

Abstract
Elevated circulating glucose resulting from complications of obesity and metabolic disease can result in the accumulation of advanced glycation end products (AGEs) of proteins, lipids, and DNA. The formation of DNA–AGEs assumes particular importance as these adducts may contribute to genetic instability and elevated cancer risk associated with metabolic disease. The principal DNA–AGE, N2-(1-carboxyethyl)-2′-deoxyguanosine (CEdG), is formed as a mixture of R and S isomers at both the polymer and monomer levels. In order to examine the miscoding potential of this adduct, oligonucleotides substituted with (R)- and (S)-CEdG and the corresponding triphosphates (R)- and (S)-CEdGTP were synthesized, and base-pairing preferences for each stereoisomer were examined using steady-state kinetic approaches. Purine dNTPs were preferentially incorporated opposite template CEdG when either the Klenow (Kf–) or Thermus aquaticus (Taq) polymerases were used. The Kf– polymerase preferentially incorporated dGTP, whereas Taq demonstrated a bias for dATP. Kf– incorporated purines opposite the R isomer with greater efficiency, but Taq favored the S isomer. Incorporation of (R)- and (S)-CEdGTP only occurred opposite dC and was catalyzed by Kf– with equal efficiencies. Primer extension from a 3′-terminal CEdG was observed only for the R isomer. These data suggest CEdG is the likely adduct responsible for the observed pattern of G transversions induced by exposure to elevated glucose or its α-oxoaldehyde decomposition product methylglyoxal. The results imply that CEdG within template DNA and the corresponding triphosphate possess different syn/anti conformations during replication which influence base-pairing preferences. The implications for CEdG-induced mutagenesis in vivo are discussed.
Increased glycolytic stress resulting from metabolic disease, or elevated levels of cytoplasmic glycolysis relative to mitochondrial oxidative phosphorylation, can result in the formation of advanced glycation end products (AGEs)1 of proteins (1), lipids (2), and DNA (3, 4). One of the major reactive aldehydes responsible for AGE formation is methylglyoxal (MG), a byproduct of the triose-phosphate isomerase reaction of glycolysis and a major carbohydrate decomposition product (5, 6). Protein AGEs have been well-studied, and they are believed to play a major role in the pathologies associated with diabetes, aging, and Alzheimer’s disease (7, 8). The pathological implications of lipid and DNA–AGE accumulation are presently unknown. The reaction of MG with DNA (Scheme 1) assumes special importance since the resulting AGEs may contribute to genetic instability and play an important role in elevating the risk for certain cancers associated with metabolic disease. Information regarding the biodistribution and pathological consequences of DNA–AGEs has been scant owing to a prior lack of quantitative methods for its determination in biological samples. Immunoaffinity methods were recently used to demonstrate elevated levels of DNA–AGEs in aorta and kidney of diabetic patients relative to normal controls, the first demonstration of a correlation between DNA–AGEs and human disease (9).
Scheme 1.

Proposed Mechanism of Formation of (R,S)-CEdG via the Addition of Methylglyoxal to N1 and N2 of 2′-dGa
aAdapted from ref 4.
Recently, we described an LC-ESI-MS/MS protocol using the isotope dilution method for quantitation of the principal DNA–AGE N2-(1-carboxyethyl)-2′-deoxyguanosine (CEdG) in biological samples. The preparation of stable isotope derivatives of (R)- and (S)-15N5-CEdG allowed for quantitative determination of their distribution in urine and in DNA hydrolysates from tissue extracts (10). Measurements in human breast tumors and adjacent tissues revealed an adduct density of ~1–10 CEdG/107 guanines, suggesting that it is a relatively common adduct in vivo. In order to explore the miscoding potential of CEdG in DNA, we synthesized site-specifically modified oligonucleotides containing stereochemically pure R or S stereoisomers and characterized the steady-state kinetics of dNTP incorporation opposite either adduct catalyzed by two relatively high fidelity DNA polymerases. Kinetic data are presented which reveal a distinct preference for CEdG/purine mispairs as well as the influence of adduct stereochemistry on mispair formation. Stereochemically pure (R)- and (S)-CEdGTP were also prepared, and their suitability as polymerase substrates was examined in order to assess whether incorporation of glycated nucleotides from the dNTP pool might contribute to the accumulation of CEdG in DNA (11). The potential implications of these results for genetic instability induced by DNA glycation are discussed.
EXPERIMENTAL PROCEDURES
Reagents
5′-Dimethoxytrityl-2-fluoro-O6-(p-nitrophenyl)-ethyl-2′-deoxyinosine 3′-[(2-cyanoethyl)-(N,N-diisopropyl)]phosphoramidite (2-F-dI-CE-phosphoramidite) and 3′-phosphate CPG support were purchased from Glen Research (Sterling, VA), dGTP was from Chem-Impex (Wood Dale, IL); Klenow enzyme was from USB (Cleveland, OH), Thermus aquaticus (Taq) polymerase was from Roche Diagnostics (Indianapolis, IN), and calf intestinal alkaline phosphatase (CIP) and associated buffers were from New England Biolabs (Ipswich, MA). All other reagents and solvents were obtained from Sigma-Aldrich (St. Louis, MO) unless otherwise indicated.
Preparation of (R)- and (S)-CEdG-Containing Oligonucleotides
Oligonucleotides were prepared using an ABI394 DNA synthesizer equipped with either standard or 2-F-dI-CE phosphoramidites (0.2 μM scale). For the preparation of CEdG-substituted oligonucleotides, F-dI-containing fully protected oligomers bound to the CPG support were suspended in an aqueous solution of 1 M d- or l-alanine in 250 mM potassium carbonate (50 °C, 40 h). Complete removal of all protecting groups was achieved by extended reaction in concentrated ammonia (50 °C, 7 days). Separation of the desired oligonucleotide from failure sequences and other impurities was achieved by ion-pairing chromatography in 100 mM triethylammonium acetate (TEAA; Fluka, Milwaukee, WI) on a 10 mm × 250 mm XBridge C18 5 μm column (Waters, Milford, MA) using a 9.0–9.5% gradient of acetonitrile (40 min, 45 °C column temperature). All purified oligonucleotides were characterized by rechromatography under the indicated conditions and analyzed by ESI-FT/MS on an LTQ-FT instrument (Thermo-Finnigan, San Jose, CA) in the Mass Spectrometry Core of the City of Hope Cancer Center (see Supporting Information). The maximum contamination by dG at the CEdG-substituted position (generated by deprotection of unreacted 2-FdI containing oligo in ammonia) was <1.4% as assessed by relative intensities of the FT-MS signals. 3′-CEdG-terminated oligonucleotides were prepared in essentially the same fashion, with the exception that synthesis was begun using 3′-phosphate CPG support, followed by the addition of the 2-F-dI-CE phosphoramidite. The resulting oligos were treated with CIP to remove the 3′-phosphate prior to final purification by HPLC.
Synthesis of (R)- and (S)-CEdGTP
To 1.8 mL of purified water in a 15 mL polypropylene tube were added 96.7 mg (168.7μmol) of dGTP, 115.9mg (1.29mmol) of d,l-glyceraldehyde, 360.5 mg of potassium phosphate monobasic, and 729.4 mg of sodium phosphate dibasic. The suspension was vortexed and placed in a water bath (65 °C, 40 min) and then agitated to yield a clear, pale yellow solution. This was allowed to react at 65 °C for an additional 220 min with periodic vortexing. The solution was stored at −80 °C until purification. An XK16/20 column (Pharmacia) was packed with DEAE-cellulose (Whatman DE32) and fitted to an HP 1100 HPLC system. Absorbance was monitored at 280 nm. A 100 μL aliquot of the reaction was injected and eluted with 5% aqueous ethanol at a flow rate of 1 mL/min. An initial 25 min desalting gradient of 0–400 mM ammonium acetate (pH 7) was followed by a gradient of 400–600 mM ammonium acetate from 25 to 140 min. A single broad peak with several small shoulders eluted from 30 to 130 min. This was collected in 20 × 5 mL fractions. Analysis by ESI-MS indicated that fractions 1–5 were predominantly inorganic salts, whereas the majority of CEdGTP eluted in fraction 18. Mass spectrometry data indicated the major impurities were consistent with CEdGDP, unreacted dGTP, and traces of CEdG tetraphosphate. This fraction was rechromatographed until all remaining traces of dGTP were removed (<0.0001% relative to CEdGTP by FT-MS). Additional purification, including separation of the diastereomers of CEdGTP, was carried out on a 10 × 250 mm XBridge column (Waters Corp., Milford, MA) using a 20 min gradient of 2–4% acetonitrile in 50 mM TEAA at 2.5 mL/min. The two diastereomers of CEdGTP were incompletely resolved, and the peaks containing them were collected in three fractions. A sample of each fraction was digested with CIP, and the configurations of the released nucleosides were determined by matching theirretention times with that of previously synthesized samples of known configuration (Supporting Information Figure S2). Fraction 1 is nearly pure R isomer and fraction 3 nearly pure S isomer. The samples were quantified by UV spectroscopy assuming a molar extinction coefficient of 13700 M– L–1.
Steady-State Kinetic Analyses of dNTP Incorporation opposite (R)- and (S)-CEdG Catalyzed by Kf–
Oligonucleotide primers were 5′-labeled by diluting 120 pmol of primer with 4 μL of water and 1 μL each of T4 polynucleotide kinase (PNK) and 10× PNK buffer together with 4 μL (8.9 pmol) of [γ-32P]ATP. Tubes were sealed and incubated (37 °C, 60 min) and then placed at 70 °C (30 min) to inactivate PNK, resulting in a 12 μM primer solution labeled to about 7.5%. To measure dNTP incorporation opposite CEdG in DNA, templates containing either (R)- or (S)-CEdG (1) were annealed to primer (7) by heating equimolar amounts of each in a volume of 70 μL to 95 °C for 5 min followed by gradual cooling to 37 °C. To this solution was added 10 μL of 1 unit/μL Klenow enzyme (USB, Cleveland, OH), resulting in a final volume of 80 μL. This gave a 2× stock solution 300 nM each in template and primer and 214 nM in Kf–. To initiate the polymerase reaction, enzyme/template/primer (ETP) stock solution (4 μL) and 2× dNTP solutions (4 μL) were combined and incubated (37 °C, 10 min). The final reaction volumes contained 50 mM Tris-HCl, pH 7.5, 10 mM MgCl2, 1 mM DTT, and 0.05 mg/mL BSA. Six concentrations of dNTPs ranging from 0.5 to 2000 μM were examined. Reactions were terminated with 16 μL of loading buffer (90% formamide, 50 mM EDTA, pH 8.0, 0.1% xylene cyanol, and 0.1% bromophenol blue). After denaturation (95 °C, 3 min) and chilling on ice, primer extension products were resolved on 20% polyacrylamide gel. Gels were run at 40 W for approximately 3.5 h, then dried, exposed using a phosphor screen (Molecular Dynamics, Sunnyvale, CA), and scanned on a Typhoon 9410 imaging station (GE Healthcare, Piscataway, NJ). Nonlinear regression analyses of data to give Vmax and KM values were carried out using Prism software (GraphPad Software, San Diego, CA).
Kf–-Catalyzed Incorporation of CEdGTP
Stock solutions of 50 mM (R)- or (S)-CEdGTP containing 50 mM MgCl2 were prepared just before use and diluted to provide the desired concentrations. Template/primer solutions were prepared by diluting a stock solution of ~73 pmol of templates (2, 3, and 4) with 48.4 pmol of labeled primer (8) and 32 μL of 10× Kf– buffer to a volume of 152 μL. After annealing, 8 μL of 0.04 unit/μL (68.4 nM) Kf– was added. The resulting ETP solutions were ~450 nM in template, 303 nM in primer, and 3.42 nM in enzyme. A range of concentrations of CEdGTP solutions were prepared in 4 μL of water. The ETP solutions were brought to 37 °C, and then 4 μL was added to each CEdGTP aliquot. Reactions were quenched at the appropriate time points and analyzed as described above.
Kf–-Catalyzed Primer Extension Reactions from 3′-CEdG
We examined the Kf–-catalyzed extension from a 3′-terminal (R)- or (S)-CEdG (oligo 5) base paired with dC (oligo 2) or dA (oligo 3) to form two contiguous G/C base pairs. A range of dCTP concentrations were prepared from a 50 mM stock solution. ETP solutions (32 μL) were prepared by diluting a volume of template stock equivalent to 14.5 pmol with water to a total volume of 22.8 μL. To this was added 10× Klenow buffer (6.4 μL) and a labeled primer solution (0.8 μL). After annealing, 1 unit/μL Kf– (2 μL) was added, and the solutions were brought to 37 °C; 4 μL of the ETP solution was then added to the dCTP solutions. Reactions were quenched and analyzed after 180 s.
Analyses of dNTP Incorporation opposite (R)- and (S)- CEdG by Taq Polymerase
ETP solution (2×) was prepared by diluting a volume of stock solution of template equivalent to 25 pmol with water to give a total volume of 55.6 μL. To this was added 10× Taq buffer (16 μL) and a labeled primer solution (2 μL of 12 μM). These were annealed (as described above) and followed by the addition of 0.5 unit/μL Taq polymerase (6.4 μL) to yield a final volume of 80 μL. The resulting 2× ETP solution is 312 nM in template, 300 nM in primer, and 2.2 nM in Taq. The three combinations of templates (1R), (1S), and control (2) with primer (7) were prepared in this fashion. For the time course reactions, ETP mixtures and 2 mM dNTP/MgCl2 solutions were prepared as above. For each experiment, 40 μL of each was mixed at 37 °C. At 10 different time points (125 min to 20 h), 5 μL of the reaction volume was removed, quenched, and analyzed by PAGE for percent extension. The incorporation of dATP opposite either (R)- or (S)-CEdG was further examined by steady-state kinetics. ETP mixtures (80 μL) were prepared as described above. A 2 mM stock solution of dATP/MgCl2 was used to prepare a range of concentrations for velocity determinations. Reactions were allowed to proceed for 60 min prior to quenching and product analyses.
RESULTS
Synthesis of (R,S)-CEdG-Containing Oligonucleotides
The synthetic strategy used to prepare stereochemically defined CEdG-containing oligonucleotides is shown in Figure 1. The N2-carboxyethyl side chain was introduced by a nucleophilic substitution reaction with d- or l-alanine on an O6-(4-nitrophenyl)-ethyl- (NPE-) protected 2-fluorodeoxyinosine (2-FdI) residue within an oligo bound to the solid-phase support. This proceeded with retention of stereochemistry and allowed for the unambiguous synthesis of oligonucleotides containing either (R)- or (S)- CEdG from d- or l-alanine, respectively. Removal of the NPE-protecting group using the standard DBU/pyridine protocol resulted in the formation of several side products which complicated purification by HPLC. Product analysis by ESI-FT/MS revealed the presence of oligomers differing from the expected m/z for full-length product by 18 and 36, suggesting basecatalyzed dehydration. Alternatively, extended reaction in aqueous ammonia removed the NPE as well as the other base protecting groups and provided products of the predicted molecular weight following purification by ion pairing chromatography. The sequences of the CEdG-containing oligonucleotides and all primers and templates used in these studies are provided in Figure 2. The stereospecific synthesis of oligonucleotides containing (R)- and (S)-CEdG not only provided templates for kinetic studies but also allowed for the absolute stereochemical assignment of CEdG nucleosides synthesized previously (10). Hydrolysis of stereochemically pure CEdG-containing oligos followed by HPLC analyses using previously described chromatographic conditions indicated that the R isomer (previously referred to as CEdG-A) eluted before the S isomer (previously referred to as CEdG-B).
FIGURE 1.

Synthesis of oligonucleotides containing (R)- or (S)-CEdG. Conditions: (a) d- or l-alanine, K2CO3, H2O/N-methylpyrrolidone, 50 °C, 48 h; (b) concentrated NH4OH, 55 °C, 4–7 days; (c) DBU/C5H5N, room temperature, 18 h. Bp indicates protected DNA bases.
FIGURE 2.

Templates (1–4) and primers (5–8) used to examine dNTP incorporation opposite (R)- and (S)-CEdG in template DNA by Kf– and Taq polymerases and incorporation of (R)- and (S)-CEdGTP opposite the standard bases by Kf–.
Steady-State Kinetics of Base Pair Formation opposite Template CEdG Catalyzed by Kf–
Template oligonucleotides containing either (R)- or (S)-CEdG (1) were annealed to primer (7) in order to examine base pairing opposite this adduct with standard dNTPs. Primer extension reactions catalyzed by Kf– were carried out over a range of dNTP concentrations, and velocity plots were constructed by quantitative integration of PAGE data. A complete set of steady-state kinetic parameters is provided in Table 1. Values for fins, defined as the ratio of apparent second-order rate constants k2app(dNTP)/k2app(dCTP) (12), were calculated for R and S isomers and were used to predict the likelihood of base substitution mutations induced by template CEdG. Values of fins >1 indicated preferential “mispair” formation, while values <1 predicted a lower probability of base pairing relative to dCTP, assuming equimolar concentrations of dNTP and dCTP in the nucleotide pool. The fins values for (R)- and (S)- CEdG in template DNA revealed a distinct bias for purine incorporation (Table 1). The preferred order of dNTP addition opposite CEdG for either isomer was dGTP > dATP > dCTP > dTTP. The bias for purine/CEdG pairing relative to dCTP base pair formation was more pronounced for the (R)-CEdG-substituted templates. In this case the incorporation of dGTP was preferred over dCTP by ~250-fold, as judged by comparison of fins values, while dATP incorporation was favored by ~30-fold. For the R isomer, the incorporation of purine triphosphates over dCTP was favored primarily as a result of lower KMs, with only a small positive contribution from kcat. The KM values for the reaction with dGTP and dATP were 85- and 20-fold smaller, respectively, relative to that measured for dCTP. The corresponding kcat values were larger by 3- and 2-fold for dGTP and dATP, respectively. For templates containing (S)-CEdG, base pairing with dGTP and dATP was preferred by ~14- and 8-fold, respectively. Incorporation of dGTP opposite (S)-CEdG was favored by a 55-fold lower KM; however, kcat relative to dCTP was lower by ~4-fold. The incorporation of dATP opposite this isomer was favored due to a 5-fold lower KM and a modest (~2-fold) increase in kcat. Incorporation of dTTP opposite (R)-CEdG was largely inhibited as a result of unfavorable KM values. This reaction was characterized by a KM which was ~2000-fold greater than that determined for dGTP, while the corresponding kcat values differed by less than 2-fold. Similar trends were observed for the reaction of dTTP with the (S)-CEdG-containing template.
Table 1.
Steady-State Kinetic Data for Base-Pairing Reactions of CEdG
| X | dNTP | Km (μM) | kcat (s–1) | k2app (kcat/KM) (M–1 s–1) | f ins |
|---|---|---|---|---|---|
| (R)-CEdG | dCTP | (2.46 ± 0.96) × 101 | (3.01 ± 0.34) × 10−4 | 1.22× 101 | 1 |
| dTTP | (5.66 ± 1.81) × 102 | (1.58 ± 0.20) × 10−3 | 2.79 | 0.229 | |
| dGTP | (2.88 ± 1.54) × 10−1 | (8.69 ± 1.03) × 10−4 | 3.02× 103 | 247.5 | |
| dATP | 1.20 ± 0.70 | (4.87 ± 0.79) × 10−4 | 4.06× 102 | 33.3 | |
| (S)-CEdG | dCTP | (2.18 ± 0.53) × 102 | (6.84 ± 0.77) × 10−3 | 3.14× 101 | 1 |
| dTTP | (4.22 ± 1.26) × 102 | (8.07 ± 1.04) × 10−4 | 1.91 | 0.061 | |
| dGTP | 3.95 ± 0.81 | (1.79 ± 0.13) × 10−3 | 4.53× 102 | 14.4 | |
| dATP | (4.87 ± 2.22) × 101 | (1.17 ± 0.41) × 10−2 | 2.40× 102 | 7.64 | |
| dC | (R)-CEdGTP | (3.75 ± 0.71) × 102 | (9.60 ± 1.30) × 10−1 | 2.56× 103 | |
| (S)-CEdGTP | (3.25 ± 0.67) × 102 | (7.80 ± 1.10) × 10−1 | 2.40× 103 | ||
| (R)-CEdGa | dATP | (8.12 ± 1.47) × 102 | (3.42 ± 0.35) × 10−3 | 4.21 | |
| (S)-CEdGa | dATP | (7.81 ± 0.55) × 102 | (5.75 ± 0.23) × 10−3 | 7.36 |
Reactions with Taq polymerase. All others catalyzed by Kf–. X = template base.
Incorporation of dNTPs opposite CEdG Catalyzed by Taq Polymerase
Primer extension reactions were also carried out with oligos (1 and 7) using the Taq enzyme. Reactions were incubated at 37 °C rather than the optimal temperature in order to prevent dissociation of the 23-mer template/primer duplex. In order to obtain experimental parameters for steady-state kinetic experiments, time course reactions were initially carried out with a fixed amount of dNTPs (2 mM) and template/primer (150 mM) (Figure 3). Incorporation of dNTPs by Taq was 10–100-fold slower than the corresponding Kf–-initiated reactions. While incorporation of purines opposite CEdG was still highly preferred, in contrast to Kf– dATP was favored over dGTP. Both dATP and dGTP incorporate preferentially opposite the S isomer (Figure 3). Similar to results obtained with Kf–, pyrimidine triphosphates were incorporated poorly opposite CEdG and with no significant stereochemical preference. Since Taq-catalyzed reactions with dATP were sufficiently processive to allow reproducible kinetic measurements, steady-state kinetic analyses were performed for templates containing (R)- and (S)-CEdG (Table 1). The ~2-fold preference for incorporation opposite the S isomer indicated by the apparent second-order rate constants was influenced primarily by kcat rather than KM.
FIGURE 3.

Progress curves for the incorporation of dNTPs opposite (R)- and (S)-CEdG in template DNA catalyzed by Taq polymerase. A control reaction showing the time course for dCTP incorporation opposite dG within the same sequence environment is provided for comparison (black dots).
Incorporation of (R,S)-CEdGTP by Kf–
Since glycation can potentially occur in the nucleoside triphosphate precursor pool, incorporation of CEdGTP during DNA synthesis represents a potential route for DNA–AGE accumulation. In order to determine the suitability of CEdGTP as a polymerase substrate during primer strand extension, we examined the steady-state kinetics of Kf–-catalyzed reactions of (R)- and (S)-CEdGTP. The triphosphates were synthesized as a stereoisomeric mixture via the reaction of dGTP with glyceraldehyde. Initial experiments used single-turnover reaction conditions and the unresolved CEdGTP stereoisomers to examine incorporation opposite dC using primer (8) and template (2). Incorporation of (R,S)-CEdGTP occurred readily opposite dC at 20 μM, while at higher concentrations, multiple additions were observed (Figure 4). An apparent second-order rate constant of ~3 × 102 M–1 s–1 was measured for this reaction. In order to provide substrates for examining the potential stereospecificity of incorporation, stereoisomers were then separated and purified by DEAE-cellulose followed by ion pairing chromatography. Stereochemical assignment was accomplished by exhaustive dephosphorylation with calf intestinal phosphatase followed by correlation with authentic (R)- and (S)-CEdG isolated from hydrolysates of oligonucleotides as described in Experimental Procedures (see Supporting Information). The incorporation of purified (R)- and (S)-CEdGTP by Kf– was examined separately using catalytic conditions (1.71 nM Kf–, 150 nM primer/template). With this increased reaction stringency, incorporation of CEdGTP only occurred opposite dC. No Kf–-catalyzed incorporation was observed opposite either dG or dA using templates 4 and 3, respectively, even at nucleotide concentrations up to 1 mM for extended reaction times (15 min). The HIV-1 reverse transcriptase similarly failed to incorporate CEdGTP opposite dA or dG under all conditions (data not shown). Moreover, the incorporation of CEdGTP opposite dC did not display any stereoisomeric preferences, as the apparent second-order rate constants (k2app) for reactions of (R)- and (S)-CEdGTP were nearly identical, ~2.5 × 103 M–1 s–1 (Table 1). Interestingly, this reaction proceeded more efficiently, as judged by k2app comparisons, than the complementary reaction involving dCTP incorporation opposite template (R)- or (S)-CEdG. This was largely due to the contribution of kcat, which at ~1 s–1 was the highest turnover value observed in our experiments.
FIGURE 4.
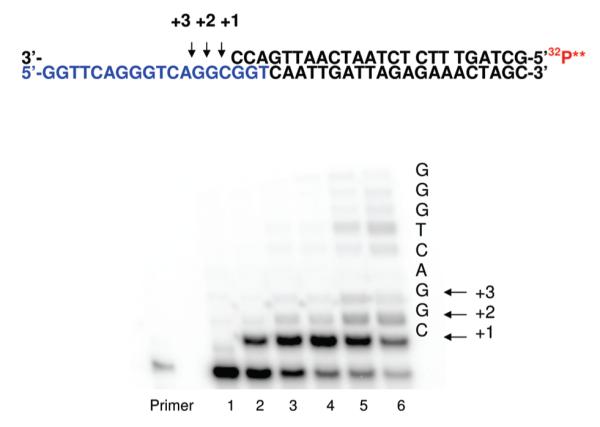
Incorporation of (R,S)-CEdGTP into template DNA by Kf– under single turnover conditions using primer (8) and template (2). For lanes 1–6, [CEdGTP] in μM: 0, 20, 50, 100, 200, and 500. Calculation of steady-state parameters from these data provided values of KM = 49.7 μM and kcat = 1.36 × 102 M–1 s–1.
Primer Extension from CEdG-Terminated Oligonucleotides by Kf–
A second important factor in assessing whether CEdGTP incorporation from the dNTP precursor pool could contribute to adduct accumulation in DNA is the efficiency by which 3′-CEdG residues are extended by polymerases. DNA primers (5) terminating in either (R)- or (S)-CEdG were base paired with dC or dA using templates(2 and 3), respectively, and examined for their ability to extend and incorporate dCTP opposite two contiguous template guanines using Kf– (Figure 5). Control reactions to examine extension from C/G and A/G terminal base pairs under identical conditions were included for comparison (Figure 5A,D). The sequential addition of two dCTPs by Kf– from terminal C/G base pairs was observed at all concentrations tested (Figure 5A, lanes 2–5). Extension from CEdG-terminated primers was only observed when paired opposite dC and only for the R isomer to yield products of length n + 2 (Figure 5B). In contrast, primers terminated with (S)-CEdG paired with template dC (Figure 5C) were resistant to extension (Figure 5C). Extension from A/C base pairs was predictably difficult, with low yields of n + 2 product detected at [dCTP] > 25 μM (Figure 5D, lanes 4 and 5). No extension was observed from terminal A/CEdG base pairs (Figure 5E,F).
FIGURE 5.
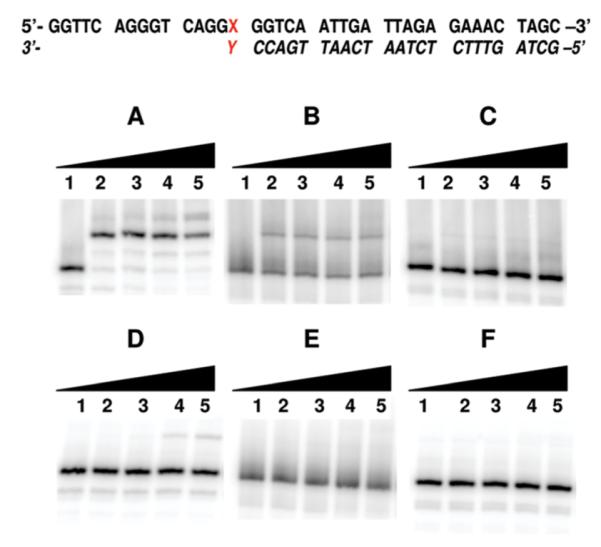
Primer extension from 3′-CEdG catalyzed by Kf–. For lanes 1–5, [dCTP] in μM: 0, 2.5, 25, 250, and 2500. (A) X = C, Y = G; (B) X = C, Y = (R)-CEdG; (C) X = C, Y = (S)-CEdG; (D) X = A, Y = G; (E) X = A, Y = (R)-CEdG; (F) X = A, Y = (S)-CEdG.
DISCUSSION
The potential pathological impact of nucleic acid glycation was first recognized 50 years ago following the report of tobacco mosaic virus inactivation due to guanine modification by glyoxal (13). Later investigations by Bucala and Shapiro on the adducts of glyoxal and MG with guanosine and 9-alkylguanine derivatives established the structure of the major reaction products with DNA and RNA (3, 14). Reaction of guanosine or guanine derivatives with glucose, glucose 6-phosphate, fructosamine, glyceraldehyde, or ribose was shown to lead to formation of N2-carboxyethylguanine derivatives, presumably through the intermediacy of MG (3, 15, 16). More recently, reaction of dsDNA with physiologically relevant concentrations of MG has been shown to result in the formation of CEdG stereoisomers as the nearly exclusive adduct (4). DNA glycation has been proposed to be a potential cause of genetic instability (17, 18), and DNA–AGE accumulation may account in part for the enhanced cancer risk associated with metabolic disease as well as type 1 and 2 diabetes (19–21). Gestational diabetes is a wellrecognized inducer of embryonic malformations, and evidence from rodent model systems has implicated glucose-induced mutagenesis as a possible mechanism (22, 23). In spite of these observations, there is little direct information available on the miscoding properties of DNA–AGEs.
To investigate the base pairing of CEdG in DNA, we prepared oligonucleotides containing uniquely substituted R or S stereoisomers for kinetic studies. Recently, an alternative synthetic approach for the preparation of CEdG-containing oligonucleotides was described (24). A significant distinction between these two routes lies in the identity of the O6 protecting group. Cao et al. used an O6-trimethylsilylethyl protecting group whereas an O6-NPE protecting group was employed in this work. Deprotection in the former case required treatment with acetic acid for 2 h, a conditionwhich can subject dG and CEdG to depurination (10). In contrast, the NPE group can be removed by prolonged reaction with NH4OH, a standard treatment for oligonucleotide deprotection. The present synthesis consisted of fewer overall steps and started with a commercially available phosphoramidite. We also prepared for the first time the corresponding nucleotides (R)- and (S)-CEdGTP in order to examine their suitability as polymerase substrates.
Purine dNTPs were preferentially incorporated opposite CEdG in template DNA by both Kf– and Taq; however, polymerase-specific purine biases were observed. The Kf– polymerase demonstrated a 7-fold preference for dGTP over dATP opposite (R)-CEdG in template DNA (Table 1). In the case of Taq for the same template/primer sequence, the progress curves for dNTP incorporation (Figure 3) indicate a clear preference for incorporation of dATP over dGTP and a bias for (S)-CEdG. The influence of adduct stereochemistry on the mutational outcome was previously demonstrated for another aldehyde adduct, the 1, N2-2-deoxyguanosine addition product of hydroxynonenal (HNE) (25). Cao et al. also observed preferential incorporation of purines opposite CEdG in DNA by Kf–; however, kinetic data were not reported (24). These results suggest that CEdG will primarily induce G transversions when acted upon by replicative polymerases.
The pattern of mispairing observed for CEdG is consistent with previous reports showing that MG-treated DNA replicated in either COS-7 or Escherichia coli cells yields a base substitution pattern consisting predominantly of G transversions (26, 27). In some respects the bypass proficiency and pattern of mispairing observed for CEdG resemble thosse of other N2-deoxyguanosine-substituted acyclic adducts (Figure 6). Similar to the adducts derived from acrolein 2, malondialdehyde 3, and acetaldehyde 4, CEdG is resistant to facile extension by Kf– (28–31). Adducts 2 and 3 exist in equilibrium with their cyclic 1,N2 counterparts, the γ-OH PdG and M1dG adducts. NMR studies have revealed that the cyclic form is preferred in single-stranded DNA and can base pair with purines by adopting a syn geometry; however, the ringopened acyclic form can pair in the normal Watson–Crick manner with dC in double-stranded DNA (32, 33). These observations have been used to explain the dramatic differences in mutation-inducing potential of adducts 2 and 3 depending upon whether they are situated in single- or double-stranded plasmids (28, 31).
FIGURE 6.
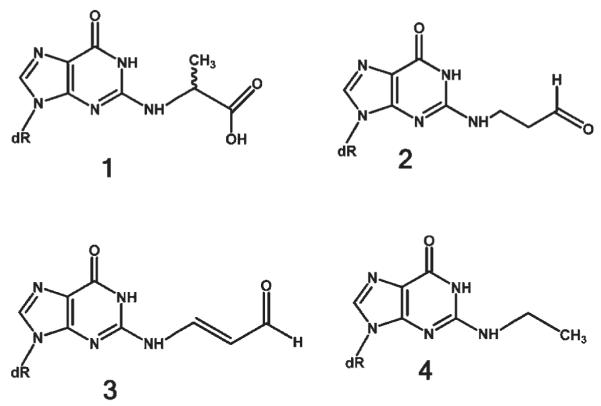
N2-substituted acyclic aldehyde adducts of 2′-deoxyguanosine: 1, (R,S)-CEdG; 2, N2-γ-(oxo)-PdG; 3, N2-3-oxoprenyl-M1dG; 4, N2-ethyl-dG. Adducts 2 and 3 are in equilibrium with the corresponding N1, N2 cyclic forms γ-OH PdG and M1dG, respectively.
We considered the possibility that a similar equilibrium could exist between cyclic and acyclic forms of CEdG and that a cyclic adduct in ssDNA templates would facilitate purine base pairing whereas adoption of an acyclic form in CEdGTP would favor dC pairing. However, in contrast to 2 and 3, the CEdG side chain terminates in a carboxylate moiety which is unlikely to cyclize at N1 since it is a relatively poor electrophile. In 13C NMR spectra of (R,S)-CEdG, only a single carboxylate resonance was observed at 175 ppm, consistent with an acyclic adduct (10). The environment-dependent variation in CEdG base-pairing preferences is also reminiscent of another N2-substituted dG adduct, the acetaldehyde-derived N2-ethyl-dG (4 in Figure 6) (30). Templated N2-ethyl-dG paired preferentially with dGTP and dCTP, whereas the corresponding N2-ethyl-dGTP paired only with dC in reactions catalyzed by Kf–. These observations suggest different syn/anti conformations at the polymerase active site, independent of cyclic/acyclic equilibration phenomena. It is possible that a syn CEdG orientation in template DNA is favored by steric interference and/or electrostatic repulsion that would occur as a result of interactions between the carboxylate side chain and amino acid residues in contact with the minor groove which are necessarily present in the anti configuration (34). Rotation to the syn purine orientation would displace the N2 side chain into the major groove. This interaction is not present when CEdGTP is the incoming nucleotide or when CEdG resides in the primer strand during replication. This minor groove steric interaction may facilitate the syn/anti isomerization of N2-substituted guanines in DNA such as N2-ethyl-dG by the lesion bypass polymerase Pol ι and improve the efficiency of bypass, as has been suggested from recent crystallographic and kinetic studies (35, 36). The role of syn/anti conformational variability about the glycosidic bond in facilitating purine/purine mismatch formation catalyzed by Kf– has also been demonstrated using deaza base analogues in conjunction with kinetic approaches (37).
Our results indicate that glycation of DNA can increase the frequency of G transversions, although the exact base substitution pattern is predicted to be minimally influenced by (a) whether glycation occurs at the dNTP or polymer level, (b) the stereochemical and purine biases of the replicative polymerase(S), and (c) the potential for stereospecific repair of (R,S)-CEdG in DNA. For example, if incorporation of CEdGTP by polymerases constitutes a significant route for the accumulation of CEdG in DNA, then replication by polymerases sharing the stereochemical bias of Kf– will result in the overrepresentation of (R)-CEdG. This is because although both CEdGTP stereoisomers can be incorporated with equivalent efficiencies (Table 1), extension can only occur from the R isomer under catalytic conditions (Figure 5). This would be predicted to substantially bias the frequency of G > C transversions due to the preference for dGTP incorporation opposite (R)-CEdG in template DNA. If the major route for DNA–AGE accumulation occurs as a result of glycation of genomic DNA, an ~1:1 distribution of R and S stereoisomers is predicted, and the base substitution pattern should follow the pattern of mispairing frequencies in Table 1 for a “Kf–-like” polymerase. Replication by polymerases with biases resembling the Taq enzyme would favor dATP incorporation and shift the base substitution pattern toward G > T transversions. However, the potential for preferential stereoisomeric repair could significantly alter the final base substitution pattern in vivo by substantially altering the ratio of (R)- and (S)- CEdG in DNA. Although plasmid DNA treated with MG resulted in an equimolar distribution of isomers as ascertained by LC-ESI-MS/MS, measurement of CEdG in urine and tissue from rodent and human specimens revealed a biased stereoisomer distribution, possibly reflecting preferential adduct repair in vivo (D. Tamae, unpublished observations). This is the subject of ongoing investigations in our laboratory.
Supplementary Material
ACKNOWLEDGMENT
The authors thank Roger Moore of the Mass Spectrometry-Proteomics Core Facility of the City of Hope for helpful discussions and advice and Yuewei Wu for technical assistance.
Footnotes
Supported in part by R01 GM59219 to J.T., the City of Hope Comprehensive Cancer Center (P30 CA33572), a California Breast Cancer Research Program predoctoral fellowship to D.T. (14GB-0162), and a Caltech/City of Hope Preceptorship Program fellowship to C.Y.
SUPPORTING INFORMATION AVAILABLE Additional data as described in the text. This material is available free of charge via the Internet at http://pubs.acs.org.
Abbreviations: AGE, advanced glycation end product; CEdG, N2-carboxyethyl-2′-deoxyguanosine; MG, methylglyoxal; LC-ESI-MS/MS, liquid chromatography–electrospray ionization-mass spectrometry/mass spectrometry; NPE, (4-nitrophenyl)ethyl; LTQ-FT, linear trap quadrupole-Fourier transform; ETP, enzyme/template/primer; Kf–, Klenow fragment exonuclease minus; Taq, Thermus aquaticus.
REFERENCES
- 1.Thornalley PJ, Battah S, Ahmed N, Karachalias N, Agalou S, Babaei-Jadidi R, Dawnay A. Quantitative screening of advanced glycation endproducts in cellular and extracellular proteins by tandem mass spectrometry. Biochem. J. 2003;375:581–592. doi: 10.1042/BJ20030763. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2.Nakagawa K, Oak JH, Higuchi O, Tsuzuki T, Oikawa S, Otani H, Mune M, Cai H, Miyazawa T. Ion-trap tandem mass spectrometric analysis of Amadori-glycated phosphatidylethanolamine in human plasma with or without diabetes. J. Lipid Res. 2005;46:2514–2524. doi: 10.1194/jlr.D500025-JLR200. [DOI] [PubMed] [Google Scholar]
- 3.Papoulis A, al-Abed Y, Bucala R. Identification of N2-(1-carboxyethyl)-guanine (CEG) as a guanine advanced glycosylation end product. Biochemistry. 1995;34:648–655. doi: 10.1021/bi00002a032. [DOI] [PubMed] [Google Scholar]
- 4.Frischmann M, Bidmon C, Angerer J, Pischetsrieder M. Identification of DNA adducts of methylglyoxal. Chem. Res. Toxicol. 2005;18:1586–1592. doi: 10.1021/tx0501278. [DOI] [PubMed] [Google Scholar]
- 5.Nemet I, Varga-Defterdarovic L, Turk Z. Methylglyoxal in food and living organisms. Mol. Nutr. Food Res. 2006;50:1105–1117. doi: 10.1002/mnfr.200600065. [DOI] [PubMed] [Google Scholar]
- 6.Phillips SA, Thornalley PJ. The formation of methylglyoxal from triose phosphates. Investigation using a specific assay for methylglyoxal. Eur. J. Biochem./FEBS. 1993;212:101–105. doi: 10.1111/j.1432-1033.1993.tb17638.x. [DOI] [PubMed] [Google Scholar]
- 7.Brownlee M. Biochemistry and molecular cell biology of diabetic complications. Nature. 2001;414:813–820. doi: 10.1038/414813a. [DOI] [PubMed] [Google Scholar]
- 8.Bar KJ, Franke S, Wenda B, Muller S, Kientsch-Engel R, Stein G, Sauer H. Pentosidine and N-(epsilon)-(carboxymethyl)-lysine in Alzheimer’s disease and vascular dementia. Neurobiol. Aging. 2003;24:333–338. doi: 10.1016/s0197-4580(02)00086-6. [DOI] [PubMed] [Google Scholar]
- 9.Li H, Nakamura S, Miyazaki S, Morita T, Suzuki M, Pischets-rieder M, Niwa T. N2-carboxyethyl-2′-deoxyguanosine, a DNA glycation marker, in kidneys and aortas of diabetic and uremic patients. Kidney Int. 2006;69:388–392. doi: 10.1038/sj.ki.5000064. [DOI] [PubMed] [Google Scholar]
- 10.Synold T, Xi B, Wuenschell GE, Tamae D, Figarola JL, Rahbar S, Termini J. Advanced glycation end products of DNA: quantification of N2-(1-carboxyethyl)-2′-deoxyguanosine in biological samples by liquid chromatography electrospray ionization tandem mass spectrometry. Chem. Res. Toxicol. 2008;21:2148–2155. doi: 10.1021/tx800224y. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Snow ET, Mitra S. Do carcinogen-modified deoxynucleotide precursors contribute to cellular mutagenesis? Cancer Invest. 1987;5:119–125. doi: 10.3109/07357908709018466. [DOI] [PubMed] [Google Scholar]
- 12.Boosalis MS, Petruska J, Goodman MF. DNA polymerase insertion fidelity. Gel assay for site-specific kinetics. J. Biol. Chem. 1987;262:14689–14696. [PubMed] [Google Scholar]
- 13.Staehelin M. Inactivation of virus nucleic acid with glyoxal derivatives. Biochim. Biophys. Acta. 1959;31:448–454. doi: 10.1016/0006-3002(59)90019-8. [DOI] [PubMed] [Google Scholar]
- 14.Shapiro R, Cohen BI, Shiuey SJ, Maurer H. On the reaction of guanine with glyoxal, pyruvaldehyde, and kethoxal, and the structure of the acylguanines. A new synthesis of N2-alkylguanines. Biochemistry. 1969;8:238–245. doi: 10.1021/bi00829a034. [DOI] [PubMed] [Google Scholar]
- 15.Nissl J, Ochs S, Severin T. Reaction of guanosine with glucose, ribose, and glucose 6-phosphate. Carbohydr. Res. 1996;289:55–56. [Google Scholar]
- 16.Ochs S, Severin T. Reaction of 2′-deoxyguanosine with glyceraldehyde. Liebigs Ann. Chem. 1994:851–853. [Google Scholar]
- 17.Bucala R, Model P, Cerami A. Modification of DNA by reducing sugars: a possible mechanism for nucleic acid aging and age-related dysfunction in gene expression. Proc. Natl. Acad. Sci. U.S.A. 1984;81:105–109. doi: 10.1073/pnas.81.1.105. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Thornalley PJ. Protecting the genome: defence against nucleotide glycation and emerging role of glyoxalase I overexpression in multidrug resistance in cancer chemotherapy. Biochem. Soc. Trans. 2003;31:1372–1377. doi: 10.1042/bst0311372. [DOI] [PubMed] [Google Scholar]
- 19.Cowey S, Hardy RW. The metabolic syndrome: A high-risk state for cancer? Am. J. Pathol. 2006;169:1505–1522. doi: 10.2353/ajpath.2006.051090. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Stevens RJ, Roddam AW, Beral V. Pancreatic cancer in type 1 and young-onset diabetes: systematic review and metaanalysis. Br. J. Cancer. 2007;96:507–509. doi: 10.1038/sj.bjc.6603571. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Bowker SL, Majumdar SR, Veugelers P, Johnson JA. Increased cancer-related mortality for patients with type 2 diabetes who use sulfonylureas or insulin. Diabetes Care. 2006;29:254–258. doi: 10.2337/diacare.29.02.06.dc05-1558. [DOI] [PubMed] [Google Scholar]
- 22.Eriksson UJ, Wentzel P, Minhas HS, Thornalley PJ. Teratogenicity of 3-deoxyglucosone and diabetic embryopathy. Diabetes. 1998;47:1960–1966. doi: 10.2337/diabetes.47.12.1960. [DOI] [PubMed] [Google Scholar]
- 23.Lee AT, Reis D, Eriksson UJ. Hyperglycemiainduced embryonic dysmorphogenesis correlates with genomic DNA mutation frequency in vitro and in vivo. Diabetes. 1999;48:371–376. doi: 10.2337/diabetes.48.2.371. [DOI] [PubMed] [Google Scholar]
- 24.Cao H, Jiang Y, Wang Y. Stereospecific synthesis and characterization of oligodeoxyribonucleotides containing an N2-(1-carboxyethyl)-2′-deoxyguanosine. J. Am. Chem. Soc. 2007;129:12123–12130. doi: 10.1021/ja072130e. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Fernandes PH, Wang H, Rizzo CJ, Lloyd RS. Sitespecific mutagenicity of stereochemically defined 1,N2-deoxyguanosine adducts of trans-4-hydroxynonenal in mammalian cells. Environ. Mol. Mutagen. 2003;42:68–74. doi: 10.1002/em.10174. [DOI] [PubMed] [Google Scholar]
- 26.Murata-Kamiya N, Kamiya H, Kaji H, Kasai H. Methylglyoxal induces G:C to C:G and G:C to T:A transversions in the supF gene on a shuttle vector plasmid replicated in mammalian cells. Mutat. Res. 2000;468:173–182. doi: 10.1016/s1383-5718(00)00044-9. [DOI] [PubMed] [Google Scholar]
- 27.Murata-Kamiya N, Kamiya H, Kaji H, Kasai H. Mutational specificity of glyoxal, a product of DNA oxidation, in the lacI gene of wild-type Escherichia coli W3110. Mutat. Res. 1997;377:255–262. doi: 10.1016/s0027-5107(97)00083-3. [DOI] [PubMed] [Google Scholar]
- 28.Yang IY, Hossain M, Miller H, Khullar S, Johnson F, Grollman A, Moriya M. Responses to the major acrolein-derived deoxyguanosine adduct in Escherichia coli. J. Biol. Chem. 2001;276:9071–9076. doi: 10.1074/jbc.M008918200. [DOI] [PubMed] [Google Scholar]
- 29.Hashim MF, Riggins JN, Schnetz-Boutaud N, Voehler M, Stone MP, Marnett LJ. In vitro bypass of malondialdehyde-deoxyguanosine adducts: differential base selection during extension by the Klenow fragment of DNA polymerase I is the critical determinant of replication outcome. Biochemistry. 2004;43:11828–11835. doi: 10.1021/bi049360f. [DOI] [PubMed] [Google Scholar]
- 30.Terashima I, Matsuda T, Fang TW, Suzuki N, Kobayashi J, Kohda K, Shibutani S. Miscoding potential of the N2-ethyl-2′-deoxyguanosine DNA adduct by the exonuclease-free Klenow fragment of Escherichia coli DNA polymerase I. Biochemistry. 2001;40:4106–4114. doi: 10.1021/bi002719p. [DOI] [PubMed] [Google Scholar]
- 31.VanderVeen LA, Hashim MF, Nechev LV, Harris TM, Harris CM, Marnett LJ. Evaluation of the mutagenic potential of the principal DNA adduct of acrolein. J. Biol. Chem. 2001;276:9066–9070. doi: 10.1074/jbc.M008900200. [DOI] [PubMed] [Google Scholar]
- 32.Mao H, Schnetz-Boutaud NC, Weisenseel JP, Marnett LJ, Stone MP. Duplex DNA catalyzes the chemical rearrangement of a malondialdehyde deoxyguanosine adduct. Proc. Natl. Acad. Sci. U.S.A. 1999;96:6615–6620. doi: 10.1073/pnas.96.12.6615. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.de los Santos C, Zaliznyak T, Johnson F. NMR characterization of a DNA duplex containing the major acroleinderived deoxyguanosine adduct gamma-OH-1,-N2-propano-2′-deoxyguanosine. J. Biol. Chem. 2001;276:9077–9082. doi: 10.1074/jbc.M009028200. [DOI] [PubMed] [Google Scholar]
- 34.Franklin MC, Wang J, Steitz TA. Structure of the replicating complex of a pol alpha family DNA polymerase. Cell. 2001;105:657–667. doi: 10.1016/s0092-8674(01)00367-1. [DOI] [PubMed] [Google Scholar]
- 35.Pence MG, Blans P, Zink CN, Hollis T, Fishbein JC, Perrino FW. Lesion bypass of N2-ethylguanine by human DNA polymerase iota. J. Biol. Chem. 2009;284:1732–1740. doi: 10.1074/jbc.M807296200. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36.Choi JY, Guengerich FP. Kinetic evidence for inefficient and error-prone bypass across bulky N2-guanine DNA adducts by human DNA polymerase iota. J. Biol. Chem. 2006;281:12315–12324. doi: 10.1074/jbc.M600112200. [DOI] [PubMed] [Google Scholar]
- 37.Kretulskie AM, Spratt TE. Structure of purine-purine mispairs during misincorporation and extension by Escherichia coli DNA polymerase I. Biochemistry. 2006;45:3740–3746. doi: 10.1021/bi052306u. [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.