

Abstract
Plasmodium falciparum causes approximately 1 million deaths annually. However increasing resistance imposes a continuous threat to existing drug therapies. We previously reported a number of potent and selective triazolopyrimidine-based inhibitors of Plasmodium falciparum dihydroorotate dehydrogenase that inhibit parasite in vitro growth with similar activity. Lead optimization of this series led to the recent identification of a preclinical candidate, showing good activity against P. falciparum in mice. As part of a backup program around this scaffold, we explored heteroatom rearrangement and substitution in the triazolopyrimidine ring and have identified several other ring configurations that are active as PfDHODH inhibitors. The imidazo[1,2-α]pyrimidines were shown to bind somewhat more potently than the triazolopyrimidines depending on the nature of the amino aniline substitution. DSM151, the best candidate in this series, binds with 4-fold better affinity (PfDHODH IC50 = 0.077 μM) than the equivalent triazolopyrimidine and suppresses parasites in vivo in the P. berghei model.
Introduction
Malaria is a persistent and successful global disease that threatens half of the world's population and kills up to a million people each year.1, 2 The developing world has urgently needed new, safe, effective and affordable antimalarial drugs since the demise of chloroquine due to the emergence of resistant P. falciparum strains.3, 4 Strides have been made to develop an array of anti-malarial agents with Artemisinin-based combination therapies (ACTs) providing a major breakthrough, succeeding in more than 90% of the malaria cases. 5–8 However, artemisinin effectiveness appears to be decreasing along Thai-Combodia border threatening these recent gains.9, 10 The limitations of current antimalarial chemotherapy underscore the need for novel drugs, ideally directed against innovative therapeutic targets. The completion of the malaria genome project11 has opened up channels to search for new targets in the parasite. Despite the identification of many essential genes and extensive efforts to understand the biology of the parasite, very few unique validated targets have been identified. Hence, the contemporary challenge is to blend our knowledge of malaria genomics and drug discovery.
Nucleic acid biosynthesis in P. falciparum differs from that in mammals. The parasite relies on purine salvage due to the absence of de novo purine synthesis12–15. The situation is reversed for pyrimidine biosynthesis where the parasite lacks salvage enzymes for preformed pyrimidine bases and nucleosides, and thus relies on the de novo biosynthesis. Plasmodium falciparum dihydroorotate dehydrogenase (PfDHODH), an essential mitochondrial flavin mononucleotide-dependent enzyme, catalyzes the fourth and rate-limiting step in de novo pyrimidine biosynthesis. 16, 17 Recent investigations have revealed that the primary task of the mitochondrial electron transport chain in the parasite is to supply oxidized ubiquinone (CoQ) to DHODH for pyrimidine synthesis, confirming the essential role of DHODH in parasite growth. 18 The efficacy of human DHODH inhibitors for the treatment of rheumatoid arthritis has been well-documented in the literature, demonstrating that DHODH is a druggable target. 19–21 Finally, X-ray structures of hDHODH and Plasmodium DHODH have highlighted an extensive variation in amino acid sequence between the human and malarial enzymes,22–25 thereby, providing the structural basis for the identification of species-specific inhibitors.
A high-throughput screen (HTS) of “drug-like” small molecules, using a colorimetric enzyme assay, led to the identification of a highly potent and selective triazolopyrimidine-based PfDHODH inhibitor 1 (DSM 1, Figure 1), (5-methyl-[1,2,4]triazolo[1,5-a]pyrimidin-7-yl)naphthalene-2-ylamine. 26–281 has potent activity against both PfDHODH (IC50 = 0.047 μM) and P. falciparum parasites in whole cell assays (EC50 = 0.079 μM for clone 3D7), without inhibiting human DHODH. However when administered orally to mice infected with P. berghei, 1 had no anti-parasite activity. The lack of efficacy of 1 was traced to rapid and possible induced metabolism and to species differences in inhibitor potency between PfDHODH and Plasmodium berghei dihydroorotate dehydrogenase (PbDHODH). 29 An integrated lead optimization program combining medicinal chemistry, enzymology and pharmacologic evaluation was utilized to identify a modified analog 2 (DSM 74, Figure 1), 5-methyl-N-(4-(trifluoromethyl)phenyl)- [1,2,4]triazolo[1,5-a]pyrimidin-7-amine, with greater metabolic stability. This compound showed good exposure in vivo after oral dosing and was able to suppress parasitemia in the P. berghei model. 29 However, 2 lacked the potency required of a clinical candidate. Subsequent X-ray structure guided lead optimization of the scaffold identified 3 (DSM265, Figure 1), 2-(1,1-difluoroethyl)-5-methyl-N-[4-(pentafluoro-λ6-sulfanyl)phenyl] [1,2,4]triazolo[1,5-a]pyrimidin-7-amine, which is a potent inhibitor of PfDHODH that is able to kill parasites in vitro and showed efficacy in a rodent model similar to chloroquine. 30, 313 is also metabolically stable and exhibits a long half-life after oral administration in rodents. Based on these strengths 3 has recently been selected as a preclinical drug development candidate.
Figure 1.

Triazolopyrimidines with active anti-malarial activity. The initial HTS hit (1), an analog with improved plasma exposure (2), and analog with optimized plasma exposure and potency (3).
As part of our back up program to identify additional compounds with clinical candidate potential we extended our explorations of the triazolopyrimidine scaffold by applying bioisosteric morphing, a hit optimization strategy to replace the core of a bioactive compound with groups of similar physical or chemical properties without loss of activity. Using this strategy we have synthesized a series of compounds with either modified bridging atoms between the triazolopyrimidine ring and the aromatic moiety (4–5, Figure 2) or heteroatom rearrangements/replacements in the triazolopyrimidine ring (6–11, Figure 3). We initially coupled each new ring system to naphthyl amine (as in 1), but then for active scaffolds we tested some additional aryl amines (12 – 16) to determine if the optimal group in this position remained similar to what we have observed for the triazolopyrimidines. Three additional heteroatom ring configurations were identified that showed submicromolar PfDHODH activity (6, 7 and 9), with the best being the imidazo[1,2-a]pyrimidines (6 and 16, Table 1) having equivalent to more potent binding affinity compared to the matched triazolopyrimidine. The imidazo[1,2-a]pyrimidine 16 was chosen for profiling of its in vivo properties, including pharmacokinetic analysis and efficacy testing in the P. berghei mouse model. Plasma exposure was lower than for the matched triazolopyrimidine, leading to reduced efficacy. The in vitro metabolism data suggested that the imidazo[1,2-a]pyrimidine scaffold is more extensively metabolized than the triazolopyrimidine scaffold in mice, suggesting additional chemistry would be required to exploit the activity of this scaffold. These studies have provided important additional insight into the developing structure activity relationships (SAR) in this promising series of anti-malarial compounds.
Figure 2.
Bioisterically transformed amide mimics 4 and 5.
Figure 3.

A. Bioisosterically transformed scaffolds 6 – 11. B. A virtual library comprising three types of rings with single isosteric changes in 1. Arrows indicate the point of isosteric change. C. Various congeners with multiple isosteric changes in 1. Arrows indicate the sites of structural diversity.
Table 1.
Activity profiles of inhibitors 4–11a
| Compd | ||||||
|---|---|---|---|---|---|---|
| Structure | Compound code names | IC50 (μM) PfDHODH | EC50 (μM) Pf3D7 | IC50 (μM) hDHODH | IC50 (μM) PbDHODH | |
| 1* |
|
DSM1 | 0.047 | 0.076 | > 100 | 0.23 |
| 2* |
|
DSM74 | 0.28 | 0.34 | > 100 | 0.38 |
| 4 |
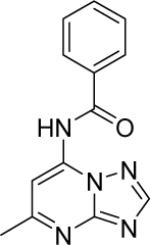
|
DSM154 | > 100 | >100 | > 100 | > 100 |
| 5 |
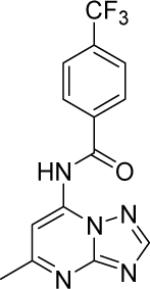
|
DSM155 | > 100 | > 50 | > 100 | > 100 |
| 6 |
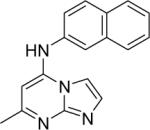
|
DSM71 | 0.15±0.01 | 0.19 (0.15 – 0.25) | > 100 | 2.8±0.28 |
| 7 |
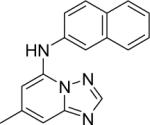
|
DSM96 | 4.8±0.7 | 5.2 (3.6 – 6.7) | n.d. | 6.3±0.80 |
| 8 |
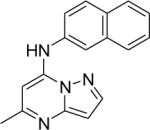
|
DSM68 | 0.4±0.09 | 0.91 (0.80 – 1.0) | > 100 | 3.3±0.4 |
| 9 |
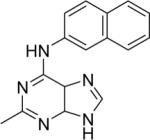
|
DSM23 | 0.44±0.17 | 2.2 (1.2 – 3.7) | > 100 | 11±3.3 |
| 10 |
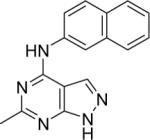
|
DSM70 | > 100 | > 12.5 | n.d. | n.d. |
| 11 |
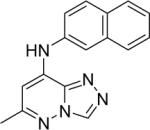
|
DSM105 | > 50 | > 5 | n.d. | >50 |
| 12 |
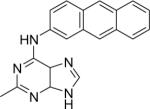
|
DSM25 | 4.8±0.38 | 3.2 (2.4 – 3.5) | n.d. | > 10 |
| 13 |
|
DSM78 | 27±6.3 | > 10 | n.d. | n.d. |
| 14 |
|
DSM79 | >25 | 3.2 (2.4 – 4.3) | n.d. | n.d. |
| 15 |
|
DSM80 | > 100 | > 25 | n.d. | n.d. |
| 16 |
|
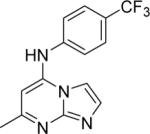
DSM151 | 0.077 ± 0.008 | 0.32 (0.30 – 0.35) | > 100 | 0.17 ± 0.018 |
Chemistry
Amide mimics
In order to test the effects of modifying the linker between the triazolopyrimidine ring and the naphthyl group, the bridging nitrogen N1 was replaced with an amide linker (Figure 2). The target inhibitors 4–5 were synthesized via a three-step sequence as depicted in Scheme 1. The first two steps were relatively straightforward, based on our reported protocol.29 Our initial attempts to perform the coupling reactions of amides with the key intermediate 7-chloro-5-methyl-[1,2,4]triazolo[1,5-a]pyrimidine (17), using traditional conditions like stirring at ambient temperature in absolute ethanol with or without the p/o base, refluxing in toluene, refluxing in absolute ethanol/dichloromethane/N,N-dimethyl formamide, with and without the p/o bases like triethyl amine /anhydrous potassium carbonate, were unsuccessful and rather afforded either the hydroxy-derivative or the ethoxy substituted products. In order to establish a robust synthetic procedure for the targets, the reaction conditions were judiciously optimized using transition metal catalyzed Buchwald-Hartwig conditions.32–35
Scheme 1.
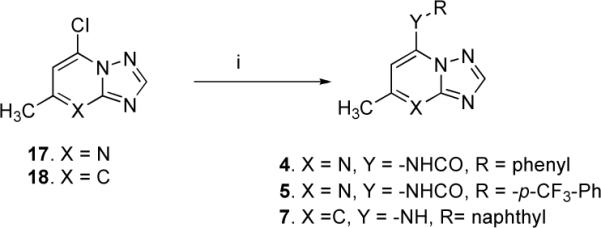
Synthetic Strategy for Inhibitors 4–5 and 7a
aReagent and conditions: (i) RCONH2 (compounds 4–5) and RNH2 (compound 7), Pd(OAc)2, BINAP NaOtBu, anhydrous toluene, 24h reflux, yield 40–50 %
We tried few combinations of 2,2'-bis(diphenylphosphino)−1,1'-binaphthyl (BINAP) as ligand, Pd(OAc)2 as a source of palladium and different reaction variables to determine the efficient catalytic system and found the amidation coupling was optimal using a Pd(OAc)2 (5 mol %)/BINAP (5 mol %) combination, sodium tertiary butoxide (5 molar equivalents) as base. Other stoichiometric bases like potassium carbonate or cesium carbonate worked less well both in terms of yields and reaction progress up to completion. A large excess of base, though almost insoluble in toluene, was essential to obtain high cross-coupling rates with dry toluene as solvent.
The reaction progress was monitored by MS and TLC screening. From a complex reaction mixture, the desired cross-coupled products 4–5 were isolated in optimal yields (40–50%) after silica gel column chromatographic purification.
Heteroatom ring rearrangements and replacements
In order to explore the SAR of the triazolopyrimidine core, we synthesized a series of derivatives to test if the heteroatoms (nitrogens) could be replaced with carbon, resulting in the corresponding heteroaromatic rings with comparable steric and electronic characteristics (Figure 3a–c). Our prime interest was the systematic evaluation of the effects of removal of individual nitrogen atoms from the triazolopyrimidine core to construct imidazo[1,2-a]pyrimidines (6), triazolo[1,5-a]pyridine (7) and pyrazolo[1,5-a]pyrimidine (8) ring systems (Figure 3b), which were further extended to the related congeneric purines (9), pyrazolo[3,4-d]pyrimidine (10) and triazolo[4,3-a]pyridazine (11) (Figure 3c). Herein we report our approaches on the synthesis of these various bioisosteres.
Scaffold hops 6–16
The imidazo[1,2-a]pyrimidine analogs (6 & 16) were readily synthesized as depicted in Scheme 2. The commercially available 2-amino-imidazole hemisulfate (19a) was condensed with ethyl acetoacetate in refluxing glacial acetic acid to afford 5-hydroxy-7-methylimidazo[1,2-a]pyrimidine (20a).36 Chlorination of 20a using phosphorus oxychloride gave the intermediate 5-chloro-7-methylimidazo[1,2-a]pyrimidine (21a). Final conjugation of 21a with 2-aminonaphthalene and 4-(trifluoromethyl)aniline afforded 6 and 16, respectively.
Scheme 2.
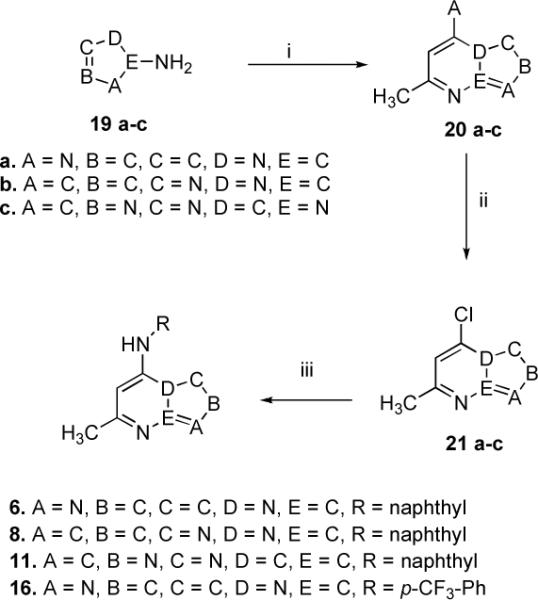
Synthesis of the Inhibitors 6, 8, 11 and 16a
aReagents and conditions : (i) CH3COCH2CO2C2H5, AcOH, reflux, 3.5–25 h, yield 70–85 %; (ii) POCl3, reflux, 40 min-2 h, yield 60–65 %; (iii) RNH2, EtOH, 12–18 h, room temp., yield 65–80 % (compounds 6,8 and 16); Pd(OAc)2, BINAP, NaOt-Bu, anhydrous toluene, 24 h reflux, yield 45 % (compound 11)
The target inhibitor 7 was obtained by coupling 5-chloro-7-methyl-[1,2,4]triazolo[1,5-a]pyridine (18, commercially available) with 2-aminonaphthalene utilizing transition-metal catalyzed conditions (Scheme 1).
Compound 8 was prepared as outlined in Scheme 2. A mixture of 1H-pyrazol-5-amine (19b) and ethyl acetoacetate was refluxed in glacial acetic acid to yield, 7-hydroxy-5-methylpyrazolo[1,5-a]pyrimidine (20b). Subsequent chlorination37, 38 (21b) and coupling with 2-amino naphthalene (Scheme 2) afforded the final adduct 8.
The congeners 9, 10, 11 and 12–15 were prepared as depicted in the Scheme 2 & 3. Regarding purine analogs 9 and 12–15, condensation of 4-amino-1H-imidazole-5-carboxamide hydrochloride salt (22a) with triethyl orthoacetate in refluxing anhydrous dimethylformamide39–41 afforded 4-(methylethoxymethylene)-aminoimidazole-5-carboxamide (23a), which was then cyclized intramolecularly at elevated temperatures to give 2-methylhypoxanthine (24a). Ensuing chlorination and coupling of 25a with variously substituted aromatic amines afforded 9 and 12–15.
Scheme 3.
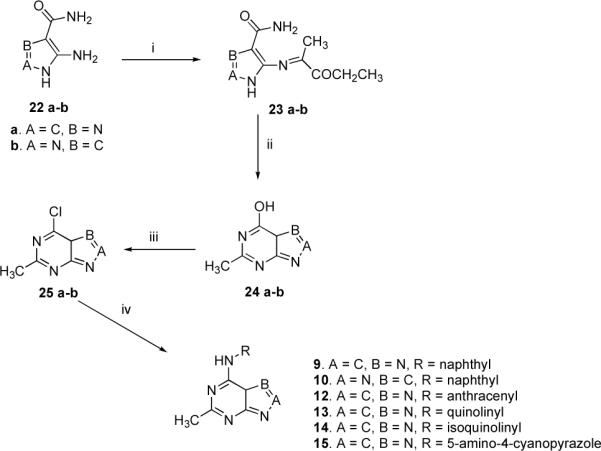
Synthesis of Inhibitors 9, 10 and 12–15a
aReagents and conditions: CH3C(OCH2CH3)3, anhydrous DMF, reflux, 15 min - 1 h, 70–80 %; (ii) heat above 200 °C for 1 h, yield 65–70 %; (iii) POCl3, 30 –45 min, reflux; (iv) RNH2, EtOH, 15–18 h, room temp., yield 68–80 % (compounds 9–12) and 78 % (compound 10)
Pyrazolo[3,4-d]pyrimidine (10) was synthesized from 5-amino-1H-pyrazole-4-carboxamide hemisulfate (22b) as delineated in Scheme 3. Refluxing 22b with triethyl orthoacetate in anhydrous DMF afforded 5-(methylethoxymethylene)-aminopyrazole-4-carboxamide (23b), intramolecular cyclization of which at elevated temperature yielded 4-hydroxy-6-methyl-1H-pyrazolo[3,4-d]pyrimidine (24b). Subsequent chlorination42 and coupling with 2-amino-naphthalene afforded 10.
The triazolopyridazine 11 was synthesized using the reaction sequence highlighted in Scheme 2. 4-amino-1,2,4-triazole (19c) interacted with ethyl acetoacetate in refluxing glacial acetic acid to produce 8-hydroxy-6-methyl-[1,2,4]triazolo[4,3-b]pyridazine (20c), which on chlorination using phosphorus oxychloride, afforded 8-chloro-6-methyl-[1,2,4]triazolo[4,3-b]pyridazine (21c).43 Various conditions using a variety of solvents, bases and temperatures were attempted and found unsuccessful. However, the reaction conditions were optimized utilizing transition metal-catalysis. As previously described (for inhibitors 4–5, Scheme 1), the catalytic combination of Pd(OAc)2 (5 mol%)/BINAP (5 mol%) was found to work well with sodium tertiary butoxide (5 molar equivalents) as base and anhydrous toluene as the solvent (Scheme 2). The reaction temperature and the reaction time were 120 °C and 24 h.
Results and Discussion
Evaluation of compound activity against DHODH and P. falciparum 3D7 cells
The functional evaluation of the target inhibitors 4–16 was carried out on the parasite and host DHODH and on P. falciparum 3D7 cells in whole cell assays (Table 1). The compounds showed a wide range of IC50 values against PfDHODH (0.077 - >100 μM) and these values showed a good correlation with activity against P. falciparum 3D7 cells (Table 1 and Figure 4). All compounds tested were inactive against human DHODH, showing that the selectivity of the series was maintained by variations in the triazolopyrimdine ring. Most compounds also showed better activity against PfDHODH than P. berghei DHODH, similar to previous reports for the series.27, 29, 31, 44 We deduce the following SAR trends by comparing the activity of these compounds against the previously described triazolopyrimidines 1 and 2.
Figure 4.
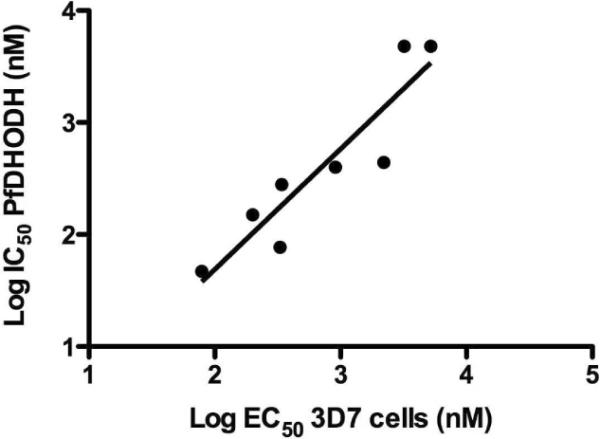
Comparison of the series activity on PfDHODH vs P. falciparum whole cell assays. The log of the PfDHODH IC50 data are plotted versus the log of the P. falciparum EC50 data (in nanomolar). The plotted data include compounds 1, 2, 4–16 described in the Table 1. Compounds for which solubility limits prevented the quantitative determination of either the IC50 or EC50 have been left off the plot. Data were fitted by linear regression analysis where r2 = 0.84 and slope = 0.93.
Replacement of the −NH linker with amide −NHCO (4 and 5) resulted in the loss of activity when an aniline or substituted aniline was adjacent to the amide bond.
Replacement of triazolopyrimidine core with imidazo[1,2-a]pyrimidine was well tolerated, and either slightly reduced or slightly enhanced binding affinity depending on the nature of the aromatic amine. 6 was 3-fold less potent than 1 (IC50 = 0.15 μM vs 0.047 μM), but 16 was 4-fold more potent than 2.
Replacement of the triazolopyrimidine core with triazolo[1,5-a]pyridine (7) resulted in a 100-fold reduction in binding affinity, thereby showing that the pyrimidine nitrogen N5 plays a pivotal role in the observed activity of the triazolopyrimidines.
Substitution of the core with pyrazolo-[1,5-a]pyrimidine (8) reduced the activity by ~ 8.5-fold demonstrating the importance of the N4 nitrogen in the five-membered ring to both the enzyme inhibition and anti parasitic activity.
The purine analog 9 involving bioisosteric replacements at two sites was ~ 9-fold less active than the comparable triazolopyrimidine compound. Additional derivatives of this scaffold were also evaluated by replacing the naphthyl group with anthracene, quinoline, isoquinoline and substituted five-membered pyrazole rings (12, 13, 14 and 15). Exchange of naphthyl with larger moiety like anthracene (12) considerably lessened the activity (~ 87-fold), which is in contrast to what was observed for this replacement in the triazolopyrimdine core structure.29 Further, incorporation of heteroatoms in the naphthyl system (13 and 14) reduced the activity as was previously observed for the triazolopyrimidine scaffold.29 The five-membered 4-cyanopyrazole (15) was inactive.
Bioisosteric modification involving four changes in the purine related analogs i.e. pyrazolo[3,4-d]pyrimidine 10 and triazolopyridazine 11 inactivated the compound.
Loss of N2 in analogs 9, 10 and 11 is also correlated to a significant reduction in activity, suggesting that N2 may also be important to the potency of the compound class. However in all cases these compounds also contained an additional nitrogen in a position not found in the triazolopyrimidines so conformation of the role of N2 through additional synthesis is still required.
Structural basis of activity differences between the triazolopyrimidines and variations in the core
The X-ray structure of key triazolopyrimidine PfDHODH inhibitors, including 1 and 2, was previously reported by our group.23 These structures showed that the triazolopyrimidine ring forms a H-bond interaction between the pyrimidine nitrogen N5 and R265, and between the bridging nitrogen N1 and H185 (Figure 5). These are the only polar interactions formed between the inhibitor and the enzyme, and site-directed mutagenesis confirmed that both interactions were essential for high affinity binding of these compounds. Analysis of the structures described in this manuscript further confirms the importance of these interactions. Compound 7, in which the pyridine nitrogen N5 is replaced with carbon is 100-fold less potent than the corresponding triazolopyrimidine (1), demonstrating that the interaction between the pyrimidine nitrogen N5 and R265 is essential for high affinity binding. In prior studies we noted that introduction of heteratoms into the naphthyl ring reduced activity for the triazolopyrimidine series.26,27 This result holds true for the purine analogs (14 and 15 compared to 9), and is explained by the X-ray structure that shows the naphthyl pocket is completely hydrophobic with no potential to contribute to H-bond interactions. Finally, the structure also provides insight into the good activity of the imidazo[1,2-a]pyrimidines 6 and 16. Nitrogen N3 is within 3.3 Å of NE1 of H185, yet this interaction cannot be productive because NE1 of H185 is an H-bond acceptor and not an H-bond donor. Replacement of N3 with carbon may thus relieve a potentially negative interaction between these groups. It also may lead to the displacement of the structural water observed to form H-bond interactions with N3, H185 and Y525. The entropic gain in releasing the water could also contribute to greater binding affinity of 16 relative to 2. It is unclear why the effect of removing N3 on improving potency was only evident when the imidazo[1,2-a]pyrimidine core was coupled with the smaller para-CF3 aniline and not when it was coupled to naphthyl amine (1 vs 6). This data suggests that the relative contribution of N3 to the electronics of the ring differs depending on the coupled aromatic amine, and that considerations beyond H-bond potential and displacement of the bound water play roles in the potency of these compounds.
Figure 5.
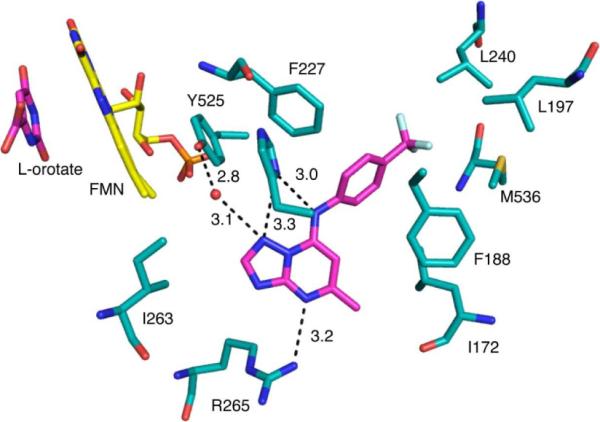
Binding site of 2 bound to PfDHODH. A limited set of residues within the 4Å shell of the bound inhibitor are displayed. 2 and orotate are displayed in pink and FMN is displayed in yellow. Hydrogen bonding distances are displayed in Å. The structure (3I6R.pdb) is displayed in PyMol.
In vitro metabolic stability and PK in mice
The most active compound in the present series 16 was advanced to metabolism and in vivo studies. In vitro metabolic analysis in human liver microsomes for 16 was compared to previous results collected for 2. Both 2 and 16 display low in vitro intrinsic clearance values (Eh) in human liver microsomes, but 16 displayed intermediate clearance in mouse microsomes suggesting that plasma exposure in mice might limit the ability of the compound to show good in vivo efficacy (Table 2). The reason 16 displays higher in vitro intrinsic clearance than 2 is unknown. To evaluate the pharmacokinetic profile of 16, mice were dosed orally with 16 at a single dose of 10 mg/kg (Figure 6A). 16 reached peak plasma concentrations of 3.9 μM after oral dosing, but plasma levels were maintained above 1 μM for only about 4 h and by 16 h concentrations were below the limit of quantitation (0.003 μM). No adverse reactions were observed following oral administration of 16 at the dose used in this study.
Table 2.
In vitro metabolism in human and mouse microsomes
| Compd | Degradation half-life (min) | In vitro CLInt (mL/min/mg protein) | Predicted EH | Putative metabolites |
|---|---|---|---|---|
| Human | ||||
| 2 * | 230 | 0.0075 | 0.29 | P+16 |
| 16 | 368 | 0.0047 | 0.21 | None detected |
| Mouse | ||||
| 16 | 51.8 | 0.034 | 0.59 | None detected |
data were previously reported in Gujjar et al. 29 Data represent single point measurements.
Figure 6.

Pharmacokinetics and in vivo efficacy of 16 in mice. A. Pharmacokinetic profile of 16 after administration of a single oral or IP dose of 10 mg/kg. Drug concentration in micromolar is plotted versus time post dose. B. Mice were infected with P. berghei on day zero and 16 was dosed orally at 50 mg/kg 1× daily for 4 days beginning 24 h after infection. As a control chloroquine was dosed at 10 mg/kg orally using the same dosing schedule.
In vivo efficacy of 16 in P. berghei mouse model
16 was evaluated for efficacy in the standard P. berghei mouse model. Mice were infected with P. berghei on day zero and dosing began 24 h after infection. Mice were dosed orally with 50 mg/kg 16, 1× daily for 4 days, and parasite blood levels were evaluated and compared to a chloroquine control (Figure 6B). 16 suppressed parasite levels by 56% as measured 24 h after the final dose. In comparison 2 was able to suppress parasite levels by 71% using the same dosing scheme and end point.29 Thus despite showing 2-4-fold better potency against PbDHODH and PfDHODH than 2 respectively, 16 is less efficacious than 2 in the mouse model. The relatively poorer plasma exposure observed in mice for 16 most likely contributes to this finding. The finding that the predicted in vitro clearance in mouse microsomes is significantly higher than human microsomes suggests that this compound would perform better against human infection than for mice.
Conclusion
In the quest for better PfDHODH antagonists, we have undertaken a study to systematically explore the importance of the number and configuration of nitrogen atoms in the triazolopyrimidine ring to the potency of the compound class. The studies show that N1, N4 and N5 are functionally important sites as the structural modification at these positions render the molecule less or completely inactive (Figure 7). N3 can be replaced by carbon leading to retained or improved potency; however the N3 nitrogen is a significant factor in the metabolic stability and favorable pharmacokinetics of 2. In particular, 16 was found to be 4-fold more potent than the equivalent triazolopyrimidine 2, however the poorer plasma exposure led to reduced efficacy against the parasite in vivo. The addition of an amide linker between the triazolopyrimidine core and the aniline also led to complete loss of activity. Overall the data have helped to define the minimal pharmacophore of the triazolopyrimidine class of PfDHODH inhibitors, and have highlighted the key importance of the pyrimidine nitrogen N5.
Figure 7.
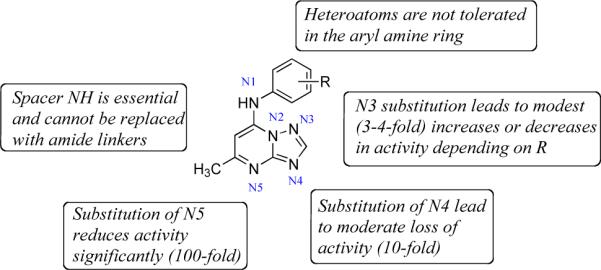
Structure activity map. Structural drawing of the lead core annotated to show where structural changes affect activity or potency.
Experimental
Protein purification and steady-state kinetic analysis
PfDHODH, PbDHODH, and hDHODH enzymes were expressed and purified as previously reported.27, 28, 45 Steady-state kinetic assays were performed as previously described.27, 28, 45 Briefly, this colorimetric assay monitors the reduction of 2,6-dichloroindophenol (DCIP; 0.12 mM) at 600 nm (ε = 18.8 mM−1cm−1), which is coupled to the reoxidation of CoQD (coenzyme QD). The assay was carried out using a solution containing 100 mM HEPES, pH 8.0, 150 mM NaCl, 10 % Glycerol, 0.1 % Triton X-100, 20 micromolar CoQD, 200 micromolar L-dihydroorotate and 120 micromolar DCIP. Reactions were initiated by addition of enzyme to a final concentration in the range of 5–10 nM while maintaining the temperature of the circulating water bath at 20 °C. The data obtained were fitted to Eq 1 using GraphPad Prism, to determine the IC50 values of the representative compounds. IC50 data were determined over a range of inhibitor concentrations using triplicate data points at each concentration and errors represent the standard error of the fit.
| Eq.1 |
P. falciparum cell culture
The malarial parasite clone 3D7 was passaged in Gibco-Invitrogen RPMI-1640 supplemented with 20% human type A+ plasma and 2% (w/v) red blood cells (obtained from Biochemed Services, Virginia).46 To study inhibition of cell proliferation, the standard [3H]-hypoxanthine uptake assay was used to measure drug-treated P. falciparum-infected-erythrocytes as described previously.47 Data were fitted to Eq. 2 to determine EC50 (n = 3 – 4 data points at each concentration). Since the standard error of the fit cannot be calculated for Eq. 2, the parasite data is reported with the 95% confidence interval from the fit.
| Eq.2 |
In vitro human microsomal metabolism
Metabolic stability was evaluated by incubating test compounds (1 μM) individually at 37°C with pooled human or mouse liver microsomes (BD Gentest, Woburn, MA) at a microsomal protein concentration of 0.4 mg/mL. The metabolic reaction was initiated by the addition of an NADPH-regenerating buffer system containing 1 mg/mL NADP, 1 mg/mL glucose-6-phosphate, 1 U/mL glucose-6-phosphate dehydrogenase and 0.67 mg/mL MgCl2, and reactions were quenched at various time points over the incubation period by the addition of ice-cold acetonitrile. The relative loss of parent compound and formation of metabolic products was monitored by LC/MS using a Waters/Micromass QTOF mass spectrometer.
Test compound concentrations were determined by comparison to a calibration curve prepared in microsomal matrix and concentration versus time data were fitted to an exponential decay function to determine the first-order rate constant for substrate depletion. This rate constant was then used to calculate the degradation half-life, in vitro intrinsic clearance (CLint,in vitro), predicted in vivo intrinsic clearance value (CLint), and predicted in vivo hepatic extraction ratio (EH) as previously described,48 and assuming predominantly hepatic cytochrome P450-mediated clearance in vivo. The scaling parameters used in these calculations included a human and mouse hepatic blood flow of 20.7 and 90 mL/min/kg, respectively, liver mass of 25.7 and 87.5 g liver/kg body weight for human and mouse, respectively, and microsomal content of 45 mg microsomal protein/g liver.49, 50
Mouse pharmacokinetics
All animal studies were performed in accordance with the Australian and New Zealand Council for the Care of Animals in Research and Training Guidelines and the study protocol was approved by the Monash University (Victorian College of Pharmacy) Animal Experimentation Ethics Committee. The pharmacokinetics of 16 were studied in non-fasted male Swiss outbred mice weighing 23–33 g. Mice had access to food and water ad libitum throughout the pre- and post-dose sampling period. Compound was administered orally by gavage (0.1 mL dose volume per mouse) in a suspension of aqueous vehicle (0.5% w/v sodium caboxymethylcellulose, 0.5% v/v benzyl alcohol and 0.4% v/v Tween 80). Samples were collected at 1, 2, 4, 7.5, 16 and 24 h post-dosing with a single blood sample from each mouse via cardiac puncture. Blood samples were transferred to heparinised tubes, centrifuged immediately, and supernatant plasma removed and stored at −20°C until analysis (within 1–2 weeks). Plasma samples were assayed by LC-MS following protein precipitation with acetonitrile and the concentration of drug in plasma was determined by comparison of the peak area to a calibration curve prepared in plasma.
P. berghei mouse in vivo efficacy testing
All animal care and experimental procedures were approved by the Institutional Animal Care and Use Committee at the University of Washington. Eight to ten week old female BALB/c mice were obtained from Jackson Laboratories (Bar Harbor, ME, USA) and maintained in a temperature/humidity controlled SPF facility with a 12 hour light/dark cycle. Plasmodium berghei isolate NK65 was obtained from MR4 (Malaria research and Reference Resource Center, Monassas, VA, USA). Erythrocytic stages of P. berghei were harvested from infected donor mice via cardiac puncture and diluted in RPMI 1640 media (ATCC, Monassas, VA, USA) supplemented with 20% heat inactivated FBS (Altanta Biologicals, Atlanta, GA, USA). Recipient mice were infected with 2×107 infected erythrocytes, 200 μL IP injection, 24 hours prior to receiving the first drug dose. Tail vein bleeds and weighing were performed daily for the duration of the experiment. All infected mice were screened prior to receiving drug or vehicle to verify an established minimum infection of 0.1% parasitemia. Inhibition of parasite growth is determined microscopically by staining a thin smear of blood obtained from the test animal using a Wright-Giemsa stain (Fisher Scientific, Houston, TX, USA). 16 was dosed in mice orally with vehicle or with 50 mg/kg/dose 1 × daily for 4 consecutive days. The parasite blood levels were evaluated and compared to the chloroquine control.
Molecular Modeling
Structures were displayed using the graphics program PyMol (DeLano, W. L. The PyMOL Molecular Graphics System (2002) on World Wide Web http://www.pymol.org).
Chemistry: General Methods
If not otherwise specified, reagents and solvents were obtained from commercial suppliers and were used without further purification. The reaction progress was monitored by thin layer chromatography (TLC) using silica gel 60 F-254 (0.25 mm) plates. Visualization was achieved with UV light and iodine vapor. Flash chromatography was carried out with silica gel (32–63 μM). The procedures for all the synthesized compounds have been provided along with relevant literature and their discussion as illustrated in the Schemes 1–3. 7-Chloro-5-methyl-[1,2,4]triazolo[1,5-a]pyrimidine (12) was synthesized as per the previously reported procedure.29 All aromatic amines or amides used in the reactions were obtained from commercial sources except naphthamide, which was synthesized according to the documented procedure.51
Analysis
1H NMR spectra were recorded on dilute solutions in CDCl3 or DMSO-d6 or MeOD-d4 at 300 MHz. Chemical shifts (δ) are reported in ppm relative to TMS and coupling constants (J) are reported in Hz. Spin multiplicities are described as s (singlet), brs (broad singlet), d (doublet), t (triplet), q (quartet) and m (multiplet). Electronspray ionization mass spectra (ESI-MS) were acquired on a Bruker Esquire liquid chromatograph-ion trap (LC-MS) mass spectrometer. Melting points (Pyrex capillary) were determined on a Mel-Temp apparatus and are not corrected. High resolution mass spectrometry (FABHRMS) data were obtained on JEOL HX-110 mass spectrometer for compound 16. Purity for all compounds in Table 1 was assessed by LC-MS. Chromatography was performed on a ZorbaxExtend C18 (80A) column using buffer A (water/5% acetonitrile/1% acetic acid) and buffer B (acetonitrile/1% acetic acid) with a 20 min gradient from 20–100% buffer B, or on an Agilent Exclipse XDB-C18 5 mm (dimensions 4.6 × 150 mm) using acetonitrile/water (0.1%formic acid), and a step gradient of 20%, 50%, 90% acetonitrile at 0, 10, 20 min respectively. Absorbance was monitored at 254 nm. Compound purity was confirmed to be >95% with the exception of compounds 5, 7 and 15 where purity was determined to be 86 – 90%. Given the poor activity of these compounds they were not of interest for further development and as the results would not be impacted by the marginal improvement in purity to bring them to 95%, additional purification was not performed.
Amide Mimics
General Procedure
To a suspension of Pd(OAc)2 (67 mg, 0.30 mmol), BINAP (185 mg, 0.30 mmol) and sodium tertiary butoxide (2.8 g, 29.70 mmol) in anhydrous toluene (25 mL), was added 7-chloro-5-methyl-[1,2,4]triazolo[1,5-a]pyrimidine (17) (1 g, 5.93 mmol) and then aromatic amides [4-(trifluoromethyl)benzamide (1.3 g, 6.56 mmol) and benzamide (0.8 g, 6.54 mmol)]. The reaction mixture was stirred for 24 h at 120 °C and the reaction was monitored by LC/MS until no starting material remained. The mixture was then filtered and the residue was washed with dichloromethane (2 × 10 mL). The dichloromethane parts were dried over anhydrous sodium sulfate and subsequently concentrated in vacuo. The crude material was purified by flash column chromatography (on silica-gel with Ethyl acetate:hexane ∷ 3:2) to afford the adducts [4 (646 mg, 43%) and 5 (918 mg, 48%)] as white solids.
N-(5-methyl-[1,2,4]triazolo[1,5-a]pyrimidin-7-yl)benzamide (4)
Mp (°C): 201. 1H NMR (300 MHz, MeOD-d4) δ (ppm): 8.50 (s, 1H), 8.09 (s, 1H), 8.06 (m, 1H), 8.03 (s, 1H), 7.74-7.59 (m, 3H), 2.72 (s, 3H). ESI-MS (m/z): 254.0 (M+).
N-(5-methyl-[1,2,4]triazolo[1,5-a]pyrimidin-7-yl)-4-trifluoromethyl)benzamide (5)
Mp (°C): 187. 1H NMR (300 MHz, MeOD-d4) δ (ppm): 8.51 (s, 1H), 8.24 (d, 2H, J = 8.1 Hz), 8.07 (s, 1H), 7.94 (d, 2H, J = 8.1 Hz), 2.74 (s, 3H); ESI-MS (m/z): 320.1 (M−) [ionizes better in negative mode].
Imidazo[1,2-a]pyrimidine derivarives
General Procedure. 5-Hydroxy-7-methylimidazo[1,2-a]pyrimidine (20a)
A solution of 2-aminoimidazole hemisulfate (19a) (1 g, 7.58 mmol) and ethyl acetoacetate (1.48 g, 11.37 mmol) in glacial acetic acid (15 mL) was refluxed for 25 h. Upon cooling and subsequent evaporation of the excess liquid under suction, a light brownish solid was obtained, filtered, washed with dichloromethane several times and dried under vacuum to give the product 20a (0.790 g, 70%).
Mp (°C): 242. 1H NMR (300 MHz, DMSO-d6) δ (ppm): 7.58 (d, 1H, J = 2.4 Hz), 7.36 (d, 1H, J = 2.4 Hz), 5.76 (s, 1H), 2.26 (s, 1H); ESI-MS (m/z): 150.1 (M+).
5-Chloro-7-methylimidazo[1,2-a]pyrimidine (20a)
A mixture of the hydroxy-intermediate 20a (0.790 g, 5.30 mmol) and phosphorus oxychloride (14 mL, 150.10 mmol) formed a clear red solution upon refluxing for 2 h. The excess phosphorus oxychloride was removed in vacuo and the residue was precipitated using dichloromethane. The precipitates were filtered, washed several times with dichloromethane and dried under vacuum to yield the chlorinated intermediate 21a (577 mg, 65%), which was used without purification for subsequent steps.
7-Methyl-N-(naphthalen-2-yl)imidazo[1,2-a]pyrimidin-5-amine (6)52
2-aminonaphthalene (490 mg, 3.42 mmol) was added to a well-stirred solution of 21a (522 mg, 3.12 mmol) in excess of absolute ethanol. The reaction mixture was heated intermittently yielding a clear solution. Stirring was then continued overnight at ambient temperature. Excess ethanol was removed by evaporation and the crude product was subjected to flash chromatographic purification (on silica-gel with dichloromethane:methanol:ammonium hydroxide ∷ 240:3:2) to yield 5 (546 mg, 70%). Mp (°C) > 250. 1H NMR (300 MHz, DMSO-d6) δ (ppm): 7.98-7.91 (m, 5H), 7.62-7.41 (m, 4H), 5.72 (s, 1H), 2.31 (s, 3H); ESI-MS (m/z): 276.1 (M+).
7-Methyl-N-(4-(trifluoromethyl)phenyl)imidazo[1,2-a]pyrimidin-5-amine (16).53
To a well-stirred solution of 21a (522 mg, 3.12 mmol) in excess absolute ethanol, was added 4-(trifluoromethyl)aniline (552 mg, 3.42 mmol) with intermittent heating yielding a clear solution. Stirring was then continued for 24 h at room temperature, excess ethanol removed by evaporation and the crude product purified by flash chromatography (on silica-gel with dichloromethane:methanol:ammonium hydroxide ∷ 240:3:2) to yield 16 (555 mg, 65%). Mp (°C): 292. 1H NMR (300 MHz, DMSO-d6) δ (ppm): 7.74 (d, 1H, J = 1.5 Hz), 7.67 (d, 2H, J = 8.7 Hz), 7.35 (d, 1H, J = 1.8 Hz), 7.26 (d, 2H, J = 8.4 Hz), 5.88 (s, 1H), 2.24 (s, 3H); ESI-MS (m/z): 293.2 (M+).
7-Methyl-N-(naphthalen-2-yl)-[1,2,4]triazolo[1,5-a]pyridin-5-amine (7)
General Procedure. To a suspension of Pd(OAc)2 (16.75 mg, 0.075 mmol), BINAP (46.44 mg, 0.075 mmol) and sodium tertiary butoxide (715 mg, 7.45 mmol) in 20 mL anhydrous toluene, was added 5-Chloro-7-methyl-[1,2,4]triazolo[1,5-a]pyridine (18, commercially obtained) (250 mg, 1.49 mmol) and then 2-amino naphthalene (235 mg, 1.64 mmol). The reaction mixture was refluxed for 24 h, subsequently cooled to room temperature and then concentrated in vacuo. The crude material was purified by flash column chromatography (on silica-gel using ethyl acetate:hexane ∷ 3:2) to afford 7 (172 mg, 42%) as creamish white solid. Mp (°C): 154. 1H NMR (300 MHz, CDCl3) δ (ppm): 8.41 (s, 1H); 7.97-7.82 (m, 4H), 7.73 (s, 1H), 7.61-7.45 (m, 3H), 7.17 (s, 1H), 6.60 (s, 1H), 2.46 (s, 3H). ESI-MS (m/z): 275.2 (M+).
Pyrazolo[1,5-a]pyrimidine
General Procedure. 7-Hydroxy-5-methylpyrazolo[1,5-a]pyrimidine (20b)
A mixture of 1H-pyrazol-5-amine (19b) (1.66 g, 20.00 mmol) and ethyl acetoacetate (2.7 mL, 2.8 g, 22.00 mmol) in glacial acetic acid (15 mL) was heated under reflux for 20 h. After the reaction mixture cooled, the excess acetic acid was removed in vacuo. The precipitated solid was then filtered, washed with ethanol and dried under vacuum to give the 20b (2.55 g, 85%) as white solid. Mp (°C): 295. 1H NMR (300 MHz, DMSO-d6) δ (ppm): 7.82 (s, 1H), 6.10 (s, 1H), 5.56 (s, 1H), 2.28 (s, 3H); ESI-MS (m/z): 150.1 (M+).
7-Chloro-5-methylpyrazolo[1,5-a]pyrimidine (21b)
A solution of phosphorus oxychloride (2.0 mL, 21.40 mmol) and 20b (149 mg, 1.00 mmol) was heated under reflux for 40 minutes. The reaction mixture was then brought to room temperature, excess reagent removed in vacuo and the residue triturated with ice-water. The chlorinated product was extracted from the aqueous mixture using dichloromethane. The organic layer was separated, dried over anhydrous sodium sulfate and then filtered. The filtrate was concentrated and purified by flash chromatography (silica-gel with ethyl acetate:hexane ∷ 3:2) to afford 21b (75 mg, 45%). Mp (°C): 38. 1H NMR (300 MHz, CDCl3) δ (ppm): 8.18 (s, 1H), 6.87 (s, 1H), 6.69 (s, 1H), 2.62 (s, 3H); ESI-MS (m/z): 168.5 (M+).
5-Methyl-N-(naphthalen-2-yl)pyrazolo[1,5-a]pyrimidin-7-amine (8)52
2-amino naphthalene (57 mg, 0.40 mmol) was added to the stirred solution of 21b (60 mg, 0.36 mmol) dissolved in absolute ethanol (20 mL). Stirring was then continued for 8 h at room temperature. Excess ethanol was removed by evaporation and the crude product was subjected to flash chromatographic purification (on silica-gel using dichloromethane:methanol:ammonium hydroxide ∷ 100:1:1) to yield 7 in quantitative yields (77 mg, 78%). Mp (°C): 136. 1H NMR (300 MHz, DMSO-d6): δ 10.01 (brs, NH exchangeable), 8.16 (s, 1H), 8.04-7.93 (m, 4H), 7.66-7.62 (m, 1H), 7.59-7.48 (m, 2H), 6.43 (s, 1H), 6.34 (s, 1H), 2.38 (s, 3H); ESI-MS (m/z): 275.1 (M+).
Triazolo[4,3-b]pyridazine
General Procedure. 8-Hydroxy-6-methyl-[1,2,4]triazolo[4,3-b]pyridazine (20c)
A well-stirred mixture of 4H-1,2,4-triazol-4-amine (500 mg, 5.95 mmol) and ethyl acetoacetate (1.16 g, 8.93 mmol) in glacial acetic acid (20 mL) was refluxed at 170 °C for 4 h. The reaction mixture was brought to room temperature and excess solvent evaporated. The residue was filtered using dichloromethane and was recrystallized from aqueous acetic acid to give 20c as white solid (750 mg, 84%). Mp (°C): 306. 1H NMR (300 MHz, DMSO-d6) δ (ppm): 9.42 (s, 1H,-OH), 6.36 (s, 1H), 2.41 (s, 3H); ESI-MS (m/z): 151.1 (M+).
8-Chloro-6-methyl-[1,2,4]triazolo[4,3-b]pyridazine (21c)
A well-stirred solution of 20c (500 mg, 3.34 mmol) in phosphorus oxychloride (10 mL, 107.01 mmol) was refluxed for 45 minutes. After completion, the reaction mixture was concentrated in vacuo to give a red residue, which was precipitated using dichloromethane to give 21c (340 mg, 62%). The chlorinated intermediate was used for the subsequent step in the crude form. Mp (°C): 181.
6-Methyl-N-(naphthalen-2-yl)-[1,2,4]triazolo[4,3-b]pyridazin-8-amine (11)
To a suspenstion of Pd(OAc)2 (22.65 mg, 0.10 mmol), BINAP (62.78 mg, 0.10 mmol) and sodium tertiary butoxide (968 mg, 10.00 mmol) in anhydrous toluene (20 mL), was added 20c (340 mg, 2.02 mmol) and then 2-naphthyl amine (318 mg, 2.22 mmol). The mixture was stirred overnight at 120 °C and then concentrated in vacuo. The crude material was purified by flash column chromatography (on silica-gel with ethyl acetate:hexane ∷11:9] to afford 11 (252 mg, 45%) as white solid. Mp (°C): 195. 1H NMR (300 MHz, DMSO-d6) δ (ppm): 10.10 (brs, NH exchangeable), 9.44 (s, 1H), 8.00-7.91 (m, 4H), 7.67-7.63 (m, 1H), 7.56-7.46 (m, 2H), 6.60 (s, 1H), 2.38 (s, 3H). ESI-MS (m/z): 276.2 (M+).
Purine analogs
General Procedure. 4-(Methylethoxymethylene)-aminoimidazole-5-carboxamide (23a)
A mixture of 5-amino-1H-imidazole-4-carboxamide hydrochloride (22a) (1g, 6.15 mmol), triethylorthoacetate (2 mL, 10.98 mmol) in anhydrous N,N-dimethyl formamide (5 mL) was heated under reflux at 180 °C for 15 minutes yielding a clear solution that was cooled. Excess solvent was removed under suction and the filtration provided 23a as light brown solid (0.949 g, 80%). Mp (°C): 248. 1H NMR (300 MHz, DMSO-d6) δ (ppm): 12.6 (brs, NH exchangeable), 7.56 (s, 1H), 7.31 (s, 2H, -CONH2), 4.16 (q, 2H), 2.29 (s, 3H), 1.28 (t, 3H); ESI-MS (m/z): 197.1 (M+).
2-Methylhypoxanthine (24a)
Intermediate 23a (1 g, 5.11 mmol) was heated at 200 °C for 30 minutes and the product was recrystallized from water to afford 24a (612 mg, 80%) as greyish solid. Mp (°C): > 300. 1H NMR (300 MHz, DMSO-d6) δ (ppm): 13.10 (brs, NH exchangeable), 12.15 (s, 1H, -OH), 8.12 (s, 1H), 2.35 (s, 3H); ESI-MS (m/z): 151.2 (M+).
6-Chloro-2-methyl-9H-purine (25a)
N,N-dimethylaniline (3.3 mL, 26.07 mmol) was added to a suspension of 24a (1g, 6.67 mmol) in phosphorus oxychloride (15 mL, 214.00 mmol) and the reaction mixture was refluxed for 1 h. Excess reagent was distilled off under reduced pressure and the syrupy residue was poured onto finely crushed ice. The aqueous solution was extracted with diethyl ether in order to remove N,N-dimethyl aniline. The ethereal solution was washed 3–4 times with cold water, dried over anhydrous sodium sulfate and distilled to afford 25a (728 mg, 65%) [Best results were obtained using sufficient volume of ether and rapidly extracting it with cold water].
Purine analogs (9, 12–15). General Procedure
Aromatic amines [2-aminonaphthalene (186.86 mg, 1.31 mmol), 2-aminoanthracene (252.10 mg, 1.31 mmol), 3-aminoquinoline (188.16 mg, 1.39 mmol), 6-aminoquinoline (188.16 mg, 1.39 mmol) and 3-amino-1H-pyrazole-4H-carbonitrile (140.92 mg, 1.30 mmol)] were added to a well-stirred solution of 25a (200 mg, 1.186 mmol) in absolute ethanol (20 mL) and stirring was continued for 15–18 h at ambient temperature. Excess of ethanol was removed by evaporation and the crude product were purified via recrystallization technique to afford 2-methyl-N-(naphthalen-2-yl)-9H-purin-6-amine (9; yield: 246 mg, 75%), N-(anthracen-2-yl)-2-methyl-9H-purin-6-amine (12; yield: 310 mg, 80%), N-(2-methyl-9H-purin-6-yl)quinolin-3-amine (13; yield: 257 mg, 78%), N-(2-methyl-9H-purin-6-yl)quinolin-6-amine (14; yield: 263 mg, 80%) and 3-(2-methyl-9H-purin-6-ylamino)-1H-pyrazole-4-carbonitrile (15; yield: 195 mg, 68%) [Recrystallization solvents‷ ethanol (for 9 & 12) and dichloromethane:methanol ‷ 2:1 (for 13–15)].
2-Methyl-N-(naphthalen-2-yl)-9H-purin-6-amine (9)52
Mp (°C) > 300. 1H NMR (300 MHz, DMSO-d6) δ (ppm): 10.45 (brs, NH exchangeable), 8.54 (s, 1H), 8.43 (s, 1H), 7.94-7.87 (m, 4H), 7.54-7.42 (m, 2H), 2.64 (s, 3H); ESI-MS (m/z): 276.2 (M+).
N-(Anthracen-2-yl)-2-methyl-9H-purin-6-amine (12)
Mp (°C) > 320 (decomp). 1H NMR (300 MHz, DMSO-d6) δ (ppm): 11.95 (bs, NH exchangeable), 8.85 (s, 1H), 8.71 (s, 1H), 8.65-8.52 (m, 2H), 8.21-7.93 (m, 4H), 7.65-7.43 (m, 2H), 2.79 (s, 3H); ESI-MS (m/z): 326.1 (M+).
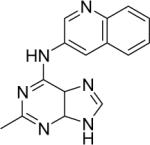
N-(2-Methyl-9H-purin-6-yl)quinolin-3-amine (13)
Mp (°C) 290. 1H NMR (300 MHz, DMSO-d6) δ (ppm): 11.98 (brs, NH exchangeable), 9.43 (s, 1H), 9.07 (s, 1H), 8.63 (s, 1H), 8.07-8.05 (m, 2H), 7.78-7.67 (m, 2H), 2.71 (s, 3H); ESI-MS (m/z): 277.1 (M+).
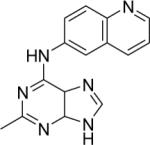
N-(2-Methyl-9H-purin-6-yl)quinolin-6-amine (14)
1H NMR (300 MHz, DMSO-d6) δ (ppm): 10.29 (brs, NH exchangeable), 8.98 (m, 1H), 8.92 (s, 1H), 8.73 (d, 1H, J = 8.1 Hz), 8.53 (s, 1H), 8.44 (m, 1H), 8.19 (d, 1H, J = 8.7 Hz), 7.77 (m, 1H), 2.70 (s, 3H, CH3). ESI-MS (m/z): 277.2 (M+).
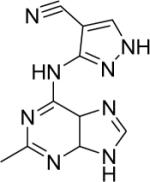
3-(2-Methyl-9H-purin-6-ylamino)-1H-pyrazole-4-carbonitrile (15)
1H NMR (300 MHz, DMSO-d6) δ (ppm): 2.62 (s, CH3), 8.58 (s, 1H), 8.63 (s, 1H). ESI-MS (m/z): 241.1 (M+).
Pyrazolo[3,4-d]pyrimidin-4-amine
General Procedure. 5-(Methylethoxymethylene)-aminopyrazole-4-carboxamide (23b)
A well-stirred solution of 5-amino-1H-pyrazole-4-carboxamide hemisulfate (22b) (1 g, 5.70 mmol) and triethylorthoacetate (1.72 mL, 9.44 mmol) in 5 mL of anhydrous dimethyl formamide was heated under reflux for 1 h at 240 °C and the resulting solution was cooled. The excess solvent was removed by evaporation with subsequent filtration providing 23b (1.38 g, 75%) as light brown solid. Mp (°C): 148 (decomp). 1H NMR (300 MHz, DMSO-d6) δ (ppm): 8.01 (s, 1H), 7.25 (d, 2H, -CONH2), 4.25 (q, 2H), 2.21 (s, 3H), 1.35 (t, 3H); ESI-MS (m/z): 197.1 (M+).
4-Hydroxy-6-methyl-1H-pyrazolo[3,4-d]pyrimidine (24b)
The intermediate 23b (1 g, 5.10 mmol) was heated at 250 °C for 2 hours and the residue was filtered using dichloromethane. The crude product was recrystallized from water to afford 24b (535 mg, 70%) as greyish solid. Mp (°C): > 300. 1H NMR (300 MHz, DMSO-d6) δ (ppm): 7.96 (s, 1H); 2.34 (s, 3H); ESI-MS (m/z): 151.1 (M+).
4-Chloro-6-methyl-1H-pyrazolo[3,4-d]pyrimidine (25b)
A mixture of 24b (500 mg, 3.33 mmol), N,N-dimethylaniline (1.7 mL) and phosphorus oxychloride (8 mL) was refluxed for 1 h till all the solid went into solution. The excess reagent was distilled under reduced pressure and the syrupy residue was poured slowly, with vigorous stirring, onto finely crushed ice. The mixture was allowed to stand for 20 minutes and the aqueous suspension was extracted with ether. The ethereal extract was washed well with water. After drying the extract over anhydrous sodium sulfate, the organic layer was distilled to yield chlorinated adduct 25b (392 mg, 70%) as yellow powder, used in this form for subsequent step.
6-Methyl-N-(naphthalen-2-yl)-1H-pyrazolo[3,4-d]pyrimidin-4-amine (10)
To a well-stirred solution of 25b (200 mg, 1.19 mmol) in 20 mL absolute ethanol, was added 2-aminonaphthalene (186.97 mg, 1.31 mmol). The stirring was continued for 12 h at ambient temperature, excess of ethanol removed by evaporation and the crude product purified via recrystallization using ethanol to afford 10 (133 mg, 78%) as light yellow powder. Mp (°C): > 300. 1H NMR (300 MHz, DMSO-d6): δ 8.46 (s, 1H), 7.93-8.00 (m, 5H), 7.50-7.59 (m, 2H), 2.64 (s, 3H); ESI-MS (m/z): 276.1 (M+).
ACKNOWLEDGEMENTS
The authors would like to recognize the late Dr. Ian Bathurst from Medicines for Malaria Venture (MMV) for his helpful support of the project. We also thank Drs. David Floyd and David Matthews of the (MMV) Expert Scientific Advisory Committee for helpful discussions, and Rajan K Paranji and Martin Sadilek for analytical support. This work was supported by the United States National Institutes of Health grants R01AI53680 (to MAP and PKR) and U01AI075594 (to MAP, PKR, SAC). MAP acknowledges support of the Welch Foundation (I-1257) and PKR also acknowledges a Grand Challenge Explorations Award from the Bill and Melinda Gates Foundation. MAP holds the Carolyn R. Bacon Professorship in Medical Science and Education and the Beatrice and Miguel Elias Distinguished Chair in Biomedical Science Beatrice and Miguel Elias Distinguished Chair in Biomedical Science.
ABBREVIATIONS USED
- PfDHODH
Plasmodium falciparum dihydroorotate dehydrogenase
- PbDHODH
Plasmodium berghei dihydroorotate dehydrogenase
- hDHODH
human dihydroorotate dehydrogenase
- HTS
high throughput screening
- BINAP
2,2'-bis(diphenylphosphino)-1,1'-binaphthyl
- CoQ
ubiquinone
- FMN
flavin mononucleotide
- ACTs
Artemisinin-based combination therapies
- SAR
structure activity relationships
- DCIP
2,6-dichloroindophenol
- TLC
thin layer chromatography
- HPLC/MS
high performance liquid chromatography/mass spectrometry
Footnotes
The authors declare no competing financial interest.
REFERENCES
- 1.Greenwood BM, Fidock DA, Kyle DE, Kappe SH, Alonso PL, Collins FH, Duffy PE. Malaria: progress, perils, and prospects for eradication. J Clin Invest. 2008;118(4):1266–76. doi: 10.1172/JCI33996. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2.Guerra CA, Gikandi PW, Tatem AJ, Noor AM, Smith DL, Hay SI, Snow RW. The limits and intensity of Plasmodium falciparum transmission: implications for malaria control and elimination worldwide. PLoS Med. 2008;5(2):e38. doi: 10.1371/journal.pmed.0050038. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.White NJ. Antimalarial drug resistance. J Clin Invest. 2004;113(8):1084–92. doi: 10.1172/JCI21682. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.White N. Drug resistance in malaria. British Medical Bulletin. 1998;54:703–715. doi: 10.1093/oxfordjournals.bmb.a011721. [DOI] [PubMed] [Google Scholar]
- 5.Olliaro P, Wells TN. The global portfolio of new antimalarial medicines under development. Clin Pharmacol Ther. 2009;85(6):584–95. doi: 10.1038/clpt.2009.51. [DOI] [PubMed] [Google Scholar]
- 6.Burrows JN, Chibale K, Wells TN. The state of the art in anti-malarial drug discovery and development. Curr Top Med Chem. 2011;11(10):1226–54. doi: 10.2174/156802611795429194. [DOI] [PubMed] [Google Scholar]
- 7.Wells TN, Alonso PL, Gutteridge WE. New medicines to improve control and contribute to the eradication of malaria. Nat Rev Drug Discov. 2009;8(11):879–91. doi: 10.1038/nrd2972. [DOI] [PubMed] [Google Scholar]
- 8.Eastman RT, Fidock DA. Artemisinin-based combination therapies: a vital tool in efforts to eliminate malaria. Nat Rev Microbiol. 2009;7(12):864–74. doi: 10.1038/nrmicro2239. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Dondorp AM, Yeung S, White L, Nguon C, Day NP, Socheat D, von Seidlein L. Artemisinin resistance: current status and scenarios for containment. Nat Rev Microbiol. 2010;8(4):272–80. doi: 10.1038/nrmicro2331. [DOI] [PubMed] [Google Scholar]
- 10.Petersen I, Eastman R, Lanzer M. Drug-resistant malaria: molecular mechanisms and implications for public health. FEBS Lett. 2011;585(11):1551–62. doi: 10.1016/j.febslet.2011.04.042. [DOI] [PubMed] [Google Scholar]
- 11.Gardner MJ, Hall N, Fung E, White O, Berriman M, Hyman RW, Carlton JM, Pain A, Nelson KE, Bowman S, Paulsen IT, James K, Eisen JA, Rutherford K, Salzberg SL, Craig A, Kyes S, Chan MS, Nene V, Shallom SJ, Suh B, Peterson J, Angiuoli S, Pertea M, Allen J, Selengut J, Haft D, Mather MW, Vaidya AB, Martin DM, Fairlamb AH, Fraunholz MJ, Roos DS, Ralph SA, McFadden GI, Cummings LM, Subramanian GM, Mungall C, Venter JC, Carucci DJ, Hoffman SL, Newbold C, Davis RW, Fraser CM, Barrell B. Genome sequence of the human malaria parasite Plasmodium falciparum. Nature. 2002;419(6906):498–511. doi: 10.1038/nature01097. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Gero A, O'Sullivan W. Purines and pyrimidines in malarial parasites. Blood Cells. 1990;16:467–484. [PubMed] [Google Scholar]
- 13.Gutteridge W, Trigg P. Incorporatin of radioactive precursors into DNA and RNA of Plasmodium knowlesi in vitro. J. Protozool. 1970;17:89–96. doi: 10.1111/j.1550-7408.1970.tb05163.x. [DOI] [PubMed] [Google Scholar]
- 14.Reyes P, Rathod P, Sanchez D, Mrema J, Rieckmann K, Heidrich H. Enzymes of purine and pyrimidine metabolism from the human malaria parasite, Plasmodium falciparum. Mol. Biochem. Parasitol. 1982;5:275–290. doi: 10.1016/0166-6851(82)90035-4. [DOI] [PubMed] [Google Scholar]
- 15.Sherman I. Biochemistry of Plasmodium. Microbiol. Reviews. 1979;43:453–495. doi: 10.1128/mr.43.4.453-495.1979. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Jones M. Pyrimidine nucleotide biosynthesis in animals: genes, enzymes, and regulation of UMP biosynthesis. Annu Rev Biochem. 1980;49:253–279. doi: 10.1146/annurev.bi.49.070180.001345. [DOI] [PubMed] [Google Scholar]
- 17.Nagy M, Lacroute F, Thomas D. Divergent evolution of pyrimidine biosynthesis between anaerobic and aerobic yeasts. Proc. Natl. Acad. Sci. USA. 1992;89:8966–8970. doi: 10.1073/pnas.89.19.8966. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Painter HJ, Morrisey JM, Mather MW, Vaidya AB. Specific role of mitochondrial electron transport in blood-stage Plasmodium falciparum. Nature. 2007;446(7131):88–91. doi: 10.1038/nature05572. [DOI] [PubMed] [Google Scholar]
- 19.Goldenberg M. Leflunomide, a novel immunomodulator for the treatment of active rheumatoid arthritis. Clinical therapeutics. 1999;21:1837–1852. doi: 10.1016/S0149-2918(00)86732-6. [DOI] [PubMed] [Google Scholar]
- 20.Hansen M, Le Nours J, Johansson E, Antal T, Ullrich A, Loeffler M, Larsen S. Inhibitor binding in a class 2 dihydroorotate dehydrogenase causes variations in the membrane-associated N-terminal domain. Protein Science. 2004;13:1031–1042. doi: 10.1110/ps.03533004. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Herrmann M, Schleyerbach R, Kirschbaum B. Leflunomide: an immunomodulatory drug for the treatment of rheumatoid arthritis and other autoimmune diseases. Immunopharmacology. 2000;47:273–289. doi: 10.1016/s0162-3109(00)00191-0. [DOI] [PubMed] [Google Scholar]
- 22.Booker ML, Bastos CM, Kramer ML, Barker RH, jr, Skerlj R, Bir Sdhu A, Deng X, Celatka C, Cortese JF, Guerrero Bravo JE, Krespo Llado KN, Serrano AE, Angulo-Barturen I, Belén Jiménez-Díaz M, Viera S, Garuti H, Wittlin S, Papastogiannidis P, Lin J, Janse CJ, Khan SM, Duraisingh M, Coleman B, Goldsmith EJ, Phillips MA, Munoz B, Wirth DF, Klinger JD, Wiegand R, Sybertz E. Novel inhibitors of Plasmodium falciparum dihydroorotate dehydrogenase with anti-malarial activity in the mouse model. J Biol Chem. 2010;285(43):33054–33064. doi: 10.1074/jbc.M110.162081. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Deng X, Gujjar R, El Mazouni F, Kaminsky W, Malmquist NA, Goldsmith EJ, Rathod PK, Phillips MA. Structural plasticity of malaria dihydroorotate dehydrogenase allows selective binding of diverse chemical scaffolds. J Biol Chem. 2009;284:26999–27009. doi: 10.1074/jbc.M109.028589. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Hurt DE, Widom J, Clardy J. Structure of Plasmodium falciparum dihydroorotate dehydrogenase with a bound inhibitor. Acta Crystallogr D Biol Crystallogr. 2006;62(Pt 3):312–23. doi: 10.1107/S0907444905042642. [DOI] [PubMed] [Google Scholar]
- 25.Liu S, Neidhardt EA, Grossman TH, Ocain T, Clardy J. Structures of human dihydroorotate dehydrogenase in complex with antiproliferative agents. Structure. 2000;8(1):25–33. doi: 10.1016/s0969-2126(00)00077-0. [DOI] [PubMed] [Google Scholar]
- 26.Phillips MA, Rathod PK, Baldwin J, Gujjar R. Dihydroorotate dehydrogenase inhibitors with selective anti-malarial activity. WO Patent 2007149211 A1, 2007. 2007
- 27.Phillips MA, Gujjar R, Malmquist NA, White J, El Mazouni F, Baldwin J, Rathod PK. Triazolopyrimidine-based dihydroorotate dehydrogenase inhibitors with potent and selective activity against the malaria parasite, Plasmodium falciparum. J Med Chem. 2008;51:3649–3653. doi: 10.1021/jm8001026. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Baldwin J, Michnoff CH, Malmquist NA, White J, Roth MG, Rathod PK, Phillips MA. High-throughput screening for potent and selective inhibitors of Plasmodium falciparum dihydroorotate dehydrogenase. J Biol Chem. 2005;280(23):21847–21853. doi: 10.1074/jbc.M501100200. [DOI] [PubMed] [Google Scholar]
- 29.Gujjar R, Marwaha A, El Mazouni F, White J, White KL, Creason S, Shackleford DM, Baldwin J, Charman WN, Buckner FS, Charman S, Rathod PK, Phillips MA. Identification of a metabolically stable triazolopyrimidine-based dihydroorotate dehydrogenase inhibitor with antimalarial activity in mice. J Med Chem. 2009;52(7):1864–72. doi: 10.1021/jm801343r. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Gujjar R, El Mazouni F, White KL, White J, Creason S, Shackleford DM, Deng X, Charman WN, Bathurst I, Burrows J, Floyd DM, Matthews D, Buckner FS, Charman SA, Phillips MA, Rathod PK. Lead optimization of aryl and aralkyl amine-based triazolopyrimidine inhibitors of Plasmodium falciparum dihydroorotate dehydrogenase with antimalarial activity in mice. J Med Chem. 2011;54(11):3935–49. doi: 10.1021/jm200265b. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Coteron JM, Marco M, Esquivias J, Deng X, White KL, White J, Koltun M, El Mazouni F, Kokkonda S, Katneni K, Bhamidipati R, Shackleford DM, Angulo-Barturen I, Ferrer SB, Jimenez-Diaz MB, Gamo FJ, Goldsmith EJ, Charman WN, Bathurst I, Floyd D, Matthews D, Burrows JN, Rathod PK, Charman SA, Phillips MA. Structure-Guided Lead Optimization of Triazolopyrimidine-Ring Substituents Identifies Potent Plasmodium falciparum Dihydroorotate Dehydrogenase Inhibitors with Clinical Candidate Potential. J Med Chem. 2011;54(15):5540–5561. doi: 10.1021/jm200592f. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Hartwig JF. Palladium-catalyzed amination of aryl halides: Mechanism and rational catalyst design. Synlett. 1997;4:329–340. [Google Scholar]
- 33.Loones KT, Maes BU, Dommisse RA, Lemiere GL. The first tandem double palladium-catalyzed aminations: synthesis of dipyrido[1,2-a:3',2'-d]imidazole and its benzo- and aza-analogues. Chem Commun (Camb) 2004;(21):2466–7. doi: 10.1039/b409161b. [DOI] [PubMed] [Google Scholar]
- 34.Urgaonkar S, Nagarajan M, Verkade JG. P[N(i-Bu)CH2CH2]3N: a versatile ligand for the Pd-catalyzed amination of aryl chlorides. Org Lett. 2003;5(6):815–8. doi: 10.1021/ol027450b. [DOI] [PubMed] [Google Scholar]
- 35.Yin J, Buchwald SL. Palladium-catalyzed intermolecular coupling of aryl halides and amides. Org Lett. 2000;2(8):1101–4. doi: 10.1021/ol005654r. [DOI] [PubMed] [Google Scholar]
- 36.Kappe T. Synthesis of some new imidazo[1,2-a]pyrimidin-5(1H)-ones as potential antineoplastic agents. J. Het. Chem. 1995;32(3):1003–1006. [Google Scholar]
- 37.Senga K, Novinson T, Wilson HR, Robins RK. Synthesis and antischistosomal activity of certain pyrazolo[1,5-a]pyrimidines. J Med Chem. 1981;24(5):610–3. doi: 10.1021/jm00137a023. [DOI] [PubMed] [Google Scholar]
- 38.Makisumi Y. Synthesis of potential anticancer agents. 5,7-Disubstituted s-triazolo[2,3-a]pyrimidines. Chem. Pharma. Bull. 1961;9:801. [Google Scholar]
- 39.Taddei D, Kilian P, Slawin AM, Derek Woollins J. Synthesis and full characterisation of 6-chloro-2-iodopurine, a template for the functionalisation of purines. Org Biomol Chem. 2004;2(5):665–70. doi: 10.1039/b312629c. [DOI] [PubMed] [Google Scholar]
- 40.Richter E, Loeffler JE, Tahlor EC. Studies in purine chenistry. VIII. A convenient synthesis of hypoxanthines and adenines. J. Am. Chem. Soc. 1960;82(12):3144–3146. [Google Scholar]
- 41.Robins RK, Dille KJ, Willits CH, Christensen B.E. Purines., II The synthesis of certain purines and the cyclization of several substituted 4,5-diaminopyrimidines. J. Am. Chem. Soc. 1953;75:263–266. [Google Scholar]
- 42.Robins RK. Potential purine antagonists. I. synthesis of some 4,6-substituted pyrazolo [3,4-d]pyrimidines. J. Am. Chem. Soc. 1956;78:784. [Google Scholar]
- 43.Steck EA, Brundage RP. Some s-triazolo[b]pyridazine. J. Am. Chem. Soc. 1959;81(23):6289–6291. [Google Scholar]
- 44.Gujjar R, El Mazouni F, White KL, White J, Creason S, Shackleford DM, Deng X, Charman WN, Bathurst I, Burrows J, Floyd DM, Matthews D, Buckner FS, Charman SA, Phillips MA, Rathod PK. Lead-optimization of aryl and aralkyl amine based triazolopyrimidine inhibitors of Plasmodium falciparum dihydroorotate dehydrogenase with anti-malarial activity in mice. J. Med. Chem. 2011 doi: 10.1021/jm200265b. in press. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 45.Baldwin J, Farajallah AM, Malmquist NA, Rathod PK, Phillips MA. Malarial dihydroorotate dehydrogenase: substrate and inhibitor specificity. J. Biol. Chem. 2002;277:41827–41834. doi: 10.1074/jbc.M206854200. [DOI] [PubMed] [Google Scholar]
- 46.Desjardins RE, Canfield CJ, Haynes JD, Chulay JD. Quantitative assessment of antimalarial activity in vitro by a semiautomated microdilution technique. Antimicrob Agents Chemother. 1979;16(6):710–8. doi: 10.1128/aac.16.6.710. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 47.Jiang L, Lee P, White J, Rathod P. Potent and selective activity of a combination of thymidine and 1843U89, a folate-based thymidylate synthase inhibitor, against Plasmodium falciparum. Antimicrob. agents and chemotherapy. 2000;44:1047–1050. doi: 10.1128/aac.44.4.1047-1050.2000. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 48.Obach RS. Prediction of human clearance of twenty-nine drugs from hepatic microsomal intrinsic clearance data: An examination of in vitro half-life approach and nonspecific binding to microsomes. Drug Metab Dispos. 1999;27(11):1350–9. [PubMed] [Google Scholar]
- 49.Davies B, Morris T. Physiological parameters in laboratory animal and humans. Pharm Res. 1993;10:1093–1095. doi: 10.1023/a:1018943613122. [DOI] [PubMed] [Google Scholar]
- 50.Houston J. Utility of in vitro drug metabolism data in predicting in vivo metabolic clearance. Biochem Pharmacol. 1994;47:1469–1479. doi: 10.1016/0006-2952(94)90520-7. [DOI] [PubMed] [Google Scholar]
- 51.Gaspari P, Banerjee T, Malachowski WP, Muller AJ, Prendergast GC, DuHadaway J, Bennett S, Donovan AM. Structure-activity study of brassinin derivatives as indoleamine 2,3-dioxygenase inhibitors. J Med Chem. 2006;49(2):684–92. doi: 10.1021/jm0508888. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 52.Phillips MA, Rathod PK, Gujjar R, Marwaha AS, Charman SA. Dihydroorotate dehydrogenase inhibitors with selective anti-malarial activity. WO Patent 2009/082691; US Patent 20090209557. 2009
- 53.Phillips MA, Rathod PK, Charman SA, Floyd D, Burrows J, Matthews G, Marwaha A, Gujar R, Coteron-Lopez J. Antimalarial agents that are inhibitors of dihydroorotate dehydrogenase. WO 2011/041304; PCT/US2010/050532, 4/07/11. 2011