

Abstract
Flagellated heterotrophic microeukaryotes have key roles for the functioning of marine ecosystems as they channel large amounts of organic carbon to the upper trophic levels and control the population sizes of bacteria and archaea. Still, we know very little on the diversity patterns of most groups constituting this evolutionary heterogeneous assemblage. Here, we investigate 11 groups of uncultured flagellates known as MArine STramenopiles (MASTs). MASTs are ecologically very important and branch at the base of stramenopiles. We explored the diversity patterns of MASTs using pyrosequencing (18S rDNA) in coastal European waters. We found that MAST groups range from highly to lowly diversified. Pyrosequencing (hereafter ‘454') allowed us to approach to the limits of taxonomic diversity for all MAST groups, which varied in one order of magnitude (tens to hundreds) in terms of operational taxonomic units (98% similarity). We did not evidence large differences in activity, as indicated by ratios of DNA:RNA-reads. Most groups were strictly planktonic, although we found some groups that were active in sediments and even in anoxic waters. The proportion of reads per size fraction indicated that most groups were composed of very small cells (∼2–5 μm). In addition, phylogenetically different assemblages appeared to be present in different size fractions, depths and geographic zones. Thus, MAST diversity seems to be highly partitioned in spatial scales. Altogether, our results shed light on these ecologically very important but poorly known groups of uncultured marine flagellates.
Keywords: diversity, heterotrophic-flagellates, MAST, pyrosequencing, stramenopiles
Introduction
Microbial eukaryotes have key roles in marine ecosystems, particularly in primary production, nutrient cycling as well as for food-web dynamics (Sherr and Sherr, 2008; Jardillier et al., 2010; Caron et al., 2012). Among heterotrophic protists, small flagellates (1–5 μm) constitute a key link between bacteria and larger protists, transferring organic carbon to upper trophic levels (Jurgens and Massana, 2008; Massana, 2011). Furthermore, heterotrophic pico- and nano-sized flagellates together with viruses are important control agents of planktonic bacteria in the oceans (Suttle, 2005; Jurgens and Massana, 2008). Pico- and nano-flagellates have been traditionally considered in bulk, but data gathered during the last decade show that they constitute an evolutionarily very diverse assemblage (Massana, 2011). Despite the large ecological importance of heterotrophic pico- and nano-flagellates, the actual diversity of the different groups as well as the distribution of diversity in space and time are among the least known within the microbial world.
In this work, we investigate a number of ecologically very relevant marine microeukaryote groups that are poorly known. These groups were collectively defined as MArine STramenopiles (MASTs; Massana et al., 2004) and are phylogenetically basal stramenopile lineages that do not belong to any other group (Figure 1). The stramenopiles constitute one of the major eukaryotic branches (Baldauf, 2003) and include a vast number of heterotrophic and autotrophic groups with large ecological importance in the oceans. Most MASTs branch near Bicosoecida, Blastocystis and Labyrinthulida (Figure 1), the earliest diverging stramenopile lineages (Riisberg et al., 2009; Tsui et al., 2009). After the initial description of MASTs, they have been regularly detected in diverse marine environmental surveys in different marine geographical areas around the world (for example, Richards and Bass, 2005; Takishita et al., 2005; Takishita et al., 2007; Not et al., 2008, 2009; Orsi et al., 2011). MAST groups are mostly composed of free-living bacterivorous flagellates, with some groups displaying algivorous preferences (MAST-6; Piwosz and Pernthaler, 2010) and other lineages being parasites (Gomez et al., 2011). The available data indicate that their sizes normally range between 2 and 8 μm, occasionally reaching 22 μm (Massana et al., 2006, 2009; Piwosz and Pernthaler, 2010). They have been found in all oceans, and altogether they may reach up to 35% of the heterotrophic flagellates (Massana et al., 2006). One group, MAST-4, presents cell abundances that are in average 9% of the heterotrophic flagellate assemblage (Massana et al., 2006), suggesting that this may be one of the most abundant heterotrophic flagellates in the oceans. However, despite having a huge abundance at a global level (estimated 1024 cells), MAST-4 shows a relatively low genetic diversity, which seems to be distributed in only five major lineages (Rodriguez-Martinez et al., 2012). Less is known about other MAST groups, although previous evidence suggests that MAST-1 and -3 are very diverse (Massana et al., 2004, 2006). Microscopic counts indicated that MAST-1 and -2 tend to have much lower abundances than MAST-4 (Massana et al., 2006). Different morphotypes of MAST-6 appear to have a large variability in their seasonal abundances (Piwosz and Pernthaler, 2010). Although most MAST groups seem to be planktonic (Massana et al., 2004), the groups MAST-9 and -12 appear to be associated to sediments, anoxic waters and/or deep-sea sediments (Massana et al., 2004; Takishita et al., 2005; Takishita et al., 2007).
Figure 1.

Schematic stramenopile phylogeny indicating the position of MAST groups. Based on a modified version of Supplementary Figure S1.
Here, we aim at moving further and explore MAST diversity using a high-throughput sequencing methodology, 454 pyrosequencing (hereafter ‘454'). We generated 454 sequence data of environmental 18S rDNA genes for six European coastal sites, considering different depths in the water column, size fractions, sediments as well as DNA and cDNA. Our main questions were: How much novel intra-group diversity is detected by 454? Will 454 saturate the diversity of all or some MAST groups? How many operational taxonomic units (OTUs) can be estimated for each group? How is MAST diversity partitioned in spatial scales? And, to what size fractions and environments MAST groups are associated and how active they are in these?
Materials and methods
Sampling, 454 sequencing and curation of the sequences
Seawater samples were collected through the BioMarKs consortium (http://www.BioMarKs.org/) in six European coastal stations: offshore Blanes (Spain), Gijon (Spain), Naples (Italy), Oslo (Norway), Roscoff (France), and Varna (Bulgaria) (see Supplementary Table S1). Water samples were taken with Niskin bottles attached to a CTD rosette at surface and deep chlorophyll maximum (DCM) depths. These samples were pre-filtered through 20-μm filters and afterwards, they were sequentially filtered through 3- and 0.8-μm polycarbonate filters (diameter: 142 mm). Filtration time did not surpass 30 min to avoid RNA degradation. Filters were flash-frozen and stored at −80 °C. Sediment samples were taken with sediment cores and small aliquots were frozen at −80 °C for downstream molecular analysis. The total number of samples considered in this study was 139 (see Supplementary Table S1).
Total DNA and RNA were extracted simultaneously from the same filter using the NucleoSpin RNA L kit (Macherey-Nagel, Düren, Germany) and quantified using a Nanodrop ND-1000 spectrophotometer (NanoDrop Technologies Inc, Wilmington, DE, USA). Extract quality was checked on a 1.5% agarose gel. To remove contaminating DNA from RNA, we used the TurboDNA kit (Ambion, Carlsbad, CA, USA). Extracted RNA was immediately reverse transcribed to DNA using the RT Superscript III random primers kit (Invitrogen, Carlsbad, CA, USA). The universal primers TAReuk454FWD1 (5′-CCAGCASCYGCGGTAATTCC-3′) and TAReukREV3 (5′-ACTTTCGTTCTTGATYRA-3′) were used to amplify the V4 region (∼380 bp) of the eukaryotic 18S rDNA (Stoeck et al., 2010). The primers were adapted for 454 using the manufacturers' specifications, and had the configuration A-adapter-tag (7 or 8 bp)-forward primer and B-adapter-reverse primer. PCRs were performed in 25 μl samples, and consisted of 1 × MasterMix Phusion High-Fidelity DNA Polymerase (Finnzymes, Espoo, Finland), 0.35 μℳ of each primer and 3% DMSO. We added a total of 5 ng of template DNA/cDNA to each PCR sample. PCRs consisted of an initial denaturation step at 98 °C for 30 s, followed by 10 cycles of 10 s at 98 °C, 30 s at 53 °C and 30 s at 72 °C, and afterwards by 15 cycles of 10 s at 98 °C, 30 s at 48 °C and 30 s at 72 °C. Amplicons were checked in a 1.5% agarose gel for successful amplification. Triplicate amplicons were pooled and purified using the NucleoSpin Extract II (Macherey-Nagel). Purified amplicons were eluted in 30 μl of elution buffer and quantified again using a Nanodrop ND-1000 spectrophotometer. The total final amount of pooled amplicons for 454 tag-sequencing was approximately 5 μg. Amplicon sequencing was carried out on a 454 GS FLX Titanium system (454 Life Sciences, Branford, CT, USA) installed at Genoscope (http://www.genoscope.cns.fr/spip/, France).
The quality of the sequences was screened, and only sequences having exact forward and reverse primer match were considered. Furthermore, the number of errors in sliding windows of 50 bp was computed, and any sequence appearing only once and containing a window having an error >1% was not considered. Errors for each sliding window were computed with the formula 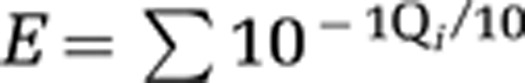 , where Qi is the quality value of the sequence at position i. Taxonomy was assigned using sequence similarity to a reference database based on SILVA 108 release (http://www.arb-silva.de/). Chimera detection was run with the UCHIME module of USEARCH (Edgar, 2010; Edgar et al., 2011), using de novo and reference-based chimera check considering the protists present in the SILVA 108 release database. Additional chimera checks were done with ChimeraSlayer (Haas et al., 2011) based on SILVA 108 release database. A summary of the quality-checked sequences of the BioMarKs data set is presented in Supplementary Table S2. Sequence data has been deposited in the MG-RAST public database (http://metagenomics.anl.gov/; data set number 4478907.3).
, where Qi is the quality value of the sequence at position i. Taxonomy was assigned using sequence similarity to a reference database based on SILVA 108 release (http://www.arb-silva.de/). Chimera detection was run with the UCHIME module of USEARCH (Edgar, 2010; Edgar et al., 2011), using de novo and reference-based chimera check considering the protists present in the SILVA 108 release database. Additional chimera checks were done with ChimeraSlayer (Haas et al., 2011) based on SILVA 108 release database. A summary of the quality-checked sequences of the BioMarKs data set is presented in Supplementary Table S2. Sequence data has been deposited in the MG-RAST public database (http://metagenomics.anl.gov/; data set number 4478907.3).
Extraction of sequences from NCBI and construction of reference databases
A seed reference stramenopile sequence data set (STR1-DB) was generated. STR1-DB (372 sequences) included all known major stramenopile groups and was used to identify other Sanger sequences in the NCBI-nr database (May 2011; about 16 million sequences) using local BLAST searches (blastn v2.2.22+, Altschul et al., 1990). With the retrieved sequences, two extra reference data sets were generated: STR4-DB (5480 sequences >700 bp) and STR5-DB (3835 sequences >1100 bp). All data sets were checked for chimeras using ChimeraSlayer (Haas et al., 2011) and the SILVA 108 database. Maximum likelihood trees were inferred for each data set using RAxMLHPC-MPI (v7.2.8; Stamatakis, 2006). These trees were used to validate the quality of the Sanger data sets as well as for mapping reads onto the phylogenies. In total, 100 trees for both topology and bootstrap were run under the model GTR+CAT/G+I. The tree with the best topology and likelihood (TREE2 from data set STR5-DB; Supplementary Figure S1) was selected and bootstrap values were inserted. Also see Supplementary Materials for further methodological details.
Mapping of stramenopile 454 reads onto Sanger-based phylogenies
Only unique (that is, non-repeated) stramenopile reads within each sample, which were also longer than 350 bp, were used for phylogenetic assignment. The reads that satisfied these conditions were 66 707 and ranged in size between 351 and 444 bp. Due to such stricter selection criterion, this number was smaller than the raw number of stramenopile reads (82 944; Supplementary Table S2). Reads were aligned to a reference data set (STR5-DB) using mothur (v1.20; Schloss et al., 2009). All 66 707 reads were inserted into a reference tree (TREE2; Supplementary Figure S1) using PPLACER v1.1 (Matsen et al., 2010). PPLACER uses maximum likelihood and Bayesian approaches to place 454 reads onto a fixed reference phylogeny. The resulting trees were visualized with Archaeopteryx v0.957 (Han and Zmasek, 2009).
Novel diversity analysis
To explore the amount of novel stramenopile diversity that was recovered by 454 sequencing, we (a) clustered independently both the 454 and Sanger data sets using different thresholds into OTUs, and (b) analyzed the distribution of similarities between 454 reads and Sanger sequences present at NCBI-nr. In approach (a), the clustering strategy was subdivided. In the first case, only 454 stramenopile reads that were unique in the entire data set were considered for OTU construction using UCLUST (Edgar, 2010) with clustering thresholds ranging between 90 and 100%. For the construction of the latter data set, we used the most stringent conditions, and entire or partial sequences that were identical to another sequence (that is, shorter or equal-length reads identical to another read) were removed. In this data set, 26 651 stramenopile reads were remaining and they were clustered. This number differs from the general BioMarKs data set (48 812 reads; Supplementary Table S2), where more relaxed parameters have been used. In the second case, a random subsample of 5480 reads (to have the same sample size as with the Sanger data set) was used for OTU clustering with the same parameters as mentioned before. Finally, the Sanger reference data set with 5480 sequences (>700 bp) was clustered with UCLUST in the same manner as with the 454 data sets. OTU-number comparisons between the Sanger (5480 sequences) and both the full (26 651 reads) and subsampled (5480 reads) 454 data sets were carried out. In approach (b), only the unique stramenopile 454 reads (26 651) were locally BLASTed against NCBI-nr (version May 2011). For each query, we retrieved only the best hit, and only hits producing alignments >300 bp with the queries were considered (26 634 reads). Our BLAST search strategy was relaxed to allow the retrieval of distant sequences (BLASTn parameters: -evalue 0.0001 -perc_identity 70). The percentage of identity with the best hits was subsequently used to construct a density distribution.
Detecting diversity patterns in MASTs using 454
A total of 27 158 MAST (1–12) reads (including the repeated ones) belonging to different samples, size fractions, depths and geographic zones were used to investigate questions on diversity and distributions of these groups. For each sampled site, DNA and cDNA reads were used to obtain insight on MAST activity. An alignment was constructed with unique MAST reads (12 919 reads, unique within samples) using mothur and the SILVA 108 database as template. A maximum likelihood phylogeny was constructed with RAxML (v7.2.8; Stamatakis, 2006) using the GTR+G+I model. This tree was used to infer diversity metrics for MAST groups and for phylogeny-based tests (phylogenetic diversity (PD), P-test and Unifrac test). PD (Faith, 1992) estimates were run in mothur and P-tests as well as Unifrac significance tests (Martin, 2002; Hamady et al., 2010) were run online (http://bmf2.colorado.edu/fastunifrac/). Rarefaction analyses were run in mothur and statistical tests were run in R (R-Development-Core-Team, 2008).
Results and discussion
In this work, we explore whether 454 recovers novel MAST diversity as well as whether diversity becomes saturated in different groups. Furthermore, we investigate how is MAST diversity partitioned in spatial scales, to what size fractions and environments MAST groups are associated and how active they are in them. Unless stated differently, results include both DNA and RNA reads.
454 reads populated Sanger-based stramenopile phylogenetic trees
Both Sanger and 454 recovered similar amounts of stramenopile diversity in terms of OTU number when considering the same number of reads (Figure 2). This agreement indicates that possible sequencing errors that may have remained in our data set after our quality controls and filtering, did not have a significant effect on our diversity estimations. When the full set of 454 sequences was considered, the extra diversity recovered by 454 became evident (Figure 2). The differences in OTU number between 454 and both the Sanger and 454 subsampled data sets decreased with the decrease of the clustering threshold. Most of the extra diversity retrieved by 454 fell within the 3% clustering threshold and thus could be associated to microdiversity (that is, highly related microorganisms based on 18S rDNA gene similarity with potentially different physiological and ecological functions). An analysis of the distribution of similarities between 454 reads and Sanger sequences deposited at NCBI-nr supported these results (Figure 3). Most of the query 454 reads (99.9%) produced alignments larger than 300 bp with the Sanger BLAST hits (that is, BLAST hits to Sanger sequences in NCBI-nr), and the majority of reads (89.3%) had a similarity to Sanger sequences >95%. Only 1.6% of the reads had Sanger BLAST hits with <90% of similarity. In summary, the backbone of the stramenopile phylogeny was recovered by Sanger sequences, with 454 recovering finer novel diversity and thus populating the Sanger core tree. These results suggest that no abundant major groups of stramenopiles are missing from this general stramenopile tree based on Sanger sequences. Any missing major group, if it exists, would have a relatively low abundance in nature or a very restricted distribution in the sampled areas. Furthermore, the agreement between 454 and Sanger data sets validated the quality of the 454 data set for downstream MAST-specific analyses.
Figure 2.

Comparison of the stramenopile diversity retrieved by Sanger and 454 sequencing. When the same number of sequences is sampled, 454 and Sanger retrieve very similar numbers of OTUs at the whole clustering threshold range. When all 454 reads are analyzed, the extra diversity retrieved by 454 becomes apparent. Note that most of the new diversity brought by 454 is located within the 3% of divergence area and thus could be associated to microdiversity. Only unique sequences have been analyzed here, and all data sets were treated equally during clustering. The complete chimera-checked 454 data set contained 26 651 sequences, while the subsampled had 5480 sequences.
Figure 3.

Distribution of the similarities (percentage of identity) between the 26 651 stramenopile reads and the Sanger stramenopile sequences at NCBI-nr (version May 2011). Only hits that aligned >300 bp to the query were considered. Note that most of the MAST diversity recovered by 454 was >98% similar to Sanger sequences already present at NCBI-nr.
Shared and distinctive phylotypes in the 454 and Sanger data sets
Reads were assigned to the MAST groups using both reference-based and phylogenetic-based approaches. The reference-based approach was based on sequence similarities to marine protists present in the SILVA 108 release database, and it will be used for the most part of this study. The phylogenetic assignment was intended to explore how reads distributed within each MAST group, a piece of information that similarity assignment could not provide. A summary of the number of Sanger sequences within each MAST group and the corresponding number of reads assigned to each group using reference-based or phylogenetic approach is presented in Table 1. Reads belonging to all MAST groups were recovered, with the exception of MAST-10, which did not present reads assigned by similarity (but it presented reads assigned phylogenetically). The number of reads assigned to each MAST group varied depending on the assignment method (Table 1). In some cases, the phylogenetic-based method assigned more reads than the reference-based counterpart and vice versa (for example, MAST-1, MAST-2, MAST-7). In other cases, both techniques tended to agree (for example, MAST-4, MAST-11, MAST-12). It has been shown that both approaches can produce different results (Koski and Golding, 2001; Porter and Golding, 2011). Phylogenetic-based approach may be more accurate, but it can be computationally very intensive. Here, we used reference-based taxonomic assignment for general intergroup comparisons and phylogenetic assignment to explore intragroup distributions of reads, as a feasible and reliable approach.
Table 1. Sanger sequences and 454 reads assigned to each MAST group using phylogenetic or sequence-similarity methods.
| Group | Sanger (NCBI-nr)a | Reference-based assignment, 454 reads | Phylogenetic assignment, 454 reads |
|---|---|---|---|
| MAST-1 | 62 | 1446 | 2756 |
| MAST-2 | 6 | 135 | 665 |
| MAST-3 | 82 | 4644 | 3913 |
| MAST-4 | 24 | 1286 | 1158 |
| MAST-6 | 2 | 327 | 133 |
| MAST-7 | 19 | 1619 | 1308 |
| MAST-8 | 3 | 619 | 1212 |
| MAST-9 | 15 | 133 | 91 |
| MAST-10 | 4 | 0 | 79 |
| MAST-11 | 1 | 164 | 129 |
| MAST-12 | 24 | 390 | 414 |
Values derived from a data set consisting in 3835 Sanger sequences >1100 bp extracted from NCBI-nr.
The analysis of the phylogenetic assignments showed that the distribution of reads within groups was variable. In one extreme, reads mapped to several nodes of the Sanger group subtrees (for example, MAST-3; Supplementary Figure S2), and in the other, they were concentrated at one specific node (for example, MAST-1; results not shown). These results were obtained after the removal of identical reads within samples, so values cannot be explained by the prevalence of a few phylotypes. Still, highly localized placements of many reads may derive from the lack of similar counterparts in the Sanger phylogeny. Some evidence supported the latter. For example, in MAST-3, 1454 reads were placed at node 168 (Supplementary Figure S2), and those reads can be clustered into 37 OTUs at 97% similarity. Therefore, the incorporation of more Sanger sequences to the tree may change the placement of these reads. Similar examples were found in other MAST groups (for example, MAST-1, -7 and -4). Conversely, there were Sanger phylotypes with no close 454 relatives, indicating that the total fine diversity of MASTs was not recovered by 454 in the used samples. This likely reflects the wider environmental and temporal sampling included in the Sanger data.
Different levels of PD between MASTs
The MAST tree constructed with only 454 reads recovered most MAST groups (Supplementary Figure S3). Still, a few incongruences were observed that were most likely due to the short length of the sequences. Despite this tree is not the most appropriate for analyzing the whole phylogeny of MASTs, it can still be used for analyzing diversity patterns within and between groups. The advantage is that it incorporates a large amount of reads and therefore it can provide more accurate estimates of diversity. Based on this tree we have estimated different amounts of PD within different MAST groups (Figure 4), indicating different amounts of evolutionary diversification. Comparing at similar sampling levels, it is apparent that MAST encompass highly diversified groups (for example, MAST-12) as well as groups showing a low diversification (for example, MAST-4). For MAST-4, it has been recently shown that it presents a very low degree of evolutionary diversification despite its huge global abundance (Rodriguez-Martinez et al., 2012).
Figure 4.

Phylogenetic diversity for the different MAST groups based on 454 reads. A rarefaction curve based on 1000 randomizations is presented, as phylogenetic diversity is dependent of sample size.
Different MASTs do represent different taxonomic levels, and this partially explains the diversity differences between groups. Nevertheless, the observed differences in diversification may be also related to variable evolutionary rates (that is, substitution rates) in different lineages and/or different times of evolutionary emergence. Most MASTs branch at the base of the stramenopile phylogeny (Figure 1), near Bicosoecida, Blastocystis and Labyrinthulida, the earliest diverging stramenopile lineages (Riisberg et al., 2009; Tsui et al., 2009). Despite the exact time of emergence of each MAST is unclear, it is evident that they constitute early branching lineages, which may have appeared more than 500 million years ago. In the paleo-oceans dominated by pelagic bacteria, heterotrophic bacterivorous flagellates like MASTs may have been among the first eukaryotic predators to emerge. Still, the low divergence observed among taxa in some groups (for example, MAST-4) may be perplexing considering their early evolution. One possible scenario would involve several extinctions along the long history of the groups besides more recent diversification events that generated the actual taxa.
Approaching the limits of MAST taxonomic diversity
The use of 454 sequencing allowed us to generate realistic estimates of OTU diversity for MASTs. For the analyzed set of samples, we estimate that we have approached to the saturation of OTU diversity in most groups when considering a clustering threshold of 98% (Figure 5; when the rarefaction curve becomes parallel to the x-axis, diversity is considered saturated). Using this threshold would decrease any inflation of diversity produced by sequencing errors that could have remained in our data set even after our strict quality controls. Therefore, 98% clustering will be used hereafter. MAST groups varied in about one order of magnitude (tens to hundreds) in their saturation levels (Figure 5). MAST-2 appeared to be the group with lowest number of OTUs and MAST-3 the group with the highest (Figure 5). This agrees with the large variation observed in PD and further emphasizes the differences between MASTs in terms of their amount of diversification. However, a comparison of the PD and the OTU rarefaction results shows that groups with many OTUs (for example, MAST-3) are not necessarily the ones with the highest PD values when compared using the same number of reads (Figures 4 and 5). This difference emerges from the different topologies of the phylogenies in each group. It is important to note that the observed saturation of diversity should be restricted to the set of samples analyzed, and more samples could add more diversity. These diversity limits are most likely among the first calculated for uncultured heterotrophic flagellates. This represents a significant contribution, as the order of magnitude of diversity for most microbial lineages is not known (Pedrós-Alió, 2006; Lopez-Garcia and Moreira, 2008).
Figure 5.

Rarefaction analysis for the different MAST groups based on 454 reads using clustering thresholds between 100 and 95% (only the curves with the thresholds 100, 99 and 98 are indicated). The final number of OTUs at 98% is indicated in brackets. Note that at 98% clustering, the curves for most groups tend to become parallel to the x-axis, indicating diversity saturation for that level.
Large heterogeneity in MAST spatial distributions
The analysis of all MAST sequences of the BioMarKs data set (excluding MAST-10) evidenced different patterns. The groups MAST-1, -3, -4, -7, -8 and -12 were well represented in the entire data set (Table 2; Supplementary Figure S4). The most abundant groups were MAST-3 (41.3% of the total MAST sequences), MAST-1 (16.0%), MAST-7 (13.7%) and MAST-4 (10.9%). Together, these groups account for 81.9% of the sequences. In particular, MAST-3 was highly represented in several samples, indicating that this group is one of the most abundant MASTs, as it has been previously suggested (Massana et al., 2004). Recent studies indicate that some MAST-3 lineages may be parasites of diatoms (Gomez et al., 2011). Most MAST groups were present in most of the sampled geographic locations (Table 2), and their total abundances varied between sites, with the higher abundances being detected in Roscoff, Naples and Gijon (Figure 6). When individual groups were analyzed in relation to geographic location, we observed that different groups predominated at different places (Supplementary Figure S5). Most of the sampled MAST assemblages in different geographic locations appeared to be different from a phylogenetic composition perspective (Unifrac test and P-test <0.05; except for assemblages from Blanes and Gijon). Furthermore, different locations presented different amounts of PD (results not shown). Such heterogeneity between geographic sites may respond to the natural temporal variability of the groups, although it may also represent environmentally associated spatial distributions. Surface and DCM samples did not present significant differences in MAST composition. However, rarefaction curves indicated that there was more PD at the DCM than at the surface (results not shown). Furthermore, phylogenetically different assemblages of MAST-1, -3, -8 and -9 appeared to be present in the DCM and subsurface samples (Unifrac test <0.05). This suggests that within some MAST groups, different strains may be exploiting different depth zones.
Table 2. MAST distributions results.
| No. of samples (n=139)a | No. of sitesb | No. of sequences | RNA:DNAc |
Size fractiond |
||||||
|---|---|---|---|---|---|---|---|---|---|---|
|
0.8–3 μm (n=39; 21 044 seq.) |
3–20 μm (n=36; 4008 seq.) |
Sediments (n=25; 1704 seq.) |
||||||||
| Averagee | s.e.f | Averagee | s.e.f | Averagee | s.e.f | |||||
| MAST-1 | 74 | 5 | 4360 | 0.66 | 13.95 | 2.64 | 18.34 | 3.64 | 8.05 | 3.70 |
| MAST-2 | 20 | 5 | 135 | 0.22 | 0.26 | 0.09 | 1.32 | 0.68 | 0.00 | 0.00 |
| MAST-3 | 98 | 6 | 11 230 | 1.68 | 26.28 | 3.61 | 40.97 | 4.33 | 14.00 | 3.06 |
| MAST-4 | 61 | 5 | 2949 | 0.69 | 13.56 | 1.76 | 2.89 | 0.67 | 0.10 | 0.09 |
| MAST-6 | 52 | 5 | 515 | 0.92 | 0.91 | 0.29 | 2.21 | 0.75 | 15.18 | 3.52 |
| MAST-7 | 68 | 5 | 3714 | 1.19 | 18.22 | 2.67 | 8.20 | 1.97 | 0.26 | 0.22 |
| MAST-8 | 93 | 6 | 1794 | 3.19 | 11.40 | 1.74 | 9.14 | 1.50 | 11.62 | 2.69 |
| MAST-9 | 49 | 5 | 366 | 0.79 | 5.85 | 2.13 | 6.59 | 3.45 | 2.07 | 1.28 |
| MAST-11 | 19 | 5 | 317 | 0.36 | 1.00 | 0.26 | 0.31 | 0.20 | 0.00 | 0.00 |
| MAST-12 | 90 | 6 | 1778 | 0.40 | 8.22 | 1.86 | 9.87 | 2.92 | 46.80 | 5.45 |
| All MAST | 122 | NA | 27 158 | NA | NA | NA | NA | NA | NA | NA |
Number of samples, where each group and all the groups were present.
Number of geographic sites, where each group was present of a total of six (Blanes, Gijon, Naples, Oslo, Roscoff and Varna).
Average RNA:DNA-read ratio.
Results for the pico (0.8–3 μm), nano (3–20 μm) and sediment fractions.
Average percentage of total MASTs represented by each MAST group in the fraction.
Standard error.
Figure 6.

Percentage of the total number of reads represented by all MASTs in samples from different geographic locations. n indicates the number of samples; *anoxic samples.
Adaptation of MAST groups to anoxic waters and sediments
Reads from different MAST groups were detected in the anoxic water column of the Black Sea (Varna sample), suggesting the presence of strains or entire groups that are adapted to low-oxygen or anoxic conditions. For example, MAST-9 was only abundant in anoxic samples (Supplementary Figure S5). Previous studies found MAST-9 associated to hydrothermal vents (Massana et al., 2004) as well as anoxic sea sediments (Takishita et al., 2005, Takishita et al., 2007). MAST-12 was also recovered from the anoxic layers of the Black Sea (Supplementary Figure S5), but it was also recovered from the oxic water column in the same as well as in other locations (for example, Oslo, Roscoff and others; Supplementary Figure S5). MAST-12, however, was significantly more abundant in sediments than in the water column (Kruskal–Wallis, P<0.05, Supplementary Figure S6). The prevalence of MAST-12 in sediments has been suggested in other studies (Massana et al., 2004). Other groups were also present in sediments (MAST-3, -6 and -8; Supplementary Figure S6). In MAST-6, the amount of reads in sediments was significantly higher than in the water column (Kruskal–Wallis, P<0.05), suggesting that this group may prefer to inhabit sediments. In MAST-3, -6, -8 and -12, there was a substantial amount of RNA in the sediments, pointing to active communities and not just the accumulation of DNA (Figure 7). Only in MAST-8, the amount of RNA in the sediments was significantly higher than DNA (Figure 7), suggesting a relatively high activity across most analyzed samples. In MAST-3, -6 and -12, there was no significant difference between DNA and RNA in the sediments, indicating moderate or regular activity levels.
Figure 7.

Percentage of the total number of MAST reads in samples from different size fractions (0.8–3.0 and 3.0–20 μm) as well as from sediments. Values for each MAST group are presented considering the used template (DNA or RNA). Note that the ratio DNA:RNA can provide an indication of activity within each size fraction or in sediments. The symbol ‘*' indicates that the difference between DNA and RNA was significant in MAST-8 (Kruskal–Wallis, P<0.05). The absence of ‘*' indicates lack of significance.
Activity levels in MASTs
The overall ratio of DNA:RNA-reads differed significantly only in the groups MAST-3, -8 and -12 (Supplementary Figure S7). In MAST-3 and -8, the amount of RNA was higher than DNA across most samples (Wilcoxon test, P<0.05), suggesting that these groups may normally display a higher activity. MAST-12 displayed the opposite pattern, and this may point to a lower activity in this group or the presence of a larger number of rDNA copies in their genomes (Zhu et al., 2005). Nevertheless, the observed DNA:RNA-reads ratios suggest similar activity levels for most MASTs in all environments, and also that their genomes probably contain a similar rDNA operon copy number. Previous studies indicate that MAST-4 has about 30 copies of the rDNA operon (Rodriguez-Martinez et al., 2009). This is considered a relatively low number, and could be associated to a comparatively small (<50 Mb) genome (Prokopowich et al., 2003). So far, there is no evidence for a large number of rDNA copies in MASTs that could have biased our analyses. The PD within the RNA fraction appeared to be lower than within the DNA (results not shown), and both fractions appeared to harbor different phylotypes (Unifrac and P-tests <0.05). This suggests that not all rDNA operons may be expressed and that some rDNA variants may be overexpressed.
Size-fractioning coupled to 454 may serve to unveil cell sizes and morphotypes of uncultured flagellates
Only in MAST-3, the proportion of reads in the 3–20-μm fraction was significantly higher than that in the 0.8–3-μm fraction (Kruskal–Wallis, P<0.05; Supplementary Figure S6). This suggests that MAST-3 may have larger cell sizes than the other MAST groups. Contrastingly, reads from MAST-4 and -7 were significantly more abundant in the 0.8–3-μm size fraction (Kruskal–Wallis, P<0.05), suggesting smaller cell sizes for these groups. In all the remaining groups, there were no significant differences between the 0.8–3- and 3–20-μm size fractions in terms of proportion of total MAST reads per sample (Supplementary Figure S6). These results agree with the limited morphological data available. For example, MAST-1 cell size ranges between 4 and 8 μm (Massana et al., 2006), and in our analyses 454 reads predominated in both 0.8–3 and 3–20 μm size fractions. MAST-4 cell sizes are about 2.3 μm (Massana et al., 2006), and in our analyses most reads fell into the 0.8–3-μm fraction. For MAST-6, two cell morphotypes have been reported, one with sizes ranging between 4 and 9 μm and another one ranging between 10 and 22 μm (Piwosz and Pernthaler, 2010). Despite the number of reads obtained in the water column for MAST-6 was low, a few more reads appeared to be present in the 3–20-μm fraction (Supplementary Figure S6). The phylogenetic composition in the 0.8–3- and 3–20-μm size fractions was compared within MAST-1, -3, -7, -8 and -12. The P-test indicated significant (P<0.05) differences between both size fractions for all groups, while the Unifrac test indicated significant (P<0.05) differences between the fractions only in MAST-3 and -8. The incongruence between tests may point to limited differences between some size fractions and more reads are needed for drawing stronger conclusions. Nevertheless, our results suggest that within some MAST groups, phylogenetically different strains may have different cell sizes.
Conclusion
To our knowledge, this is the first study investigating the diversity patterns of MAST groups using 454 pyrosequencing. Our results indicated that MAST groups harbored very different levels of taxonomic and PD. Despite not all MAST diversity known from Sanger sequences was recovered by 454, the latter technique recovered novel intra-group diversity. The large number of sequences provided by 454 sequencing allowed us to approach to the diversity limits of each group. The number of OTUs (clustered at 98% and near the saturation levels) for MAST groups ranged from tens to hundreds. This diversity presented a very heterogeneous spatial distribution, and therefore we can conclude that not all MASTs are equally represented at each geographic location at any particular time. Our results allowed us to label each group with respect to the typical cell size and habitat preference. Regarding cell size, some groups appeared to be mostly picoplankters (MAST-4, -7 and -11), and others included pico- and nanoplankters (MAST-1, -3 and -8), being the latter two well represented in sediments. In addition, MAST-6 and -12 preferred to inhabit sediments and MAST-9 preferred anoxic waters. We did not evidence large differences in activity, although some groups appeared more active in specific environments and size fractions. Our data also suggested that different MAST assemblages may be present in different size fractions, but more data is needed to confirm this observation. Altogether, our results serve to pave the road for future more holistic studies focusing on these groups of uncultured flagellates, which have key roles in marine ecosystems.
Acknowledgments
Financial support for this work has been provided by a Marie Curie Intra-European Fellowship grant (PIEF-GA-2009-235365) to RL and by projects BioMarKs (2008-6530, ERA-net Biodiversa, EU) and FLAME (CGL2010-16304, MICINN, Spain) to RM. Large-scale computing resources were provided by the Canarian Institute of Astrophysics (www.iac.es), through the Barcelona Supercomputer Center and the Spanish Network of Supercomputing (grants BCV-2010-3-0003 and 2011-2-0003/3-0005 to RL and RM). We thank the BioMarKs consortium for undertaking the sampling and performing the initial laboratory processing of the samples, in particular Sarah Romac. We thank Hiroyuki Ogata and Jean-Michel Claverie for the implementation of bioinformatics tools through a BioMarKs grant and a project from the French National Research Agency (ANR-08-BDVA-003) to Jean-Michel Claverie. Javier del Campo is thanked for providing curated Sanger sequences of Ochrophyta. Berit Kaasa at the University of Oslo is thanked for running the nutrient analyses. We thank the three reviewers and the editor who helped to improve this work.
Footnotes
Supplementary Information accompanies the paper on The ISME Journal website (http://www.nature.com/ismej)
Supplementary Material
References
- Altschul SF, Gish W, Miller W, Myers EW, Lipman DJ. Basic local alignment search tool. J Mol Biol. 1990;215:403–410. doi: 10.1016/S0022-2836(05)80360-2. [DOI] [PubMed] [Google Scholar]
- Baldauf SL. The deep roots of eukaryotes. Science. 2003;300:1703–1706. doi: 10.1126/science.1085544. [DOI] [PubMed] [Google Scholar]
- Caron D, Countway P, Jones A, Kim D, Schnetzer A. Marine protistan diversity. Ann Rev Mar Sci. 2012;4:6.1–6.27. doi: 10.1146/annurev-marine-120709-142802. [DOI] [PubMed] [Google Scholar]
- Edgar RC. Search and clustering orders of magnitude faster than BLAST. Bioinformatics. 2010;26:2460–2461. doi: 10.1093/bioinformatics/btq461. [DOI] [PubMed] [Google Scholar]
- Edgar RC, Haas BJ, Clemente JC, Quince C, Knight R. UCHIME improves sensitivity and speed of chimera detection. Bioinformatics. 2011;27:2194–2200. doi: 10.1093/bioinformatics/btr381. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Faith D. Conservation evaluation and phylogenetic diversity. Biol Conserv. 1992;61:1–10. [Google Scholar]
- Gomez F, Moreira D, Benzerara K, Lopez-Garcia P. Solenicola setigera is the first characterized member of the abundant and cosmopolitan uncultured marine stramenopile group MAST-3. Environ Microbiol. 2011;13:193–202. doi: 10.1111/j.1462-2920.2010.02320.x. [DOI] [PubMed] [Google Scholar]
- Haas BJ, Gevers D, Earl AM, Feldgarden M, Ward DV, Giannoukos G, et al. Chimeric 16S rRNA sequence formation and detection in Sanger and 454-pyrosequenced PCR amplicons. Genome Res. 2011;21:494–504. doi: 10.1101/gr.112730.110. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hamady M, Lozupone C, Knight R. Fast UniFrac: facilitating high-throughput phylogenetic analyses of microbial communities including analysis of pyrosequencing and PhyloChip data. ISME J. 2010;4:17–27. doi: 10.1038/ismej.2009.97. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Han MV, Zmasek CM. PhyloXML: XML for evolutionary biology and comparative genomics. BMC Bioinformatics. 2009;10:356. doi: 10.1186/1471-2105-10-356. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Jardillier L, Zubkov MV, Pearman J, Scanlan DJ. Significant CO2 fixation by small prymnesiophytes in the subtropical and tropical northeast Atlantic Ocean. ISME J. 2010;4:1180–1192. doi: 10.1038/ismej.2010.36. [DOI] [PubMed] [Google Scholar]
- Jurgens K, Massana R.2008Protistan Grazing on Marine Bacterioplankton2nd edn.Wiley-Blackwell: Hoboken, NJ [Google Scholar]
- Koski LB, Golding GB. The closest BLAST hit is often not the nearest neighbor. J Mol Evol. 2001;52:540–542. doi: 10.1007/s002390010184. [DOI] [PubMed] [Google Scholar]
- Lopez-Garcia P, Moreira D. Tracking microbial biodiversity through molecular and genomic ecology. Res Microbiol. 2008;159:67–73. doi: 10.1016/j.resmic.2007.11.019. [DOI] [PubMed] [Google Scholar]
- Martin AP. Phylogenetic approaches for describing and comparing the diversity of microbial communities. Appl Environ Microbiol. 2002;68:3673–3682. doi: 10.1128/AEM.68.8.3673-3682.2002. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Massana R. Eukaryotic picoplankton in surface oceans. Ann Rev Microbiol. 2011;65:91–110. doi: 10.1146/annurev-micro-090110-102903. [DOI] [PubMed] [Google Scholar]
- Massana R, Castresana J, Balague V, Guillou L, Romari K, Groisillier A, et al. Phylogenetic and ecological analysis of novel marine stramenopiles. Appl Environ Microbiol. 2004;70:3528–3534. doi: 10.1128/AEM.70.6.3528-3534.2004. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Massana R, Terrado R, Forn I, Lovejoy C, Pedros-Alio C. Distribution and abundance of uncultured heterotrophic flagellates in the world oceans. Environ Microbiol. 2006;8:1515–1522. doi: 10.1111/j.1462-2920.2006.01042.x. [DOI] [PubMed] [Google Scholar]
- Massana R, Unrein F, Rodriguez-Martinez R, Forn I, Lefort T, Pinhassi J, et al. Grazing rates and functional diversity of uncultured heterotrophic flagellates. ISME J. 2009;3:588–596. doi: 10.1038/ismej.2008.130. [DOI] [PubMed] [Google Scholar]
- Matsen FA, Kodner RB, Armbrust EV. pplacer: linear time maximum-likelihood and Bayesian phylogenetic placement of sequences onto a fixed reference tree. BMC Bioinformatics. 2010;11:538. doi: 10.1186/1471-2105-11-538. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Not F, del Campo J, Balague V, de Vargas C, Massana R. New insights into the diversity of marine picoeukaryotes. PLoS One. 2009;4:e7143. doi: 10.1371/journal.pone.0007143. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Not F, Latasa M, Scharek R, Viprey M, Karleskind P, Balague V, et al. Protistan assemblages across the Indian Ocean, with a specific emphasis on the picoeukaryotes. Deep Sea Res Part I Oceanogr Res Papers. 2008;55:1456–1473. [Google Scholar]
- Orsi W, Edgcomb V, Jeon S, Leslin C, Bunge J, Taylor GT, et al. Protistan microbial observatory in the Cariaco Basin, Caribbean. II. Habitat specialization. ISME J. 2011;5:1357–1373. doi: 10.1038/ismej.2011.7. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Pedrós-Alió C. Marine microbial diversity: can it be determined. Trends Microbiol. 2006;14:257–263. doi: 10.1016/j.tim.2006.04.007. [DOI] [PubMed] [Google Scholar]
- Piwosz K, Pernthaler J. Seasonal population dynamics and trophic role of planktonic nanoflagellates in coastal surface waters of the Southern Baltic Sea. Environ Microbiol. 2010;12:364–377. doi: 10.1111/j.1462-2920.2009.02074.x. [DOI] [PubMed] [Google Scholar]
- Porter TM, Golding GB. Are similarity- or phylogeny-based methods more appropriate for classifying internal transcribed spacer (ITS) metagenomic amplicons. New Phytologist. 2011;192:775–782. doi: 10.1111/j.1469-8137.2011.03838.x. [DOI] [PubMed] [Google Scholar]
- Prokopowich CD, Gregory TR, Crease TJ. The correlation between rDNA copy number and genome size in eukaryotes. Genome. 2003;46:48–50. doi: 10.1139/g02-103. [DOI] [PubMed] [Google Scholar]
- R-Development-Core-Team. R: A Language and Environment for Statistical Computing. R Foundation for Statistical Computing: Vienna, Austria; 2008. [Google Scholar]
- Richards TA, Bass D. Molecular screening of free-living microbial eukaryotes: diversity and distribution using a meta-analysis. Curr Opin Microbiol. 2005;8:240–252. doi: 10.1016/j.mib.2005.04.010. [DOI] [PubMed] [Google Scholar]
- Riisberg I, Orr RJ, Kluge R, Shalchian-Tabrizi K, Bowers HA, Patil V, et al. Seven gene phylogeny of heterokonts. Protist. 2009;160:191–204. doi: 10.1016/j.protis.2008.11.004. [DOI] [PubMed] [Google Scholar]
- Rodriguez-Martinez R, Labrenz M, del Campo J, Forn I, Jurgens K, Massana R. Distribution of the uncultured protist MAST-4 in the Indian Ocean, Drake Passage and Mediterranean Sea assessed by real time quantitative PCR. Environ Microbiol. 2009;11:397–408. doi: 10.1111/j.1462-2920.2008.01779.x. [DOI] [PubMed] [Google Scholar]
- Rodriguez-Martinez R, Rocap G, Logares R, Romac S, Massana R.2012Low evolutionary diversification in a widespread and abundant uncultured protist (MAST-4) Mol Biol Evol(in press). [DOI] [PubMed]
- Schloss PD, Westcott SL, Ryabin T, Hall JR, Hartmann M, Hollister EB, et al. Introducing mothur: open-source, platform-independent, community-supported software for describing and comparing microbial communities. Appl Environ Microbiol. 2009;75:7537–7541. doi: 10.1128/AEM.01541-09. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sherr E, Sherr B.2008Understanding roles of microbes in marine pelagic food webs: a brief historyIn: Kirchman DL (ed.).Microbial Ecology of the Oceans Wiley-Blackwell: Hoboken, NJ; 27–44. [Google Scholar]
- Stamatakis A. RAxML-VI-HPC: maximum likelihood-based phylogenetic analyses with thousands of taxa and mixed models. Bioinformatics. 2006;22:2688–2690. doi: 10.1093/bioinformatics/btl446. [DOI] [PubMed] [Google Scholar]
- Stoeck T, Bass D, Nebel M, Christen R, Jones MD, Breiner HW, et al. Multiple marker parallel tag environmental DNA sequencing reveals a highly complex eukaryotic community in marine anoxic water. Mol Ecol. 2010;19 (Suppl 1:21–31. doi: 10.1111/j.1365-294X.2009.04480.x. [DOI] [PubMed] [Google Scholar]
- Suttle CA. Viruses in the sea. Nature. 2005;437:356–361. doi: 10.1038/nature04160. [DOI] [PubMed] [Google Scholar]
- Takishita K, Miyake H, Kawato M, Maruyama T. Genetic diversity of microbial eukaryotes in anoxic sediment around fumaroles on a submarine caldera floor based on the small-subunit rDNA phylogeny. Extremophiles. 2005;9:185–196. doi: 10.1007/s00792-005-0432-9. [DOI] [PubMed] [Google Scholar]
- Takishita K, Yubuki N, Kakizoe N, Inagaki Y, Maruyama T. Diversity of microbial eukaryotes in sediment at a deep-sea methane cold seep: surveys of ribosomal DNA libraries from raw sediment samples and two enrichment cultures. Extremophiles. 2007;11:563–576. doi: 10.1007/s00792-007-0068-z. [DOI] [PubMed] [Google Scholar]
- Tsui CK, Marshall W, Yokoyama R, Honda D, Lippmeier JC, Craven KD, et al. Labyrinthulomycetes phylogeny and its implications for the evolutionary loss of chloroplasts and gain of ectoplasmic gliding. Mol Phylogenet Evol. 2009;50:129–140. doi: 10.1016/j.ympev.2008.09.027. [DOI] [PubMed] [Google Scholar]
- Zhu F, Massana R, Not F, Marie D, Vaulot D. Mapping of picoeucaryotes in marine ecosystems with quantitative PCR of the 18S rRNA gene. FEMS Microbiol Ecol. 2005;52:79–92. doi: 10.1016/j.femsec.2004.10.006. [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.