

Introduction
Recent evidence suggests that signals transmitted by receptor tyrosine kinases (RTK) and G-protein coupled receptors (GPCR) are integrated to promote efficient growth factor stimulation of cellular responses (Waters et al., 2004). The important feature of this model is that agents that disrupt GPCR function (e.g. pertussis toxin (PTX) and the C-terminal tail of GRK2, which sequesters Gβγ subunits) block the growth factor-stimulated activation of various effector modules, such as p42/p44 mitogen activated protein kinase (p42/p44 MAPK) (Luttrell et al., 1995; Fedorov et al., 1998; Conway et al., 1999; Alderton et al., 2001; Waters et al., 2003). This invokes a role for GPCR and places the G-protein down-stream from the RTK. There is now a body of evidence which supports this type of model in mammalian cells. For instance, the IGF-1 and FGF receptors use the G-protein, Gi to stimulate activation of p42/p44 MAPK in fibroblasts and skeletal muscle, respectively (Luttrell et al., 1995; Fedorov et al., 1998). We have also reported that the platelet derived growth factor (PDGF)-induced activation of c-Src and p42/p44 MAPK can be reduced by PTX and CT-GRK2 in airway smooth muscle (ASM) cells and HEK 293 cells (Conway et al., 1999; Alderton et al., 2001; Waters et al., 2003) and that the overexpression of Giα2 enhances the stimulation of p42/p44 MAPK by PDGF, associated with a PDGFβ receptor kinase-catalyzed tyrosine phosphorylation of Giα2 (Alderton et al., 2001). The tyrosine phosphorylation of endogenous Giα2 might prevent reformation of the inactive Gαβγ complex, thereby prolonging the lifetime of active G-protein subunits, including Gβγ. The integrative signal mechanism is distinct from the transactivation of RTK by GPCR agonists, which involves stimulation of the tyrosine phosphorylation of the RTK.
S1P1 receptor-PDGFβ receptor signaling complex
The S1P1 receptor, which binds sphingosine 1-phosphate (S1P), was first identified by Lee et al. (1998). To date, five closely related GPCR termed S1P1–5 have been characterized as high affinity S1P receptors (Hla and Maciag, 1990; Okazaki et al., 1993; MacLennan et al., 1994; Graler et al., 1998; Glickman et al., 1999; Yamazaki et al., 2000). They are integral membrane proteins that exhibit approximately 50% amino-acid sequence identity. Recent data suggests that the S1P1 and S1P3 receptor are involved in S1P-induced cell migration, while the S1P2 receptor inhibits cell migration (Takuwa, 2002). We have reported that the PDGFβ receptor and S1P1 receptor form a complex in HEK 293 cells and ASM cells (Alderton et al., 2001; Waters et al., 2003). The formation of the PDGFβ receptor-S1P1 receptor complex is not increased by PDGF or S1P (Alderton et al., 2001; Waters et al., 2003), suggesting that the PDGFβ receptor and/or a tethering protein is limiting for formation of the complex. The key feature of the model is that the close proximity association between the PDGFβ receptor and the S1P1 receptor permits the use of activated G-protein subunits (made available by the constitutively active or S1P-stimulated S1P1 receptor) by the PDGFβ receptor to induce signal transmission in response to PDGF. Signal integration by the PDGFβ receptor-S1P1 receptor complex occurs because c-Src is recruited to the PDGFβ receptor-S1P1 receptor complex in response to PDGF and is activated by a S1P1/Gi-dependent mechanism (Conway et al., 1999; Waters et al., 2005). This results in a c-Src-catalysed tyrosine phosphorylation of Grb-2 associated binder, Gab1 (Rakhit et al., 2000; Waters et al., 2005), which is followed by recruitment of phosphoinositide 3-kinase 1a (PI3K1a)-dynamin II to tyrosine phosphorylated Gab1 (Rakhit et al., 2000; Waters et al., 2005). The recruited dynamin II functions to pinch off endocytic vesicles containing the PDGFβ receptor-S1P1 receptor complex in a PI3K-dependent manner, which are then internalized. We have also shown that β-arrestin (which functions to load GPCR complexes into clathrin coated pits prior to endosome formation and is also an adaptor protein for c-Src) plays a critical role as over-expression of the clathrin binding domain of β-arrestin (319-418) reduced the PDGF- and S1P-induced activation of p42/p44 MAPK in HEK 293 cells (Waters et al., 2005) and β-arrestin I is associated with the PDGFβ receptor-S1P1 receptor complex in these cells (Alderton et al., 2001). p42/p44 MAPK is recruited to the PDGFβ receptor-S1P1 receptor complex in endosomes and is activated (Waters et al., 2003, 2005). See Scheme 1 for a summary of this model. Others have shown that GPCR-dependent activation of p42/p44 MAPK requires β-arrestin and that activation of MEK1 by c-Raf can be blocked by inhibitors of clathrin-mediated GPCR endocytosis in cells (Daaka et al., 1998). Therefore, in conjunction with our findings, this suggests that c-Raf-MEK1 is internalized with RTK–GPCR complexes to regulate p42/p44 MAPK that subsequently associates with the RTK–GPCR complex.
Scheme 1.

Schematic demonstrating complex formation between S1P1 receptor and PDGFβ receptor enables PDGF-stimulated recruitment of c-Src and subsequent activation by Gβγ subunits (made available by constitutively active or S1P-stimulated S1P1 receptor). This leads to β-arrestin I-mediated endocytosis of the PDGFβ receptor-S1P1 receptor complex along with attendant signaling molecules that allow activation of p42/p44 MAPK in endosomes in response to PDGF. This pool of cytoplasmic activated endosomal p42/p44 MAPK appears to regulate MLC20 phosphorylation by MLCK leading to migratory responses to PDGF.
Constitutive activation of S1P1 receptor and PDGFβ receptor signal transmission
We have characterized a compound called SB649146 (obtained from Glaxo SmithKline (USA), who identified it as an apparent S1P1 receptor antagonist). We found that this compound, in fact, has unusual properties and functions as a protean agonist of the S1P1 receptor. We have used SB649146 to obtain evidence that the endogenous constitutively active S1P1 receptor regulates cell migration in response to PDGF. In this regard, we have reported that SB649146, which exhibits exquisite specificity for the S1P1 receptor, decreased the PDGF- or S1P-induced stimulation of p42/p44 MAPK and blocked ASM cell migration in response to PDGF (Waters et al., 2006). These results were confirmed by anti-sense mediated down-regulation of the S1P1 receptor, which reduced activation of p42/p44 MAPK in response to both S1P and PDGF (Waters et al., 2003). SB649146 does not interact directly with the PDGFβ receptor, but instead exerts its effect by reducing the constitutive activity of the S1P1 receptor in the complex with the PDGFβ receptor (Waters et al., 2006). Therefore, SB649146 functions by reducing the availability of active Gi subunits that can be used by the PDGFβ receptor. SB649146 does not directly inhibit Gi and does not modulate PDGFβ receptor signal transmission by this mechanism (Waters et al., 2006). Additional evidence to support the possibility that constitutively active S1P1 receptor enhances PDGFβ receptor signal transmission was demonstrated by results which showed that mutant recombinant S1P1 receptors (R120A, E121A), which are deficient in their ability to bind S1P but are constitutively active (Parrill et al., 2000), increase PDGF-stimulated p42/p44 MAPK activation to a level comparable with the wild type recombinant S1P1 receptor (Waters et al., 2006). SB649146 is a protean agonist (Kenakin, 2001; Gbahou et al., 2003) as its action is dependent upon the existence of more than one active GPCR conformation. This was evidenced by the ability of SB649146 to reduce constitutive basal S1P1 receptor-stimulated GTPγS binding (inverse agonism), yet weakly stimulate the p42/p44 MAPK pathway (partial agonism) on its own (Waters et al., 2006). GPCR can exist in an inactive conformation (R) and multiple active conformations (e.g. R*1 and R*2). R*1 and R*2 are proposed to exhibit different efficacy in terms of coupling to Gi. However, these conformations of the receptor cannot stimulate p42/p44 MAPK unless ligand is present to recruit c-Src. In Scheme 2, R*1 is designated with high efficacy while R*2 has low efficacy for coupling to G-protein. The PDGFβ receptor interacts with the high efficacy R*1 form, because SB649146 reduced the PDGF-induced activation of p42/p44 MAPK. In contrast, R*2 is dissociated from the PDGFβ receptor. The binding of SB649146 to R*2 stabilizes this receptor conformation resulting in the stimulation of the p42/p44 MAPK pathway with low efficacy. However, binding of SB649146 to R*2 will also reduce the amount of R*1 that can engage the PDGFβ receptor signaling system by mass action. Therefore, the binding of SB649146 to R*2 and, thus, conversion of high efficacy R*1 to low efficacy R*2 by mass action provides a logical explanation for the ability of SB649146 to reduce basal S1P1 receptor-stimulated GTPγS binding and to decrease PDGF-stimulated activation of p42/p44 MAPK. These findings also imply that the protean agonist SB649146 causes redistribution of the S1P1 receptor between the PDGFβ receptor-R*1 S1P1 receptor complex and free R*2 S1P1 receptor pools. We have been able to detect the R*1 and R*2 conformations of the S1P1 receptor by immunofluorescent imaging of intact cells. SB649146 reduces the PDGF-stimulated internalization of the PDGFβ receptor-S1P1 receptor complex and this provides a mechanism by which SB649146 decreases the activation of p42/p44 MAPK by PDGF. The pool of S1P1 receptor that is internalized with the PDGFβ receptor in response to PDGF therefore corresponds to the R*1 conformation. SB649146 also stimulates the internalization of a second pool of the S1P1 receptor, typical of its ability to function as a protean agonist. Internalization of the S1P1 receptor occurs without accompanying PDGFβ receptor, and therefore, this second pool of S1P1 receptor corresponds to the R*2 conformation.
Scheme 2.
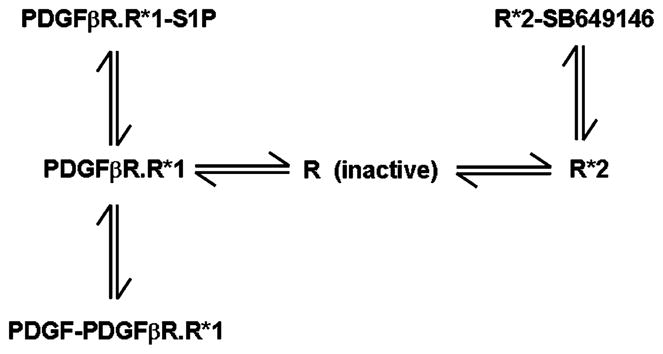
Schematic of the effect of protean agonism of the S1P1 receptor with SB649146 and the consequential effect on the engagement of S1P1 receptor with the PDGFβ receptor signaling system.
S1P may preferentially bind to R*1 conformation of the S1P1 receptor in the complex with the PDGFβ receptor. This conclusion is based upon four findings. Firstly, overexpression of PDGFβ receptor enhances S1P-stimulation of p42/p44 MAPK. Secondly, PDGFβ receptor kinase inhibitors (e.g. tyrphostin AG1296) reduce S1P stimulation of p42/p44 MAPK. Thirdly, S1P induces internalization of the PDGFβ receptor and S1P1 receptor and these receptors are co-localized in the same endocytic vesicles (Waters et al., 2003). Fourthly, S1P does not induce dissociation of the PDGFβ receptor-S1P1 receptor complex. The interpretation of this finding is complex. S1P could potentially bind to a third conformation of the S1P1 receptor termed R*3, which is dissociated from the PDGFβ receptor. In theory, the binding of S1P to R*3 should cause dissociation of the PDGFβ receptor-R*1 S1P1 receptor complex by mass action. However, in order for S1P to fail to induce dissociation of the PDGFβ receptor-R*1 S1P1 receptor complex, the amount of R*3 must be very low or non-existent. Alternatively, the amount of the R*3 conformation might be large, but that the binding of S1P to R*1 and R*3 occurs with equal affinity, thereby having no effect on the position of the equilibrium between these forms. We favor the model in which R*3 is negligible or non-existent given that S1P responses are enhanced in cells over-expressing the PDGFβ receptor and reduced by tyrphostin AG1296. Therefore, the effects of S1P are likely to be mediated by the R*1 S1P1 receptor that forms the complex with the PDGFβ receptor. It follows from this model, that the binding of SB649146 to the R*2 conformation and the subsequent reduction in the amount of R*1 by mass action provides a logical explanation for the inhibitory effect of SB649146 on the S1P-stimulated activation of p42/p44 MAPK (Waters et al., 2006).
SB649146 had no effect on basal GTPγS binding in HEK 293 cell membranes expressing recombinant S1P2 or S1P3 receptors suggesting that SB649146 exhibits exquisite specificity for the S1P1 receptor (Waters et al., 2006). Moreover, SB649146 did not affect basal GTPγS binding to Gi in membranes expressing recombinant Lysophosphatidic acid (LPA)1 receptor. Therefore, SB649146 does not directly inhibit Gi and does not modulate PDGFβ receptor signal transmission by this mechanism. Finally, SB649146 does not reduce the EGF- or PMA-induced activation of p42/p44 MAPK in ASM cells and a direct effect of SB649146 on the PDGFβ RTK itself has been excluded (Waters et al., 2006).
These findings are the first to report that protean/inverse agonist compounds which affect GPCR function reduce growth factor-induced RTK signaling, fundamentally broadening their mechanism of action.
LPA receptors
LPA binds to a family of GPCR. Three of these are members of the endothelial differentiation gene family (termed EDG receptors, and now renamed LPA1–3), whereas GPR23 (LPA4) is a member of the purinergic GPCR cluster (Fukushima and Chun, 2001; An et al., 1997). The LPA1 receptor is coupled to effectors via the heterotrimeric G-protein Gi to inhibit adenylyl cyclase and to stimulate activation of the p42/p44 MAPK pathway linked to cellular response (An et al., 1997).
Trk A receptor-LPA1 receptor signaling complex
We have previously reported that the nerve growth factor (NGF)-dependent activation of the p42/p44 MAPK pathway is reduced by PTX in PC12 cells (Rakhit et al., 2001), suggesting that the Trk A receptor (binds NGF) may use a GPCR-mediated pathway to signal to p42/p44 MAPK. This model is supported by data showing that the NGF-dependent activation of p42/p44 MAPK is enhanced in cells over-expressing β-arrestin I (Rakhit et al., 2001). More recently, we have demonstrated that the constitutively active LPA1 receptor provides Gβγ subunits which are used by the Trk A receptor to enhance activation of p42/p44 MAPK in response to NGF (Moughal et al., 2004). Four lines of evidence support this model. Firstly, the Trk A receptor was co-immunoprecipitated with the myc-tagged LPA1 receptor from PC12 cell lysates using anti-myc tag antibody, suggesting that these proteins form a complex. Neither NGF nor LPA promote formation of the complex, suggesting that the Trk A receptor and/or a tethering protein is limiting for the formation of the complex. Secondly, anti-sense mediated down regulation of the LPA1 receptor reduced NGF- and LPA-stimulated activation of p42/p44 MAPK. Thirdly, Ki16425, a selective protean agonist of the LPA1 receptor reduced LPA- and NGF-induced stimulation of p42/p44 MAPK, and weakly stimulated p42/p44 MAPK on its own. In support of a functional interaction between these receptors, the treatment of cells with Ki16425 inhibited NGF-stimulated neurite outgrowth (differentiation). Fourthly, over-expression of the CT-GRK-2 peptide, which sequesters Gβγ, reduced the NGF-induced activation of p42/p44 MAPK. In contrast, the stimulation of the LPA1 receptor with LPA lead to a predominant Giα2-mediated Trk A-independent activation of p42/p44 MAPK. This was evident from data showing that the over-expression of Giα2 enhanced LPA-, but not NGF-induced activation of p42/p44 MAPK.
A similar transition model for LPA1 receptor conformations (in comparison with the S1P1 receptor, see Scheme 3) can be described which explain our findings. As before, R*1 is designated with high efficacy while R*2 has low efficacy for coupling to Gi. The R*1 conformation of the LPA1 receptor is associated with the Trk A receptor, while the R*2 is dissociated from Trk A receptor. Therefore, binding of Ki16425 to R*2 stimulates p42/p44 MAPK with low efficacy and reduces the amount of R*1 by mass action, thereby decreasing the NGF-induced activation of p42/p44 MAPK. There are two major differences with the model that describes S1P1 receptor interaction with the PDGFβ receptor. Firstly, the LPA-induced activation of p42/p44 MAPK is unaffected by Trk A receptor kinase inhibitors. In contrast, S1P stimulation of this protein kinase pathway is reduced by PDGFβ receptor kinase inhibitors. Secondly, LPA induces a significant reduction in the amount of LPA1 receptor that is associated with the Trk A receptor, while S1P has no effect on dissociation of the PDGFβ receptor-R*1 S1P1 receptor complex. We have therefore proposed that LPA binds to a third conformation, R*3 which is dissociated from Trk A receptor. We have also concluded from these results that the amount of R*3 that binds LPA must be relatively high (determined by the equilibrium constant for transition of R→R*3) in order for LPA to induce dissociation of the Trk A receptor-R*1 LPA1 receptor complex. In addition, the binding of Ki16425 to R*2 provides a logical explanation for the ability of Ki16425 to reduce LPA-induced activation of p42/p44 MAPK mediated by the R*3 conformation.
Scheme 3.
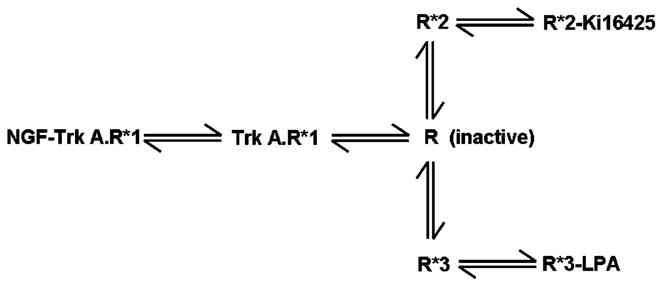
Schematic of the effect of protean agonism of the LPA1 receptors with Ki16425 and the consequential effect on the engagement of LPA1 receptor with the Trk A receptor signaling system.
The distinct conformational states of the LPA1 receptor may also use different G-protein subunits to induce signal transduction. Our data support a model in which R*1 uses Gβγ subunits to enhance NGF-induced activation of p42/p44 MAPK. Indeed, complex formation between the two receptors might enable a close proximity interaction of Gβγ subunits with the Trk A receptor signaling machinery. In contrast, the putative R*3 conformation of the LPA1 receptor, which binds LPA, uses Giα2 subunits to stimulate p42/p44 MAPK. Therefore, the LPA-induced dissociation of LPA1 receptor from the Trk A receptor-LPA1 receptor complex is in line with the concept that binding of LPA to R*3 reduces the concentration of R*1 engaged with Trk A receptor by mass action. These findings account for the LPA-induced switch in the functioning of the LPA1 receptor from using Gβγ to Giα2.
We have shown that Ki16425 does not modulate Trk A receptor signaling via direct inactivation of Gi, nor does it modulate the activity of intermediate adaptor proteins/kinases involved in the regulation of p42/p44 MAPK. In addition, Ki16425 does not reduce activation of the p42/p44 MAPK pathway by EGF in PC12 cells or EGF or PDGF in ASM cells, suggesting that Ki16425 does not interact with other RTK signaling modules.
Integrative RTK–GPCR signaling and cancer
We have defined two model systems for RTK–GPCR signal integration that regulate fundamental biological processes such as cell migration and differentiation. These findings may have implication for cancer as there is a growing body of evidence that S1P and LPA are important factors in maintaining the survival of cancer cells. There are now several reports demonstrating that sphingosine kinase (SK), the enzyme that catalyzes formation of S1P from sphingosine has an important role in breast cancer cells, suggesting that S1P may regulate survival, proliferation and migration of these cells (Nava et al., 2002; Wang et al., 1999). Ectopic expression of SK1 increased S1P levels, estrogen-dependent tumorigenesis, and blocked apoptosis of MCF-7 cells induced by anti-cancer drugs, sphingosine and TNF-α (Wang et al., 1999). SK1 is also required for EGF-induced migration, proliferation and survival of MCF-7 breast cancer cells (Sarker et al., 2005), suggesting that S1P may enhance some of the effects of EGF in these cells. S1P also stimulates BCC growth through activation of the serum response element (SRE) and indirectly by enhancing the IGF-II synthesis and function (Goetzl et al., 1999). Interestingly, a study describing a similar model of RTK–GPCR integrative signaling has been very recently described and demonstrated that the chemokine GPCR, CXCR4 and IGF-1R and G-protein subunits Giα2 and Gβ are constitutively associated in a complex in breast cancer MDA-MB-231 cells (Akekawatchai et al., 2005). This complex regulates migratory signaling in these cells. The finding is particularly interesting given that chemokines may underlie an inflammatory basis of breast cancer progression and metastasis.
Receptor complex formation may be a fundamental paradigm as there is increasing evidence for its importance. For instance, HER2/neu (also known as c-erbB-2) gene encodes a 185 kDa transmembrane growth RTK, which is the preferred heterodimerisation partner of the EGF receptor (Graus-Porta et al., 1997). HER2 may act as an epithelial cell amplifier of stroma-derived growth factor signals, such as EGF, by delaying EGF dissociation from its receptor, enhancing coupling of EGF receptor to the mitogen-activated protein kinase pathway, and impeding the rate of EGF receptor downregulation. Thus, HER2 is a master regulator of a signaling network that drives epithelial cell proliferation. Future research will be directed toward the potential for HER2 to form complexes with GPCR and in particular S1P receptors.
LPA is also increased in ovarian cancer (Xu et al., 1998) and the expression of autotaxin (lysophospholipase D), the enzyme that catalyzes formation of LPA is strongly expressed in colon cancer (Yano et al., 2003). Moreover, LPA-induced epithelial ovarian cancer cell invasion and migration in vitro is mediated by the VEGFR-2, evidenced by data showing that the selective VEGFR-2 inhibitor, SU1498 blocked LPA-stimulated invasion and migration of these cells (So et al., 2005).
Conclusion
Our findings suggest that an integrated network between GPCR and RTK arises from the proximity interaction between the two receptors at the cell surface. The ability of S1P and LPA receptors to enhance RTK-mediated signaling via a mechanism involving functional complex formation between RTK and GPCR might be important in terms of the efficiency with which growth factors stimulate cell growth and metastasis in cancer.
Summary
We describe here formation of a novel functional signaling complex between RTK and GPCRπ. This permits the use of activated G-protein subunits by the RTK in response to growth factor and that are made available by the constitutive activity of the GPCR or by binding of ligand to the latter. Moreover, β-arrestin associates with the receptor complex and participates in growth factor-dependent recruitment of c-Src, whereupon the kinase is activated by Gβγ subunits. This enables signal relay to down-stream effectors such as p42/p44 mitogen-activated protein kinases. The novel RTK–GPCR complex is involved in regulating important cellular responses, such as growth and cell migration, and dysfunction of this complex might play a significant role in hyperplasic disease states.
Acknowledgments
This work was supported by grants from the BBSRC to NJP and SP and CA92160 and HL 61469 from NIH to GT.
References
- Akekawatchai C, Holland JD, Kochetkova M, Wallace JC, McColl SH. Transactivation of CXCR4 by IGF-1R in human MDA-MB-231 breast cancer epithelial cells. J Biol Chem. 2005;280:39701–8. doi: 10.1074/jbc.M509829200. [DOI] [PubMed] [Google Scholar]
- Alderton F, Rakhit S, Kong KC, Palmer T, Sambi B, Pyne S, et al. Tethering of the platelet-derived growth factor beta receptor to G-protein-coupled receptors. A novel platform for integrative signaling by these receptor classes in mammalian cells. J Biol Chem. 2001;276:28578–85. doi: 10.1074/jbc.M102771200. [DOI] [PubMed] [Google Scholar]
- An S, Dickens MA, Bleu T, Hallmark OG, Goetzl EJ. Molecular cloning of the human Edg2 protein and its identification as a functional cellular receptor for lysophosphatidic acid. Biochem Biophys Res Comm. 1997;231:619–22. doi: 10.1006/bbrc.1997.6150. [DOI] [PubMed] [Google Scholar]
- Conway A-M, Rakhit S, Pyne S, Pyne NJ. Platelet-derived-growth-factor stimulation of the p42/p44 mitogen-activated protein kinase pathway in airway smooth muscle: role of pertussis-toxin-sensitive G-proteins, c-Src tyrosine kinases and phosphoinositide 3-kinase. Biochem J. 1999;337:171–7. [PMC free article] [PubMed] [Google Scholar]
- Daaka Y, Luttrell L, Ahn S, Della Rocca GJ, Ferguson SG, Caron MG, et al. Essential role for G protein-coupled receptor endocytosis in the activation of mitogen-activated protein kinase. J Biol Chem. 1998;273:685–8. doi: 10.1074/jbc.273.2.685. [DOI] [PubMed] [Google Scholar]
- Fedorov YV, Jones NC, Olwin BB. Regulation of myogenesis by fibroblast growth factors requires beta–gamma subunits of pertussis toxin-sensitive G proteins. Mol Cell Biol. 1998;18:5780–7. doi: 10.1128/mcb.18.10.5780. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Fukushima N, Chun J. The LPA receptors. Prostaglandins Other Lipid Mediators. 2001;64:21–32. doi: 10.1016/s0090-6980(01)00105-8. [DOI] [PubMed] [Google Scholar]
- Gbahou F, Roulea A, Morisset S, Parmentier R, Crochet S, Lin JS, et al. Protean agonist at histamine H3 receptors in vivo and in vitro. Proc Natl Acad Sci (USA) 2003;100:11086–91. doi: 10.1073/pnas.1932276100. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Glickman M, Malek RL, Kwitek-Black AE, Jacob HJ, Lee NH. Molecular cloning, tissue-specific expression, and chromosomal localisation of a novel nerve growth factor-regulated G-protein-coupled receptor, nrg-1. Mol Cell Neurosci. 1999;14:141–52. doi: 10.1006/mcne.1999.0776. [DOI] [PubMed] [Google Scholar]
- Goetzl EJ, Dolezalova H, Kong Y, Zeng L. Dual mechanism for lysophospholipid induction of proliferation of human breast cancer cells. Cancer Res. 1999;59:4732–7. [PubMed] [Google Scholar]
- Graler MH, Bernhardt G, Lipp M. A lymphoid tissue-specific receptor, EDG6, with potential immune modulatory functions mediated by extracellular lysophospholipids. Genomics. 1998;53:164–9. doi: 10.1007/978-3-642-60162-0_17. [DOI] [PubMed] [Google Scholar]
- Graus-Porta D, Beerli RR, Daly JM, Hynes NE. ErbB-2, the preferred heterodimerization partner of all ErbB receptors, is a mediator of lateral signaling. EMBO J. 1997;16:1647–55. doi: 10.1093/emboj/16.7.1647. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hla T, Maciag T. An abundant transcript induced in differentiating human endothelial cells encodes a polypeptide with structural similarities to G-protein-coupled receptors. J Biol Chem. 1990;265:9308–13. [PubMed] [Google Scholar]
- Kenakin T. Inverse, protean, and ligand-selective agonism: matters of receptor conformation. FASEB J. 2001;15:598–611. doi: 10.1096/fj.00-0438rev. [DOI] [PubMed] [Google Scholar]
- Lee M-J, Van Brocklyn JR, Thangada S, Liu CH, Hand AR, Menzeleev R, et al. Sphingosine-1-phosphate as a ligand for the G protein-coupled receptor EDG-1. Science. 1998;279:1552–5. doi: 10.1126/science.279.5356.1552. [DOI] [PubMed] [Google Scholar]
- Luttrell LM, Van Biesen T, Hawes BE, Koch WJ, Touhara K, Lefkowitz RJ. G(betagamma) subunits mediate mitogen-activated protein kinase activation by the tyrosine kinase insulin-like growth factor 1 receptor. J Biol Chem. 1995;270:16495–8. doi: 10.1074/jbc.270.28.16495. [DOI] [PubMed] [Google Scholar]
- MacLennan AJ, Browe CS, Gaskin AA, Lado DC, Shaw G. Cloning and characterisation of a putative G-protein coupled receptor potentially involved in development. Mol Cell Neurosci. 1994;5:201–9. doi: 10.1006/mcne.1994.1024. [DOI] [PubMed] [Google Scholar]
- Moughal N, Waters C, Sambi B, Pyne S, Pyne NJ. Nerve growth factor signaling involves interaction between the Trk A receptor and LPA receptor 1 systems: nuclear translocation of the LPA receptor 1 and Trk A receptors in pheochromocytoma 12 cells. Cell Signal. 2004;16:127–36. doi: 10.1016/j.cellsig.2003.08.004. [DOI] [PubMed] [Google Scholar]
- Nava VE, Hobson P, Murthy S, Milstein S, Spiegel S. Sphingosine kinase type 1 promotes oestrogen dependent tumorigenesis of breast cancer MCF-7 cells. Exp Cell Res. 2002;281:115–27. doi: 10.1006/excr.2002.5658. [DOI] [PubMed] [Google Scholar]
- Okazaki H, Ishizaka N, Sakurai T, Kurokawa K, Goto K, Kumada M, et al. Molecular cloning of a novel putative G protein-coupled receptor expressed in the cardiovascular system. Biochem Biophys Res Comm. 1993;190:1104–9. doi: 10.1006/bbrc.1993.1163. [DOI] [PubMed] [Google Scholar]
- Parrill AB, Wang D, Bautista DL, Van Brocklyn J, Lorinez Z, Fischer DJ, et al. Identification of EDG1 receptor residues that recognise sphingosine 1-phosphate. J Biol Chem. 2000;275:39379–84. doi: 10.1074/jbc.M007680200. [DOI] [PubMed] [Google Scholar]
- Rakhit S, Pyne S, Pyne NJ. The platelet-derived growth factor receptor stimulation of p42/p44 mitogen-activated protein kinase in airway smooth muscle involves a G-protein-mediated tyrosine phosphorylation of Gab1. Mol Pharmacol. 2000;58:413–20. doi: 10.1124/mol.58.2.413. [DOI] [PubMed] [Google Scholar]
- Rakhit S, Pyne S, Pyne NJ. Nerve growth factor stimulation of p42/p44 mitogen-activated protein kinase in PC12 cells: role of G(i/o), G protein-coupled receptor kinase 2, beta-arrestin I, and endocytic processing. Mol Pharmacol. 2001;60:63–70. doi: 10.1124/mol.60.1.63. [DOI] [PubMed] [Google Scholar]
- Sarker S, Maceyka M, Hait NC, Paugh SW, Senkala H, Milstein S, et al. Sphingosine kinase 1 is required for migration, proliferation and survival of MCF-7 human breast cancer cells. FEBS Lett. 2005;579:5313–7. doi: 10.1016/j.febslet.2005.08.055. [DOI] [PubMed] [Google Scholar]
- So J, Wang FQ, Navari J, Schreher J, Fishman DA. LPA-induced epithelial ovarian cancer (EOC) in vitro invasion and migration are mediated by VEGF receptor 2 (VEGF-R2) Gynecol Oncol. 2005;97:870–8. doi: 10.1016/j.ygyno.2005.03.004. [DOI] [PubMed] [Google Scholar]
- Takuwa Y. Subtype-specific differential regulation of Rho family G proteins and cell migration by the Edg family sphingosine-1-phosphate receptors. Biochim Biophys Acta. 2002;1582:112–20. doi: 10.1016/s1388-1981(02)00145-2. [DOI] [PubMed] [Google Scholar]
- Wang F, Van Brocklyn JR, Edsall L, Nava VE, Spiegel S. Sphingosine 1-phosphate inhibits motility of human cancer cells independently of cell surface receptors. Cancer Res. 1999;59:6185–91. [PubMed] [Google Scholar]
- Waters C, Sambi B, Kong KC, Thompson D, Pitson SM, Pyne S, et al. Sphingosine 1-phosphate and plateletderived growth factor (PDGF) act via PDGF beta receptor-sphingosine 1-phosphate receptor complexes in airway smooth muscle cells. J Biol Chem. 2003;278:6282–90. doi: 10.1074/jbc.M208560200. [DOI] [PubMed] [Google Scholar]
- Waters C, Pyne S, Pyne NJ. The role of G-protein coupled receptors and associated proteins in receptor tyrosine kinase signal transduction. Semin Cell Dev Biol. 2004;15:309–23. doi: 10.1016/j.semcdb.2003.12.020. [DOI] [PubMed] [Google Scholar]
- Waters C, Connell M, Pyne S, Pyne NJ. c-Src is involved in regulating signal transmission from PDGFβ receptor-GPCR signal complexes in mammalian cells. Cell Signal. 2005;17:263–77. doi: 10.1016/j.cellsig.2004.07.011. [DOI] [PubMed] [Google Scholar]
- Waters C, Long J, Gorshkova I, Fujiwara Y, Connell M, Belmonte K, et al. Cell migration activated by platelet-derived growth factor receptor is blocked by an inverse agonist of the sphingosine 1-phosphate receptor-1. FASEB J. 2006;20:509–11. doi: 10.1096/fj.05-4810fje. [DOI] [PubMed] [Google Scholar]
- Xu Y, Shen Z, Wiper DW, Wu M, Morton RE, Elson P, et al. Lysophosphatidic acid as a potential biomarker of ovarian and other gynecologic cancer. JAMA. 1998;280:719–23. doi: 10.1001/jama.280.8.719. [DOI] [PubMed] [Google Scholar]
- Yamazaki Y, Kon J, Sato K, Tomura H, Sato M, Yoneya T, et al. EDG6 as a putative sphingosine 1-phosphate receptor coupling to Ca2+ signaling pathway. Biochem Biophys Res Comm. 2000;268:583–9. doi: 10.1006/bbrc.2000.2162. [DOI] [PubMed] [Google Scholar]
- Yano Y, Hayashi Y, Sano K, Shinmaru H, Kuroda Y, Yokozaki H, et al. Expression and localization of ectonucleotide pyrophosphatase/phosphodiesterase-I-3 (E-NPP3/CD203c/PD-Iβ/B10/gp130RB13-6) in human colon carcinoma. Int J Mol Med. 2003;12:763–6. [PubMed] [Google Scholar]