

Abstract
Cyclic phosphatidic acid (1-acyl-sn-glycerol-2,3-cyclic phosphate; cPA) is a naturally occurring analog of lysophosphatidic acid (LPA) with a variety of distinctly different biological activities from those of LPA. In contrast to LPA, a potent inducer of tumor cell invasion, palmitoyl-cPA inhibits FBS- and LPA-induced transcellular migration and metastasis. To prevent the conversion of cPA to LPA we synthesized cPA derivatives by stabilizing the cyclic phosphate ring; to prevent the cleavage of the fatty acid we generated alkyl ether analogs of cPA. Both sets of compounds were tested for inhibitory activity on transcellular tumor cell migration. Carba derivatives, in which the phosphate oxygen was replaced with a methylene group at either the sn-2 or the sn-3 position, showed much more potent inhibitory effects on MM1 tumor cell transcellular migration and the pulmonary metastasis of B16-F0 melanoma than the natural pal-cPA. The antimetastatic effect of carba-cPA was accompanied by the inhibition of RhoA activation and was not due to inhibition of the activation of LPA receptors.
Keywords: Cyclic phosphatidic acid, Lysophosphatidic acid, LPA, Invasion, Metastasis
1. Introduction
The Physarum phosphatidic acid with a cyclic phosphate at sn-2 and sn-3 positions of the glycerol backbone, originally designated as PHYLPA, was the first isolated cyclic phosphatidic acid (cPA) species with a marked reversible inhibitory activity on the eukaryotic DNA polymerase α-family [1,2] and cell proliferation [3]. Subsequent to the discovery of PHYLPA, we also isolated phosphatidates with a cyclic phosphate ring from human serum and some other organisms [4]. cPA has diverse but distinct biological activities [5], including anti-proliferative action [1–3], induction of stress fiber formation [6], regulation of differentiation and survival of neuronal cells [7], and inhibition of tumor cell invasion and metastasis in vitro [8], and in vivo [8,9].
To understand the molecular mechanism of tumor cell invasion, a cellular barrier-based migration assay system was developed by Akedo et al. [10]. In this system, the tumor cells are seeded on a mesothelial cell monolayer and the number of invasion foci underneath the monolayer is counted and regarded to be a measure of transcellular tumor cell migration of the in vitro. This assay represents a reliable model of peritoneal dissemination, and the invasive capacity of cancer cells determined with this system correlates well with their invasiveness in vivo in the peritoneal cavity. Using this model, it has been shown that a highly invasive clone of the AH130 rat ascites hepatoma, MM1, requires serum for the transcellular migration. Among the constituents of serum, 1-oleoyl-lysophosphatidic acid (1-oleoyl-2-hydroxy-sn-glycerol-3-phosphate, LPA) could completely substitute for serum in restoring transcellular migration [11]. Using this assay system [8] cPA, in sharp contrast to LPA, showed an inhibitory effect on transcellular migration. Among some naturally occurring cPA species, palmitoyl-cPA (cPA 16:0) was the most potent inhibitor of tumor cell transcellular migration. cPA 16:0 also suppressed experimental pulmonary metastasis of B16 mouse melanoma cells injected into the tail vein of mice [8] and the bombesin-induced peritoneal metastasis of azoxymethane-induced intestinal cancers [9]. These results raise the possibility for a potential use of cPA in cancer therapy.
However, hydrolysis of the cyclic phosphate ring will lead to the formation of LPA, which is well known to augment tumor invasion and metastasis. Furthermore, cleavage of the fatty acid by phospholipases may limit the effective half-life of the cPA analogs. In the present study, we report the synthesis and characterization of some metabolically stabilized derivatives of cPA on tumor cell invasion and metastasis. Among these novel compounds carba derivatives of cPA (ccPA), in which the phosphate oxygen was replaced with a methylene group at either the sn-2 or the sn-3 position, showed much more potent inhibitory effect on the transcellular migration of MM1 cancer cells in vitro and the pulmonary metastasis of B16 melanoma cells than the natural cPA 16:0.
2. Materials and methods
2.1. Chemical synthesis of cPA derivatives designed to stabilize fatty acid moiety
1-O-palmityl-cPA (alkyl-cPA 16:0) and carbamoyl-palmityl-cPA (CBM-cPA 16:0) were prepared by the cyclic phosphorylation of the corresponding 1-O-hexadecyl-sn-glycerol or 1-O-carbamoyl-sn-glycerol, respectively [12].
Alkyl-cPA 16:0: white powder, 1H-NMR (CD3OD); δ 0.90 (3H, t, J=6.7), 1.20–1.38 (27H, m), 1.56 (2H, m), 3.48–3.56 (3H, m), 3.61 (1H, dd, J=10.5, 6.0 Hz), 3.93 (1H, dt, J=8.8, 7.2 Hz), 4.23 (1H, ddd, J =13.0, 8.9, 6.2 Hz), 4.50 (1H, m).
CBM-cPA 16:0: 1H-NMR (CD3OD); δ 0.89 (3H, t, J=6.7 Hz), 1.26–1.38 (25H, m), 1.46–1.52 (2H, m), 3.07 (2H, t, J=7.0 Hz), 3.97 (1H, dt, J=9.1, 7.0 Hz), 4.15 (2H, d, J =4.8 Hz), 4.23 (1H, ddd, J=12.5, 9.1, 6.4 Hz), 4.54 (1H, m); IR (KBr); cm−1 3360, 2920, 2851, 1691, 1533, 1469, 1238, 1143, 802, 719, 615.
2.2. Chemical synthesis of cPA derivatives designed to stabilize cyclic phosphate ring (ccPA)
2-O-carba-cPA (2ccPA) was prepared as shown in Fig. 1 [13].
Fig. 1.

Chemical synthesis of 2ccPA. See text for details.
2.2.1. Synthesis of (2,2-dimethyl-[1,3]dioxan-5-ylmethyl)-phosphonic acid dimethyl ester 2
Trimethyl phosphite (8.6 ml) was added to the iodide 1 prepared by the method of Dubois et al. [14]. (1.12 g, 4.62 mmol), and the mixture was heated under reflux at 130 °C for 14 h. Additional 17.2 ml of trimethyl phosphite was added, and the mixture was further refluxed for 6 h. The reaction mixture was left to cool, and was subjected to vacuum distillation to remove the residual trimethyl phosphite. The product was purified by silica gel column chromatography (CHCl3/MeOH (15:1)) to obtain (2,2-dimethyl-[1,3]dioxan-5-ylmethyl)-phosphonic acid dimethyl ester 2 (986 mg, 90%). The phosphonic acid (2) obtained was directly used in the following reaction.
1H-NMR (270 MHz CDCl3); δ 1.42 (s, 6H, IP), 1.81 (dd, 2H, J=18.87, 6.93 Hz, CH2P(O)(OMe)2), 1.92–2.21 (m, 1H, H-2), 3.66 (dd, 2H, J=12.05, 7.10 Hz, H-3), 3.78 (d, 6H, J =11.22 Hz, P(O)(OCH3)2), 4.02 (dd, 2H×1/2, J=11.87, 1.32 Hz, H-3), 4.17 (dd, 2H×1/2, J=14.68, 7.09 Hz, H-3).
2.2.2. Synthesis of (2-methoxy-2-oxo-[1,2]oxaphospholan-4-yl)methanol 3
Phosphonic acid dimethyl ester 2 (76.4 mg, 0.32 mmol) was dissolved in a mixture of toluene (3.8 ml) and methanol (0.13 ml), and p-toluenesulfonic acid hydrate (14.0 mg (0.23 eq)) was added. The mixture was heated at reflux for 3 h. The solution was left to cool, and the solvent was removed under reduced pressure. The product was purified by silica gel column chromatography using a CHCl3/MeOH (15:1) solvent, to obtain cyclic phosphonic ester 3(40.3 mg, 76%).
1H-NMR (300 MHz CDCl3); δ 1.73–2.12 (m, 2H, H-4), 2.69–2.87 (m, 1H, H-2), 3.66 (d, 2H, J=6.43 Hz, H-1), 3.78 (dd, 3H, J =0.55, 11.02 Hz, OCH3), 3.83–4.40 (m, 2H, H-3).
2.2.3. Synthesis of cyclic phosphonate 4
Cyclic phosphonic ester 3 (8.4 mg, 0.051 mmol) was dissolved in dichloromethane (3 ml). Dimethylaminopyridine (DMAP; 1.9 mg, 0.3 eq), oleic acid (18.6 mg, 1.3 eq), and 1-(3-dimethylaminopropyl)-3-ethylcarbodiimide hydrochloride (WSC; 19.4 mg, 2 eq) were added to the solution at 0 °C. The reaction mixture was stirred at room temperature for 1 day. The reaction solution was diluted with MeOH (2 ml) and washed with water, and the organic layer was extracted with ethyl acetate. The organic layer was dried over sodium sulfate and the solvent was removed under reduced pressure. The crude product was purified by silica gel column chromatography using a benzene/ethyl acetate (1:1) solvent to isolate cyclic phosphonate 4A (15.6 mg, 72%).
In a similar manner, cyclic phosphonate 3 was reacted with the appropriate fatty acids to yield cyclic phosphonate 4B (16:0; 89.7 mg, 51%) and 4C (16:1; 89.6 mg, 35%), respectively.
Cyclic phosphonate 4A: 1H-NMR (500 MHz CDCl3); δ 0.88 (t, 3H, J=6.87, H-18′), 1.26–1.30 (m, 20H, H-4′–7′, H-12′–17′), 1.60–1.63 (m, 2H, H-3′), 1.66–1.77 (m, 2H×1/2, H-4), 1.99–2.03 (m, 4H, H-8′, H-11′), 2.04–2.13 (m, 2H×1/2, H-4), 2.32 (t, 2H, J =7.48 Hz, H-2′), 2.82–2.99 (m, 1H, H-2), 3.80 (dd, 3H, J=11.14, 4.12 Hz, OCH3), 3.76–4.35 (m, 4H, H-1, H-3), 5.31–5.38 (m, 2H, H-9′, H-10′).
Cyclic phosphonate 4B: 1H-NMR (400 MHz CDCl3); δ 0.88 (t, 3H, J=6.83 Hz, H-16′), 1.25–1.28 (m, 24H, H-4′-15′), 1.59–1.63 (m, 2H, H-3′), 1.67–1.78 (m, 2H×1/2, H-4), 1.98–2.13 (m, 2H×1/2, H-4), 2.32 (t, 2H, J=7.55 Hz, H-2′), 2.83–2.92 (m, 1H, H-2), 3.80 (dd, 3H, J=11.23, 3.16 Hz, OCH3), 3.94–4.36 (m, 4H, H-1, H-3).
Cyclic phosphonate 4C: 1H-NMR (500 MHz CDCl3); δ 0.88 (t, 3H, J=6.83 Hz, H-16′), 1.28–1.30 (m, 16H, H-4′–7′, H-12′–15′), 1.60–1.61 (m, 2H, H-3′), 1.65–1.78 (m, 2H×1/2, H-4), 2.01–2.02 (m, 4H, H-8′, H-11′), 2.05–2.13 (m, 2H×1/2, H-4), 2.32 (t, 2H, J=7.43 Hz, H-2′), 2.84–2.96 (m, 1H, H-2), 3.80 (dd, 3H, J=11.09, 3.30 Hz, OCH3), 3.72–4.36 (m, 4H, H-1, H-3), 5.30–5.39 (m, 2H, H-9′, H-10′).
2.2.4. Synthesis of 2-O-carba-oleoyl-cPA 5A
Cyclic phosphonate 4A (33.3 mg, 0.077 mmol) was dissolved in dichloromethane (4 ml), and TMSBr (35.5 mg, 3 eq) was added at −15 °C. The mixture was stirred for 4.5 h. The reaction mixture was poured into ice water (20 ml), and the product was extracted with cold ether (10 ml). The organic layer was dried over sodium sulfate and the solvent was removed under reduced pressure. The crude product was purified by silica gel column chromatography first using a hexane/ethyl acetate (2:1) and subsequently using a CHCl3/MeOH (5:1) to obtain 2-O-carba-oleoyl-cPA 5A (12.1 mg, 38%).
In a similar manner, cyclic phosphonate 4B and 4C were converted to ccPA 5B(16:0; 3.5 mg, 10%) and ccPA 5C (16:1; 3.4 mg, 8%), respectively.
2ccPA 5A: 1H-NMR (500 MHz CDCl3–CD3OD); δ 0.88 (t, 3H, J=6.88 Hz, H-18′), 1.26–1.30 (m, 20H, H-4′–7′, H-12′–17′), 1.60–1.61 (m, 2H, H-3′), 1.70–1.77 (m, 2H×1/2, H-4), 1.99–2.03 (m, 4H, H-8′, H-11′), 2.04–2.09 (m, 2H×1/2, H-4), 2.31 (t, 2H, J=7.48 Hz, H-2′), 2.90–2.91 (m, 1H, H-2), 3.92–4.29 (m, 4H, H-1, H-3), 5.31–5.38 (m, 2H, H-9′, H-10′). HRMS calcd for C22H41O5P (M+) 416.2692 found 416.2683.
2ccPA 5B: 1H-NMR (400 MHz CDCl3–CD3OD); δ 0.88 (t, 3H, J=6.71 Hz, H-16′), 1.26 (m, 24H, H-4′–15′), 1.59–1.63 (m, 2H, H-3′), 1.70–1.80 (m, 2H×1/2, H-4), 2.04–2.13 (m, 2H×1/2, H-4), 2.31 (t, 2H, J=7.63 Hz, H-2′), 2.86–2.96 (m, 1H, H-2), 3.91–4.32 (m, 4H, H-1, H-3).
2ccPA 5C: 1H-NMR (500 MHz CDCl3–CD3OD); δ 0.88 (t, 3H, J=6.85 Hz, H-16′), 1.26–1.30 (m, 16H, H-4′–7′, H-12′–15′), 1.61 (m, 2H, H-3′), 1.70–1.78 (m, 2H×1/2, H-4), 1.99–2.03 (m, 4H, H-8′, H-11′), 2.05–2.12 (m, 2H×1/2, H-4), 2.31 (t, 2H, J=7.65 Hz, H-2′), 2.85–2.95 (m, 1H, H-2), 3.91–4.31 (m, 4H, H-1, H-3), 5.31–5.38 (m, 2H, H-9′, H-10′).
2.2.5. Isolation of 2ccPA
To the solution of 5 in diethyl ether was added a 0.05 M NaOH aqueous solution in a separating funnel. The aqueous extracts were freeze-dried and the sodium salt 2ccPA was obtained as a white powder.
The synthesis of 3-O-carba-cPA (3ccPA) is shown in Fig. 2 [15].
Fig. 2.

Chemical synthesis of 3ccPA. See text for details.
2.2.6. Synthesis of (3R)-1-dimethylphosphono-4-benzyloxy-3-butanol 7
To a solution of methylphosphonic acid dimethyl ester (2.6 ml, 24.0 mmol) in THF (40 ml) was added n-butyllithium (1.65 M solution in hexane) (14.5 ml, 24.0 mmol) at −78°C, and the reaction mixture was stirred for 0.5 h at −78 °C. To this mixture was added BF3 etherate (3.04 ml, 24.0 mmol), followed by a solution of (R)-benzylglycidyl ether 6 (1.83 ml, 12.0 mmol) in THF (10 ml). The reaction mixture was stirred for 2 h at −78 °C and then warmed to −20 °C and stirred for 2 h. The reaction mixture was quenched by the addition of saturated NH4Cl, extracted with ether (100 ml×6) and washed with saturated NaCl (70 ml). The combined organic layer was dried over anhydrous MgSO4, and the solvent was removed under reduced pressure. The residue was purified by column chromatography on silica gel (eluted with CHCl3/MeOH (30:1)) to give 7 (4.7 g, 68%).
[α]24D −9.8° (C=4.2, CHCl3); 1H-NMR (270 MHz CDCl3); δ 1.5–2.1 (4H, m), 3.0 (1H, br s), 3.34 (1H, dd, J=10.8, 5.4 Hz), 3.47 (1H, dd, J=10.8, 2.7 Hz), 3.74 (6H, d, J=10.5 Hz), 3.78–3.83 (1H, m), 4.54 (2H, s), 7.2–7.4 (5H, m); IR (cm−1 neat): 3407, 2952, 2854, 1454; MS (m/z): 289 (M+H)+; HRMS calcd for C13H22O5P (M+H)+ 289.1203 found 289.1214.
2.2.7. Synthesis of (5R)-2-methoxy-5-benzyloxymethyl-1,2-oxaphospholane 2-oxide 8
To a solution of 7 (440 mg, 1.53 mmol) in toluene (20 ml) was added a pyridinium p-toluenesulfonate (1.15 g, 4.58 mmol), and the reaction mixture was heated at reflux for 3 h. The mixture was cooled and diluted with ethyl acetate (100 ml). The mixture was washed with saturated NaCl (70 ml), and the organic layer was dried over anhydrous MgSO4. The solvent was removed under reduced pressure, and the residue was purified by column chromatography on silica gel (300 Mesh) (eluted with CHCl3/MeOH (30:1)) to give 8 (254 mg, 0.99 mmol, 65%).
1H-NMR (270 MHz CDCl3); δ 1.72–2.4 (4H, m), 3.52–3.66 (2H, m), 3.76 (3H×0.5, d, J=11.0 Hz), 3.78 (3H×0.5, d, J =11.0 Hz), 4.58 (2H×0.5, s), 4.59 (2H×0.5, s), 7.2–7.4 (5H, m); IR (cm−1 neat): 2950, 2856, 1454; MS (m/z): 257 (M+H)+, 256 (M)+; HRMS calcd for C12H17O4P (M)+ 256.0865 found 256.0861.
2.2.8. Synthesis of (5R)-2-methoxy-5-hydroxymethyl-1,2-oxaphospholane 2-oxide 9
To a solution of 8 (252 mg, 0.98 mmol) in ethanol (5 ml) was added 20% Pd (OH)2/C (25 mg), and the mixture was stirred under H2 at room temperature for 1 day. The catalyst was removed by filtration and the filtrate was evaporated under reduced pressure. The residue was purified by column chromatography on silica gel (eluted with CHCl3/MeOH (20:1)) to give 9 (143 mg, 0.86 mmol, 88%).
1H-NMR (270 MHz CDCl3); δ 1.50–2.70 (4H, m), 3.55–3.68 (2H, m), 3.80 (3H, d, J=11.0 Hz), 4.30–4.40 (1H×0.5, m), 4.40–4.55 (1H×0.5, m); IR (cm−1 neat): 3403, 3368, 2954, 2854, 1454, 1415; MS (m/z): 167 (M+H)+; HRMS calcd for C5H12O4P (M+H)+ 167.0471 found 167.0470.
2.2.9. Synthesis of (5R)-2-methoxy-5-O-palmitoyl-methyl-1,2-oxaphospholane 2-oxide 10B
To a solution of 9 (40 mg, 0.24 mmol) in CH2Cl2 (1 ml) was added successively dimethylaminopyridine (DMAP) (9 mg, 0.06 mmol), triethylamine (50 μl, 0.36 mmol), and palmitoylchloride (94 μl, 0.31 mmol) at 0 °C. The reaction mixture was stirred for 1 day at room temperature. The reaction mixture was diluted with ethyl acetate (25 ml) and washed by saturated NaCl (20 ml). The organic layer was dried over anhydrous MgSO4 and evaporated under reduced pressure. The residue was purified by column chromatography on silica gel (eluted with CHCl3/MeOH (40:1)) to give 10B (79.5 mg, 0.20 mmol, 82%).
In a similar manner, cyclic phosphonate 9 was reacted with appropriate fatty acid chlorides to obtain cyclic phosphonate 10A (18:1; 85%) and 10C (16:1; 83%), respectively.
(5R)-2-Methoxy-5-O-oleoyl-methyl-1,2-oxaphospholane 2-oxide 10A: 1H-NMR (270 MHz CDCl3); δ 0.88 (3H, t, J=7 Hz), 1.23–1.26 (20H, m), 1.63 (2H, t, J=7 Hz), 1.76–2.40 (10H, m), 3.79 (3H, d, J=10.5 Hz), 4.09 (1H×0.5, dd, J=11.0, 6.0 Hz), 4.14 (1H×0.5, dd, J =11.0, 6.0 Hz), 4.24 (1H×0.5, dd, J=11.0, 4.7 Hz), 4.29 (1H×0.5, dd, J=11.0, 4.7 Hz), 4.35–4.45 (1H×0.5, m), 4.47–4.50 (1H×0.5, m), 5.25–5.45 (2H, m); IR (cm−1 neat): 2925, 2854, 1741, 1457, 1417; MS (m/z): 430 (M)+; HRMS calcd for C23H44O5P (M)+ 430.2845 found 430.2848.
(5R)-2-Methoxy-5-O-palmitoyl-methyl-1,2-oxaphospholane 2-oxide 10B: 1H-NMR (270 MHz CDCl3); δ 0.88 (3H, t, J=7 Hz), 1.33–1.35 (24H, m), 1.63 (2H, t, J=7 Hz), 1.80–2.45 (6H, m), 3.80 (3H, d, J=11.0 Hz), 4.13 (1H, dd, J=11.0, 6.0 Hz), 4.30 (1H, dd, J=11.0, 4.7 Hz), 4.36–4.48 (1H, m); IR (cm−1 neat): 2915, 2850, 1735, 1463, 1413; MS (m/z): 404 (M)+; HRMS calcd for C21H41O5P (M)+ 404.2692 found 404.2697.
(5R)-2-Methoxy-5-O-palmitoleoyl-methyl-1,2-oxaphospholane 2-oxide 10C: 1H-NMR (270 MHz CDCl3); δ 0.88 (3H, t, J=7 Hz), 1.29–1.31 (16H, m), 1.45–2.40 (12H, m), 3.80 (3H, d, J=11.0 Hz), 4.09 (1H×0.5, dd, J=11.0, 6.0 Hz), 4.14 (1H×0.5, dd, J=11.0, 6.0 Hz), 4.24 (1H×0.5, dd, J=11.0, 4.7 Hz), 4.29 (1H×0.5, dd, J=11.0, 4.7 Hz), 4.35–4.45 (1H×0.5, m), 4.47–4.50 (1H×0.5, m), 5.25–5.45 (2H, m); IR (cm−1 neat): 2927, 2854, 1739, 1457, 1417; MS (m/z): 402 (M)+; HRMS calcd for C21H39O5P (M)+ 402.2533 found 402.2540.
2.2.10. Synthesis of 3-O-carba-cPA 11B
To a solution of 10B (40 mg, 99.0 μmol) in CH2Cl2 (1 ml) was added bromotrimethylsilane (TMSBr) (13 μl, 0.30 mmol), and the reaction mixture was stirred for 1 h at room temperature. The reaction mixture was condensed under reduced pressure, and the residue was purified by column chromatography on silica gel (eluted with CHCl3/MeOH (10:1)) to give 11B (30.1 mg, 77.2 μmol, 78%).
In a similar manner, cyclic phosphonates 11A and 11C were treated with TMSBr to obtain cyclic phosphonate 11A (18:1; 73%) and 11C (16:1; 75%), respectively.
(5R)-2-hydroxy-5-O-oleoyl-methyl-1,2-oxaphospholane 2-oxide 11A: [α]24D +9.4° (C=3.1, CHCl3); 1H-NMR (270 MHz CDCl3); δ 0.88 (3H, t, J=7 Hz), 1.24–1.26 (20H, m), 1.63 (2H, t, J=7.0 Hz), 1.70–2.40 (10H, m), 4.09 (1H, dd, J=11.0, 6.0 Hz), 4.29 (1H, dd, J=11.0, 4.7 Hz), 4.44–4.55 (1H, m), 5.25–5.45 (2H, m); IR (cm−1 neat): 2919, 2854, 1735, 1456, 1417; FABMS (m/z): 416 (M+H)+.
(5R)-2-hydroxy-5-O-palmitoyl-methyl-1,2-oxaphospholane 2-oxide 11B: [α]24D +13.7° (C=1.7, CHCl3); 1H-NMR (270 MHz CDCl3); δ 0.89 (3H, t, J=7 Hz), 1.27–1.29 (24H, m), 1.63 (2H, t, J =7.0 Hz), 1.70–2.45 (6H, m), 4.13 (1H, dd, J=11.0, 6.0 Hz), 4.30 (1H, dd, J=11.0, 4.7 Hz), 4.36–4.48 (1H, m); IR (cm−1 neat): 2917, 2852, 1726, 1461; FABMS (m/z): 391 (M+H)+; HRMS calcd for C20H40O5P (M+H)+ 391.2614 found 391.2612.
(5R)-2-hydroxy-5-O-palmitoleoyl-methyl-1,2-oxaphospholane 2-oxide 11C: [α]24D +12.0° (C=1.42, CHCl3); 1H-NMR (270 MHz CDCl3); δ 0.88 (3H, t, J=7 Hz), 1.29–1.31 (16H, m), 1.45–2.40 (12H, m), 4.09 (1H, dd, J=11.0, 6.0 Hz), 4.29 (1H, dd, J =11.0, 4.7 Hz), 4.38–4.60 (1H, m), 5.30–5.40 (2H, m); IR (cm−1 neat): 2925, 2854, 1739, 1456, 1417; FABMS (m/z): 389 (M+H)+. HRFABMS calcd for C20H38O5P (M+H)+ 389.2457 found 389.2456.
2.2.11. Isolation of 3ccPA
To the solution of 11 in diethyl ether was added a 0.05 M NaOH aqueous solution in a separating funnel. The aqueous extracts were freeze-dried and the sodium salt 3ccPA was obtained as a white powder.
2.3. Reagents
Bovine serum albumin (BSA; fraction V, fatty acid free) and 1-oleoyl-sn-lysophosphatidic acid (LPA 18:1) were purchased from Sigma-Aldrich (St. Louis, MO). LPA and cPA were dissolved in PBS containing 0.1% (w/v) BSA.
2.4. Cells and culture conditions
Rat mesothelial cells were isolated from the mesentery of Donryu rats (Nippon Bio-Supp. Center, Tokyo) and cultured in Eagle’s MEM supplemented with amino acids and vitamins (Nissui, Tokyo) and 10% FBS. After the mesothelial cells became confluent, the monolayer was used for the invasion assay. MM1 cells, a highly-invasive clone of the AH130 rat hepatoma cell line, were cultured in suspension using modified MEM supplemented with 10% FBS. The mouse melanoma B16-F0 cells were cultured in DMEM supplemented with 10% FBS.
2.5. Transcellular migration assay for in vitro invasion
The assay procedure was essentially the same as previously described [10]. Briefly, tumor cells were seeded over a mesothelial cell monolayer and cultured in medium containing the test materials. After 20 h, the supernatant was removed and the monolayer was fixed with 10% formalin in PBS. The number of penetrated individual tumor cells and tumor cell colonies, called invasion foci, was determined using an inverted phase-contrast microscope. The invasive capacity was calculated based on the number of invasion foci/cm2.
2.6. RhoA activation assay
MM1 cells were washed and starved in serum-free medium for 3 h. Cells were stimulated with 25 μM LPA 18:1 in the absence or presence of cPA. The cells were harvested with centrifugation and lysed in cold lysis buffer (50 mM Tris, pH 7.5, 1% Triton X-100, 0.5% sodium deoxycholate, 0.1% SDS, 500 mM NaCl, 10 mM MgCl2, 10 μg/ml each of leupeptin and aprotinin, and 1 mM PMSF). The cell lysates were incubated at 4 °C for 45 min with glutathione beads (GE Healthcare, Piscataway, NJ) coupled with bacterially expressed fusion protein containing the Rho-binding domain of Rhotekin (GST-RBD, provided by Dr. M. Schwartz, Scripps Research Institute, La Jolla, CA). The beads were washed and the samples were analyzed by SDS-PAGE and the amount of RhoA bound to its target was visualized using Western blots with a monoclonal antibody to RhoA (Santa Cruz Biotechnology, Santa Cruz, CA). The blots were developed with a peroxidase-conjugated secondary antibody using the ECL chemiluminescence system (GE Healthcare, Piscataway, NJ) and detected using LAS-3000 (Fuji Film, Tokyo). Rho activity was determined as the amount of RBD-bound Rho versus total Rho in the lysate determined by densitometric analysis of blots by using an Image Gauge (Fuji Film, Tokyo).
2.7. Measurement of [Ca2+]i
The rat hepatoma (RH7777) cells do not respond to LPA with changes in [Ca2+]i and have been used to establish lines stably expressing each of the LPA1/2/3 receptors. The characterization of these cell lines and the procedure of cell labeling and monitoring has been described previously [16].
2.8. Experimental pulmonary metastasis
Harvested B16-F0 melanoma cells were suspended in PBS at a density of 5×106 viable cells/ml. 0.1 ml cell suspension was injected into the tail vein with or without 1–8 μg ccPA per mouse. After 14 days the animals were euthanized, autopsy was carried out, and the metastatic foci of the melanoma in the lungs were counted. Five or six mice were exposed to each treatment and the data represent the mean±SD. Dunnett’s multiple comparison test and Aspin–Welch’s t-test were used for the statistical treatment of these data.
3. Results
3.1. Effect of synthetic cPA derivatives on FBS- and LPA-dependent transcellular tumor cell migration
Fig. 3 shows the molecular structure of four novel cPA 16:0 derivatives we synthesized. Two derivatives, the ether-linked alkyl-cPA 16:0 and the carbamoyl derivative CBM-cPA 16:0, were designed to prevent the cleavage of the fatty acid by phospholipases. The other two cPA derivatives, 2-O-carba-palmitoyl-cPA (2ccPA 16:0) and 3-O-carba-palmitoyl-cPA (3ccPA 16:0), were designed to prevent the opening of the cyclic phosphate ring by phospholipases and phosphodiesterases. The analogs were tested for their activity in the MM1 transcellular tumor migration assay in vitro. cPA derivatives were tested at 25 μM on serum-induced MM1 cell transcellular migration (Fig. 4). In this assay, three cPA derivatives, CBM-cPA 16:0, 2ccPA 16:0, and 3ccPA 16:0 showed significant inhibitory activity of the serum-induced transcellular migration. Next, we tested the effect of these derivatives on LPA-induced (25 μM) transcellular migration in the same assay. Among the four derivatives tested, a different pattern has emerged in that particularly the 2ccPA showed the most potent inhibitory effect (Fig. 5).
Fig. 3.

Structures of the cPA derivatives used in the present study.
Fig. 4.
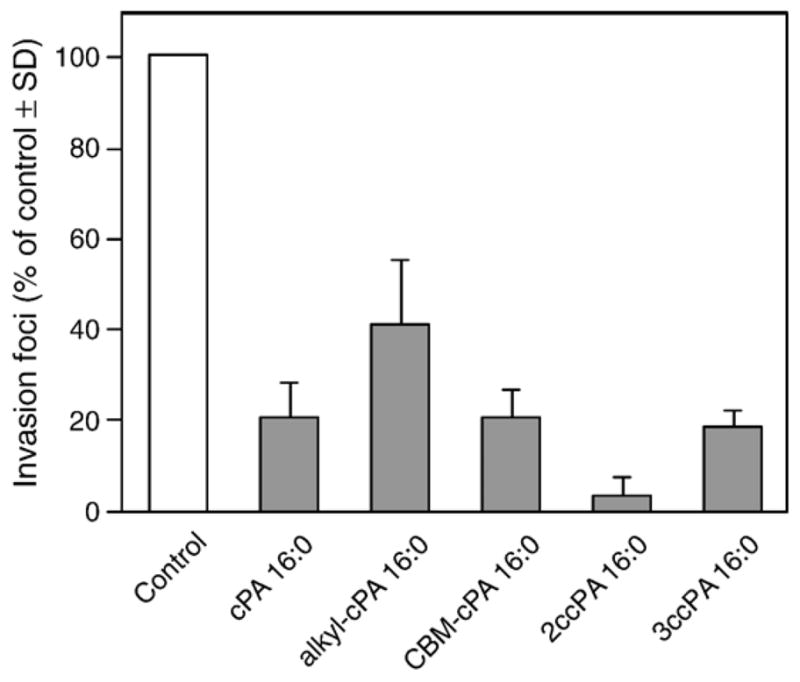
Effect of cPA on serum-induced invasion in vitro. MM1 cells (1×105 cells/ml) were seeded on the mesothelial cell monolayer with or without cPA (25 μM) in the presence of 10% FBS. The number of invasion foci is expressed as the mean (% of control)±SD of at least three experiments.
Fig. 5.
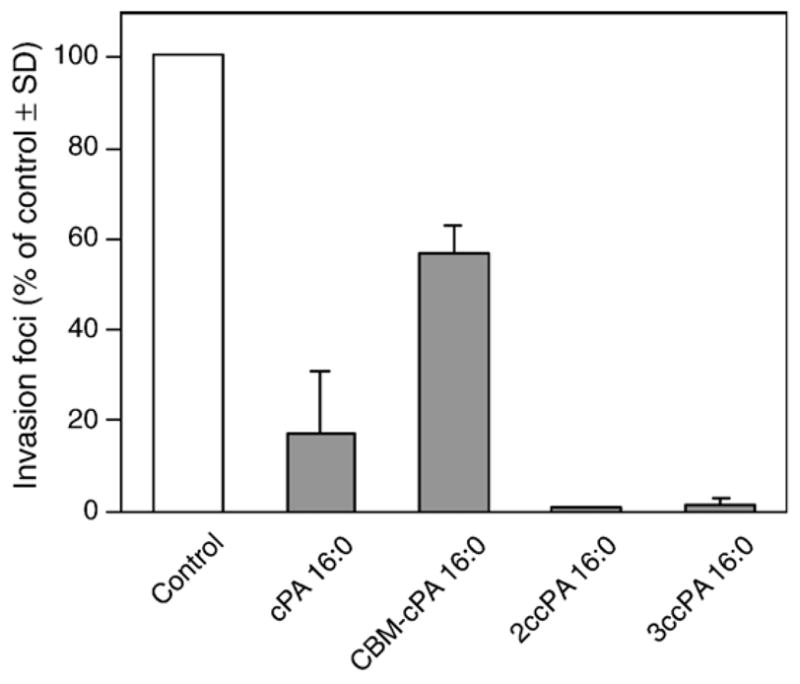
Effect of cPA on LPA-induced invasion in vitro. MM1 cells were washed with serum-free medium and 1.5×105 cells/ml were seeded on mesothelial cell monolayer with or without the 25 μM cPA. MM1 cell invasion was initiated by the addition of 25 μM LPA. The number of invasion foci is expressed as the mean (% of control)±SD of at least three experiments.
To further characterize the structure–activity relationship of the inhibitory effect of ccPA derivatives, a set with different fatty acyl chains was synthesized and their inhibitory activity was compared using the MM1 transcellular migration assay (Fig. 6). Our previous studies on naturally occurring cPAs showed that saturated fatty acid-substituted cPA were more potent inhibitors of transcellular migration than those derivatives with unsaturated fatty acids. As reported before [8], 12.5 μM cPA 16:1, 18:1, 16:0 inhibited transcellular migration by 23%, 57%, 86%, respectively. However, in the case of ccPAs, those with the unsaturated fatty acids, oleate or palmitoleate, inhibited transcellular tumor cell migration more strongly than palmitoyl ccPA.
Fig. 6.

The dose–response relationship of 2ccPA inhibition of LPA-induced invasion of MM1 cells in vitro. Cells were washed with serum-free medium and seeded at a density of 1.5×105 cells/ml on the mesothelial cell monolayer with or without the indicated ccPA and invasion was elicited by the addition of 25 μM LPA. The number of invasion foci is expressed as the mean (% of control)±SD of at least three determinations.
Cyclic PA elicits anti-proliferative activity in fibroblasts cultured in a chemically defined medium, but in the presence of serum, almost no anti-proliferative activity is observed [3]. In the range of concentrations that effectively suppressed trans-cellular migration, naturally occurring cPA 16:0 showed no effect on the proliferation of MM1 cells in the presence of serum [8]. We also tested the effect of ccPA on proliferation of MM1 cells in the presence of FBS. ccPA at concentrations that potently inhibited transcellular migration had no effect on the proliferation of MM1 cells (Table 1).
Table 1.
Effect of carba-cPA on FBS/LPA-induced transcellular migration and proliferation of MM1 cells
| Carba-cPA | Transcellular migration inhibition (%)a |
Proliferation (control %)b |
||
|---|---|---|---|---|
| FBS-induced | LPA-induced | 24 h | 72 h | |
| 2ccPA 16:0 (10 μM) | 82.8 | 92.8 | 104.5 | 94.7 |
| 2ccPA 16:1 (10 μM) | 78.7 | 98.6 | 104.1 | 100.8 |
| 2ccPA 18:1 (10 μM) | 89.3 | 96.0 | 113.3 | 102.4 |
| 3ccPA 16:0 (25 μM) | 81.9 | 98.9 | 94.4 | 93.2 |
| 3ccPA 16:1 (25 μM) | 86.9 | 99.9 | 102.1 | 98.8 |
| 3ccPA 18:1 (25 μM) | 90.1 | 99.9 | 105.1 | 112.7 |
The number of cells was counted at 24 and 72 h.
MM1 cells were seeded on mesothelial cell monolayer with or without indicated carba-cPAs.
MM1 cells were cultured in the presence of 10% FBS with or without carba-cPAs.
3.2. Effect of ccPA on LPA-induced activation of RhoA
LPA elicits the transient activation of RhoA which is an essential event in transcellular tumor cell migration [17]. When applied together, cPA 16:0 dose-dependently inhibited LPA-induced activation of RhoA (Fig. 7). 2ccPA 16:1 showed a similar potency to the inhibitory effect of cPA 16:0. Both cPA analogs applied at as low as a twentieth fraction of LPA concentration significantly inhibited RhoA activation.
Fig. 7.

Effect of 2ccPA 16:1 on LPA-induced RhoA activation. MM1 cells were stimulated with 25 μM LPA 18:1 in the presence of cPA or ccPA at indicated concentrations. RhoA activity is expressed as the mean (% of control)±SD of three experiments.
3.3. ccPA does not inhibit the LPA receptors
LPA receptors, particularly LPA1, have been reported to be associated with tumor cell invasion [18]. To determine if the anti-invasive effect of the ccPA derivatives was mediated through inhibition of the LPA receptors, we examined the effect of 2ccPA 16:1 on the activation of LPA1, LPA2, and LPA3 expressed individually in RH7777 cells [19]. 2ccPA 16:1 did not elicit or inhibit LPA-induced Ca2+ transients in these cells suggesting that ccPA achieve their inhibitory effect on LPA-induced transcellular migration through a molecular target different from these LPA receptors (Fig. 8).
Fig. 8.
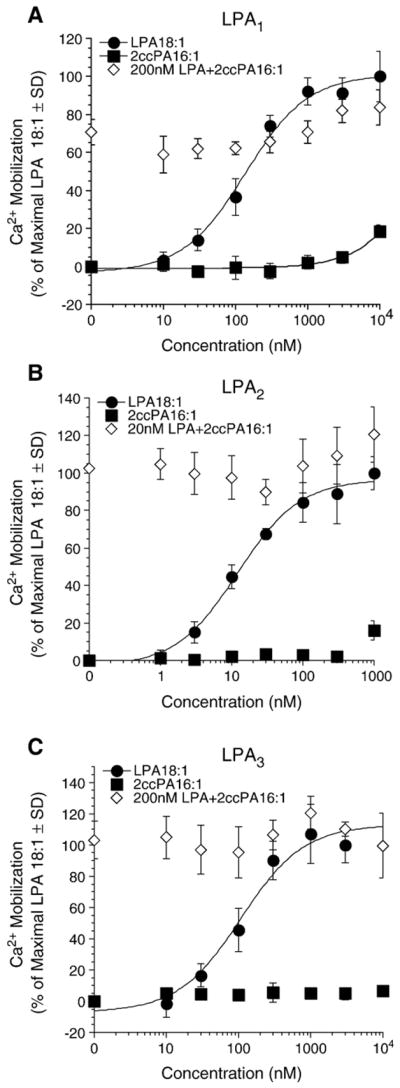
Effect of 2ccPA 16:1 on LPA 18:1-induced Ca2+ mobilization in RH7777 cells stably expressing LPA receptors. Intracellular Ca2+ transients (mean±SD) were measured in response to the application of increasing concentrations of LPA 18:1 alone (filled circles), 2ccPA16:1 alone (filled squares), or mixed with either 200 nM or 20 nM (in case of LPA2) LPA18:1 (open diamond). 100% represents the maximal Ca2+ mobilization elicited by LPA 18:1 at the individual LPA receptors, LPA1 (A), LPA2 (B) or LPA3 (C).
3.4. Inhibition of B16 melanoma cell metastasis by ccPA
We have previously shown that cPA 16:0 exerts an inhibitory effect on experimental pulmonary metastasis [8]. Here we tested the effect of ccPAs using the B16-F0 mouse melanoma metastasis model. A single injection of 1–8 μg ccPA per animal concomitantly with the inoculation of B16-F0 cells into C57BL/6 mice strongly suppressed pulmonary metastasis as shown in Fig. 9A and B. Both 2ccPAs and 3ccPAs inhibited pulmonary metastasis significantly higher than the naturally occurring cPA 16:0. The mean of pulmonary tumor nodules in vehicle-treated animals varied between the experiments ranging from 49±20 (the lowest) to 274±113 (the highest). Regardless the actual number of foci ccPA and cPA elicited a highly significant reduction in the number of metastases (Fig. 9). In addition to metastatic nodules in the lungs, we recorded the number of animals with visible tumor foci outside the lungs in the thoracic and abdominal cavities, and ccPA not only decreased the number of pulmonary tumor nodules but also reduced the number of mice with extrapulmonary metastases (data not shown). We also determined the body weight as an indicator of the general health condition of B16 melanoma-inoculated mice and compared it to vehicle-injected mice with or without ccPAs. No difference in body weights was found between any of the groups studied during the test period, indicating that ccPAs in the concentration range applied were not toxic to the animals.
Fig. 9.

Effect of 2ccPA (A) or 3ccPA (B) on the number of lung tumor nodules on post-inoculation day 14. Columns and vertical bars represent the mean±SD of 5 mice (A) or 6 mice (B). ***P<0.001, **P<0.01 vs. the vehicle group based on Dunnett’s multiple comparison test. #P<0.05 vs. the vehicle group based on Aspin–Welch’s t-test.
4. Discussion
In the present study, we report the synthesis of stabilized cPA analogs. Because the 2,3-cyclic phosphate ring can be hydrolyzed by phosphodiesterases and phospholipases [20–22], that lead to LPA formation, we designed carba-derivatives to prevent ring opening of cPA. We demonstrate that the ccPA derivatives exert potent inhibitory effect on serum- and LPA-induced transcellular migration of MM1 cancer cells (Figs. 4 and 5) without any effect on cell proliferation (Table 1). In agreement with the results of the in vitro assays, ccPA also inhibited experimental pulmonary metastasis more strongly than the naturally occurring cPA (Fig. 9) without any toxic effect on the B16 melanoma inoculated mice.
The novel ccPA derivatives maintained a potent inhibitory effect on transcellular tumor cell migration and experimental metastasis exceeding that of cPA 16:0. However, the inhibitory effect of ccPAs substituted with unsaturated fatty acids was more potent than that of derivatives with saturated fatty acids. This structure–activity relationship is opposite to the effect of natural cPAs. The molecular target(s) of cPA and ccPA remain to be identified, but the differences in the structure–activity relationship suggest that these targets are not identical. Nonetheless, both targets recognize the cyclic phosphate ring and distinguish the fatty acid/alcohol substituent.
LPA application within 15 min transiently activates RhoA in MM1 cells [17] and we found that cPA 16:0 inhibits LPA-induced RhoA activation [23]. Although we expected that ccPA will inhibit RhoA activation more potently than natural cPA but, unexpectedly, the extent of inhibition was almost similar as cPA 16:0 (Fig. 7). We do not yet have an explanation for this effect but one must consider the limitations of the pull-down assay. We have tested the expression of LPA1/2/3/4 transcripts using RT-PCR analysis, which revealed LPA1 and LPA2 in MM1 cells (data not shown). To rule out that ccPA accomplished its inhibition on LPA-induced RhoA activation by directly inhibiting LPA receptors we examined whether ccPA exerted an inhibitory effect on the Ca2+ responses evoked by the individual receptor subtypes. Our data with LPA1/2/3 argue against this possibility since the most potent derivative in the invasion assay did not inhibit any of these receptors (Fig. 8). Some reports suggested that LPA-induced invasion and migration is mediated by LPA1 [18,24–26]. We and others have shown that cPA weakly activates LPA1 [16,27,28]. These results taken together suggest that cPA may not simply compete with LPA for the activation of these receptors. We recently reported that ccPA inhibits autotaxin-induced cancer cell invasion [29]. Since addition of LPC 18:1, the substrate of autotaxin, does not induce transcellular migration in MM1 cells [11], the lysophospholipase D activity of autotaxin generated by these cells does not appear to produce sufficient LPA 18:1 to elicit cell migration. Therefore, the mechanism of inhibition described in this study does not solely depend on autotaxin inhibition. But, the fact that ccPA inhibits both LPA production by ATX and some events downstream of the LPA receptors, including RhoA activation, point to a multiplicity of targets and at the same time enhance the possibility for the use of ccPA in cancer treatment. Further studies focusing on the molecular target(s) as well as the mechanism of inhibition by ccPA of cancer cell invasion and metastasis are under way in our laboratories and will increase the feasibility of ccPA derivatives in the treatment of cancer metastasis.
Acknowledgments
This work was supported by grants from Japan Society for the Promotion of Science (KM), The Naito Foundation (KM), Hayashi Memorial Foundation for Female Natural Scientists (KM), and NIH (HL 61469 and CA92160 to GT).
References
- 1.Murakami-Murofushi K, Shioda M, Kaji K, Yoshida S, Murofushi H. Inhibition of eukaryotic DNA polymerase alpha with a novel lysophosphatidic acid (PHYLPA) isolated from myxoamoebae of Physarum polycephalum. J Biol Chem. 1992;267:21512–21517. [PubMed] [Google Scholar]
- 2.Takahashi Y, Shimada Y, Shioda M, Yoshida S, Murofushi H, Murakami-Murofushi K. Isolation of a new species of Physarum lysophosphatidic acid, PHYLPA, and its effect on DNA polymerase activity. Cell Struct Funct. 1993;18:135–138. doi: 10.1247/csf.18.135. [DOI] [PubMed] [Google Scholar]
- 3.Murakami-Murofushi K, Kaji K, Kano K, Fukuda M, Shioda M, Murofushi H. Inhibition of cell proliferation by a unique lysophosphatidic acid, PHYLPA, isolated from Physarum polycephalum: signaling events of antiproliferative action by PHYLPA. Cell Struct Funct. 1993;18:363–370. doi: 10.1247/csf.18.363. [DOI] [PubMed] [Google Scholar]
- 4.Kobayashi T, Tanaka-Ishii R, Taguchi R, Ikezawa H, Murakami-Murofushi K. Existence of a bioactive lipid, cyclic phosphatidic acid, bound to human serum albumin. Life Sci. 1999;65:2185–2191. doi: 10.1016/s0024-3205(99)00483-x. [DOI] [PubMed] [Google Scholar]
- 5.Murakami-Murofushi K, Uchiyama A, Fujiwara Y, Kobayashi T, Kobayashi S, Mukai M, Murofushi H, Tigyi G. Biological functions of a novel lipid mediator, cyclic phosphatidic acid. Biochim Biophys Acta. 2002;1582:1–7. doi: 10.1016/s1388-1981(02)00131-2. [DOI] [PubMed] [Google Scholar]
- 6.Fischer DJ, Liliom K, Guo Z, Nusser N, Virag T, Murakami-Murofushi K, Kobayashi S, Erickson JR, Sun G, Miller DD, Tigyi G. Naturally occurring analogs of lysophosphatidic acid elicit different cellular responses through selective activation of multiple receptor subtypes. Mol Pharmacol. 1998;54:979–988. doi: 10.1124/mol.54.6.979. [DOI] [PubMed] [Google Scholar]
- 7.Fujiwara Y, Sebok A, Meakin S, Kobayashi T, Murakami-Murofushi K, Tigyi G. Cyclic phosphatidic acid elicits neurotrophin-like actions in embryonic hippocampal neurons. J Neurochem. 2003;87:1272–1283. doi: 10.1046/j.1471-4159.2003.02106.x. [DOI] [PubMed] [Google Scholar]
- 8.Mukai M, Imamura F, Ayaki M, Shinkai K, Iwasaki T, Murakami-Murofushi K, Murofushi H, Kobayashi S, Yamamoto T, Nakamura H, Akedo H. Inhibition of tumor invasion and metastasis by a novel lysophosphatidic acid (cyclic LPA) Int J Cancer. 1999;81:918–922. doi: 10.1002/(sici)1097-0215(19990611)81:6<918::aid-ijc13>3.0.co;2-e. [DOI] [PubMed] [Google Scholar]
- 9.Ishihara R, Tatsuta M, Iishi H, Baba M, Uedo N, Higashino K, Mukai M, Ishiguro S, Kobayashi S, Murakami-Murofushi K. Attenuation by cyclic phosphatidic acid of peritoneal metastasis of azoxymethane-induced intestinal cancers in Wistar rats. Int J Cancer. 2004;110:188–193. doi: 10.1002/ijc.20069. [DOI] [PubMed] [Google Scholar]
- 10.Akedo H, Shinkai K, Mukai M, Mori Y, Tateishi R, Tanaka K, Yamamoto R, Morishita T. Interaction of rat ascites hepatoma cells with cultured mesothelial cell layers: a model for tumor invasion. Cancer Res. 1986;46:2416–2422. [PubMed] [Google Scholar]
- 11.Imamura F, Horai T, Mukai M, Shinkai K, Sawada M, Akedo H. Induction of in vitro tumor cell invasion of cellular monolayers by lysophosphatidic acid or phospholipase D. Biochem Biophys Res Commun. 1993;193:497–503. doi: 10.1006/bbrc.1993.1651. [DOI] [PubMed] [Google Scholar]
- 12.Kobayashi S, Tokunoh R, Shibasaki M, Shinagawa R, Murakami-Murofushi K. Synthesis of 1-O-acylglycerol 2,3-cyclic phosphate: determination of the absolute structure of PHYLPA, a specific inhibitor of DNA polymerase α. Tetrahedron Lett. 1993;34:4047–4050. [Google Scholar]
- 13.Murofushi K, Mutsuko M, Kobayashi S, Murofushi H. 03/104246 Patent WO.
- 14.Dubois J, Fourès C, Bory S, Falcou S, Gaudry M, Marquet A. Synthesis of 5,5′-dihydroxyleucine and 4-fluoro 5,5′-dihydroxyleucine, the reduction products of 4-carboxyglutamic and 4-carboxy-4-fluoroglutamic acids. Tetrahedron. 1991;47:1001–1012. [Google Scholar]
- 15.Mukai M, Kobayashi S, Murofushi H, Murofushi K. 02/094286 Patent WO.
- 16.Fujiwara Y, Sardar V, Tokumura A, Baker D, Murakami-Murofushi K, Parrill A, Tigyi G. Identification of residues responsible for ligand recognition and regioisomeric selectivity of lysophosphatidic acid receptors expressed in mammalian cells. J Biol Chem. 2005;280:35038–35050. doi: 10.1074/jbc.M504351200. [DOI] [PubMed] [Google Scholar]
- 17.Mukai M, Nakamura H, Tatsuta M, Iwasaki T, Togawa A, Imamura F, Akedo H. Hepatoma cell migration through a mesothelial cell monolayer is inhibited by cyclic AMP-elevating agents via a Rho-dependent pathway. FEBS Lett. 2000;484:69–73. doi: 10.1016/s0014-5793(00)02129-3. [DOI] [PubMed] [Google Scholar]
- 18.Yamada T, Sato K, Komachi M, Malchinkhuu E, Tobo M, Kimura T, Kuwabara A, Yanagita Y, Ikeya T, Tanahashi Y, Ogawa T, Ohwada S, Morishita Y, Ohta H, Im DS, Tamoto K, Tomura H, Okajima F. Lysophosphatidic acid (LPA) in malignant ascites stimulates motility of human pancreatic cancer cells through LPA1. J Biol Chem. 2004;279:6595–6605. doi: 10.1074/jbc.M308133200. [DOI] [PubMed] [Google Scholar]
- 19.Durgam GG, Virag T, Walker MD, Tsukahara R, Yasuda S, Liliom K, van Meeteren LA, Moolenaar WH, Wilke N, Siess W, Tigyi G, Miller DD. Synthesis, structure–activity relationships, and biological evaluation of fatty alcohol phosphates as lysophosphatidic acid receptor ligands, activators of PPARgamma, and inhibitors of autotaxin. J Med Chem. 2005;48:4919–4930. doi: 10.1021/jm049609r. [DOI] [PubMed] [Google Scholar]
- 20.Friedman P, Haimovitz R, Markman O, Roberts MF, Shinitzky M. Conversion of lysophospholipids to cyclic lysophosphatidic acid by phospholipase D. J Biol Chem. 1996;271:953–957. doi: 10.1074/jbc.271.2.953. [DOI] [PubMed] [Google Scholar]
- 21.Lee S, Lynch KR. Brown recluse spider (Loxosceles reclusa) venom phospholipase D (PLD) generates lysophosphatidic acid (LPA) Biochem J. 2005;391:317–323. doi: 10.1042/BJ20050043. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Tsuda S, Okudaira S, Moriya-Ito K, Shimamoto C, Tanaka M, Aoki J, Arai H, Murakami-Murofushi K, Kobayashi T. Cyclic phosphatidic acid is produced by autotaxin in blood. J Biol Chem. 2006;281:26081–26088. doi: 10.1074/jbc.M602925200. [DOI] [PubMed] [Google Scholar]
- 23.Mukai M, Iwasaki T, Tatsuta M, Togawa A, Nakamura H, Murakami-Murofushi K, Kobayashi S, Imamura F, Inoue M. Cyclic phosphatidic acid inhibits RhoA-mediated autophosphorylation of FAK at Tyr-397 and subsequent tumor-cell invasion. Int J Oncol. 2003;22:1247–1256. [PubMed] [Google Scholar]
- 24.Shida D, Kitayama J, Yamaguchi H, Okaji Y, Tsuno NH, Watanabe T, Takuwa Y, Nagawa H. Lysophosphatidic acid (LPA) enhances the metastatic potential of human colon carcinoma DLD1 cells through LPA1. Cancer Res. 2003;63:1706–1711. [PubMed] [Google Scholar]
- 25.Hama K, Aoki J, Fukaya M, Kishi Y, Sakai T, Suzuki R, Ohta H, Yamori T, Watanabe M, Chun J, Arai H. Lysophosphatidic acid and autotaxin stimulate cell motility of neoplastic and non-neoplastic cells through LPA1. J Biol Chem. 2004;279:17634–17639. doi: 10.1074/jbc.M313927200. [DOI] [PubMed] [Google Scholar]
- 26.Shida D, Kitayama J, Yamaguchi H, Hama K, Aoki J, Arai H, Yamashita H, Mori K, Sako A, Konishi T, Watanabe T, Sakai T, Suzuki R, Ohta H, Takuwa Y, Nagawa H. Dual mode regulation of migration by lysophosphatidic acid in human gastric cancer cells. Exp Cell Res. 2004;301:168–178. doi: 10.1016/j.yexcr.2004.08.008. [DOI] [PubMed] [Google Scholar]
- 27.An S, Bleu T, Zheng Y, Goetzl EJ. Recombinant human G protein-coupled lysophosphatidic acid receptors mediate intracellular calcium mobilization. Mol Pharmacol. 1998;54:881–888. doi: 10.1124/mol.54.5.881. [DOI] [PubMed] [Google Scholar]
- 28.Erickson JR, Wu JJ, Goddard JG, Tigyi G, Kawanishi K, Tomei LD, Kiefer MC. Edg-2/Vzg-1 couples to the yeast pheromone response pathway selectively in response to lysophosphatidic acid. J Biol Chem. 1998;273:1506–1510. doi: 10.1074/jbc.273.3.1506. [DOI] [PubMed] [Google Scholar]
- 29.Baker DL, Fujiwara Y, Pigg KR, Tsukahara R, Kobayashi S, Murofushi H, Uchiyama A, Murakami-Murofushi K, Koh E, Bandle RW, Byun HS, Bittman R, Fan D, Murph M, Mills GB, Tigyi G. Carba analogs of cyclic phosphatidic acid are selective inhibitors of autotaxin and cancer cell invasion and metastasis. J Biol Chem. 2006;281:22786–22793. doi: 10.1074/jbc.M512486200. [DOI] [PMC free article] [PubMed] [Google Scholar]