions with RNA. The position of E284 of KREPB4 and E236 of KREPB5 in the PUF domain aligns with residues that form hydrogen bonds with RNA and mediate base specificity in canonical PUF proteins (Edwards et al. 2001; Wang et al. 2002; Cheong and Hall 2006). The new alignment therefore predicts that KREPB4 and KREPB5 lack both universally conserved catalytic residues of the RNase III motif, and indicates the presence of a PUF domain in KREN1, KREN2, and KREN3 in the place previously identified in the editing endonucleases as a dsRBM (Worthey et al. 2003).
FIGURE 1.

Alignment of sequence motifs from KREPB4, KREPB5, KREN1, KREN2, and KREN3, highlighting residues mutated in this study with outlined boxes. Amino acid numbers for each protein are indicated. Inset shows mutations examined, with arrows indicating those that showed a growth defect when exclusively expressed (see Fig. 2). Residues required for RNase III catalytic activity denoted by an open triangle. (A) The U1-like zinc finger motif (C2H2 shaded in gray) is conserved in KREPB4 but degenerates in KREPB5. (B) The RNase III signature motif depicts universally conserved catalytic Asp (gray shading) that is a Glu in both KREPB4 and KREPB5. The α-helical region responsible for dimerization with another RNase III domain to form the catalytic valley is noted. (C) C-terminal portion of RNase III motif and adjacent PUF domain. The universally conserved catalytic Glu residue is conserved in KREN1-3 but degenerates in KREPB4 and KREPB5 in this alignment. The single PUF motif (region denoted by bar) contains a conserved His residue (gray shading) in all 63 kinetoplastids examined, which is consistent with the base-stacking amino acid in known PUF domains. The Glu residue (denoted by solid triangle) in KREPB4 and KREPB5 was previously (Worthey et al. 2003) aligned with the C-terminal RNase III catalytic Glu but is consistent with being one of two base-pairing amino acids in the PUF motif in the current alignment. For alignment details, see Materials and Methods.
Functional analysis of amino acid changes in KREPB4 and KREPB5
Because KREPB4 and KREPB5 are essential, mutations that prevent their function are lethal when exclusively expressed. Cell lines in which the expression of KREPB4 or KREPB5 can be down-regulated have been previously generated in our laboratory, and provided a background in which to test mutant alleles (Wang et al. 2003; Babbarwal et al. 2007). Down-regulation of KREPB4 was accomplished by tetracycline-inducible RNAi targeting T. brucei KREPB4 in procyclic form (PF) cells (Babbarwal et al. 2007). To avoid RNAi silencing, wild-type or mutant KREPB4 alleles from either Trypanosoma cruzi (60.1% nucleotide identity, longest stretch of perfect homology 15 bp) or Leishmania major (43.9% nucleotide identity, longest stretch of perfect homology 8 bp) were constitutively expressed from the β-tubulin locus in the PF KREPB4 RNAi cell line; therefore, RNAi-mediated silencing of T. brucei KREPB4 results in exclusive expression of the introduced allele in the β-tubulin locus. Addition of a TAP-tag on the transgenic KREPB4 protein was used to confirm expression by Western analysis (Supplemental Fig. 2). The wild-type T. cruzi KREPB4 allele functionally complemented for the loss of T. brucei KREPB4 expression (Fig. 2A), but the L. major allele did not, despite apparent stabilization of the expressed L. major KREPB4 protein after repression of T. brucei KREPB4 (Supplemental Fig. 3). Robust cell growth was observed when the E274A mutant T. cruzi KREPB4 allele was exclusively expressed, even though this mutation is predicted to disrupt hydrogen bonding to RNA by the PUF domain (Fig. 1) and additionally inactivate the RNase III motif predicted by the previous alignment of Worthey et al. (2003). In contrast, the C26A/C29A mutation designed to disrupt the predicted zinc finger failed to support cell growth when exclusively expressed. Down-regulation of KREPB5 was accomplished by removing tetracycline-induced expression of an ectopic T. brucei KREPB5 allele in bloodstream form (BF) cells that had the endogenous KREPB5 alleles eliminated by homologous recombination (Wang et al. 2003). Thus, constitutive expression of wild-type or mutant T. brucei KREPB5 alleles from β-tubulin locus in these cells leads to exclusive expression after removal of tetracycline. As with KREPB4, expression of transgenic KREPB5 was confirmed by Western analysis probing for TAP-tag (Supplemental Fig. 2). Constitutive expression of wild-type KREPB5 permitted normal growth in the absence of tetracycline-induced expression of KREPB5, as did versions with mutations orthologous to those for KREPB4 above: E236A that disrupts the putative PUF/RNase III domain, or C14A that disrupts the zinc finger domain (Fig. 2B). Additional mutations to the RNase III signature motif of KREPB5 that were designed to disrupt dimerization based on the Aquifex aeolicus RNase III structure (Gan et al. 2006) were similarly analyzed in this cell line (Fig. 2C; Supplemental Fig. 2B). The G121R mutant did not rescue the growth phenotype, while the F114S mutant permitted almost normal growth. The E122A, S123R, or H127D mutants permitted growth indistinguishable from the wild type.
FIGURE 2.

Growth analysis of exclusively expressed mutant versions of KREPB4 or KREPB5 reveals amino acids essential for function. (A) Repression (R) of KREPB4 by inducing RNAi in parental PF cell line causes a large growth defect compared to uninduced cells with endogenous KREPB4 expression (E). Exclusive expression of wild-type KREPB4 from T. cruzi (Tc WT) allows cells to grow well after repression of endogenous KREPB4 by RNAi. While exclusive expression demonstrates that the E284A mutation permits near-normal growth (Tc E284A (R)), the C51A/C54A mutation prevents KREPB4 function (Tc C51A/C54A (R)). (B) Repression (R) of KREPB5 in the parental BF conditional null cell line leads to growth defect compared with cells expressing wild-type KREPB5 (E). Exclusive expression of wild-type and mutant KREPB5 proteins permits normal growth (WT (R), C14A (R), or E236A (R)). (C) Exclusive expression of additional KREPB5 mutant proteins reveals essential amino acids. Control parental BF KREPB5 conditional null cell line demonstrates growth defect when KREPB5 is repressed (R) compared to expressed (E). While exclusive expression of some KREPB5 proteins permits normal growth (WT (R), E122A (R), S123R (R), H127D (R)) others produced slight (F114S (R)) or major (G121R (R)) growth defects. As all cell lines grew normally with tetracycline-regulated KREPB5 expressed, growth curves for cells that express mutant alleles have been omitted for clarity.
Analysis of editosomes purified via TAP-tagged KREPB4 and KREPB5
The protein composition of complexes and subcomplexes isolated via TAP-tagged KREPB4 and KREPB5 was assessed using a combination of gradient fractionation, Western blot, SYPRO Ruby- or silver-stained gels, and mass spectrometry. Samples from single-step purifications (binding to IgG Sepharose followed by TEV elution) from cells expressing TAP-tagged wild-type T. brucei KREPB4 or KREPB5, or C51A/C54A mutant KREPB4 were fractionated on 10%–30% glycerol gradients, and fractions were analyzed by Western blot for editosome components KREPA1, KREPA2, KREL1, and KREPA3 (Fig. 3A). Fractionation of TEV eluates from KREPB4 E284A, KREPB5 E236A, and KREPB5 C14A mutants were highly similar to wild-type proteins and were therefore not pursued further (Supplemental Fig. 4). In contrast to wild-type proteins, which have significant ∼20S complexes (fractions 15–20) typical of intact editosomes, the C51A/C54A KREPB4 mutant had a distinct profile with the majority of signal present in fractions 5–10. The subcomplexes isolated by this mutant KREPB4 had predominant signals for KREPA2 and KREL1, with little if any KREPA1 or KREPA3. Probing these blots with anti-calmodulin binding peptide (CBP) revealed that the TAP-tagged protein is present throughout the gradient, which is typical for such transgenically expressed editosome proteins and may represent nonspecific aggregates (Schnaufer et al. 2003). For both wild-type KREPB4 and KREPB5, gradient fractions 1–6, 7–12 (∼10S), or 15–20 (∼20S) were pooled for subsequent purification by calmodulin affinity, while fractions 5–10 and 15–20 were pooled for the C51A/C54A KREPB4 mutant. Samples were reanalyzed by Western blot probing for editosome components KREPA1, KREPA2, KREL1, KREPA3, KRET2, KREPA6, and KREN1 (Fig. 3B). All surveyed proteins were detected as expected in wild-type ∼20S complexes for both KREPB4 and KREPB5. Profiles for mutant and wild-type KREPB4 again show the predominant association of KREPA2 and KREL1 in subcomplexes and indicate relatively low amounts of editosome proteins in the ∼20S pool from C51A/C54A KREPB4 mutant. KREPA6 was also present in these subcomplexes, in amounts comparable to ∼20S wild-type complexes. KREN1 is notably present in C51A/C54A KREPB4 subcomplexes as well. Wild-type KREPB5 subcomplexes appeared to be lacking KREPA1 and KRET2 relative to the other tested proteins. TAPs without intervening gradient fractionation of the TEV eluate were also performed on all of the TAP-tagged cell lines, and isolated proteins were assessed by Western blot (Fig. 3C). Most purifications yielded robust signals for KREPA1, KREPA2, KREL1, and KREPA3, indicating the presence of both insertion and deletion subcomplex; however, the C51A/C54A KREPB4 mutant predominantly isolated a subcomplex of the editosome containing KREPA2 and KREL1, and the G121R KREPB5 mutant failed to isolate detectable amounts of editosome proteins, despite being expressed and detected in TEV eluates (Supplemental Fig. 2C). The low amount of CBP signal for the G121R mutant protein implies protein lability during TAP purification in the absence of associated editosome proteins. Variations in the epitopes present in the C-terminal tags of the transgenic KREPB5 proteins produces proteins of different sizes when these blots were probed with anti-CBP antibody (see Materials and Methods).
FIGURE 3.

Western analyses of TAP isolated complexes using tagged KREPB4 or KREPB5. (A) Western analyses of 10%–30% glycerol gradient fractions of TEV eluates for KREPB4 wild-type (top panel), KREPB4 C51A/C54A mutant (middle panel), and KREPB5 wild-type (bottom panel) that were simultaneously probed with monoclonal antibodies against KREPA1, KREPA2, KREL1, and KREPA3 (top part in each panel) to show editosome components. The same blots were also probed with anti-CBP antibody (bottom part in each panel) to show tagged KREPB4 or KREPB5. Note the KREPB4 C51A/C54A mutant predominantly purifies a subcomplex of ∼10S. (B) Western analyses of complexes from pooled gradient fractions (indicated) that were further purified by calmodulin affinity. Blots were probed for editosome components KREPA1, KREPA2, KREL1, KREPA3, KRET2, KREPA6, and KREN1. (C) Western analyses of complexes sequentially purified by placing TEV eluates directly onto calmodulin affinity resin. These calmodulin eluates were used in subsequent editing activity assays. Note that the G121R mutation to KREPB5 substantially prevents copurification of other editosome proteins. Variation in the size of tagged proteins in anti-CBP Western results from the presence or absence of Myc-His or V5 epitopes.
Mass spectrometry of TAP-isolated KREPB4 and KREPB5 complexes detected most of the ∼20S editosome proteins, with components of the deletion subcomplex (KREX2, KREPA2, and KREL1) present in subcomplex samples (Table 1; Supplemental Table 3). The only proteins known to directly associate with the ∼20S editosome that were absent from KREPB4 wild-type samples are those rarely detected in TAP-isolated complexes: KREH1, MEAT1, KREPB9, and KREPB10 (Panigrahi et al. 2006; Aphasizheva et al. 2009; Lerch et al. 2012). Similar results were observed with wild-type KREPB5 samples, with KREPA5 additionally not detected. Components of the insertion subcomplex (KRET2, KREPA1, KREL2) were underrepresented in KREPB4 and KREPB5 subcomplexes, which is consistent with Western analyses in Figure 3B that showed little if any KREPA1 or KRET2 in isolated subcomplexes. Fewer editosome proteins were detected in KREPB4 C51A/C54A samples, but components of the deletion subcomplex were again observed. Interestingly, KREN1 was observed with multiple peptides in KREPB4 C51A/C54A samples, while KREN2 and KREN3 were not detected, and only KREN2 was detected in KREPB5 subcomplexes (albeit by a single peptide). Samples representative of pooled gradient fractions analyzed by mass spectrometry were also resolved on 10% SDS-PAGE gels and stained with SYPRO Ruby or silver (Fig. 4). The protein composition of the KREPB4 and KREPB5 wild-type complexes was similar, although certain bands were more prominent in each sample. The protein composition of the KREPB5 complex was also largely similar to KREN1, KREN2, and KREN3 complexes after TAPs without intervening gradient fractionation of TEV eluates (Supplemental Fig. 5). The profile of the C51A/C54A KREPB4 mutant complex showed a significant reduction in the number of bands, which is consistent with the reduced number of editosome proteins detected by mass spectrometry and Western analysis. Together these data show that TAP-isolated KREPB4 and KREPB5 complexes show the typical composition of ∼20S editosomes, and indicate that these proteins appear to be most stably associated with the deletion subcomplex.
TABLE 1.
Editosome proteins detected by mass spectrometry of TAP-tagged wild-type KREPB4 (B4-WT), C51A/C54A mutant KREPB4 (B4-C51A/C54A), or wild-type KREPB5 (B5-WT) from pooled glycerol gradient fractions of TEV eluates that were further purified by calmodulin affinity chromatography (fraction numbers indicated) or from sequential IgG and calmodulin affinity purifications (IgG-Cal)
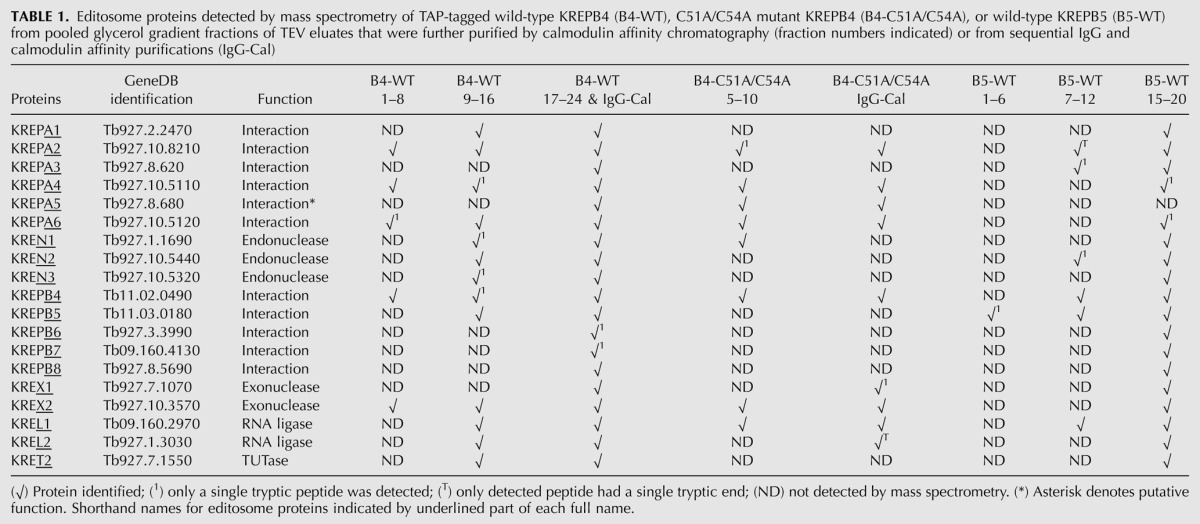
FIGURE 4.

Protein-stained SDS-PAGE shows protein complexity of samples analyzed by mass spectrometry. Representative gels analyzing calmodulin eluates of pooled glycerol gradient fractions (listed above each lane) for KREPB4 wild type (left panel, SYPRO Ruby staining), KREPB4 C51A/C54A (middle panel, silver staining), and KREPB5 wild type (right panel, silver staining). Sizes (in kiloDaltons) corresponding to molecular-weight markers are shown to the left of each panel.
Editing activities of editosomes purified via TAP-tagged, mutated KREPB4 and KREPB5
In vitro analysis of editosomes with mutations in KREPB4 or KREPB5 shows that mutation of the zinc finger in KREPB4 prevents cleavage and reduces precleaved insertion activities, but in vitro editing activities persist when mutations to potentially catalytic glutamate residues are present. Calmodulin eluates for wild-type and mutant KREPB4 and KREPB5 TAP-purifications (Fig. 3C) were analyzed for precleaved and cleavage editing activities to determine whether the introduced mutations prevented catalytic functions of the editosome. Because the G121R mutation to KREPB5 prevented isolation of editosome (Fig. 3C), it was not examined in these assays. The RNA substrates in precleaved assays mimic RNA editing substrates that have already undergone endonucleolytic cleavage. Precleaved editing assays analyzing complexes isolated via wild-type and mutant KREPB4 showed that while deletion editing activities of U removal and ligation are retained (Fig. 5B, left panel), the insertion editing activities of the C51A/C54A zinc finger mutant are impaired (Fig. 5A, left panel). For wild-type and mutant KREPB4 complexes, product corresponding to ligated input RNAs was significantly greater than edited product that had U's inserted by KRET2. These results are consistent with the predominant presence of components of the deletion subcomplex in these isolates observed by Western blot and mass spectrometry. Precleaved deletion and insertion assays showed that complexes isolated via wild-type and mutant KREPB5 retain these editing activities (Fig. 5, right panels). Cleavage assays show that only the KREPB4 C51A/C54A mutant lacks insertion and deletion endonucleolytic activities (Fig. 6). Endonucleolytic activity is typically localized to ∼20S and larger complexes and is absent from subcomplexes, so the lack of activity observed with the C51A/C54A mutant is consistent with the nature of the isolated complexes. Complexes that contain the E284A mutation to KREPB4 or the C14A, F114S, E122A, or E236A mutations to KREPB5 retain both insertion and deletion cleavage activities observed with wild-type alleles.
FIGURE 5.

TAP-isolated KREPB4 and KREPB5 complexes retain precleaved editing activity. Editing activity on input RNA is only observed in the presence of guide RNA (+g), not in its absence (−g). Positive control activity (20S+) was obtained using a ∼20S fraction from gradient fractionated mitochondrial or whole-cell lysate. Asterisks in schematics denote location of radiolabel. Arrows indicate edited product. (A) Precleaved insertion assay of complexes isolated via KREPB4 wild type (WT), C51A/C54A, and E284A (left panel) or via KREPB5 wild type, C14A, E236A, V5-tagged wild type (WT-V5), F114S, and E122A (right panel). (B) Precleaved deletion cleavage assay of complexes described above.
FIGURE 6.

Mutation of putative zinc finger of KREPB4 prevents normal cleavage activities in TAP-isolated complexes. Asterisks in the schematics denote location of radiolabel. Cleavage product (arrows) is only observed in the presence of guide RNA (+g), not in its absence (−g). Positive control activity (20S+) was obtained using a ∼20S fraction from gradient fractionated mitochondrial or whole cell lysate. The reference ladder was produced by RNase T1 (T1). (A) Insertion cleavage assay of complexes isolated via KREPB4 wild type (WT), C51A/C54A, and E284A (left panel) or via KREPB5 wild type, C14A, E236A, V5-tagged wild type (WT-V5), F114S, and E122A (right panel). (B) Deletion cleavage assay of complexes described above. Note the lack of cleavage product by complexes with the C51A/C54A mutation in KREPB4.
DISCUSSION
The data we present here identify conserved domains that are critical for the function of KREPB4 and KREPB5 and therefore are critical for editosome function and parasite survival. The lethality associated with exclusively expressed C51A/C54A mutant KREPB4 demonstrates that the conserved zinc finger is essential for function. In contrast, the degenerate zinc finger in KREPB5 is not required for function, as exclusive expression of the C14A mutant permits normal growth. While the G121R mutation to the RNase III motif of KREPB5 prevents function and association with editosome proteins, other mutations to this motif preserve normal function. We also show that the PUF domain of KREPB4 and KREPB5 is conserved in the editing endonucleases in a new sequence analysis that suggests a previously published alignment incorrectly indicated that residues essential for RNase III function were conserved in KREPB4 and KREPB5 (E284 and E236, respectively). We further demonstrate that KREPB4 and KREPB5 have preferential association with components of the deletion subcomplex, providing clues toward the functional architecture of the editosome. Together, these data reveal functional domains of KREPB4 and KREPB5 and suggest that both proteins lack endonucleolytic activity.
Different alignments of related editosome proteins KREPB4, KREPB5, KREN1, KREN2, and KREN3 produce different predictions for functional domains in KREPB4 and KREPB5. In the previously published identification of the RNase III motif in these proteins, a combination of several algorithms, including hidden Markov modeling (HMM), were used to predict domains and alignments (Worthey et al. 2003). This early analysis used three sequences (from T. brucei 927, T. cruzi, and L. major) of each protein, which provided a limited diversity of sequence information. The new alignment presented here includes additional sequences from Leishmania braziliensis, Leishmania infantum, Leishmania mexicana, Leishmania tarentolae, Trypanosoma brucei 427, Trypanosoma gambiense, Trypanosoma congolense, Trypanosoma vivax, and Trypanosoma evansi. This expanded alignment with 63 sequences results in a relative shift between the KREPB4/5 and KREN1/2/3 C-terminal halves compared to the original alignment, with a number of consequences (Supplemental Fig. 1). First, the new alignment indicates that neither of the two catalytic residues that are universally conserved in other RNase III proteins are retained in KREPB4 and KREPB5, strongly suggesting that these proteins lack endonucleolytic capacity. In contrast, the endonucleolytic abilities of these proteins remained ambiguous in the original alignment, as residues E284 of KREPB4 and E236 of KREPB5 were both potential catalytic residues in the RNase III motif as well as RNA-binding residues in the PUF domain. The second consequence of the new alignment is that the PUF domain no longer overlaps with the degenerate RNase III motif, avoiding the unprecedented combination of these domains in KREPB4 and KREPB5. The third observation from the new alignment is that the editing endonucleases—KREN1, KREN2, and KREN3—have conserved sequences consistent with the presence of a PUF domain in the region previously identified as a dsRBM in the endonucleases. The conclusion that KREN1, KREN2, and KREN3 lack a dsRBM is therefore supported by the combination of the presence PUF domain in this region, the weak sequence conservation to the canonical dsRBM sequence in the original alignment, and the inability of motif-finding algorithms to detect the dsRBM in these endonucleases. Although both PUF and dsRBM domains bind RNA, the manner in which they bind is qualitatively distinct, as PUF domains confer nucleotide specificity while dsRBMs bind RNA nonspecifically (Wang et al. 2002; Wickens et al. 2002; Gan et al. 2005, 2006). Thus, the identification of PUF domains in the endonucleases may have implications regarding the mechanism of substrate specificity.
Mutational analysis of KREPB4 and KREPB5 also indicates that these proteins lack catalytic activity. For KREN1, KREN2, and KREN3, we previously demonstrated that mutation of the universally conserved catalytic aspartate or glutamate in the RNase III motif (highlighted in Fig. 1B,C) prevents endonuclease function and is lethal when exclusively expressed (Carnes et al. 2005, 2008; Trotter et al. 2005). The functional consequences of replacing wild-type KREPB4 or KREPB5 with various mutant versions were similarly assessed. The interpretation of the results from exclusive expression of the E274A and E236A mutant versions (of TcKREPB4 and TbKREPB5, respectively) depends on the alignment, and both will be considered. If the previously published alignment is correct and E274 in TcKREPB4 and E236 in TbKREPB5 correspond to conserved catalytic residues of the RNase III motif, then mutation of these residues to alanine would be expected to prevent any existing endonucleolytic function and, additionally, have the potential to alter PUF domain binding. In contrast, if the new alignment is correct, mutation of these amino acids could only impact PUF domain binding to RNA (Wang et al. 2002). Our results show that mutation of E274A in TcKREPB4 and E236A in TbKREPB5 does not prevent normal function in vivo, making the possibility of endonucleolytic ability unlikely and suggesting either that this residue is not critical for PUF domain function or that the PUF domain itself is not essential. Furthermore, complexes isolated via TAP-tagged TbKREPB4-E284A or TbKREPB5-E236A mutants retained endonucleolytic function in vitro (Fig. 6), consistent with the conclusion that these amino acids are not part of a catalytically active RNase III domain. Both alignments agree for the remaining mutant alleles examined. The other potentially catalytic amino acid that is universally conserved in other RNase III enzymes is semi-conserved in TbKREPB4 and TbKREPB5: a relatively similar glutamate replacing the standard aspartate, E164 and E122, respectively. Our experiments show that the E122A mutation to KREPB5 has no measurable effect on function in vivo (Fig. 2) or in vitro (Figs. 5, 6), providing further evidence that the degenerate RNase III motif lacks direct catalytic activity.
While the mutations made to potential catalytic residues had no apparent effect on function, other mutations prevented function and thereby identified essential protein domains. Mutation of the zinc finger in KREPB4 prevented function in vivo (Fig. 2) and prevented cleavage in vitro (Fig. 6), results that reflect the disruption to the integrity of the complex (Fig. 3). The subcomplexes isolated by C51A/C54A mutant KREPB4 also had impaired precleaved insertion in vitro (Fig. 5), consistent with the preferential association with the deletion subcomplex at the expense of the insertion subcomplex. The presence of KREN1 in the C51A/C54A KREPB4 subcomplexes demonstrated by Western and mass spectrometry analyses is intriguing, since KREN1 has been shown to preferentially interact with the insertion subcomplex (Carnes et al. 2011). A direct interaction between KREN1 and KREPB4 would be consistent with these results and provide a means to bridge the deletion and insertion subcomplexes. Unfortunately, a direct comparison with KREN2 was not possible due to the lack of a suitable antibody. Together, these results suggest that the zinc finger in KREPB4 is required to maintain or promote protein interactions between the deletion and insertion subcomplexes, in addition to the RNA-binding role predicted by domain homology. The zinc finger in KREPB4 was identified by homology with spliceosome protein U1C from U1 small nuclear ribonucleoprotein particle (U1 snRNP) (Worthey et al. 2003). The U1 snRNP recognizes the 5′ splice site and is involved in assembly of the spliceosome. Base-pairing of the 5′ end of U1 snRNA to the 5′ splice site is facilitated by U1C in a manner that requires other U1 snRNP proteins (Heinrichs et al. 1990; Nelissen et al. 1994). Thus, the function of the U1-like zinc finger in KREPB4 may be to facilitate or recognize mRNA/gRNA duplexes such as editing sites, perhaps requiring interactions with other editosome proteins to do so. While the conserved zinc finger motif in KREPB4 is essential, the degenerate zinc finger motif of KREPB5 is dispensable. This represents one of the few characterized functional differences between KREPB4 and KREPB5 in these highly similar proteins. Since the editing endonucleases KREN1-3 possess intact zinc finger motifs, our results predict that these are of functional significance. For KREPB5, the F114S mutation led to a moderate growth defect, and the G121R mutation prevented growth when exclusively expressed (Fig. 2), indicating the importance of these amino acids in vivo. Mutations in the degenerate RNase III signature motif were chosen in an attempt to alter dimerization of KREPB5 with any of the known endonucleases and were facilitated by examination of homologous residues in the RNase III crystal structure 2EZ6 from Aquifex aeolicus (Gan et al. 2006). This structure shows that the α-helical “signature motifs” of two RNase III proteins lie anti-parallel next to each other along the center of the dimerization domain. The highly conserved G homologous to G121 in KREPB5 lies in a region where any larger amino acid would be predicted to create steric interference that would prevent normal alignment of the α-helix with another RNase III motif. Because the G121R mutant KREPB5 does not isolate intact editosomes, one possible interpretation is that KREPB5 requires dimerization with an endonuclease for proper incorporation into the editosome. Alternatively, improper positioning of the α-helix caused by G121R mutation may destabilize overall protein structure, which subsequently leads to the inability to normally interact with editosome proteins. The F114 residue at the end of the α-helix is predicted to interact with the catalytic Asp residue (see Fig. 1B, open triangle) of the other protein in the RNase III dimer. The F114S substitution is likely to cause conformational changes affecting residues critical for metal binding and catalysis. Thus, mutation of this residue in KREPB5 might alter either the dimerization with or catalytic efficiency of the opposing RNase III domain. A relatively small decrease in catalytic efficiency caused by F114S would be consistent with the relatively small growth defect and could explain the absence of an observed defect in cleavage in vitro, since the dynamic range of these assays is limited.
Exclusive expression of KREPB4 from T. cruzi, but not L. major, was sufficient to support growth in T. brucei, mirroring the relatedness of the protein sequences. The T. brucei KREPB4 protein is 55.8% identical to the T. cruzi ortholog but is only 24.6% identical to the L. major ortholog. The fact that the steady-state abundance of LmKREPB4 appeared to increase after the endogenous TbKREPB4 was repressed by RNAi suggests that the Leishmania protein can interact with T. brucei editosome proteins if it doesn't have to compete with TbKREPB4. Something in the nature of these interactions must be functionally impaired, however, since LmKREPB4 cannot support editosome function in T. brucei.
Mass spectrometry and Western data reveal that KREPB4 and KREPB5 preferentially associate with components of the deletion subcomplex, which provides additional clues to enigmatic aspects of editosome architecture. Recent work has similarly shown that KREPB6, KREPB7, and KREPB8 preferentially associate with the deletion subcomplex, while the endonucleases KREN1, KREN2, and KREN3 preferentially associate with the insertion subcomplex (Carnes et al. 2011; Guo et al. 2012). Prior to the current study, little was known about how KREPB4 and KREPB5 interact with other editosome components. Leishmania KREPB5 appeared to be present as a single copy in each editosome, and direct binding of KREPB5 with KREPA3 was revealed by yeast two-hybrid and Escherichia coli coexpression/copurification analyses (Li et al. 2009; Schnaufer et al. 2010). KREPA3 interactions appear to bridge the insertion and deletion subcomplexes (Guo et al. 2008, 2012; Schnaufer et al. 2010). The preferential association of KREPB4 and KREPB5 with components of the deletion subcomplex combined with the known association of KREPB5 with KREPA3 suggests that KREPB4 and KREPB5 might similarly bridge the gap between insertion and deletion subcomplexes by interacting with the endonucleases that associate with the insertion subcomplex. The presence of KREN1 in the C51A/C54A mutant subcomplexes would be consistent with this hypothesis if it reflects a direct interaction between KREN1 and KREPB4.
The hypothesis that the characterized editing endonucleases, KREN1–3, form intermolecular heterodimers with KREPB4 and/or KREPB5 has not been directly confirmed but continues to gain support by the accumulation of circumstantial evidence. First, all known RNase III domains function as dimers, including several dicer enzymes that form intramolecular heterodimers. Second, experiments have indicated that a single copy of either KREN1, KREN2, or KREN3 is present in particular editosome, ruling out homodimerization (Carnes et al. 2011). Evidence presented here indicates that KREPB4 and KREPB5 lack essential endonucleolytic residues or that these residues are not required for function, consistent with the proposed role as noncatalytic partners with the characterized endonucleases.
MATERIALS AND METHODS
Alignments
Amino acid sequences for KREPB4, KREPB5, KREN1, KREN2, and KREN3 from multiple kinetoplastid species were aligned using MUSCLE (Edgar 2004). TriTrypDB gene identifiers for the 63 sequences used are in Supplemental Table 1, with the exception of the T. brucei evansi STIB805 sequences, which were obtained from unpublished work (J Carnes, C Hertz-Fowler, A Ivens, A Jackson, DH Lai, M Lewis, J Lukeš, ZR Lun, A Schnaufer, K Stuart, et al., unpubl.). Conserved domains within these protein sequences were identified using Motif Scan (http://myhits.isb-sib.ch/cgi-bin/motif_scan) (Pagni et al. 2004).
Plasmid constructs and cell lines
Details of cloning, site directed mutagenesis, and oligo sequences are in Supplemental Materials. To create PF cell lines expressing C-terminal TAP-tagged proteins under tetracycline regulation, PCR-amplified coding sequences from T. brucei 427 genomic DNA were cloned into expression plasmids pLEW79-TAP (Wirtz et al. 1999; Panigrahi et al. 2003) or pLEW-MHTAP (Jensen et al. 2007) using HindIII and BamHI restriction sites; the only difference between these plasmids is the presence of tandem Myc and His epitopes preceding the TAP tag. Some constructs included a V5 epitope tag (Flaspohler et al. 2010). NotI-digested TAP-tag plasmids were transfected into PF 29.13 cells, and transgenic lines were selected by phleomycin resistance and tetracycline-dependent expression of the tagged protein confirmed by Western blot. To create cell lines for exclusive expression of KREPB4 or KREPB5 mutant genes, coding sequences were cloned into the HindIII and BamHI sites of pHD1344tub. Plasmid pHD1344tub was created by PCR amplifying the tubulin targeting sequence from pHD323 (Hotz et al. 1997) with oligos 5205 and 5206, digesting with SacI and XmaI and cloning into the same sites in pHD1344 (Haile et al. 2003). NotI-digested pHD1344tub plasmids expressing TcKREPB4 alleles were transfected into the PF 29.13-derived KREPB4 RNAi cell line (Babbarwal et al. 2007), and similarly, KREPB5 alleles were transfected into BF 427-derived KREPB5 conditional null cells (Wang et al. 2003), with transgenic lines selected by puromycin resistance and expression of the tagged protein confirmed by Western.
Growth of cells in vitro
BF cells were grown in HMI-9 with 10% FBS. PF cells were grown in SDM-79 with 10% FBS. For growth curve analyses, cell density was measured by Coulter counter, with BF cultures reseeded at about 2 × 105 cells/mL in 5 mL every day and PF cultures reseeded at about 1 × 106 cells/mL in 5 mL every 2 d.
TAP-tag purifications and mass spectrometry
Editosomes were isolated from PF 29.13 cells expressing tandem affinity purification (TAP) tagged versions of WT or mutant KREPB4 or KREPB5 by induction with 500 ng/mL tetracycline as previously described (Rigaut et al. 1999; Schnaufer et al. 2003). Briefly, approximately 2 × 1010 cells were harvested and lysed in 20 mL of IPP150, 1% Triton X-100, and complete protease inhibitors (Roche) at 4°C, and then clarified by centrifugation at 10,000g. Purification of editosomes via TAP-tagged KREPB4 or KREPB5 used sequential IgG and calmodulin affinity chromatography as previously described (Rigaut et al. 1999). Proteins in editosome samples isolated by tagged KREPB4 and KREPB5 were denatured with 8 M urea, diluted 1:8, and digested in-solution with trypsin. The resulting peptides were fractionated and analyzed by tandem mass spectrometry (LC-MS/MS) as described (Panigrahi et al. 2001a,b).
Fractionation on glycerol gradients
Fractionation of TEV eluates on 10%–30% glycerol gradients was performed as previously described (Schnaufer et al. 2003). After fractionation, glycerol gradients were divided into 0.5 mL fractions from the top, flash frozen in liquid nitrogen, and stored at −80°C.
SDS-PAGE and Western analyses
SDS-PAGE loading buffer was added to samples containing purified protein complexes and resolved on 10% SDS-PAGE gels (Criterion Tris-HCl, Bio-Rad). Two methods were used for staining. SYPRO Ruby was used following the manufacturer's protocol (Molecular Probes) and visualized with an Alpha Innotech AlphaImager EP or a Storm PhosphorImager (GE Healthcare). Silver staining was carried out using the SilverQuest kit (Invitrogen) and recorded using a white-light trans-illuminator and a digital camera. For Western analysis, protein samples were separated by electrophoresis on 10% SDS-PAGE gels, transferred to Immobilon-P membranes (Fisher), and probed using monoclonal antibodies against KREPA1, KREPA2, KREL1, and KREPA3 as previously described (Panigrahi et al. 2001a). Blots were sequentially stripped and reprobed using rabbit polyclonal primary antibodies against CBP at 1:2000 (GenScript), KREPA6 at 1:2000 or KRET2 at 1:2000 (Schnaufer et al. 2003) along with goat-anti-rabbit conjugated HRP secondary antibody at 1:2000. Blots were developed with ECL kit (Pierce) per the manufacturer's instructions. Positive control ∼20S samples from purified PF mitochondria (IsTaR 1.7a strain) or PF 427 whole-cell lysates were generated as previously described (Stuart et al. 2004; Carnes et al. 2005).
In vitro enzymatic assays
For precleaved editing and endonuclease cleavage reactions, protein complexes purified via sequential IgG and calmodulin affinity columns were incubated with 32P-labeled substrate RNAs at 28°C for 3 h, and the RNA was then extracted and analyzed by electrophoresis on 11% polyacrylamide 7 M urea gels that were dried and visualized by PhosphorImager (GE Healthcare). A6-derived substrate assays follow standard protocols described in detail elsewhere (Kable et al. 1996; Seiwert et al. 1996; Cruz-Reyes et al. 2001; Carnes et al. 2005).
A6-derived insertion endonuclease
Cleavage of 70 nucleotides (nt) A6-eES1 pre-mRNA with gA6[14] gRNA was performed as described (Carnes et al. 2005).
A6-derived deletion endonuclease
Cleavage of 73 nt A6short/TAG.1 pre-mRNA with D34 gRNA was performed as described (Carnes et al. 2005).
A6-derived precleaved editing
Standard precleaved deletion and insertion editing were assayed as previously described using 5′-labeled U5 5′CL and U5 3′CL with gA6[14]PC-del and 5′-labeled 5′CL18 and 3′CL13pp with gPCA6-2A RNAs, respectively (Igo et al. 2000, 2002).
SUPPLEMENTAL MATERIAL
Supplemental material is available for this article.
ACKNOWLEDGMENTS
We thank Bryan Jensen and Marilyn Parsons (Seattle BioMed) for the gift of pLEW-GFP-S-TAP, Wim Hol (University of Washington) for discussions, Fred Buckner (University of Washington) for the gift of T. cruzi DNA, and Carey Wickham and Brian Panicucci (Seattle BioMed) for technical assistance. The project described was supported by grant no. R01AI014102 from the National Institute of Allergy and Infectious Diseases.
Footnotes
Article published online ahead of print. Article and publication date are at http://www.rnajournal.org/cgi/doi/10.1261/rna.035048.112.
REFERENCES
- Aphasizhev R, Aphasizheva I 2011. Mitochondrial RNA processing in trypanosomes. Res Microbiol 162: 655–663 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Aphasizheva I, Ringpis GE, Weng J, Gershon PD, Lathrop RH, Aphasizhev R 2009. Novel TUTase associates with an editosome-like complex in mitochondria of Trypanosoma brucei. RNA 15: 1322–1337 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Babbarwal VK, Fleck M, Ernst NL, Schnaufer A, Stuart KD 2007. An essential role of KREPB4 in RNA editing and structural integrity of the editosome in Trypanosoma brucei. RNA 13: 737–744 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Carnes J, Trotter JR, Ernst NL, Steinberg AG, Stuart K 2005. An essential RNase III insertion editing endonuclease in Trypanosoma brucei. Proc Natl Acad Sci 102: 16614–16619 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Carnes J, Trotter JR, Peltan A, Fleck M, Stuart K 2008. RNA editing in Trypanosoma brucei requires three different editosomes. Mol Cell Biol 28: 122–130 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Carnes J, Zelaya-Soares C, Wickham C, Stuart K 2011. Endonuclease associations with three distinct editosomes in Trypanosoma brucei. J Biol Chem 286: 19320–19330 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Cheong CG, Hall TM 2006. Engineering RNA sequence specificity of Pumilio repeats. Proc Natl Acad Sci 103: 13635–13639 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Cruz-Reyes J, Zhelonkina A, Rusche L, Sollner-Webb B 2001. Trypanosome RNA editing: simple guide RNA features enhance U deletion 100-fold. Mol Cell Biol 21: 884–892 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Edgar RC 2004. MUSCLE: a multiple sequence alignment method with reduced time and space complexity. BMC Bioinformatics 5: 113.. doi: 10.1186/1471-2105-5-113. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Edwards TA, Pyle SE, Wharton RP, Aggarwal AK 2001. Structure of Pumilio reveals similarity between RNA and peptide binding motifs. Cell 105: 281–289 [DOI] [PubMed] [Google Scholar]
- Flaspohler JA, Jensen BC, Saveria T, Kifer CT, Parsons M 2010. A novel protein kinase localized to lipid droplets is required for droplet biogenesis in trypanosomes. Eukaryot Cell 9: 1702–1710 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Gan J, Tropea JE, Austin BP, Court DL, Waugh DS, Ji X 2005. Intermediate states of ribonuclease III in complex with double-stranded RNA. Structure 13: 1435–1442 [DOI] [PubMed] [Google Scholar]
- Gan J, Tropea JE, Austin BP, Court DL, Waugh DS, Ji X 2006. Structural insight into the mechanism of double-stranded RNA processing by ribonuclease III. Cell 124: 355–366 [DOI] [PubMed] [Google Scholar]
- Guo X, Ernst NL, Stuart KD 2008. The KREPA3 zinc finger motifs and OB-fold domain are essential for RNA editing and survival of Trypanosoma brucei. Mol Cell Biol 28: 6939–6953 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Guo X, Carnes J, Ernst NL, Winkler M, Stuart K 2012. KREPB6, KREPB7, and KREPB8 are important for editing endonuclease function in Trypanosoma brucei. RNA 18: 308–320 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Haile S, Estévez AM, Clayton C 2003. A role for the exosome in the in vivo degradation of unstable mRNAs. RNA 9: 1491–1501 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hajduk S, Ochsenreiter T 2010. RNA editing in kinetoplastids. RNA Biol 7: 229–236 [DOI] [PubMed] [Google Scholar]
- Heinrichs V, Bach M, Winkelmann G, Luhrmann R 1990. U1-specific protein C needed for efficient complex formation of U1 snRNP with a 5′ splice site. Science 247: 69–72 [DOI] [PubMed] [Google Scholar]
- Hotz H-R, Hartmann C, Huober K, Hug M, Clayton C 1997. Mechanisms of developmental regulation in Trypanosoma brucei: a polypyrimidine tract in the 3′-untranslated region of a surface protein mRNA affects RNA abundance and translation. Nucleic Acids Res 25: 3017–3025 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Igo RP Jr, Palazzo SS, Burgess MLK, Panigrahi AK, Stuart K 2000. Uridylate addition and RNA ligation contribute to the specificity of kinteoplastid insertion RNA editing. Mol Cell Biol 20: 8447–8457 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Igo RP Jr, Weston D, Ernst N, Panigrahi AK, Salavati R, Stuart K 2002. Role of uridylate-specific exoribonuclease activity in kinetoplastid RNA editing. Eukaryot Cell 1: 112–118 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Jensen BC, Kifer CT, Brekken DL, Randall AC, Wang Q, Drees BL, Parsons M 2007. Characterization of protein kinase CK2 from Trypanosoma brucei. Mol Biochem Parasitol 151: 28–40 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kable ML, Seiwert SD, Heidmann S, Stuart K 1996. RNA editing: a mechanism for gRNA-specified uridylate insertion into precursor mRNA. Science 273: 1189–1195 [DOI] [PubMed] [Google Scholar]
- Larkin MA, Blackshields G, Brown NP, Chenna R, McGettigan PA, McWilliam H, Valentin F, Wallace IM, Wilm A, Lopez R, et al. 2007. Clustal W and Clustal X version 2.0. Bioinformatics 23: 2947–2948 [DOI] [PubMed] [Google Scholar]
- Lerch M, Carnes J, Acestor N, Guo X, Schnaufer A, Stuart K 2012. Editosome accessory factors KREPB9 and KREPB10 in Trypanosoma brucei. Eukaryot Cell 11: 832–843 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Li F, Ge P, Hui WH, Atanasov I, Rogers K, Guo Q, Osato D, Falick AM, Zhou ZH, Simpson L 2009. Structure of the core editing complex (L-complex) involved in uridine insertion/deletion RNA editing in trypanosomatid mitochondria. Proc Natl Acad Sci 106: 12306–12310 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Macrae IJ, Doudna JA 2007. Ribonuclease revisited: structural insights into ribonuclease III family enzymes. Curr Opin Struct Biol 17: 138–145 [DOI] [PubMed] [Google Scholar]
- Nelissen RL, Will CL, van Venrooij WJ, Luhrmann R 1994. The association of the U1-specific 70K and C proteins with U1 snRNPs is mediated in part by common U snRNP proteins. EMBO J 13: 4113–4125 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Pagni M, Ioannidis V, Cerutti L, Zahn-Zabal M, Jongeneel CV, Falquet L 2004. MyHits: a new interactive resource for protein annotation and domain identification. Nucleic Acids Res 32: W332–W335 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Panigrahi AK, Gygi S, Ernst N, Igo RP Jr, Palazzo SS, Schnaufer A, Weston D, Carmean N, Salavati R, Aebersold R, et al. 2001a. Association of two novel proteins, TbMP52 and TbMP48, with the Trypanosoma brucei RNA editing complex. Mol Cell Biol 21: 380–389 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Panigrahi AK, Schnaufer A, Carmean N, Igo RP Jr, Gygi SP, Ernst NL, Palazzo SS, Weston DS, Aebersold R, Salavati R, et al. 2001b. Four related proteins of the Trypanosoma brucei RNA editing complex. Mol Cell Biol 21: 6833–6840 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Panigrahi AK, Schnaufer A, Ernst NL, Wang B, Carmean N, Salavati R, Stuart K 2003. Identification of novel components of Trypanosoma brucei editosomes. RNA 9: 484–492 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Panigrahi AK, Ernst NL, Domingo GJ, Fleck M, Salavati R, Stuart KD 2006. Compositionally and functionally distinct editosomes in Trypanosoma brucei. RNA 12: 1038–1049 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Rigaut G, Shevchenko A, Rutz B, Wilm M, Mann M, Seraphin B 1999. A generic protein purification method for protein complex characterization and proteome exploration. Nat Biotechnol 17: 1030–1032 [DOI] [PubMed] [Google Scholar]
- Schnaufer A, Ernst N, Palazzo SS, O'Rear J, Salavati R, Stuart K 2003. Separate insertion and deletion sub-complexes of the Trypanosoma brucei RNA editing complex. Mol Cell 12: 307–319 [DOI] [PubMed] [Google Scholar]
- Schnaufer A, Wu M, Young-jun P, Nakai T, Deng J, Proff R, Hol WGJ, Stuart KD 2010. A protein-protein interaction map of trypanosome ∼20s editosomes. J Biol Chem 285: 5282–5295 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Seiwert SD, Heidmann S, Stuart K 1996. Direct visualization of uridylate deletion in vitro suggests a mechanism for kinetoplastid RNA editing. Cell 84: 831–841 [DOI] [PubMed] [Google Scholar]
- Sievers F, Wilm A, Dineen D, Gibson TJ, Karplus K, Li W, Lopez R, McWilliam H, Remmert M, Soding J, et al. 2011. Fast, scalable generation of high-quality protein multiple sequence alignments using Clustal Omega. Mol Syst Biol 7: 539 doi: 10.1038/msb.2011.75 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Stuart K, Panigrahi AK, Schnaufer A 2004. Identification and characterization of trypanosome RNA editing complex components. Methods Mol Biol 265: 273–291 [DOI] [PubMed] [Google Scholar]
- Stuart KD, Schnaufer A, Ernst NL, Panigrahi AK 2005. Complex management: RNA editing in trypanosomes. Trends Biochem Sci 30: 97–105 [DOI] [PubMed] [Google Scholar]
- Trotter JR, Ernst NL, Carnes J, Panicucci B, Stuart K 2005. A deletion site editing endonuclease in Trypanosoma brucei. Mol Cell 20: 403–412 [DOI] [PubMed] [Google Scholar]
- Wang X, McLachlan J, Zamore PD, Hall TM 2002. Modular recognition of RNA by a human pumilio-homology domain. Cell 110: 501–512 [DOI] [PubMed] [Google Scholar]
- Wang B, Ernst NL, Palazzo SS, Panigrahi AK, Salavati R, Stuart K 2003. TbMP44 is essential for RNA editing and structural integrity of the editosome in Trypanosoma brucei. Eukaryot Cell 2: 578–587 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wickens M, Bernstein DS, Kimble J, Parker R 2002. A PUF family portrait: 3′UTR regulation as a way of life. Trends Genet 18: 150–157 [DOI] [PubMed] [Google Scholar]
- Wirtz E, Leal S, Ochatt C, Cross GAM 1999. A tightly regulated inducible expression system for conditional gene knock-outs and dominant-negative genetics in Trypanosoma brucei. Mol Biochem Parasitol 99: 89–101 [DOI] [PubMed] [Google Scholar]
- Worthey EA, Schnaufer A, Mian IS, Stuart K, Salavati R 2003. Comparative analysis of editosome proteins in trypanosomatids. Nucleic Acids Res 31: 6392–6408 [DOI] [PMC free article] [PubMed] [Google Scholar]