

Abstract
Frontotemporal lobar degeneration with TDP-43 inclusions (FTLD-TDP) is a fatal neurodegenerative disease with no available treatments. Mutations in the progranulin gene (GRN) causing impaired production or secretion of progranulin are a common Mendelian cause of FTLD-TDP; additionally, common variants at chromosome 7p21 in the uncharacterized gene TMEM106B were recently linked by genome-wide association to FTLD-TDP with and without GRN mutations. Here we show that TMEM106B is neuronally expressed in postmortem human brain tissue, and that expression levels are increased in FTLD-TDP brain. Furthermore, using an unbiased, microarray-based screen of >800 microRNAs (miRs), we identify microRNA-132 as the top microRNA differentiating FTLD-TDP and control brains, with <50% normal expression levels of three members of the microRNA-132 cluster (microRNA-132, microRNA-132*, and microRNA-212) in disease. Computational analyses, corroborated empirically, demonstrate that the top mRNA target of both microRNA-132 and microRNA-212 is TMEM106B; both microRNAs repress TMEM106B expression through shared microRNA-132/212 binding sites in the TMEM106B 3′UTR. Increasing TMEM106B expression to model disease results in enlargement and poor acidification of endo-lysosomes, as well as impairment of mannose-6-phosphate-receptor trafficking. Finally, endogenous neuronal TMEM106B colocalizes with progranulin in late endo-lysosomes, and TMEM106B overexpression increases intracellular levels of progranulin. Thus, TMEM106B is an FTLD-TDP risk gene, with microRNA-132/212 depression as an event which can lead to aberrant overexpression of TMEM106B, which in turn alters progranulin pathways. Evidence for this pathogenic cascade includes the striking convergence of two independent, genomic-scale screens on a microRNA:mRNA regulatory pair. Our findings open novel directions for elucidating miR-based therapies in FTLD-TDP.
Introduction
The neurodegenerative dementia frontotemporal lobar degeneration with TDP-43 inclusions (FTLD-TDP) is a sporadic and familial neurodegenerative disease causing progressive impairment in language, behavioral control, or both (McKhann et al., 2001; Baker et al., 2006). One of the major forms of presenile dementia (Ratnavalli et al., 2002), FTLD-TDP is characterized by ubiquitinated inclusions comprised primarily of the HIV TAR-DNA binding protein of 43 kDa, or TDP-43 (Arai et al., 2006; Neumann et al., 2006). These TDP-43 inclusions are also found in the motor neuron disease amyotrophic lateral sclerosis (ALS) (Arai et al., 2006; Neumann et al., 2006). Mutations in the TDP-43 gene (TARDBP) are rare genetic causes of both FTLD-TDP (Benajiba et al., 2009) and ALS (Gitcho et al., 2008), suggesting that FTLD-TDP and ALS might be two phenotypic ends of one disease spectrum (Chen-Plotkin et al., 2010a). Additionally, mutations in the progranulin gene (GRN)—a secreted growth factor (He and Bateman, 2003)—are a major Mendelian cause of FTLD-TDP (Baker et al., 2006; Cruts et al., 2006) and account for ∼10% of all cases (Gass et al., 2006). Deficiency of progranulin production (Baker et al., 2006; Cruts et al., 2006) or secretion (Mukherjee et al., 2008; Shankaran et al., 2008) appears to be the disease mechanism in these autosomal-dominant FTLD-TDP cases (Cruts and Van Broeckhoven, 2008).
To identify additional risk factors for FTLD-TDP, we previously conducted a genome-wide association study (GWAS) and showed that chromosome 7p21 variants within the gene TMEM106B confer increased risk of FTLD-TDP, with an odds ratio of 1.6 (Van Deerlin et al., 2010), and this association has been replicated (van der Zee et al., 2011). Intriguingly, decreased plasma progranulin levels correlate with TMEM106B risk genotypes (Finch et al., 2011), and, in ALS patients, TMEM106B genotypes associated with FTLD-TDP increase the risk of developing dementia (Vass et al., 2011). While these observations correlate with TMEM106B genotype, they do not provide mechanistic evidence that TMEM106B is the causative 7p21 genetic signal observed in the GWAS. Furthermore, very little is known about TMEM106B, a 274 aa, predicted single transmembrane domain protein, with no yeast ortholog and homology only to two other uncharacterized members of the TMEM106 family.
Here, we investigate the genetic regulation and pathophysiological function of TMEM106B, both of which were previously unknown. We demonstrate that TMEM106B is elevated in FTLD-TDP brains. We further show that TMEM106B is normally repressed by microRNA (miR)-132 and miR-212, which are significantly decreased in FTLD-TDP. Finally, we demonstrate that TMEM106B overexpression in turn disrupts endosomal–lysosomal pathways, sequesters progranulin in TMEM106B-positive late endosomes or lysosomes, and increases intracellular levels of progranulin. We thus establish TMEM106B mechanistically as the 7p21 genetic risk factor for FTLD-TDP and elucidate pathophysiological steps that may be amenable to targeted intervention in an otherwise fatal disease.
Materials and Methods
Human brain samples
Frontal cortex samples from 12 FTLD-TDP cases (5 with GRN mutations and 7 without GRN mutations) and 6 neurologically normal controls of either sex (see Table 1 for details) were obtained from the University of Pennsylvania Center for Neurodegenerative Disease Research Brain Bank. Total RNA was isolated and evaluated for quality control parameters as previously described (Chen-Plotkin et al., 2008), with the exception that a column purification step was not used, to retain small RNAs. Protein was sequentially extracted from a subset of frontal cortex samples. Informed consent was obtained for postmortem studies.
Table 1.
Human brain samples
| Group | N | Gender | Age at death (years) Median (IQR) |
|---|---|---|---|
| FTLD-TDP with GRN mutations | 5 | 2M/3F | 68 (65–76) |
| c.26C>A (A9D) | |||
| c.911G>A (W304X) | |||
| c.1252C>T (R418X)—2 cases | |||
| c.1477C>T (R493X) | |||
| FTLD-TDP without GRN mutations | 7 | 3M/4F | 68 (56–73) |
| Neurologically normal controls | 6 | 4M/2F | 71 (60–75) |
Characteristics of postmortem brain samples used for this study. All GRN genetic variants used in this study are believed to be pathogenic (http://www.molgen.ua.ac.be/admutations/). M, Male; F, female. Nomenclature follows cDNA sequence NM_002087.2.
Of note, some of the frontal cortex samples used for mRNA quantitation were previously reported in our GWAS study (Van Deerlin et al., 2010); these data were included here so that sets of data from multiple brain regions included the same samples.
microRNA screening and qRT-PCR validation
One microgram of total RNA from each individual brain sample, as well as 1 μg of a pooled reference sample, was hybridized to the miRCURY LNA array version 11.0 (Exiqon) for microRNA quantitation. No microRNA enrichment was needed, as concentrations of miRs were high. Statistical analyses of miR expression were performed using open source R software packages available from Bioconductor and specifically the limma package for two-color arrays. Microarray quality control was performed as previously described (Chen-Plotkin et al., 2008); no outlier chips were identified for elimination. Raw data were RMA normalized (Wettenhall and Smyth, 2004), and median values for each microRNA were used to compare groups using pairwise contrasts within an ANOVA model correcting for gender and age. R-scripts for these analyses are available on request. Promising candidate microRNAs found by array screening to differ in disease were evaluated using qRT-PCR with TaqMan microRNA assays from Applied Biosystems (AB Assay ID 000457, AB Assay ID 002132, AB Assay ID 000515); microRNAs were normalized to either the geometric mean of two housekeeping small RNAs (human let-7a, AB Assay ID 000377; human RNU38b, AB Assay ID 001004) or a neuron-specific microRNA (human miR-124a, AB Assay ID 000446).
qRT-PCR was also used for transcript quantitation for TMEM106B and other mRNAs of interest as previously described (Chen-Plotkin et al., 2008). Relative quantification was performed (delta–delta method) using β-actin and/or PPIA as our reference housekeeping gene after verification that these transcripts remain constant in the experimental conditions used.
SNP genotyping
DNA was extracted from brain samples and genotyping was performed using TaqMan chemistry-based allelic discrimination assays as previously described (Van Deerlin et al., 2010).
MicroRNA target prediction and verification
The TargetScan program (release 5.1, http://www.targetscan.org) was used to predict targets of specific microRNAs and specific microRNA regulators of TMEM106B. To verify the predicted regulation of TMEM106B by miR-132 or miR-212, we performed three types of assays.
First, in an endogenous TMEM106B assay, HEK293 cells were plated 1 d before transfection at 2 × 105 per well in 12-well plates. The following day, 75 pmol of miR mimic (miR-132, Applied Biosystems PM10166; miR-212, Applied Biosystems PM10340; miR-124 Applied Biosystems PM 10691) were transfected using Lipofectamine 2000 (Invitrogen) according to manufacturer's instructions in serum-free conditions. Medium was replaced after 4–6 h to standard 10% FBS-containing medium. Appropriate amounts of microRNA mimics were determined empirically by testing a range of concentrations. Forty-eight hours after transfection, cells were harvested, and total RNA was isolated using the miRNeasy kit (Qiagen) before quantitation by qRT-PCR.
Second, TMEM106B constructs containing the full 3′ untranslated region (3′UTR), or lacking various portions of the 3′UTR were cloned into a commercially available construct (TMEM106B construct, Origene SC113580). For miR-132/212 Site 1 and 2 deletions, the seed match region (Fig. 4A, red nucleotides on the 3′ end of sequences shown) was deleted. Four micrograms of the relevant TMEM106B construct was transfected into each well of a 6-well plate. RNA and protein were harvested at 48 h after transfection. For protein studies, cells were lysed in RIPA buffer with protease inhibitors and PMSF on ice, and lysates were centrifuged (16,000 × g, 4°C, 15 min) to remove debris, before downstream immunoblotting.
Figure 4.
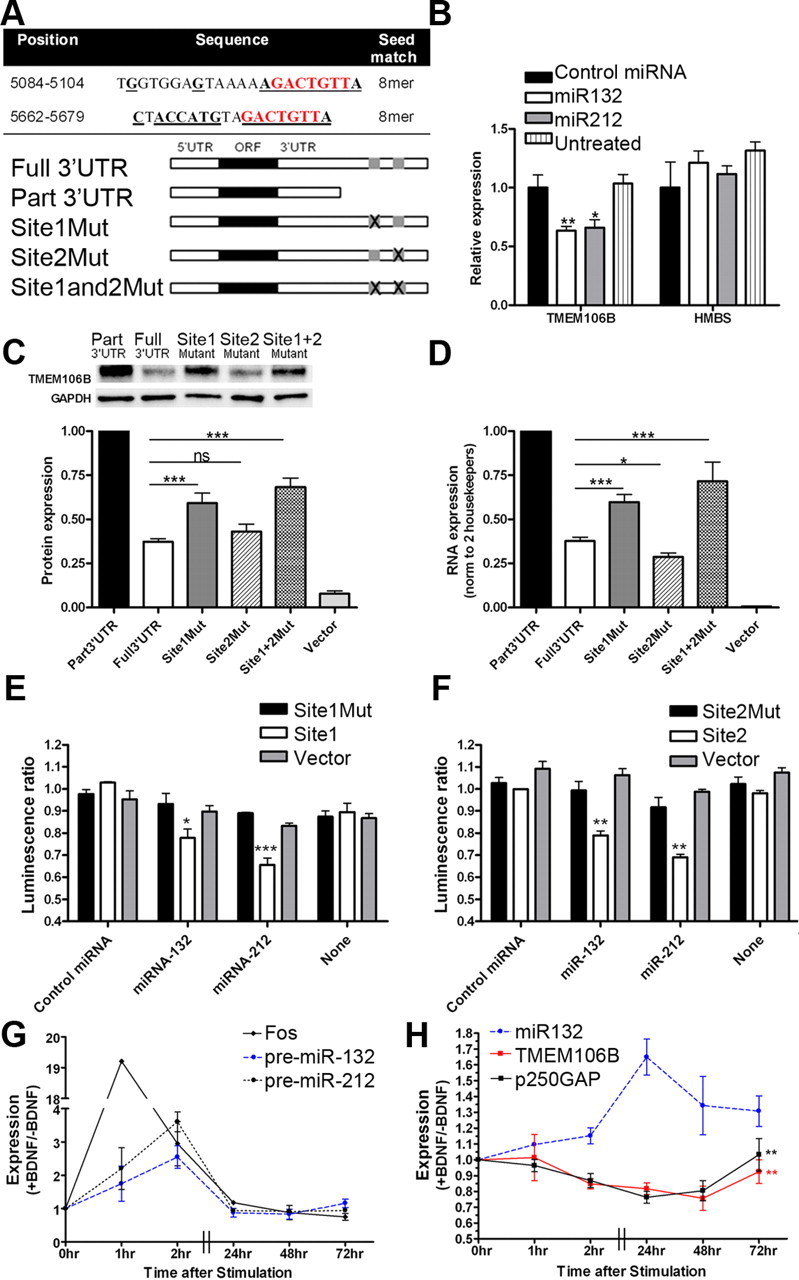
TMEM106B is regulated by miR-132 and miR-212. A, miR-132 and miR-212 are predicted to target the same mRNAs through a common seed region (nucleotides in red). The TMEM106B 3′UTR has two 8mer miR-132/miR-212 target sites at positions 5084 and 5662 (NM_001134232.1). Bolded, underlined nucleotides indicate regions of perfect complementarity between the TMEM106B 3′UTR and miR-132, which may have greater affinity than miR-212 for Site 2 of the TMEM106B 3′UTR through a stretch of complementarity outside the seed region. Constructs containing TMEM106B with the intact 3′UTR (full 3′UTR), or truncations or targeted deletions removing one or both miR-132/miR-212 sites, were used for experiments shown in C and D. B, Endogenous TMEM106B mRNA transcript is significantly reduced with addition of miR-132 or miR-212 mimics (mean ± SEM from four independent transfections). C, Compared with the truncated construct (part 3′UTR), protein levels of the TMEM106B construct containing both miR-132/212 binding sites (full 3′UTR) was decreased by 65%. Deletion of miR-132/212 Site 1 restored TMEM106B protein levels by ∼40%, while deletion of miR-132/212 Site 2 had minimal effect. Targeted deletions of both miR-132/212 binding sites (Site1Mut and Site2Mut) resulted in even greater restoration of TMEM106B expression. Representative immunoblot (top) and quantitation (mean ± SEM of five independent replicates) are shown. For TMEM106B constructs, only the 75 kDa band was apparent for cell lysates, which were always kept on ice. D, Corresponding TMEM106B mRNA levels (mean ± SEM of five independent replicates) for the same constructs. E, F, Luciferase reporters were generated containing TMEM106B 3′UTR miR-132/212 binding Site 1 (E) or Site 2 (F) with 5–10 flanking base pairs. Transfection of miR-132 or miR-212 resulted in significant decreases in reporter activity (mean ± SEM from three independent replicates) when either site was intact. No change was seen for reporters containing scrambled versions of the miR-132/miR-212 binding sites (Site1Mut and Site2Mut) or for reporters without miR-132 binding sites (Vector Laboratories). Luminescence ratio is the ratio between the firefly luciferase reporter under 3′UTR control and the constitutively active renilla luciferase reporter, controlling for differences in transfection efficiency. G, H, BDNF was applied to neuronally differentiated SHSY5Y cells to induce CREB-responsive genes. Pre-miR-132 and pre-miR-212, as well as the canonical CREB-responsive gene fos, were induced by BDNF within 1–2 h (Panel G). Mature miR-132 was maximally induced 24 h after BDNF treatment (H). Expression of the known miR-132 target gene p250GAP, as well as TMEM106B, decreased with BDNF treatment, with maximal repression at 48 h for TMEM106B. miR-132 levels had a significant effect on both TMEM106B (p = 0.009) and p250GAP (p = 0.006) expression. Ratios of BDNF-treated to nontreated conditions are shown for a minimum of four separate transfections. All panels: *p < 0.05, **p < 0.01, ***p < 0.001.
Third, luciferase reporter constructs were created adding 35–40 bp of TMEM106B 3′UTR sequence including and flanking the relevant miR-132/212 binding sites to the 3′UTR regulatory region of a commercially available dual-luciferase reporter (pmiRGLO Vector, Promega). In addition, mutated versions of these same constructs, containing the same 35–40 bp in a scrambled order to abrogate miR-132/212 binding, were also cloned into the luciferase reporters in a similar manner. For miR-132/212 Site 1, the following sequence (flanking regions plus miR binding sites, with miR binding site in bold) was inserted: CTAGTGAGATGGTGGAGTAAAAAGACTGTTAAACATTGCA. For miR-132/212 Site 1 mutated sequence, the following sequence (flanking regions plus scrambled miR binding sites, with scrambled miR binding site in bold) was inserted: CTAGTGAGATAGGGGTGAATACAATGAGCTTAACATTGCA. For miR-132/212 Site 2, the following sequence (flanking regions plus miR binding sites, with miR binding site in bold) was inserted: CTAGTATTACTACCATGTAGACTGTTATAGTTTGCA. For miR-132/212 Site 2 mutated sequence, the following sequence (flanking regions plus scrambled miR binding sites, with scrambled miR binding site in bold) was inserted: CTAGTATTAGTACTACGTACAGGTATTTAGTTTGCA. These constructs were then cotransfected into HEK293 cells with microRNA mimics as described above. Fifty nanograms of the relevant luciferase reporter construct was cotransfected. Twenty-four hours after transfection, lysates were prepared according to instructions from the Dual Luciferase Assay kit (ProMega) and read on a Berthold LB941 TriStar vTI Multimode Reader.
TMEM106B antibodies
An N-terminus peptide corresponding to amino acid residues 4–19 (SLSHLPLHSSKEDAYDC) was synthesized and used to immunize rabbits, and site-specific IgG was affinity purified with an N-terminus peptide column. The resulting antibody was named N2077. Antibody specificity was verified with immunoblots and immunofluorescence microscopy (Fig. 1). Peptide preabsorption was performed with a 5:1 (by weight) ratio of immunizing peptide to N2077 antibody, incubated overnight at 4°C, with rotation.
Figure 1.
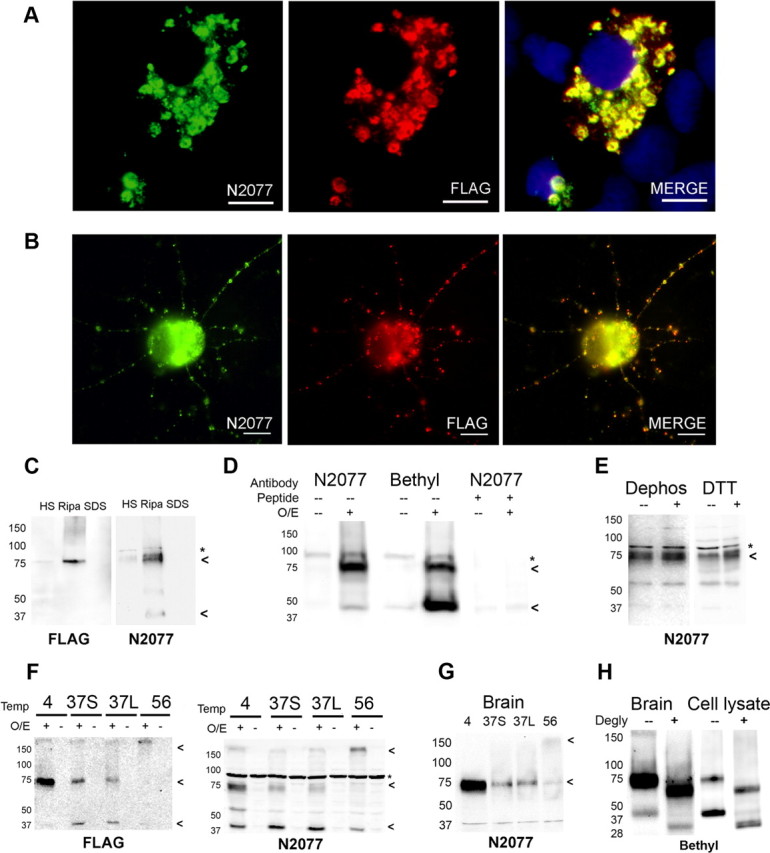
TMEM106B antibody and protein characterization. We raised a novel polyclonal antibody (N2077) recognizing amino acids 4–19 of TMEM106B. A, B, HEK293 cells (A) or murine primary cortical neurons (B) were transfected with FLAG-tagged TMEM106B. Double-label immunofluorescence microscopy demonstrates that anti-FLAG antibody recognizes the same cellular structures as anti-TMEM106B antibody (N2077), demonstrating the specificity of our antibody in a cell biological context. C, HEK293 cells transfected with FLAG-tagged TMEM106B were sequential extracted into high-salt (HS), RIPA, and 2% SDS (SDS) buffers. A RIPA-soluble 75 kDa band was recognized by both anti-FLAG antibody and anti-TMEM106B antibody (N2077), demonstrating the specificity of our antibody in a biochemical context. D, TMEM106B overexpression in HEK293 cells resulted in the appearance of bands at 75 and 40 kDa, detected by both N2077 (first two columns) and a commercially available N-terminus antibody (Bethyl, middle two columns). Both bands disappeared when N2077 was preabsorbed with immunizing peptide (last two columns). O/E indicates whether TMEM106B was overexpressed (+) or not (−). Peptide indicates whether the antibody was preabsorbed with peptide immunogen (+) or not (−). E, Neither dephosphorylation with lambda phosphatase nor treatment with the reducing agent DTT changed the electrophoretic mobility of TMEM106B. Dephos, Dephosphorylated; DTT, DTT treated; (−), control; (+), treated condition. F, G, TMEM106B shows unusual heat sensitivity at temperatures above 4°C, even in the presence of protease inhibitors. In cell lysates from HEK293 cells transfected with FLAG-tagged TMEM106B (F), TMEM106B appeared primarily as a 75 kDa band when samples were kept on ice (4). When heated at 37°C for 30 min (37S) or 45 min (37L), the 75 kDa band faded, and a 40 kDa species began to appear. At higher temperatures (e.g., 56°C for 15 min, lane labeled 56), both the 40 and 75 kDa bands were lost and a >150 kDa aggregate appeared at the top of the gel. O/E indicates whether TMEM106B was overexpressed (+) or not (−). Blots were probed with both anti-FLAG and N2077 antibodies, demonstrating the specificity of the bands. Similar heat sensitivity was seen for TMEM106B extracted from human brain tissue from normal individuals (G). In contrast to HEK293 cell lysates, however, the 40 kDa band was never prominent in human brain samples. For all panels, samples were extracted into RIPA buffer, and equal amounts of protein were loaded into all lanes. H, TMEM106B from human brain samples (left) or overexpressed in HEK293 cells (right) was deglycosylated with PNGase F after short pretreatment at 37°C. The 75 and 40 kDa bands observed before deglycosylation (−) collapsed to lower-molecular-weight species of ∼60 and 31 kDa, respectively, after PNGase F treatment (+). Note that the predicted molecular weight of TMEM106B is 31 kDa. Blots probed with the Bethyl TMEM106B antibody. All immunoblots: arrowheads indicate TMEM106B species. *Nonspecific band.
In addition, we tested commercially available antibodies against TMEM106B raised against N-terminus amino acids 1–50 (Bethyl Laboratories) and against the C terminus of the protein (Proteintech). Neither antibody demonstrated specificity in immunocytochemical experiments (data not shown), but the N-terminus antibody recognized the same bands as N2077 on immunoblot (Fig. 1D) and was used to confirm key biochemical findings.
Cell culture and transfection
Primary cortical and hippocampal neurons were prepared from embryonic day 18 (E18) to E20 C57BL/6 mice, plated, and maintained as previously described (Tseng et al., 2006). In addition, HEK293 and SHSY5Y cells were maintained and used for experiments. Where indicated, cells were transfected with the respective constructs using Lipofectamine 2000 (Invitrogen) 1 d after plating (50–70% confluency) according to manufacturer instructions; transfections were performed under serum-free conditions. SHSY5Y cells were neuronally differentiated before their use in experiments as previously described (Remenyi et al., 2010). Neuro2A cells were neuronally differentiated directly after transfection for 48 h before their use by incubation in neurobasal media (catalog #1208, Invitrogen) with B27 (catalog #1209, Invitrogen) together with complete DMEM in a 1:1 ratio in an adaption of a previously described protocol (Ishikura et al., 2005).
Treatment of cells with lysosomal and proteosomal inhibitors
Cells were treated with the vacuolar ATPase inhibitor bafilomycin A1, lysosomal protease inhibitors Pepstatin A or Leupeptin, proteosomal inhibitor MG132, or control DMSO as previously described (Capell et al., 2011), with the exception that Leupeptin was not used in a mix but rather alone at a concentration of 1 μm.
Immunofluorescence microscopy
Cells were fixed (2% paraformaldehyde, 15 min, room temperature), washed five times with PBS, and then blocked for 60 min at room temperature in blocking buffer (3% BSA, 0.05% saponin, in PBS). Primary antibodies were diluted in blocking buffer, and coverslips were incubated overnight at 4°C. The next day, coverslips were washed four times with blocking buffer before incubation with secondary antibody. Secondary antibodies were diluted in blocking buffer, and coverslips were incubated for 1 h at room temperature protected from light. Following this incubation, coverslips were washed five times with blocking buffer, once with PBS, and then mounted on slides (ProLong Gold, Invitrogen).
The following antibodies and conditions were used: TMEM106B: N2077 antibody (see “TMEM106B antibodies”) was used at 1 μg/ml; FLAG: M2 antibody (Sigma) was used at 1 μg/ml; Progranulin: goat anti-human GRN (R&D Systems) was used at 1 μg/ml, and sheep anti-mouse GRN (R&D Systems) was used at 1 μg/ml; TDP-43: N171 antibody (mouse monoclonal antibody generated at University of Pennsylvania, Philadelphia, PA) (Lippa et al., 2009) was used at 1 μg/ml; mannose-6-phosphate receptor (M6PR, cation independent): mouse anti-human M6PR (catalog #ab2733, Abcam) was used at 2 μg/ml; LAMP-1: mouse anti-human H4A3 (DSHB) and mouse anti-human CD107A (catalog #555798, BD Biosciences) were used at 1 μg/ml; rat anti-mouse 1D4B (DSHB) was used at 1 μg/ml; and GM130: mouse GM130 antibody recognizing human, mouse, and rat GM130 (BD Biosciences) was used at 1.25 μg/ml. All secondary antibodies were Alexa Fluor antibodies (Invitrogen) used at 1:1000, with the exception of an AMCA anti-mouse IgG (Vector Laboratories) that was used at 1:100.
Lysosensor and Lysotracker staining
The weak base dyes Lysosensor DND-189 (Invitrogen) and Lysotracker DND-99 (Invitrogen) were used to assess the acidity of intracellular organelles, since they both fluoresce intensely at low pH, and more weakly or not at all at higher pH. Live cells were incubated with Lysosensor (1 μm, 30 min, 37°C) or with Lysotracker (100 nm, 2 h, 37°C). Lysosensor-stained cells were imaged live immediately in 4°C PBS. Lysotracker-stained cells were imaged live in 4°C PBS or fixed and stained for immunofluorescence microscopy as above.
Quantification of LAMP-1+ organelle size, Lysosensor/Lysotracker mean fluorescence intensity, and progranulin staining appearance
LAMP-1+ organelle size.
An average of 12 fields containing a representative heterogeneous mixture of cells with and without TMEM106B overexpression after transient transfection was captured at 40× magnification after staining for LAMP-1 and TMEM106B. The diameter of LAMP-1+ organelles was measured for an average of three TMEM106B-overexpressing and three TMEM106B-non-overexpressing cells per field, and average diameters (±SEM) were calculated for cells with and without TMEM106B overexpression. TMEM106B-overexpressing cells occasionally contained very large vacuolar LAMP-1+ organelles; these were not included in the quantitation.
Lysosensor/Lysotracker mean fluorescence intensity.
An average of 11 fields containing a representative heterogeneous mixture of cells with and without TMEM106B overexpression after transient transfection was captured at 40× magnification after staining for TMEM106B and the indicated pH-sensitive dye. The cytoplasmic compartment was outlined as a region of interest for measures of mean fluorescence intensity (MFI) in the Lysosensor or Lysotracker channel. An average of two TMEM106B-overexpressing and three TMEM106B-non-overexpressing cells per field was quantitated for MFI, and average MFIs (±SEM) were calculated for cells with and without TMEM106B overexpression.
Progranulin staining appearance.
Six fields containing a representative heterogeneous mixture of cells with and without TMEM106B overexpression after transient transfection were imaged at 20× magnification after staining for TMEM106B and progranulin. The presence or absence of intense puncta of progranulin staining was then scored for an average of 14 TMEM106B-overexpressing cells and >50 TMEM106B-non-overexpressing cells per field.
Protein preparation and immunoblotting
Where indicated, sequential biochemical fractionation of cell lysates or postmortem human brain samples was performed. Samples were sequentially extracted in buffers of increasing strength, as follows: high-salt (10 mm Tris, 500 mm NaCl, 2 mm EDTA, 1 mm DTT, 10% sucrose, pH 7.5), RIPA (50 mm Tris, 150 mm NaCl, 5 mm EDTA, 0.5% sodium deoxycholate, 1% NP-40, 0.1% SDS, pH 8.0), and 2% SDS. Since most TMEM106B is extractable in RIPA with a small fraction in high-salt, we extracted directly into RIPA for routine biochemical experiments unless otherwise indicated. Immunoblotting was performed as previously described (Neumann et al., 2006).
ELISA
Sandwich ELISAs were used for measurement of progranulin and the control secreted protein IGFBP-2 from cell culture lysates and conditioned medium. Specifically, progranulin quantification was performed using a commercially available ELISA (Human progranulin ELISA kit, AdipoGen). IGFBP-2 quantification was also performed using a commercially available ELISA (Human IGFBP-2 ELISA kit, RayBiotech).
For the progranulin secretion assay, 50 μl samples of conditioned medium were removed from each cell culture well (containing a total of 1 ml of medium) at designated time intervals after transfection. At the last time point (48 h), a 50 μl conditioned medium sample was collected, the remaining medium was removed, cells were washed once with PBS, and cell lysates were then extracted in RIPA buffer as described in the preceding section.
Statistical tests
Two-tailed t tests were used, unless confirming a specific directionality of change (e.g., qRT-PCR verification of microarray results), in which case one-tailed t tests were used. Statistical analysis of microarray data was performed as described in the MicroRNA screening and qRT-PCR validation section. For BDNF treatment experiments in SHSY5Y cells, linear regressions were used to evaluate the correlation of miR levels with target mRNA levels, adjusting for time point of measurements and the interaction of miR effect with time. For repeated measurements of extracellular progranulin over time, repeated-measures two-way ANOVA was used to evaluate the effect of increased TMEM106B expression.
Results
TMEM106B shows increased expression in FTLD-TDP brain
TMEM106B is a minimally characterized protein. Thus, we first sought to investigate its expression in normal and FTLD-TDP brain tissue. We thus raised an affinity-purified polyclonal antibody, N2077, which specifically recognizes an N-terminus peptide sequence unique to TMEM106B.
In both HEK293 cells (Fig. 1A) and murine primary cortical neurons (Fig. 1B) transfected with a FLAG-tagged TMEM106B construct, staining patterns for N2077 and anti-FLAG antibody showed nearly perfect overlap. Similarly, immunoblots performed on sequentially extracted HEK293 cell lysates transfected with FLAG-tagged TMEM106B constructs showed the same band when probed with N2077 or antibodies against the tag (Fig. 1C), again demonstrating that the N2077 antibody recognizes TMEM106B. Furthermore, both N2077 and the anti-FLAG antibodies recognize TMEM106B in RIPA-extracted samples, consistent with the predicted transmembrane character of TMEM106B. Finally, preabsorption of N2077 with the peptide immunogen resulted in the disappearance of TMEM106B immunobands on immunoblotting (Fig. 1D), as well as the disappearance of TMEM106B staining within cellular structures on immunofluorescence (data not shown). Together, these results demonstrate the specificity of the N2077 antibody for TMEM106B in both biochemical and cell biological contexts.
Unexpectedly, however, the molecular weight of the main species recognized by both N2077 and antibodies directed against the FLAG tag was ∼75 kDa, with a fainter band occasionally seen at 40 kDa, while the predicted molecular weight of TMEM106B is 31 kDa. We accordingly tested other commercially available antibodies against TMEM106B; only one recognized immunobands that increased with overexpression of TMEM106B, and this antibody demonstrated the same 75 and 40 kDa bands (Fig. 1D), with possibly greater affinity for the 40 kDa band than N2077.
To investigate the apparent discrepancy between predicted and observed molecular weights of TMEM106B, we manipulated the protein in various ways. Dephosphorylating or reducing cell lysates with DTT did not change the molecular weight (Fig. 1E). Incubating the protein at 37°C or above, however, demonstrated that the 75 kDa species recognized by both N2077 and antibodies against the TMEM106B construct tag was very sensitive to heat, even in the presence of protease inhibitors. Specifically, when cell lysates containing TMEM106B were kept at 37°C for 30 min, several changes were observed in the immunoblot pattern (Fig. 1F). First, >50% of the protein was lost. Second, the 40 kDa band became more prominent. Third, when cell lysates containing TMEM106B were heated to ≥56°C, both the 75 and 40 kDa bands were lost, and a high-molecular-weight species was seen at the top of the gel, again consistent with the transmembrane character of TMEM106B. Human brain homogenates behaved similarly, with the exception that the 40 kDa band was much less prominent (Fig. 1G).
Finally, TMEM106B has recently been reported to be glycosylated, with at least five glycosylation sites C terminal to the predicted transmembrane domain (Lang et al., 2012). We confirmed this finding and extended it to brain tissue by deglycosylating cell lysates and human brain homogenates. Specifically, we found that if we combined treatment with N-glycosidase F with short pretreatment with heat, the 40 kDa band (prominent in cell lysates and visible although less prominent in human brain homogenates) collapsed to the expected 31 kDa weight of TMEM106B, while the 75 kDa band collapsed to ∼60 kDa (Fig. 1H). Together, these data suggest that TMEM106B is glycosylated and detergent soluble, and exists primarily in a heat-sensitive protein complex migrating at an apparent molecular weight of 75 kDa. Accordingly, all subsequent biochemical experiments performed for quantitation were done with the samples kept at 4°C.
Having demonstrated the specificity of our N2077 antibody, we used it to evaluate the expression of TMEM106B in human frontal cortex brain tissue (n = 5 each of normal cases for FTLD-TDP without GRN mutations, FTLD-TDP with GRN mutations). TMEM106B was abundantly expressed in human brain tissue, with a cytoplasmic perikaryal localization in neurons from normal individuals (Fig. 2A), reminiscent of the expression pattern of TMEM106B in murine primary cortical neurons (Fig. 1B). Intriguingly, TMEM106B appeared more widely distributed in the cytoplasm of neurons from individuals with FTLD-TDP, and particularly FTLD-TDP with GRN mutations [GRN(+) FTLD-TDP; Fig. 2A]. In these individuals, TMEM106B expression extended beyond the cell body into neuronal processes, filling the cytoplasm of the cells. Of note, none of the commercially available TMEM106B antibodies produced a clear staining pattern in human brain tissue (data not shown), whereas perikaryal neuronal staining of N2077 was robust and disappeared upon preabsorption of the antibody with the peptide immunogen (Fig. 2B).
Figure 2.
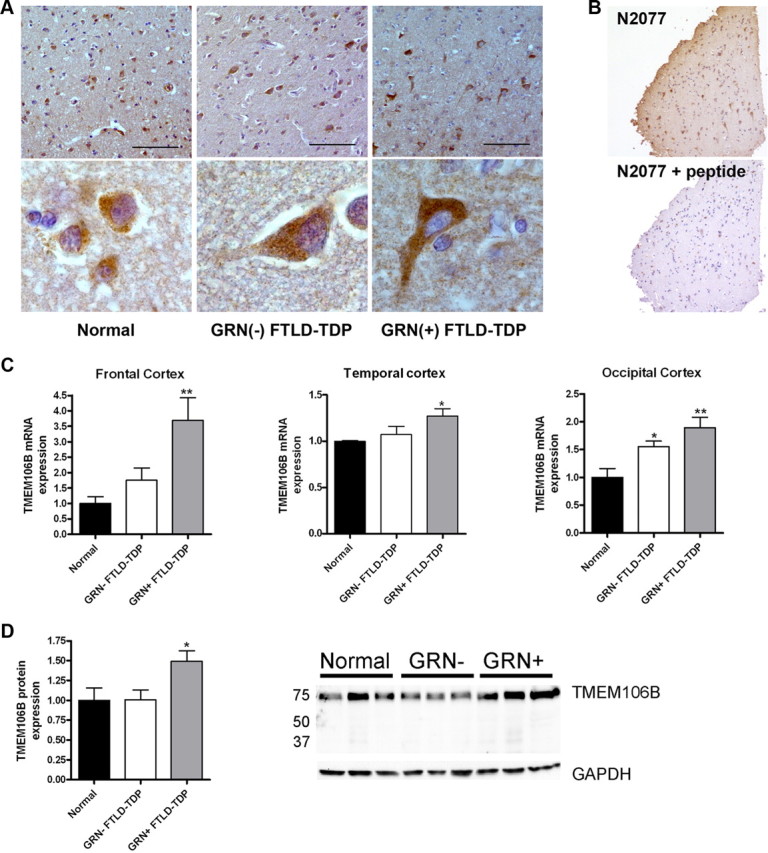
TMEM106B expression is increased in FTLD-TDP. A, Immunohistochemical staining was performed with N2077 anti-TMEM106B antibody on frontal cortex brain sections from age-matched controls (Normal), GRN(−) FTLD-TDP, and GRN(+) FTLD-TDP). GRN(+) FTLD-TDP patients had more diffuse TMEM106B staining, extending throughout the cell body and into neuronal processes. Representative lower-magnification images (top) and higher-magnification images of typical neurons from the same field (bottom) are shown. Scale bar, 100 μm. B, Neuronal staining (top) was abolished with preabsorption of N2077 with the immunizing peptide (bottom). Scale bar, 200 μm. C, Total mRNA was isolated from neurologically normal controls (n = 6), GRN(−) FTLD-TDP (n = 7), and GRN(+) FTLD-TDP (n = 5), and TMEM106B transcript expression was measured by qRT-PCR in multiple brain regions. Compared with both normal controls and to GRN(−) FTLD-TDP, GRN(+) FTLD-TDP had significantly higher levels of TMEM106B expression. Means ± SEM are shown. *p < 0.05, **p < 0.01. D, Frontal cortex protein was RIPA extracted. Equal amounts of total protein from neurologically normal controls (n = 4), GRN(−) FTLD-TDP (n = 3), and GRN(+) FTLD-TDP (n = 4) were loaded, and immunoblots were probed for TMEM106B. Corroborating our mRNA findings, GRN(+) FTLD-TDP brain showed higher levels of TMEM106B protein expression. Quantification (mean ± SEM) includes all available samples; representative subset immunoblot is also shown.
These immunohistochemical findings, as well as preliminary data reported in our prior GWAS (Van Deerlin et al., 2010), suggested that TMEM106B expression is increased in FTLD-TDP. We therefore quantified TMEM106B expression in FTLD-TDP brain.
At the mRNA level, qRT-PCR demonstrated that TMEM106B brain expression was lowest in normal individuals (n = 6), higher in FTLD-TDP patients without GRN mutations [GRN(−) FTLD-TDP, n = 7] and highest in GRN(+) FTLD-TDP (n = 5; see Table 1 for patient and control characteristics). These trends were observed in multiple areas of the brain (Fig. 2C), with the most striking differences seen in frontal cortex, corroborating our previously published report (Van Deerlin et al., 2010) with a wider range of samples. For a subset of patients, never-thawed frontal cortex samples were available for protein quantitation, and quantitation of immunoblots performed on these frontal cortex samples corroborated the mRNA finding that TMEM106B expression is increased in GRN(+) FTLD-TDP (Fig. 2D). It is worth noting that human brain TMEM106B expression appears to be much higher than in HEK293 cells, which show negligible expression by immunoblot in the absence of overexpression. In addition, we have previously shown that the GRN(+) FTLD-TDP brains used in this study do not have more severe TDP-43 pathology than the GRN(-) FTLD-TDP brains (Chen-Plotkin et al., 2010b), suggesting that general mechanisms of TDP-43 mediated pathology and/or neurodegeneration cannot account for the observed differences in TMEM106B expression.
Thus, TMEM106B appears to be abundantly expressed in human brain tissue, with particularly high levels of expression in GRN(+) FTLD-TDP brain, and possible modest increases in GRN(-) FTLD-TDP brain as well.
The microRNA-132 cluster is decreased in FTLD-TDP brain
We next sought to identify genetic modifiers of TMEM106B expression, which might be responsible for the observed increase in expression in FTLD-TDP. To do this, and to simultaneously pursue effectors of a distinct mRNA expression signature previously demonstrated in FTLD-TDP (Chen-Plotkin et al., 2008), we performed global miR expression profiling. miRs—small noncoding RNAs of ∼22 bp—have emerged in recent years as major regulators of gene expression, binding to complementary sequences in the 3′UTR of specific mRNAs and repressing expression by targeting mRNAs for degradation or blocking translation (Filipowicz et al., 2008). In addition, miR dysfunction has been implicated in a growing number of diseases, including the neurodegenerative disorders Alzheimer's disease (Hébert et al., 2008; Wang et al., 2008), Parkinson's disease (Kim et al., 2007), and spinocerebellar ataxia (Lee et al., 2008).
Utilizing a microarray platform testing all known human miRs (n = 836), we compared frontal cortex tissue from FTLD-TDP patients (n = 12) and controls (n = 6, see Table 1 for patient and control characteristics). Three miRs in the miR-132 cluster—miR-132, miR-132*, and miR-212—all emerged within the 11 miRs showing significantly dysregulated expression in FTLD-TDP (Fig. 3A). A fourth putative member of the miR-132 cluster—miR-212*—is not in the microRNA database miRBase 17 (http://www.mirbase.org/index.shtml) and was not represented on the microarray.
Figure 3.

The microRNA 132/212 cluster is decreased in FTLD-TDP. A, Frontal cortex samples from normal controls (n = 6) and patients with FTLD-TDP (n = 12) were evaluated by microarray for differentially expressed microRNAs. Eleven microRNAs demonstrated nominally significant differences in FTLD-TDP compared with controls, with microRNA-132 (miR-132) demonstrating the most significant association (p = 0.0001), and a decrease of ∼50% in FTLD-TDP. B, Confirmation of microarray screening results by qRT-PCR showed significant 65–75% reductions in miR-132, miR-132*, and miR-212 for FTLD-TDP frontal cortex samples from patients with (GRN+, n = 5) and without (GRN−, n = 7) progranulin mutations, compared with neurologically normal controls (n = 6). Absolute levels of miR-132, however, were 50–100 times higher than levels of either miR-132* or miR-212. Relative expression (mean ± SEM) of microRNAs are shown on a log2 scale, calibrated to one normal sample's miR-132 expression value. *p < 0.05, **p < 0.01. C, Three of the 11 microRNAs with differential expression in FTLD-TDP are known to arise from processing of the same CREB-responsive primary transcript. Specifically, a primary transcript on chromosome 17 gives rise to pre-miR-132 and pre-miR-212. Pre-miR-132 is then further processed to yield mature miR-132 and miR-132*, while pre-miR-212 yields mature mir-212. D, MiR-132 (x-axis), miR-132* (y-axis, red), and miR-212 (y-axis, blue) show highly correlated expression levels across all 18 human samples, suggesting that the observed decrease of all three microRNAs may be due to decreased expression of the shared primary transcript. For each miRNA, values are normalized to one sample within that group to account for the much higher expression levels of miR-132.
The top miR thus identified was miR-132 (p = 0.0001, Fig. 3A), a CREB-activated miR previously reported to play diverse roles in neuronal differentiation (Vo et al., 2005; Magill et al., 2010). In addition, by corroborative qRT-PCR, miR-132, miR-132*, and miR-212 all showed <50% expression in both GRN(−) FTLD-TDP and GRN(+) FTLD-TDP compared with normal controls (qRT-PCR; Fig. 3B). This decreased expression relative to normal controls persisted in both GRN(−) FTLD-TDP and GRN(+) FTLD-TDP subgroups, removing the possibility that one subgroup is driving the effect; a significant difference also persisted when quantitation was normalized to the brain-expressed miR miR-124, removing the possibility that neuronal loss associated with FTLD-TDP is driving the effect (data not shown). However, absolute levels of miR-132 were ∼100 times higher than miR-132* and miR-212 in all groups (Fig. 3B).
MiR-132, miR-132*, and miR-212 are all generated from a shared primary miR transcript (pri-miR) on chromosome 17; this pri-miR is then processed into two pre-miRs, which in turn become the three mature miRs (Fig. 3C). Interestingly, miR-132, miR-132*, and miR-212 levels were highly correlated on an individual-to-individual basis (R2 values >90%; Fig. 3D), suggesting that the observed decrease of all three miRs may result from decreased expression of the pri-miR transcript in FTLD-TDP.
MiR-132 and miR-212 are dual repressors of TMEM106B through shared binding sites in the 3′UTR
Having identified the miR-132 cluster by genomic-scale screening for FTLD-TDP-dysregulated miRs, we next performed computational analyses of miR-132 cluster targets using the program TargetScan (Lewis et al., 2003) (release 5.1, http://www.targetscan.org). Strikingly, the top computationally predicted target of both miR-132 and miR-212 was TMEM106B, ranked 1 of 283 predicted mRNA targets (Table 2). Specifically, miR-132 and miR-212 share one recognition sequence, and two sites within the TMEM106B 3′UTR contain this “seed match” (Fig. 4A, nucleotides in red). Indeed, on an individual-by-individual basis, regardless of disease status, frontal cortex levels of both miR-132 and miR-212 showed significant inverse associations with frontal cortex levels of TMEM106B (linear regression one-tailed p value = 0.03 for miR-132, 0.05 for miR-212).
Table 2.
Top ten mRNA targets of miR-132/212, as predicted by Targetscan
| Rank | Target gene | Gene name | Total context score |
|---|---|---|---|
| 1 | TMEM106B | Transmembrane protein 106B | −0.96 |
| 2 | ZNF516 | Zinc finger protein 516 | −0.89 |
| 3 | LEMD3 | LEM domain containing 3 | −0.81 |
| 4 | CDC2L6 | Cell division cycle 2-like 6 | −0.81 |
| 5 | GMFB | Glia maturation factor, beta | −0.79 |
| 6 | ETNK1 | Ethanolamine kinase 1 | −0.79 |
| 7 | TIMM9 | Translocase of inner mitochondrial membrane 9 homolog | −0.75 |
| 8 | SLC26A7 | Solute carrier family 26, member 7 | −0.71 |
| 9 | CCDC88A | Coiled-coil domain containing 88a | −0.69 |
| 10 | MIA3 | Melanoma inhibitory activity family, member 3 | −0.68 |
The total context score reflects the likelihood of true targeting and is related to the number of predicted binding sites, the quality of each binding site, and the genomic context. A more negative value suggests a stronger probability of true targeting. Of note, miR-132 and miR-212 have the same target binding sequence, thus accounting for the shared mRNA targets.
To determine whether the computationally predicted relationship between miR-132/212 and TMEM106B could be demonstrated experimentally, we transfected each miR into HEK293 cells. Transfection of either miR-132 or miR-212 resulted in a 40% decrease of endogenous mRNA levels of TMEM106B, suggesting that these miRs are bona fide negative regulators of TMEM106B (Fig. 4B).
We next evaluated the site-specific contributions of the two predicted miR-132/212 binding sites within the TMEM106B 3′UTR (Fig. 4A, Sites 1 and 2) on miR-132/212 regulation of TMEM106B. First, we used a “subtraction” strategy—removing miR-132/212 binding sites from the native TMEM106B 3′UTR by truncations and by site-directed deletions (Fig. 4A). We expressed these TMEM106B 3′UTR mutants in HEK293 cells and assayed their expression levels under the regulation of endogenous miR-132/212.
Expression of a TMEM106B construct lacking the predicted 3′UTR miR-132/212 binding sites resulted in high TMEM106B protein expression (Fig. 4C). Inclusion of the 3′ end of the 3′UTR in the full 3′UTR-containing TMEM106B construct resulted in a 65% decrease of TMEM106B protein (Fig. 4C) and mRNA levels (Fig. 4D), compared with the construct without miR-132/212 binding sites—demonstrating the repressive function of this portion of the 3′UTR. Moreover, a targeted deletion eliminating only the seed region of miR-132/212 Site 1 from the full 3′UTR construct resulted in significant restorations of TMEM106B protein (Fig. 4C) and mRNA (Fig. 4D) levels, while a targeted deletion eliminating only the seed region of miR-132/212 Site 2 resulted in negligible change in TMEM106B levels. Finally, a construct containing targeted deletions of both miR-132/212 Site 1 and miR-132/212 Site 2 seed regions resulted in the largest restorations (>50%) of TMEM106B expression at the protein (Fig. 4C) and mRNA (Fig. 4D) levels. Thus, miR-132/212 Site 1 and miR-132/212 Site 2 are necessary in mediating the repression of TMEM106B by endogenous microRNAs, with greater repressor activity from Site 1.
Second, we used an “addition” strategy—selectively adding the full miR-132/212 binding Site 1 or 2 to luciferase reporter constructs in the 3′ regulatory region. Cotransfection of luciferase reporter constructs with miR-132 or miR-212 into HEK293 cells resulted in a significant reduction in luciferase reporter activity for both the Site 1 construct (Fig. 4E) and the Site 2 construct (Fig. 4F). This reduction was not seen when cells were treated with an irrelevant microRNA or with no microRNAs, or when mir-132/212-site-containing reporters were substituted with either empty vector (Fig. 4E,F, gray bars) or a luciferase reporter construct inserting the same base pairs in a scrambled order that would abolish microRNA binding (Fig. 4E,F, black bars). Thus, either miR-132/212 Binding Site 1 or miR-132/212 Binding Site 2 of the TMEM106B 3′UTR is sufficient to confer regulatory activity by miR-132 and miR-212.
We next evaluated whether miR-132/212 activation by a physiologic stimulus in a neuronal cell would demonstrate the same regulatory effect of miR-132/212 on TMEM106B.
Previous studies have shown that in neurons, miR-132 and miR-212 are CREB-activated miRs (Impey et al., 2004; Magill et al., 2010) and that treatment with BDNF, which activates CREB-mediated transcription, also increases miR-132 and miR-212 expression. Because the miR-132/212 binding sites in the TMEM106B 3′UTR are not conserved beyond primates, we used retinoic acid-induced neuronally differentiated human SHSY5Y cells. BDNF treatment in SHSY5Y cells resulted in a rapid (1–2 h) increase in both the known CREB-responsive gene fos and precursor forms of miR-132 and miR-212, consistent with CREB-activated transcription of these two miRs (Fig. 4G). We observed a subsequent rise in mature forms of miR-132 and miR-212, peaking at 24 h after BDNF treatment. Concomitant with the rise in miR-132, TMEM106B transcript levels fell, with a maximum effect 48 h after BDNF treatment (Fig. 4H). Reduction in TMEM106B expression was significantly associated with rise in miR-132, and similar in magnitude to observed reduction in the known miR-132 target gene p250GAP (Wayman et al., 2008; Remenyi et al., 2010), consistent with miR-132 repression of TMEM106B in a neuronal setting (Fig. 4H). Similar trends were seen for miR-212, but total levels were 5–10 times lower than miR-132 in SHSY5Y cells and negative correlation with TMEM106B and p250GAP expression was not significant for miR-212 (data not shown). Together, these results provide empirical confirmation of the computationally predicted regulation of TMEM106B by miR-132 and miR-212.
Neuronal TMEM106B is localized to late endosomes or lysosomes
Having established that the miR-132 cluster is decreased in FTLD-TDP, that TMEM106B is increased in FTLD-TDP, and that miR-132 and miR-212 can both repress TMEM106B expression, we turned our attention to the biology of this previously uncharacterized protein. Using immunofluorescence microscopy, we first evaluated the subcellular localization of TMEM106B using the N2077 antibody. Of note, the commercially available antibodies against TMEM106B did not show colocalization with antibodies against various tags upon expression of tagged constructs, so we did not use them for cell biological experiments. FLAG-tagged TMEM106B in HEK293 cells (Fig. 5A) colocalized with the late endosomal/lysosomal marker LAMP-1, supporting previous reports using immortalized cell lines that TMEM106B may be localized to lysosomes (Lang et al., 2012). We extended these findings by demonstrating that endogenous TMEM106B in nontransfected murine primary cortical neurons (Fig. 5B) also colocalized with LAMP-1, as well as Lysotracker, an indicator of acidic organelles (Fig. 5C). In contrast, there was little overlap in staining between TMEM106B and Golgi markers (Fig. 5D) or between TMEM106B and TDP-43 (Fig. 5E). Similar late endosomal/lysosomal staining patterns were seen for murine primary hippocampal neurons, and for primary hippocampal and primary cortical neurons from rats (data not shown). Thus, neuronal TMEM106B is localized primarily in late endosomes or lysosomes.
Figure 5.
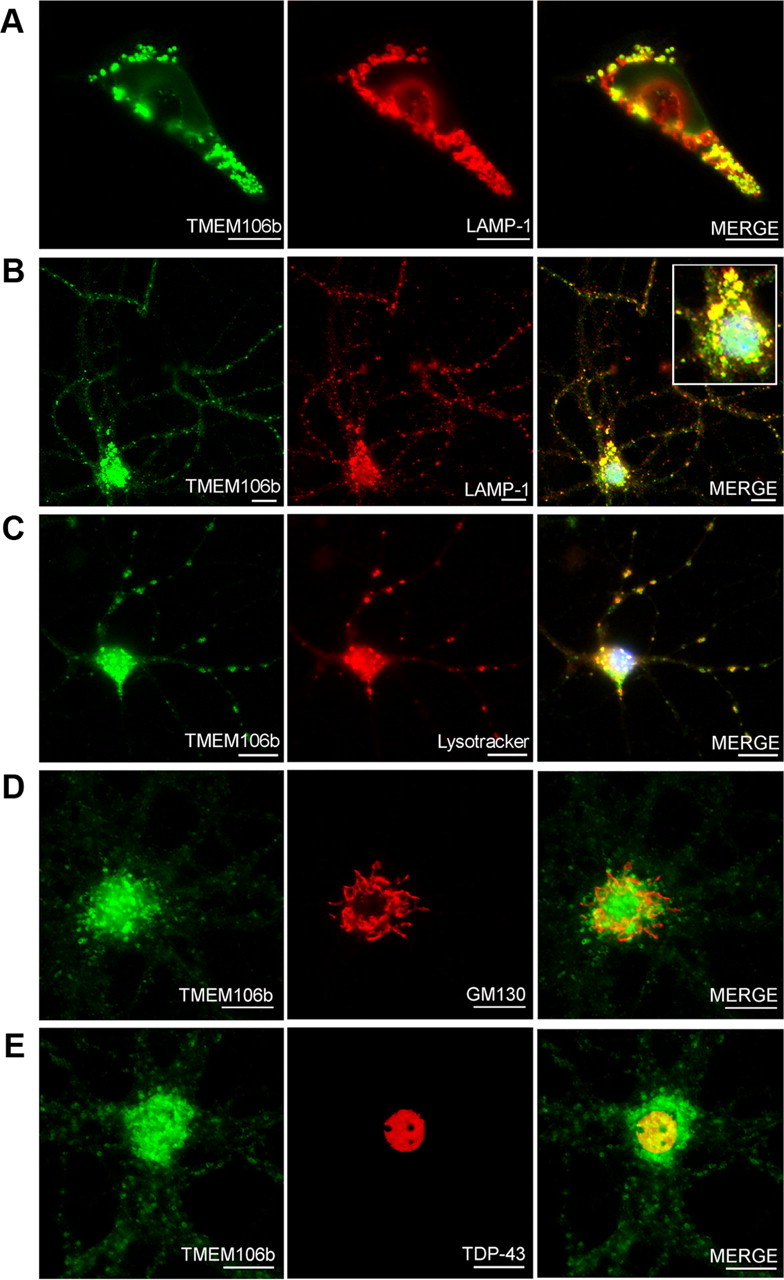
TMEM106B is associated with late endosomes/lysosomes in neurons. TMEM106b antibody N2077 was used for immunofluorescence microscopy. Scale bars, 10 μm. A, Structures staining with TMEM106B antibody (green) also expressed the late endosomal/lysosomal marker LAMP-1 (red) in HEK293 cells transiently transfected with TMEM106B. B, In nontransfected primary murine cortical neurons, endogenous TMEM106B (green) colocalized with LAMP-1 (red) in cell processes, and in the cell body (inset). C, In nontransfected primary murine cortical neurons, TMEM106B (green) colocalized with a marker for acidic organelles, Lysotracker (red), corroborating the association of TMEM106B with late endosomes/lysosomes. D, TMEM106B did not colocalize with the cis-Golgi marker GM130. E, TMEM106B did not colocalize with TDP-43.
Overexpression of TMEM106B results in abnormalities in the endosomal–lysosomal pathway
Having established the late endosomal/lysosomal subcellular localization of TMEM106B, we next evaluated the effect of overexpression of TMEM106B on the endosomal–lysosomal pathway. To do this, we transiently overexpressed TMEM106B and performed double-label immunofluorescence for TMEM106B and LAMP-1. As shown in Figure 6A, HeLA cells overexpressing TMEM106B (arrowhead) demonstrated enlargement of LAMP-1+/TMEM106B+ organelles, while neighboring cells with normal levels of TMEM106B did not. We observed this general enlargement of LAMP-1+ organelles upon TMEM106B overexpression in multiple cell lines, including HEK293 cells, HeLA cells, and the mouse neuronal cell line Neuro2A, with virtually no “normal-sized” LAMP-1+ organelles (Fig. 6B) seen. Additionally, we occasionally observed the formation of very large cytoplasmic vacuolar structures ∼5 μm in diameter; these structures showed LAMP-1 and TMEM106B staining colocalized at the limiting membrane (Fig. 6A, bottom).
Figure 6.
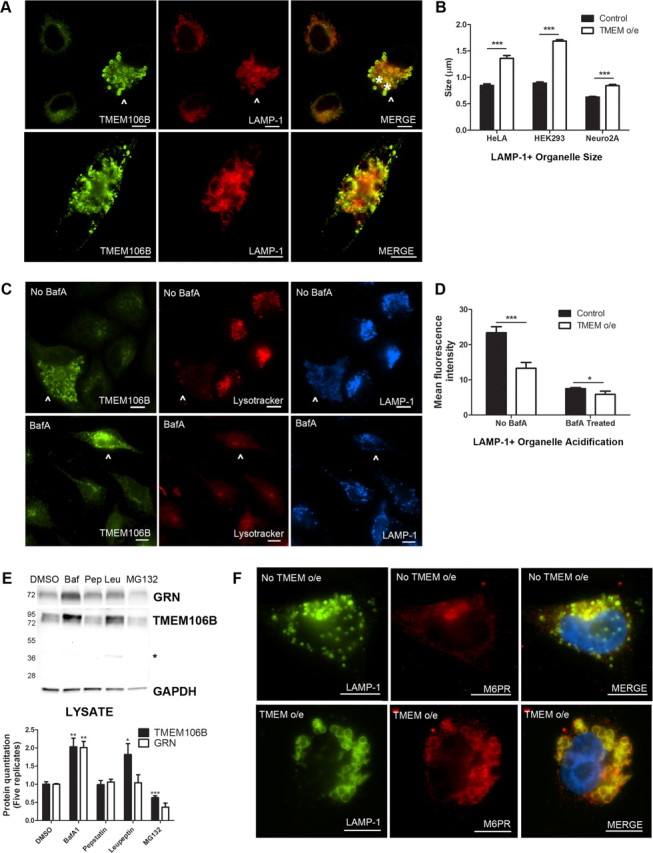
Overexpression of TMEM106B results in abnormalities in the endosomal–lysosomal pathway. A, In HeLA cells overexpressing TMEM106B (arrowhead), LAMP-1+ organelles demonstrate a general increase in size, compared with neighboring cells not overexpressing TMEM106B. In addition, overexpression of TMEM106B also results in occasional formation of large vacuolar structures ∼5 μm in diameter (asterisks indicate two vacuolar structures in top panel, also pictured throughout the cytoplasm of cell in bottom panel). While these large vacuolar structures occur only occasionally with TMEM106B overexpression (the more typical finding is enlarged ∼1.5 μm LAMP-1+ organelles), they are not seen in the absence of TMEM106B overexpression. B, Similar results were obtained in HeLAs, HEK293 cells, and in the neuronal cell line Neuro2A. Size quantitation (mean ± SEM) was performed by measuring LAMP-1+ organelle diameter on >10 40× fields containing a mixture of cells with and without TMEM106B overexpression. Because the large vacuolar structures are only occasionally seen, they were not included in the quantitation. C, HeLA cells overexpressing TMEM106B (arrowhead) showed less intense staining with Lysotracker, a dye which demonstrates greater fluorescence at lower pH, than neighboring cells not overexpressing TMEM106B (top). This effect was abrogated by treatment of cells with bafilomycin A1, an inhibitor of the vacuolar ATPase, which resulted in diminished Lysotracker fluorescence for all cells (bottom). D, Quantitation of mean fluorescence intensity for cells overexpressing TMEM106B demonstrated that Lysotracker staining was significantly less intense than in neighboring cells with normal levels of TMEM106B expression. Quantitation (mean ± SEM) was performed on >10 40× fields containing a mixture of cells with and without TMEM106B overexpression. E, Immunoblot analysis of HeLA cells treated with the vacuolar ATPase inhibitor bafilomycin A1 (Baf) showed increased intracellular levels of TMEM106B and progranulin. Treatment with the lysosomal protease inhibitor leupeptin (Leu) increased levels of TMEM106B but did not affect levels of progranulin. Treatment with the lysosomal protease inhibitor pepstatin A (Pep) did not affect either protein, while treatment with the proteasome inhibitor MG132 decreased TMEM106B levels. Representative immunoblot (top) and quantitation of five replicate immunoblots (mean ± SEM, bottom) are shown. Asterisk indicates TMEM106B 40 kDa band only seen with leupeptin treatment. F, Under normal conditions, the cation-independent M6PR does not colocalize with LAMP-1. In cells overexpressing TMEM106B, M6PR colocalizes with LAMP-1 at the limiting membrane of enlarged LAMP-1+ organelles. *p < 0.05, **p < 0.01, ***p < 0.001. For all immunofluorescence panels, TMEM106B staining was performed with N2077. Scale bar, 10 μm.
Since large vacuolar structures have been described upon treatment of cells with alkalizing agents such as the lysosomotropic drug chloroquine (Brown et al., 1984), we sought to determine whether these large LAMP-1+/TMEM106B+ organelles were acidified. To do this, we used the pH-sensitive dyes Lysotracker and Lysosensor, both weak bases that fluoresce intensely at low pH and weakly (or not at all) at higher pH. As shown in Figure 6C, HeLA cells overexpressing TMEM106B (arrowhead) contained large LAMP-1+/TMEM106B+ organelles, which were not as acidic as the LAMP-1+ organelles in neighboring cells with normal levels of TMEM106B expression. Indeed, the Lysotracker mean fluorescence intensity of the cytoplasmic compartment of cells overexpressing TMEM106B was significantly lower than that of cells expressing normal levels of TMEM106B, indicating a general failure to acidify organelles (Fig. 6D). The effect of TMEM106B overexpression was reminiscent of the cellular alkalization induced by bafilomycin A1 (BafA1), a selective inhibitor of the vacuolar ATPase that acidifies lysosomes (Fig. 6C, bottom, D). Similar results were obtained in HEK293 cells, and in both HeLAs and HEK293 cells live-imaged with the Lysosensor dye (data not shown).
Alkalization of cells with BafA1 was recently reported to result in increased TMEM106B expression (Lang et al., 2012). We confirmed this finding in HeLA cells (Fig. 6E) and HEK293 cells (data not shown). Thus, not only can TMEM106B overexpression impair endosomal–lysosomal acidification, but impairing the acidification of these organelles can in turn result in increased expression of TMEM106B.
We sought to determine whether these enlarged, poorly acidified LAMP-1+ organelles were improperly acidified lysosomes, or late endosomes unable to mature into lysosomes. To do so, we performed double-label immunofluorescence for LAMP-1 and the cation-independent M6PR, which is not present on lysosomes, under conditions of TMEM106B overexpression. In HeLA cells at steady state, the M6PR was found primarily in the trans-Golgi network (TGN) and did not colocalize extensively with LAMP-1 (Fig. 6F, top). However, in cells overexpressing TMEM106B, LAMP-1 and the M6PR colocalized extensively in enlarged organelles (Fig. 6F, bottom), suggesting that there may be a block in endosomal–lysosomal fusion, and, further, that retrograde transport from the late endosome to the TGN is affected.
Together, our data suggest that overexpression of TMEM106B results in endosomal–lysosomal dysfunction as demonstrated by dramatic abnormalities in late endosome-lysosome morphology and acidification, and M6PR trafficking. Moreover, blocking endosomal–lysosomal function could itself prevent TMEM106B degradation, causing its levels to increase, further exacerbating these defects.
Overexpression of TMEM106B alters the appearance and compartmentalization of progranulin
Since progranulin has been previously linked to lysosomes through proteomic identification of lysosomal contents (Kollmann et al., 2005), we investigated whether TMEM106B and progranulin are found together in lysosomes. Indeed, double-label immunofluorescence showed that TMEM106B partially colocalized with progranulin in murine primary cortical neurons (Fig. 7A). Moreover, endogenous neuronal TMEM106B and progranulin appeared most colocalized in neuronal processes, within LAMP-1+ organelles (Fig. 7B).
Figure 7.
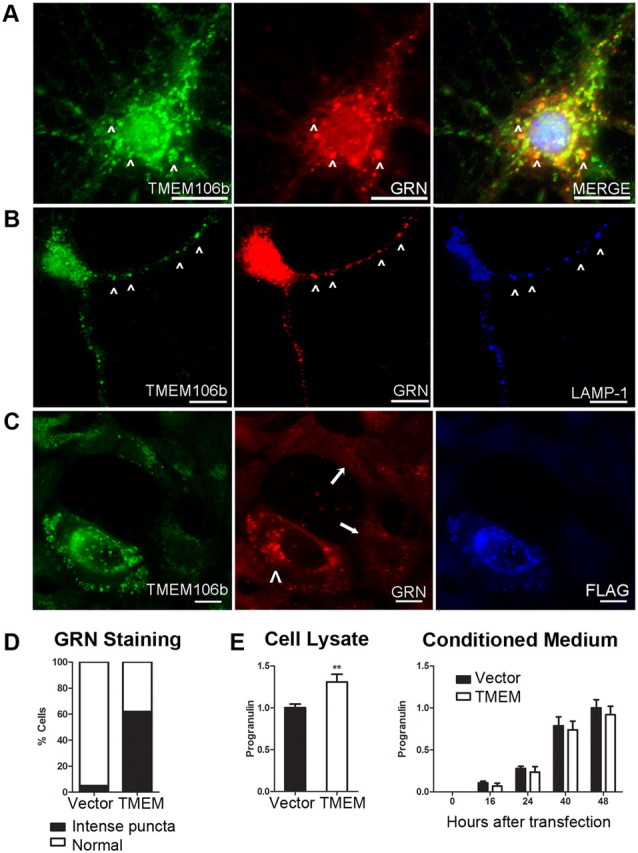
Overexpression of TMEM106B alters the compartmentalization of progranulin. A–C, Immunofluorescence microscopy performed on cells stained for TMEM106B (N2077 antibody) and progranulin. Scale bar, 10 μm. A, B, Endogenous TMEM106B (green) in nontransfected primary murine cortical neurons colocalized with progranulin (GRN, red) in the cell body (A, arrowheads), and in processes (B, arrowheads). TMEM106B and GRN colocalized within late endosomes or lysosomes, as indicated by LAMP-1 staining (B, blue). C, Progranulin (GRN, red) appearance changed under conditions of TMEM106B overexpression. Progranulin formed intensely stained cytoplasmic puncta variably colocalizing with TMEM106B (green) only in HEK293 cells overexpressing FLAG-tagged TMEM106B (arrowhead). In the absence of TMEM106B overexpression, progranulin staining was much less intense (arrows). D, More than 60% of cells overexpressing TMEM106B showed intense cytoplasmic puncta of progranulin, compared with <5% of cells with normal levels of TMEM106B expression. Assessment of progranulin staining pattern performed on six 20× fields of HEK293 cells containing a mixture of cells with and without TMEM106B overexpression. E, Intracellular (left) and extracellular/secreted (right) pools of progranulin were measured by ELISA under conditions of TMEM106B overexpression (TMEM, white bars) versus vector transfection in HEK293 cells. Progranulin measurements (mean ± SEM for five experiments) were normalized to total protein in the cell lysate, to account for differential rates of cell growth. Overexpression of TMEM106B resulted in a 30% increase in intracellular progranulin, with a trend toward decreased extracellular progranulin. Intracellular progranulin is shown measured at 48 h after transfection of TMEM106B. Extracellular progranulin is shown at baseline and indicated time periods after transfection of TMEM106B. **p < 0.01.
Defects in the production or secretion of progranulin are known to cause FTLD-TDP and among the cases with progranulin mutations, we observed increased mRNA and protein expression of TMEM106B (Fig. 2). Furthermore, progranulin has been reported to internalize via endocytosis to lysosomes using sortilin-1 as a plasma membrane receptor (Hu et al., 2010), and our data suggested that TMEM106B overexpression may impair the endosomal–lysosomal pathway. Finally, impairment of endosomal–lysosomal acidification, shown here to be an effect of TMEM106B overexpression, has been reported by others (Capell et al., 2011) to result in increased progranulin levels, a finding we confirmed (Fig. 6E). Thus, we asked what impact increased TMEM106B expression might have on progranulin.
Intriguingly, overexpression of TMEM106B in HEK293 cells (Fig. 7C, arrowhead) and HeLA cells resulted in striking changes in the appearance of progranulin (Fig. 7C), in addition to the enlargement of LAMP-1+ organelles. Indeed, ∼60% of cells overexpressing TMEM106B contained intense puncta that colocalized with progranulin, while <5% of cells with normal levels of TMEM106B expression showed this pattern (Fig. 7D). In neighboring, nontransfected cells with normal levels of TMEM106B expression (Fig. 7C, arrows), progranulin intracellular staining was much less intense, exhibiting the more diffuse pattern previously reported in the literature (Shankaran et al., 2008).
These observations suggested that increased expression of TMEM106B might alter the trafficking of progranulin. We therefore tested the hypothesis that TMEM106B overexpression would affect levels of intracellular or extracellular progranulin. As shown in Figure 7E, intracellular progranulin levels significantly increased by ∼30% with overexpression of TMEM106B in HEK293 cells. In contrast, extracellular progranulin measured in the conditioned medium remained relatively constant, showing a nonsignificant slight decrease in the setting of TMEM106B overexpression. Normalization of progranulin measurements to those of another secreted protein, IGFBP-2, did not alter these results and in fact increased the apparent differences. Thus, overexpression of TMEM106B increases levels of intracellular progranulin.
Together, these data demonstrate that aberrant overexpression of TMEM106B affects the distribution and intracellular levels of progranulin, suggesting that the two proteins may act in the same pathogenic pathway in FTLD-TDP.
Discussion
Following our recent GWAS reporting the uncharacterized gene TMEM106B as a risk factor for FTLD-TDP, here we describe the genetic regulation of TMEM106B and its relationship to progranulin. Specifically, we show that TMEM106B is highly expressed in human brain tissue, with particularly increased expression in GRN(+) FTLD-TDP. We further demonstrate that FTLD-TDP (with and without GRN mutations) shows <50% normal expression of three members of the miR-132 cluster: miR-132, miR-132*, and miR-212. Strikingly, miR-132 and miR-212 repress TMEM106B through two specific sites in the TMEM106B 3′UTR. Having observed increased TMEM106B expression in FTLD-TDP, and uncovered a possible upstream cause in miR-132/212, we then explored the consequences of TMEM106B overexpression. Overexpression of TMEM106B, which shows steady-state neuronal localization to late endo-lysosomes and colocalizes in these organelles with progranulin, disrupts late endo-lysosome function and consequently, both the appearance and compartmentalization of progranulin. Together, the data presented in the current study are compatible with a model (Fig. 8) that illustrates how decreased levels of miR-132/212 lead to increased TMEM106B expression, perturbation of progranulin pathways, and increased risk of developing FTLD-TDP.
Figure 8.

Hypothetical model of causes and effects of TMEM106B overexpression in FTLD-TDP. Our data are compatible with a model whereby decreased levels of miR-132/212 result in increased TMEM106B expression. As a result, increased TMEM106B expression leads to (1) endosomal–lysosomal dysfunction, which may in turn further increase levels of TMEM106B, and also to (2) perturbation of progranulin pathways, thereby increasing the risk of developing FTLD-TDP. Black arrows indicate steps evidenced by the current study, gray arrows indicate steps reported in the literature, and yellow arrows indicate hypothetical steps.
There have been conflicting data regarding the levels of TMEM106B expression in FTLD-TDP. In our initial article (Van Deerlin et al., 2010), we reported that FTLD-TDP patients show increased expression of TMEM106B mRNA in frontal cortex. Others, however, have not found significant differences in TMEM106B mRNA expression in FTLD-TDP patients versus controls (van der Zee et al., 2011). In the present study, we demonstrate again that TMEM106B expression is increased in disease states. First, we directly measure human brain TMEM106B at the mRNA and protein levels, quantifying transcript levels in multiple brain regions; we consistently observe higher levels of TMEM106B in GRN(+) FTLD-TDP. In addition, we present a second, independent line of evidence suggesting that TMEM106B expression is aberrantly elevated in FTLD-TDP. Specifically, we find that miR-132 and miR-212, both computationally predicted and experimentally shown to negatively regulate TMEM106B, are significantly reduced in GRN(−) and GRN(+) FTLD-TDP brain. The expected effect of a reduction in these miRs would be de-repression, or overexpression, of their target mRNA TMEM106B.
In terms of miR-132/212 regulation of TMEM106B, while both miRs can decrease TMEM106B expression, miR-132 may be the dominant miR. Specifically, miR-132 levels are orders of magnitude higher than miR-212 levels in human brain tissue, and only miR-132 elevation was significantly associated with decrease of TMEM106B in neuronal cells.
The miR-132 cluster—comprised of miR-132, miR-132*, miR-212, and a fourth potential member, miR-212*—has been previously shown to be important in neuronal development and in the neurobiology of addictive behavior. Much of the literature has focused on miR-132 and miR-212, which are also the only two miRs in the cluster that show potential binding to TMEM106B. Specifically, introduction of miR-132 in primary neuronal cultures has been reported to stimulate neurite outgrowth through specific effector mRNAs (Vo et al., 2005), and knockdown of the miR-132 cluster in newborn hippocampal neurons decreases the length and arborization of dendrites (Magill et al., 2010). Increasing striatal expression of miR-212 through lentiviral delivery, on the other hand, has been reported to decrease addictive cocaine ingestion behavior in rats, while inhibition of miR-212 has an opposing effect (Hollander et al., 2010). Both miR-132 and miR-212 appear to be regulated by the transcription factor CREB (Impey et al., 2004), but the connection, if any, between the neuronal phenotypes observed with miR-132 manipulation and the whole organism behaviors observed with miR-212 manipulation are unclear. In any case, miR-132 and miR-212 have been postulated to be the downstream effectors of the phenotypes—including increased neuronal survival—associated with CREB activation (Wayman et al., 2008; Magill et al., 2010). Thus, the greatly decreased levels of miR-132 cluster expression observed in FTLD-TDP may have deleterious effects on neurons beyond the de-repression of TMEM106B.
In addition to demonstrating the miR-132/212 regulation of TMEM106B, our study adds to a growing body of evidence that TMEM106B and progranulin may be mechanistically related. That is, TMEM106B genotype has been reported to correlate with plasma levels of progranulin in normal controls (Finch et al., 2011), and with age at onset in GRN(+) FTLD-TDP (Cruchaga et al., 2011). Evidence from the current study to support a link between TMEM106B and progranulin includes the following: (1) increased protein and mRNA expression of TMEM106B in GRN(+) FTLD-TDP cases; (2) abnormal localization of TMEM106B in neuronal processes in GRN(+) FTLD-TDP cases; (3) colocalization of TMEM106B and progranulin in late endo-lysosomes; and (4) increased accumulation of intracellular progranulin when TMEM106B is overexpressed. Indeed, we provide the first demonstration of a relationship between TMEM106B and progranulin in a model system that can be directly manipulated. Of note, a prior study (Lang et al., 2012) reported no change in progranulin levels with overexpression of TMEM106B, a result that contradicts our present result. Various methodological differences, such as our use of ELISA to measure progranulin, may explain the discrepancy.
At a more general level, the data presented here strongly support TMEM106B as a risk gene for FTLD-TDP and the source of the 7p21 signal found by GWAS. First, there is the striking fact that both miR-132 and TMEM106B emerged as single-best hits from independent genomic-scale screens in FTLD-TDP. Their convergence as a microRNA:mRNA regulatory pair greatly adds to our confidence that both are real signals and also argues for the centrality of the miR-132/212: TMEM106B pathway in this disease. Second, we link TMEM106B to effects on progranulin—haploinsufficiency of which is a known pathogenic mechanism in FTLD-TDP. In doing so, our data suggest a clear direction for further mechanistic studies of TMEM106B. It is worth noting that establishment of whether a GWAS signal is “true” and, additionally, what the causative gene behind a signal might be, is not always straightforward. Indeed, while additional cohorts of clinical FTLD (van der Zee et al., 2011) and clinical-pathologic FTLD/FTLD-TDP cases (Finch et al., 2011) have replicated the association of FTLD with rs1990622 genotype, some clinical cohorts have not demonstrated this association (Rollinson et al., 2011), possibly because only ∼50% of FTLD clinical cases have underlying FTLD-TDP.
Future directions suggested by the data presented here include a detailed analysis of the biochemistry of TMEM106B. Specifically, we have shown that TMEM106B is glycosylated and heat sensitive, with both 75 and 40 kDa species—collapsing to ∼60 and 31 kDa, respectively, after deglycosylation—apparent by immunoblotting. However, it is presently unclear whether TMEM106B, with a predicted molecular weight of 31 kDa, migrates at the higher molecular weight because it self-associates into a dimer or because it is complexed to another unknown protein. Understanding the native complex in which TMEM106B occurs may be important in determining its molecular function in normal and disease states.
Additionally, the exact molecular mechanisms by which increased expression of TMEM106B affects progranulin are at present unclear and would be an important direction for future studies. Several hypotheses are compatible with our data. First, increased TMEM106B might cause intracellular retention of progranulin, an intriguing possibility since decreased secretion of progranulin is a known mechanism of FTLD-TDP pathogenesis (Mukherjee et al., 2008; Shankaran et al., 2008). We note here that while we did not see a significant drop in secreted progranulin with TMEM106B overexpression, the trend toward decreased extracellular progranulin was present at every time point, and technical limitations such as the high turnover rate in culture medium may have precluded our ability to see a significant change in levels of secreted progranulin. Second, increased TMEM106B might increase the internalization of progranulin from the extracellular space, in the same way that sortilin-1 overexpression increases progranulin's internalization and sorting into lysosomes (Hu et al., 2010). Third, increased TMEM106B may impair the maturation of lysosomes, as suggested by our observation of enlarged, poorly acidified organelles expressing both LAMP-1 and the M6PR upon TMEM106B overexpression. This defect may in turn alter the dynamics of progranulin internalization and turnover, as others have shown (Capell et al., 2011), and we have confirmed that blocking lysosomal acidification can increase levels of progranulin.
In conclusion, we demonstrate here the microRNA regulation of TMEM106B and its effects on the endosomal–lysosomal pathway and the FTLD-TDP-associated protein progranulin. Our findings thus open up novel directions for elucidating miR-based mechanisms of neurodegeneration in FTLD-TDP and related TDP-43 proteinopathies that could become targets for drug discovery in these currently untreatable diseases.
Footnotes
This work was supported by grants from the NIH (AG-033101, AG-17586), the Koller Foundation for ALS Research, the Burroughs Wellcome Fund Career Award for Medical Scientists (A.S.C.P.), and the Benaroya Fund (A.S.C.P.). V.M.-Y.L. is the John H. Ware III Chair in Alzheimer's Disease Research, and J.Q.T. is the William Maul Measey-Truman G. Schnabel Jr. Chair of Geriatric Medicine and Gerontology. We thank Joshua Plotkin for valuable discussions; Judy Qiang, Margaret Maronski, and Marc Dichter for technical assistance; and many patients and their families for contributing samples for this study.
The authors declare no competing financial interests.
References
- Arai T, Hasegawa M, Akiyama H, Ikeda K, Nonaka T, Mori H, Mann D, Tsuchiya K, Yoshida M, Hashizume Y, Oda T. TDP-43 is a component of ubiquitin-positive tau-negative inclusions in frontotemporal lobar degeneration and amyotrophic lateral sclerosis. Biochem Biophys Res Commun. 2006;351:602–611. doi: 10.1016/j.bbrc.2006.10.093. [DOI] [PubMed] [Google Scholar]
- Baker M, Mackenzie IR, Pickering-Brown SM, Gass J, Rademakers R, Lindholm C, Snowden J, Adamson J, Sadovnick AD, Rollinson S, Cannon A, Dwosh E, Neary D, Melquist S, Richardson A, Dickson D, Berger Z, Eriksen J, Robinson T, Zehr C, et al. Mutations in progranulin cause tau-negative frontotemporal dementia linked to chromosome 17. Nature. 2006;442:916–919. doi: 10.1038/nature05016. [DOI] [PubMed] [Google Scholar]
- Benajiba L, Le Ber I, Camuzat A, Lacoste M, Thomas-Anterion C, Couratier P, Legallic S, Salachas F, Hannequin D, Decousus M, Lacomblez L, Guedj E, Golfier V, Camu W, Dubois B, Campion D, Meininger V, Brice A. TARDBP mutations in motoneuron disease with frontotemporal lobar degeneration. Ann Neurol. 2009;65:470–473. doi: 10.1002/ana.21612. [DOI] [PubMed] [Google Scholar]
- Brown WJ, Constantinescu E, Farquhar MG. Redistribution of mannose-6-phosphate receptors induced by tunicamycin and chloroquine. J Cell Biol. 1984;99:320–326. doi: 10.1083/jcb.99.1.320. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Capell A, Liebscher S, Fellerer K, Brouwers N, Willem M, Lammich S, Gijselinck I, Bittner T, Carlson AM, Sasse F, Kunze B, Steinmetz H, Jansen R, Dormann D, Sleegers K, Cruts M, Herms J, Van Broeckhoven C, Haass C. Rescue of progranulin deficiency associated with frontotemporal lobar degeneration by alkalizing reagents and inhibition of vacuolar ATPase. J Neurosci. 2011;31:1885–1894. doi: 10.1523/JNEUROSCI.5757-10.2011. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Chen-Plotkin AS, Geser F, Plotkin JB, Clark CM, Kwong LK, Yuan W, Grossman M, Van Deerlin VM, Trojanowski JQ, Lee VM. Variations in the progranulin gene affect global gene expression in frontotemporal lobar degeneration. Hum Mol Genet. 2008;17:1349–1362. doi: 10.1093/hmg/ddn023. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Chen-Plotkin AS, Lee VM, Trojanowski JQ. TAR DNA-binding protein 43 in neurodegenerative disease. Nat Rev Neurol. 2010a;6:211–220. doi: 10.1038/nrneurol.2010.18. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Chen-Plotkin AS, Xiao J, Geser F, Martinez-Lage M, Grossman M, Unger T, Wood EM, Van Deerlin VM, Trojanowski JQ, Lee VM. Brain progranulin expression in GRN-associated frontotemporal lobar degeneration. Acta Neuropathol. 2010b;119:111–122. doi: 10.1007/s00401-009-0576-2. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Cruchaga C, Graff C, Chiang HH, Wang J, Hinrichs AL, Spiegel N, Bertelsen S, Mayo K, Norton JB, Morris JC, Goate A. Association of TMEM106B gene polymorphism with age at onset in granulin mutation carriers and plasma granulin protein levels. Arch Neurol. 2011;68:581–586. doi: 10.1001/archneurol.2010.350. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Cruts M, Gijselinck I, van der Zee J, Engelborghs S, Wils H, Pirici D, Rademakers R, Vandenberghe R, Dermaut B, Martin JJ, van Duijn C, Peeters K, Sciot R, Santens P, De Pooter T, Mattheijssens M, Van den Broeck M, Cuijt I, Vennekens K, De Deyn PP, et al. Null mutations in progranulin cause ubiquitin-positive frontotemporal dementia linked to chromosome 17q21. Nature. 2006;442:920–924. doi: 10.1038/nature05017. [DOI] [PubMed] [Google Scholar]
- Cruts M, Van Broeckhoven C. Loss of progranulin function in frontotemporal lobar degeneration. Trends Genet. 2008;24:186–194. doi: 10.1016/j.tig.2008.01.004. [DOI] [PubMed] [Google Scholar]
- Filipowicz W, Bhattacharyya SN, Sonenberg N. Mechanisms of post-transcriptional regulation by microRNAs: are the answers in sight? Nat Rev Genet. 2008;9:102–114. doi: 10.1038/nrg2290. [DOI] [PubMed] [Google Scholar]
- Finch N, Carrasquillo MM, Baker M, Rutherford NJ, Coppola G, Dejesus-Hernandez M, Crook R, Hunter T, Ghidoni R, Benussi L, Crook J, Finger E, Hantanpaa KJ, Karydas AM, Sengdy P, Gonzalez J, Seeley WW, Johnson N, Beach TG, Mesulam M, et al. TMEM106B regulates progranulin levels and the penetrance of FTLD in GRN mutation carriers. Neurology. 2011;76:467–474. doi: 10.1212/WNL.0b013e31820a0e3b. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Gass J, Cannon A, Mackenzie IR, Boeve B, Baker M, Adamson J, Crook R, Melquist S, Kuntz K, Petersen R, Josephs K, Pickering-Brown SM, Graff-Radford N, Uitti R, Dickson D, Wszolek Z, Gonzalez J, Beach TG, Bigio E, Johnson N, et al. Mutations in progranulin are a major cause of ubiquitin-positive frontotemporal lobar degeneration. Hum Mol Genet. 2006;15:2988–3001. doi: 10.1093/hmg/ddl241. [DOI] [PubMed] [Google Scholar]
- Gitcho MA, Baloh RH, Chakraverty S, Mayo K, Norton JB, Levitch D, Hatanpaa KJ, White CL, 3rd, Bigio EH, Caselli R, Baker M, Al-Lozi MT, Morris JC, Pestronk A, Rademakers R, Goate AM, Cairns NJ. TDP-43 A315T mutation in familial motor neuron disease. Ann Neurol. 2008;63:535–538. doi: 10.1002/ana.21344. [DOI] [PMC free article] [PubMed] [Google Scholar]
- He Z, Bateman A. Progranulin (granulin-epithelin precursor, PC-cell-derived growth factor, acrogranin) mediates tissue repair and tumorigenesis. J Mol Med (Berl) 2003;81:600–612. doi: 10.1007/s00109-003-0474-3. [DOI] [PubMed] [Google Scholar]
- Hébert SS, Horré K, Nicolaï L, Papadopoulou AS, Mandemakers W, Silahtaroglu AN, Kauppinen S, Delacourte A, De Strooper B. Loss of microRNA cluster miR-29a/b-1 in sporadic Alzheimer's disease correlates with increased BACE1/beta-secretase expression. Proc Natl Acad Sci U S A. 2008;105:6415–6420. doi: 10.1073/pnas.0710263105. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hollander JA, Im HI, Amelio AL, Kocerha J, Bali P, Lu Q, Willoughby D, Wahlestedt C, Conkright MD, Kenny PJ. Striatal microRNA controls cocaine intake through CREB signalling. Nature. 2010;466:197–202. doi: 10.1038/nature09202. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hu F, Padukkavidana T, Vægter CB, Brady OA, Zheng Y, Mackenzie IR, Feldman HH, Nykjaer A, Strittmatter SM. Sortilin-mediated endocytosis determines levels of the frontotemporal dementia protein, progranulin. Neuron. 2010;68:654–667. doi: 10.1016/j.neuron.2010.09.034. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Impey S, McCorkle SR, Cha-Molstad H, Dwyer JM, Yochum GS, Boss JM, McWeeney S, Dunn JJ, Mandel G, Goodman RH. Defining the CREB regulon: a genome-wide analysis of transcription factor regulatory regions. Cell. 2004;119:1041–1054. doi: 10.1016/j.cell.2004.10.032. [DOI] [PubMed] [Google Scholar]
- Ishikura N, Clever JL, Bouzamondo-Bernstein E, Samayoa E, Prusiner SB, Huang EJ, DeArmond SJ. Notch-1 activation and dendritic atrophy in prion disease. Proc Natl Acad Sci U S A. 2005;102:886–891. doi: 10.1073/pnas.0408612101. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kim J, Inoue K, Ishii J, Vanti WB, Voronov SV, Murchison E, Hannon G, Abeliovich A. A MicroRNA feedback circuit in midbrain dopamine neurons. Science. 2007;317:1220–1224. doi: 10.1126/science.1140481. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kollmann K, Mutenda KE, Balleininger M, Eckermann E, von Figura K, Schmidt B, Lübke T. Identification of novel lysosomal matrix proteins by proteome analysis. Proteomics. 2005;5:3966–3978. doi: 10.1002/pmic.200401247. [DOI] [PubMed] [Google Scholar]
- Lang CM, Fellerer K, Schwenk BM, Kuhn PH, Kremmer E, Edbauer D, Capell A, Haass C. Membrane orientation and subcellular localization of transmembrane protein 106B (TMEM106B), a major risk factor for frontotemporal lobar degeneration. J Biol Chem. 2012;287:19355–19365. doi: 10.1074/jbc.M112.365098. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lee Y, Samaco RC, Gatchel JR, Thaller C, Orr HT, Zoghbi HY. miR-19, miR-101 and miR-130 co-regulate ATXN1 levels to potentially modulate SCA1 pathogenesis. Nat Neurosci. 2008;11:1137–1139. doi: 10.1038/nn.2183. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lewis BP, Shih IH, Jones-Rhoades MW, Bartel DP, Burge CB. Prediction of mammalian microRNA targets. Cell. 2003;115:787–798. doi: 10.1016/s0092-8674(03)01018-3. [DOI] [PubMed] [Google Scholar]
- Lippa CF, Rosso AL, Stutzbach LD, Neumann M, Lee VM, Trojanowski JQ. Transactive response DNA-binding protein 43 burden in familial Alzheimer disease and Down syndrome. Arch Neurol. 2009;66:1483–1488. doi: 10.1001/archneurol.2009.277. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Magill ST, Cambronne XA, Luikart BW, Lioy DT, Leighton BH, Westbrook GL, Mandel G, Goodman RH. microRNA-132 regulates dendritic growth and arborization of newborn neurons in the adult hippocampus. Proc Natl Acad Sci U S A. 2010;107:20382–20387. doi: 10.1073/pnas.1015691107. [DOI] [PMC free article] [PubMed] [Google Scholar]
- McKhann GM, Albert MS, Grossman M, Miller B, Dickson D, Trojanowski JQ. Clinical and pathological diagnosis of frontotemporal dementia: report of the Work Group on Frontotemporal Dementia and Pick's Disease. Arch Neurol. 2001;58:1803–1809. doi: 10.1001/archneur.58.11.1803. [DOI] [PubMed] [Google Scholar]
- Mukherjee O, Wang J, Gitcho M, Chakraverty S, Taylor-Reinwald L, Shears S, Kauwe JS, Norton J, Levitch D, Bigio EH, Hatanpaa KJ, White CL, Morris JC, Cairns NJ, Goate A. Molecular characterization of novel progranulin (GRN) mutations in frontotemporal dementia. Hum Mutat. 2008;29:512–521. doi: 10.1002/humu.20681. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Neumann M, Sampathu DM, Kwong LK, Truax AC, Micsenyi MC, Chou TT, Bruce J, Schuck T, Grossman M, Clark CM, McCluskey LF, Miller BL, Masliah E, Mackenzie IR, Feldman H, Feiden W, Kretzschmar HA, Trojanowski JQ, Lee VM. Ubiquitinated TDP-43 in frontotemporal lobar degeneration and amyotrophic lateral sclerosis. Science. 2006;314:130–133. doi: 10.1126/science.1134108. [DOI] [PubMed] [Google Scholar]
- Ratnavalli E, Brayne C, Dawson K, Hodges JR. The prevalence of frontotemporal dementia. Neurology. 2002;58:1615–1621. doi: 10.1212/wnl.58.11.1615. [DOI] [PubMed] [Google Scholar]
- Remenyi J, Hunter CJ, Cole C, Ando H, Impey S, Monk CE, Martin KJ, Barton GJ, Hutvagner G, Arthur JS. Regulation of the miR-212/132 locus by MSK1 and CREB in response to neurotrophins. Biochem J. 2010;428:281–291. doi: 10.1042/BJ20100024. [DOI] [PubMed] [Google Scholar]
- Rollinson S, Mead S, Snowden J, Richardson A, Rohrer J, Halliwell N, Usher S, Neary D, Mann D, Hardy J, Pickering-Brown S. Frontotemporal lobar degeneration genome wide association study replication confirms a risk locus shared with amyotrophic lateral sclerosis. Neurobiol Aging. 2011;32:758.e1–e7. doi: 10.1016/j.neurobiolaging.2010.12.005. [DOI] [PubMed] [Google Scholar]
- Shankaran SS, Capell A, Hruscha AT, Fellerer K, Neumann M, Schmid B, Haass C. Missense mutations in the progranulin gene linked to frontotemporal lobar degeneration with ubiquitin-immunoreactive inclusions reduce progranulin production and secretion. J Biol Chem. 2008;283:1744–1753. doi: 10.1074/jbc.M705115200. [DOI] [PubMed] [Google Scholar]
- Tseng HC, Ruegg SJ, Maronski M, Messam CA, Grinspan JB, Dichter MA. Injuring neurons induces neuronal differentiation in a population of hippocampal precursor cells in culture. Neurobiol Dis. 2006;22:88–97. doi: 10.1016/j.nbd.2005.10.007. [DOI] [PubMed] [Google Scholar]
- Van Deerlin VM, Sleiman PM, Martinez-Lage M, Chen-Plotkin A, Wang LS, Graff-Radford NR, Dickson DW, Rademakers R, Boeve BF, Grossman M, Arnold SE, Mann DM, Pickering-Brown SM, Seelaar H, Heutink P, van Swieten JC, Murrell JR, Ghetti B, Spina S, Grafman J, et al. Common variants at 7p21 are associated with frontotemporal lobar degeneration with TDP-43 inclusions. Nat Genet. 2010;42:234–239. doi: 10.1038/ng.536. [DOI] [PMC free article] [PubMed] [Google Scholar]
- van der Zee J, Van Langenhove T, Kleinberger G, Sleegers K, Engelborghs S, Vandenberghe R, Santens P, Van den Broeck M, Joris G, Brys J, Mattheijssens M, Peeters K, Cras P, De Deyn PP, Cruts M, Van Broeckhoven C. TMEM106B is associated with frontotemporal lobar degeneration in a clinically diagnosed patient cohort. Brain. 2011;134:808–815. doi: 10.1093/brain/awr007. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Vass R, Ashbridge E, Geser F, Hu WT, Grossman M, Clay-Falcone D, Elman L, McCluskey L, Lee VM, Van Deerlin VM, Trojanowski JQ, Chen-Plotkin AS. Risk genotypes at TMEM106B are associated with cognitive impairment in amyotrophic lateral sclerosis. Acta Neuropathol. 2011;121:373–380. doi: 10.1007/s00401-010-0782-y. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Vo N, Klein ME, Varlamova O, Keller DM, Yamamoto T, Goodman RH, Impey S. A cAMP-response element binding protein-induced microRNA regulates neuronal morphogenesis. Proc Natl Acad Sci U S A. 2005;102:16426–16431. doi: 10.1073/pnas.0508448102. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wang WX, Rajeev BW, Stromberg AJ, Ren N, Tang G, Huang Q, Rigoutsos I, Nelson PT. The expression of microRNA miR-107 decreases early in Alzheimer's disease and may accelerate disease progression through regulation of beta-site amyloid precursor protein-cleaving enzyme 1. J Neurosci. 2008;28:1213–1223. doi: 10.1523/JNEUROSCI.5065-07.2008. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wayman GA, Davare M, Ando H, Fortin D, Varlamova O, Cheng HY, Marks D, Obrietan K, Soderling TR, Goodman RH, Impey S. An activity-regulated microRNA controls dendritic plasticity by down-regulating p250GAP. Proc Natl Acad Sci U S A. 2008;105:9093–9098. doi: 10.1073/pnas.0803072105. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wettenhall JM, Smyth GK. limmaGUI: a graphical user interface for linear modeling of microarray data. Bioinformatics. 2004;20:3705–3706. doi: 10.1093/bioinformatics/bth449. [DOI] [PubMed] [Google Scholar]