

Abstract
Group I p21-activated kinases (PAKs) are important effectors of the small GTPases Rac and Cdc42, which regulate cell motility/migration, survival, proliferation and gene transcription. Hyperactivation of these kinases have been reported in many tumor types, making PAKs attractive targets for therapeutic intervention. PAKs are activated by growth factor-mediated signaling and are negatively regulated by the tumor suppressor NF2/Merlin. Thus, tumors characterized by NF2 inactivation would be expected to show hyperactivated PAK signaling. Based on this rationale, we evaluated the status of PAK signaling in malignant mesothelioma (MM), an aggressive neoplasm that is resistant to current therapies and shows frequent inactivation of NF2. We demonstrate that group I PAKs are activated in most MMs and MM cell lines and that genetic or pharmacological inhibition of PAKs is sufficient to inhibit MM cell proliferation and survival. We also identify downstream effectors and signaling pathways that may contribute mechanistically to PAK-related tumorigenesis. Specifically, we show that inhibition of PAK results in attenuation of AKT and Raf-MAPK signaling and decreased tumor cell viability. Collectively, these data suggest that pharmacological inhibition of group I PAKs may have therapeutic efficacy in tumors characterized by PAK activation.
Keywords: p21-activated kinase (PAK), malignant mesothelioma, small molecule inhibitor, targeted therapy, NF2/Merlin, AKT, mitogen-activated protein kinases (MAPK), Raf-1
Introduction
The p21-activated kinases (PAKs) are serine/threonine kinases activated by the small GTPases Cdc42 and Rac in response to a variety of cell stimuli (1–3). The PAK family is grouped into two classes; group I, which includes PAK1-3, and group II, which contains PAK4-6 (4–7). When inactivated, group I PAKs are thought to exist as an autoinhibitory homodimer, whereby the PAK inhibitory domain (PID) of one PAK monomer binds to the kinase domain of a second monomer, thereby inactivating catalytic activity (8). PAKs are activated via binding of their p21-GTPase-binding domain (PBD) to small GTPases (8). Binding to small GTPases through the PBD relieves the interaction between the PID and kinase domains, permitting phosphorylation at PAK threonine 423 within the kinase domain, preventing reformation of the autoinhibitory dimer state (8). Once serine 423 is phosphorylated, PAKs undergo autophosphorylation of serine 141 and other serine residues within the C-terminal PID/PBD domains to keep PAKs catalytically active by preventing autoinhibitory homodimerization (7). Additionally, phosphoinositides were recently demonstrated to be essential co-factors for PAK1 activation (9). Group II PAKs, with the exception of PAK5, lack a recognizable PID domain and, thus, were initially thought to be constitutively active in cells (7). However, recent studies have demonstrated that autophosphorylation of group II PAKs occurs during growth-factor receptor-mediated signaling (7).
Once activated, PAKs can phosphorylate a myriad of downstream effectors regulating cellular processes such as motility, proliferation and survival. Initially discovered through their interaction with Cdc42 and Rac, PAKs were later shown to signal downstream of these small GTPases to regulate cytoskeletal dynamics and cell motility via direct phosphorylation of substrates such as paxillin, cortactin, LIMK and myosin light-chain kinase (MLCK) (2). More recently, other group I PAK substrates have been elucidated and shown to regulate other cellular processes. PAK1 can directly phosphorylate both Raf-1 and MEK1, thereby positively regulating mitogen-activated protein kinase (MAPK) signaling to promote cell proliferation and survival (10). PAKs were further shown to be important mediators and regulators of growth-factor receptor-induced MAPK activation (11). Additionally, PAKs are required for oncogenic Ras- and ErbB2-regulated MAPK signaling and tumorigenesis (12, 13).
Previous work has revealed that Merlin, the product of the neurofibromatosis type 2 tumor suppressor gene (NF2), is a substrate of Group I PAKs (3, 14). Phosphorylation of Merlin at Serine 518 by PAK1/2 thwarts the inhibitory effect of Merlin and, thus, constitutes an alternate means to abolish this tumor suppressor axis in proliferating cells (3, 14). Interestingly, Merlin was also shown to negatively regulate PAKs by directly interacting with the PBD domain and preventing recruitment of PAKs to focal adhesions (15). Moreover, the NF2/Merlin status of a cell was found to inversely correlate with PAK activity (15). The latter finding suggests that in tumor cells with inactivation of NF2, PAK activity exists in a hyperactivated state, potentially contributing to the proliferation, survival and invasiveness of cancer cells.
Malignant mesotheliomas (MMs) are aggressive, diffuse neoplasms arising from the serosal lining of the pleura, peritoneal or pericardial cavities. Although exposure to asbestos is considered a causal event in most MM patients, tumor latency following initial exposure is typically 20–40 years (16, 17). During this time, affected mesothelial cells acquire multiple genetic alterations contributing to MM pathogenesis. One such somatic genetic change that occurs in approximately 50% of MMs is biallelic inactivation of the NF2 tumor suppressor gene by a combination of point mutation and allelic loss (18). Concordantly, targeted-deletion of one copy of Nf2 in mice was sufficient to accelerate MM formation in mice exposed to asbestos (19, 20). Thus, inactivation of the NF2 tumor suppressor gene is thought to be an important event in the pathogenesis of many MMs.
Although loss of NF2 in MM and other tumor types, including schwannoma, meningioma, melanoma, and renal cell carcinoma, contributes to tumorigenesis, restoration of NF2 expression as a therapy is unattainable currently due to difficulties of long-term gene expression and immune responses associated with viral-mediated gene therapy (21–24). Thus, targeting pathways that are normally negatively regulated by NF2, and whose activity or signaling becomes aberrant when this tumor suppressor is inactivated, may represent a more achievable treatment strategy. In this investigation, we evaluated PAKs as potential targets for therapeutic intervention in MM. We determined that PAK1 and PAK2 are phosphorylated and activated in most human and murine MM tumor specimens and cell lines tested. We also demonstrate that genetic or pharmacological inhibition of PAK signaling is sufficient to inhibit MM cell viability, proliferation and survival. Furthermore, we show that hyperactivated PAK signal to a variety of downstream effectors, including the AKT and Raf-MAPK signaling axes, contribute to tumor cell survival and proliferation. Collectively, these findings provide strong preclinical evidence supporting group I PAK-targeted therapy as a potential intervention for the treatment of MM and other neoplasms.
Materials and Methods
Immunohistochemistry
Slides of formalin-fixed, paraffin-embedded samples of human and murine MM specimens were antigen-retrieved with citrate and incubated overnight with anti-phospho-PAK1/2/3 (pSer141 – 1:100) (Sigma Aldrich). Sections were stained with DAB and counterstained with hematoxylin. A tissue microarray (TMA) of human MM specimens was obtained through Fox Chase Cancer Center’s Histopathology Core Facility. To demonstrate antibody specificity, murine MM tissue was treated or not with lambda phosphatase (NEB) for 3 hours and then subjected to IHC analysis with anti-phospho-PAK1/2/3.
Primary cell cultures
Primary mouse MM cells were isolated from ascitic fluid and/or peritoneal lavage, as described elsewhere (19). Patient-derived MM cell lines were established and characterized as previously reported (25, 26).
2-D gel electrophoresis and immunoblot analysis
Briefly, actively proliferating placebo-treated or IPA-3-treated tumor cells were harvested and lysed in a non-denaturing buffer (7 M urea, 4% CHAPS). Protein extracts were separated in the first dimension by isoelectric focusing (IEF) on 7 cm/pH 4–7 IEF strips (Biorad) for 2 hr at 8,000 V-hr. IEF strips were then reduced in SDS-buffer and embedded into the top well of a 4–12% gradient SDS-PAGE gel for separation in the second dimension, and then proteins were transferred to nitrocellulose membrane. Antibodies specific for total PAK1 and PAK1/2/3 (Cell Signaling) were used to probe the membrane and determine where specific PAK isoforms ran on an SDS-PAGE gel.
siRNA against PAK1 and PAK2
Stealth™ siRNA pools against human PAK1 and 2 (Invitrogen) were nucleofected into human Meso 22 cells using an Amaxa™ Cell Line Kit R and program T20 of a Nucleofector™ System (Lonza AG). After 48 hr, the cells were harvested, protein was extracted, and immunoblot analysis was performed.
Lentiviral shRNA virus production and infection of MM cell lines
The pLKO.1 shGFP, shPAK1A, shPAK1B, shPAK2A and shPAK2B vectors were purchased from the RNAi Consortium through Sigma-Aldrich. For all experiments, 70% confluent 293 HEK cells (10 cm plates) were transfected with 5 μg of each of the vectors individually plus 3.75 μg and 1.25 μg of psPAX2 packaging and pMD2G envelope vectors, respectively. After transfecting for 24 hours, the media was removed and fresh media was added to the 293 cells. Media was then collected 24 and 48 hours later, and the virus-containing media supplemented with 8 μg/mL of polybrene was used to infect ME12 and Meso 22 cell lines. 24 hours after infection, the ME12 and Meso 22 cells were selected in media containing 2 μg/mL puromycin and passaged under continuous selection for at least 2 passages before use in experiments.
Immunoblotting
Immunoblots were prepared with 50 μg of protein lysate/sample, as previously described (27). Antibodies against phospho-PAK (P-PAK1/2/3, pSer141 – Sigma Adlrich), total PAK1/2/3, P-AKT (Ser-473), total AKT, p44/42 MAPK (Erk1/2) (137F5) (Cell Signaling), P-ERK1/2 (E4), and β-actin (Santa Cruz Biotechnology) were used for immunoblot analysis.
pcDNA-PID vector construction
The untagged PID domain of PAK1 was subcloned from a pBMN-PID-GFP plasmid via restriction digestion with XhoI and BamHI. The gel-purified fragment was ligated into pcDNA3.1 (pcDNA-PID) and subsequently confirmed via restriction enzyme digestion analysis and DNA sequencing.
Clonogenic Assays
Two human MM cell lines (Meso 22, ME12) were nucleofected with pcDNA or pcDNA-PID vectors (10 μg/nucleofection) using an Amaxa™ Cell Line Kit R and program T20 of the Nucleofector™ System. A third human MM line (Meso-17) was transfected with pcDNA or pcDNA-PID vectors (20 μg/transfection) using the Xfect™ Transfection Reagent (Clontech). Cells were harvested 24 or 72 hr post-nucleofection/transfection for immunoblot analysis and cell viability assays, respectively. For stable clonogenic assays, cells were nucleofected/transfected with pcDNA, pcDNA-PID, pcDNA-PID + pcDNA-HA-myr-AKT1, or pcDNA-PID + myc-Raf1(BXB) for 48 hr and then selected with 400 μg/mL of G418 (neomycin) for 2 weeks. Colonies were fixed with 4% paraformaldehyde, stained with Diff-Quik (Fisher Scientific), and then counted.
For shRNA knockdown clonogenic assays, MM cells were infected and selected with puromycin (2 ug/mL) for at least two passages. Cells were then counted, and 500 cells were plated per well in 6-well plates. After 1–2 weeks, the resulting cell colonies were fixed with 4% paraformaldehyde and stained with 0.2% crystal violet in 20% ethanol.
Cell Number Assay
To evaluate cell proliferation, ME12 and Meso 22 cells stably expressing shGFP, shPAK1A, shPAK1B, shPAK2A or shPAK2B (2 × 104 cells each, at passage 3 of selection) were seeded on a 12-well plate in triplicate (Day 1). Cells were counted each day for 3 days and plotted as a line graph with standard deviations.
IPA-3 treatment of MM cell lines
MM cells were treated with varying concentrations of IPA-3, a small molecule inhibitor of PAK or DMSO control, for 24, 48 or 72 hr for immunoblot analysis of antibody arrays, or for cell viability, cell cycle, and apoptosis assays.
Cell viability assays
MM cells were each seeded onto 96-well plates at a density of 5,000 cells per well. After 24 hr, cells were treated with 25 μM IPA-3 or DMSO vehicle control for 72 hr. MTS reagent was added and absorbance was determined at 490 nm as a read out of cell viability. MM cells were nucleofected with pcDNA or pcDNA-PID vectors (10 μg/nucleofection); 24 hr post-nucleofection, cells were seeded onto 96-well plates at a density of 5,000 cells per well. MTS reagent was added to the plate 72 hr post-nucleofection, and absorbance was determined at 490 nm as a read out of cell viability. As a control, ME12 and Meso 22 cells were also treated with control compound PIR3.5 at increasing concentrations and analyzed for cell viability via MTS assay.
Cell cycle analysis
MM cells were treated with 25 μM IPA-3 or DMSO vehicle control for 24 hr, harvested, and fixed in cold 70% ethanol overnight. The next day, cells were pelleted and washed in PBS before being treated with RNase in PBS (200 μg/mL) for 30 min at room temperature. Cells were then pelleted and stained with propidium iodide (10 μg/mL). DNA content was determined on a FACscan Flow Cytometer (Beckton Dickinson) and quantitated using FlowJo analysis software.
Apoptosis assay
Tumor cells were treated with 25 μM IPA-3 or vehicle control for 48 hr. The cells were harvested and analyzed for apoptosis using the Cell Death Detection ElisaPLUS Kit (Roche Applied Biosciences). All experiments were performed in triplicate in replicate independent experiments.
Antibody array analysis
Human PhosphoKinase PathwayScan Antibody Arrays were purchased from R&D Systems. MM cell lines were treated with 25 μM IPA-3 of vehicle control or nucleofected with pcDNA or pcDNA-PID as described above. Cells were harvested and lysed for protein extraction according to the manufacturer’s protocol. For each treatment condition, 500 μg of total protein extract was incubated with membrane A or membrane B overnight at 4° C. Membranes were developed as recommended by the manufacturer.
Results
Group I PAKs are activated in MM tumors and cell lines
In order to evaluate the phosphorylation and activation status of group I PAKs in MM specimens, we used a three-pronged approach. First, we used a phospho-specific antibody against serine 141 of PAK1/2/3 to assess PAK phosphorylation via immunohistochemistry (IHC) on a tissue microarray (TMA) spotted with archival human MM tumors. All 15 pleural MM tumor specimens tested stained more positively for active PAK than did normal pleural lining surrounding the lung (Fig. 1A). Concordantly, asbestos-induced murine MM tumors also stained positively for active PAK (Fig. 1B). Lamda phosphatase treatment of murine MM tissues abolished phospho-PAK1/2/3 staining, demonstrating the specificity of the antibody used in our IHC analyses (Supplemental Fig. 3). Secondly, we evaluated the phosphorylation status of group I PAKs in human MM cell lines by immunoblotting using the same antibody employed for the IHC analysis. As shown in Figure 1C, hyperactivation of PAK was found in most human and murine MM cell lines tested. In all, PAK phosphorylation was observed in ~80% of human and murine MM cell lines tested in which NF2-expression was lost. It should be noted that not all MM cell lines demonstrated strong PAK phosphorylation, even though all were NF2-deficient, suggesting that other pathways that may control PAK activity in tumor cells.
Figure 1.

PAKs are activated in MM specimens and cell lines. A) Immunohistochemical-staining of human MM and normal lung pleura with a phospho-specific antibody against Ser141 of PAK1/2/3 (P-PAK1/2/3). B) Immunohistochemical staining of asbestos-induced MMs from wild-type and Nf2+/− mice using P-PAK1/2/ antibody. In figure 1B, NF2 should be mouse designation Nf2. C) Immunoblot analysis of P-PAK1/2/3, PAK1, and total PAK and β-actin in human MM cell lines (top panel) and P-PAK1/2/3, PAK PAK1/2/3 and β-actin MM cell lines from asbestos-treated wild-type and Nf2+/− mice (bottom panel). D) 2-dimensional immunoblot analysis of PAK1, PAK1/2/3 and P-PAK1/2/3 levels in human MM cell lines.
The antibody used for both IHC and immunoblot analyses recognizes phosphorylated forms of all three group I PAKs. To determine if a specific group I PAK is consistently activated in MM, we used a third approach to assess specific group I PAK phosphorylation: 2-D gel electrophoresis/immunoblot analysis. Seven human MM cell line extracts were separated by isoelectric focusing (IEF) followed by second-dimension SDS PAGE electrophoresis. Proteins from the gels were transferred to nitrocellulose and then blotted with antibodies against PAK1, PAK1/2/3 and P-PAK1/2/3 antibodies. As shown in Figure 1D and Supplemental Figure 1, PAK1 was expressed and appeared to be phosphorylated based on the presence of multiple forms migrating toward the more acidic isoelectric point (pI). In addition, immunoblot analysis with the PAK1/2/3 antibody revealed a second, slower-migrating isoform of group I PAKs that was also phosphorylated (Fig. 1D and Supplemental Fig. 1). For further clarification, we used RNAi against PAK1 or PAK2, which revealed that the slower migrating band is PAK1 and the faster migrating band is PAK2 (Supplemental Fig. 2). Immunoblot analysis of the 2D western blot with an antibody against phosphorylated PAK1/2/3 (P-PAK1/2/3) appeared to recognize PAK 2 with higher affinity than PAK1 (Fig. 1D and Supplementary Figs. 1 and 2). Together, these data demonstrate that PAK1 and PAK2 are phosphorylated and active in human MM cells.
Expression of a dominant-negative PAK-PID is sufficient to inhibit PAK phosphorylation and tumor cell viability and proliferation
We next tested whether or not PAK activity is required for MM cell viability. In order to inhibit all three group I PAKs simultaneously, we expressed the PAK1 kinase inhibitory domain (PID – a.a. 83–149 – which inhibits all group I PAKs) exogenously in MM cells. Transient expression of PID of MM cell lines by nucleofection for 72 hr in ME12 and Meso 22 cells or by transfection for 24 hr in Meso 17 cells resulted in decreased phospho-PAK levels, based on immunoblot analysis (Fig. 2A and Supplemental Fig. 4). Total PAK levels were also decreased in ME12 and Meso 22 cells after 72 hrs, but were unchanged when PID was expressed for 24 hr (Fig. 2A and Supplemental Fig. 6). To test whether PAK activity is required for tumor cell viability, MM cells were transiently nucleofected/transfected with PID and cell viability was evaluated 72 hr later. As shown in Figure 2B, expression of PID was sufficient to inhibit cell viability by at least 2-fold in ME12 and Meso 22 cells. Similar results were observed in transfected Meso 17 cells (Supplemental Fig. 4B). In light of the fact that transfection efficiencies are generally low in MM cell lines, we decided to stably-express PID and evaluate its effect on cell proliferation/survival via colony formation assay. MM cells were nucleofected or transfected with PID or control pcDNA plasmids and then selected with G418 beginning 24 hr post-nucleofection/transfection and lasting 2 weeks, at which point the cells were fixed and stained. As shown in Figure 2B and Supplemental Fig. 4B, colony formation was undetectable in all three MM cell lines expressing the PID sequence; however, colony formation was robust in the neomycin-selected control cells transfected with pcDNA. Together, these data suggest that group I PAK activity is required for MM cell survival and proliferation.
Figure 2.
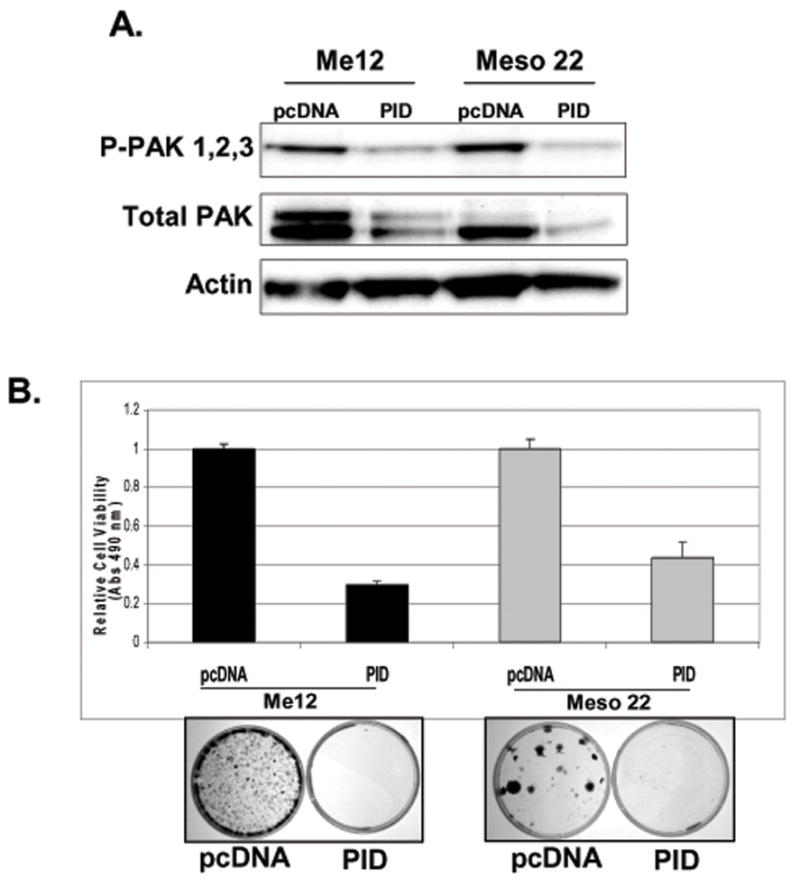
Dominant-negative PID expression inhibits cell viability and PAK phosphorylation in human MM cell lines. A) Immunoblot analysis of P-PAK1/2/3, total PAK1/2/3 and β-actin levels 72-hr post-nucleofection of pcDNA or pcDNA-PID in Me12 and Meso 22 cells. B) MTS cell viability assay 72-hr post-nucleofection of pcDNA or pcDNA-PID in Me12 and Meso 22 cells. C) G418-resistent colonies 2-weeks post-nucleofection of pcDNA or pcDNA-PID in Me12 and Meso 22 cells.
Knockdown of PAK1 and PAK2 decreases MM cell proliferation
In a complementary experiment, we addressed whether expression of PAK1 and/or PAK2, the two predominant group I PAKs expressed in MM, are required for MM cell proliferation. Using lentiviruses expressing shRNAs against PAK1, PAK2 or GFP (control), we infected ME12 and Meso 22 cell lines and selected with puromycin to make stable cell lines. As shown in Figure 3A, we were able to knock down both PAK1 and PAK2 effectively with two different shRNAs to each isoform, when compared to shGFP control cells. The stable cell lines were then seeded for cell proliferation and clonogenic assays. In Figure 3B, knock down of PAK1 had a modest effect on MM cell proliferation, with the exception of shPAK1A knockdown, which essentially blocked Meso 22 cell proliferation. Knockdown of PAK2 consistentlty caused proliferation arrest in ME12 cells, whereas only one shRNA against PAK2 (shPAK2B) caused obvious changes in Meso 22 cell proliferation (Fig. 3B). Similar results were found in clonogenic assays in which cells were sparsely seeded and allowed to grow for a week, followed by subsequent staining with crystal violet to document colony formation (Fig. 3C). ME12 cell lines grew much slower that Meso 22 cells in this assay, such that ME12 colonies were not large enough for visualization with crystal violet. To circumvent this issue, the ME12 cell lines seeded and counted for cell doubling assays were allowed to grow for a week and stainable colonies were obtained for analysis (Fig. 3C, left panel). Together, these data and our results with the PID construct (Fig. 2) implicate PAK1 and PAK2 in the regulation of MM cell proliferation.
Figure 3.

Stable knockdown of PAK1 or PAK2 inhibits MM cell proliferation. A) Immunoblot analysis demonstrating knockdown of PAK1 (top panel) and PAK2 (middle panel, bottom band). B) Plot of cell number (×104) over time (days) for ME12 and Meso 22 cells infected with lentivirus expressing shGFP (control), shPAK1A, shPAK1B, shPAK2A, and shPAK2B. C) Clonogenic assay demonstrating colony formation in ME12 and Meso 22 cells infected with lentivirus expressing shGFP (control), shPAK1A, shPAK1B, shPAK2A and shPAK2B.
A novel small molecule inhibitor of PAKs (IPA-3) inhibits PAK1 and PAK2 phosphorylation, MM cell viability, proliferation and survival
Although genetic attenuation of PAK activity via exogenous expression of a dominant-negative PID sequence and knockdown with shRNA was sufficient to inhibit MM cell viability and proliferation, targeted expression of PID or shRNA expression in cancer cells is not a feasible or rational cancer therapy. On the other hand, small molecule inhibitors of kinases have been effectively exploited as targeted therapies to inhibit cancer cell proliferation and survival. However, many of the small molecule compounds targeting kinases are actually ATP-competitive inhibitors and, as such, can be promiscuous and target multiple kinases due to the fact that ATP-binding sites in kinases are evolutionarily conserved (8). Recently, a small molecule inhibitor of PAK1, and to lesser extent PAK2/3/5, was discovered that effectively inhibits PAK activation by locking or stabilizing PAK homodimers in an autoinhibitory state via covalent binding to the PID sequence of PAKs (28, 29). Additionally, this inhibitor, dubbed inhibitor of PAK activation 3 (IPA-3), was demonstrated to be cell permeable and capable of inhibiting PDGF-mediated activation of MAPK signaling, similar to genetic perturbation of PAK1 (11, 28).
To test whether IPA-3 can inhibit MM cell viability akin to exogenous expression of the PID sequence of PAK1, we tested the effect of increasing concentrations of IPA3, or DMSO control, on MM cell viability. As shown in Figure 4A, addition of IPA-3 for 72 hr inhibited MM cell viability in a dose-dependent manner, with an IC50 of approximately 25 μM. Treatment of MM cell lines with control compound PIR3.5 produced little change in cell viability, suggesting the effect observed with IPA-3 is mediated through inhibition of group I PAKs (Supplemental Fig. 5). To evaluate whether IPA-3 inhibits PAK phosphorylation, we treated ME12 and Meso 22 cells for 72 hr and evaluated PAK1/2/3 phosphorylation via immunoblot analysis (Fig. 4B). IPA-3-treated MM cells showed a marked reduction in PAK phosphorylation and slightly decreased total PAK levels (Fig. 4B). Although IPA-3 was initially identified as an inhibitor of PAK1, we also found that IPA-3-treated ME12 cells displayed decreases in both PAK1 and PAK2 phosphorylation with both forms shifting from more acidic (or phosphorylated) to more basic based on 2-D gel electrophoresis immunoblot analysis (Fig. 4C). Together, these data suggest that IPA-3 treatment can inhibit group I PAK phosphorylation and MM cell viability as effectively as over-expression of PID.
Figure 4.

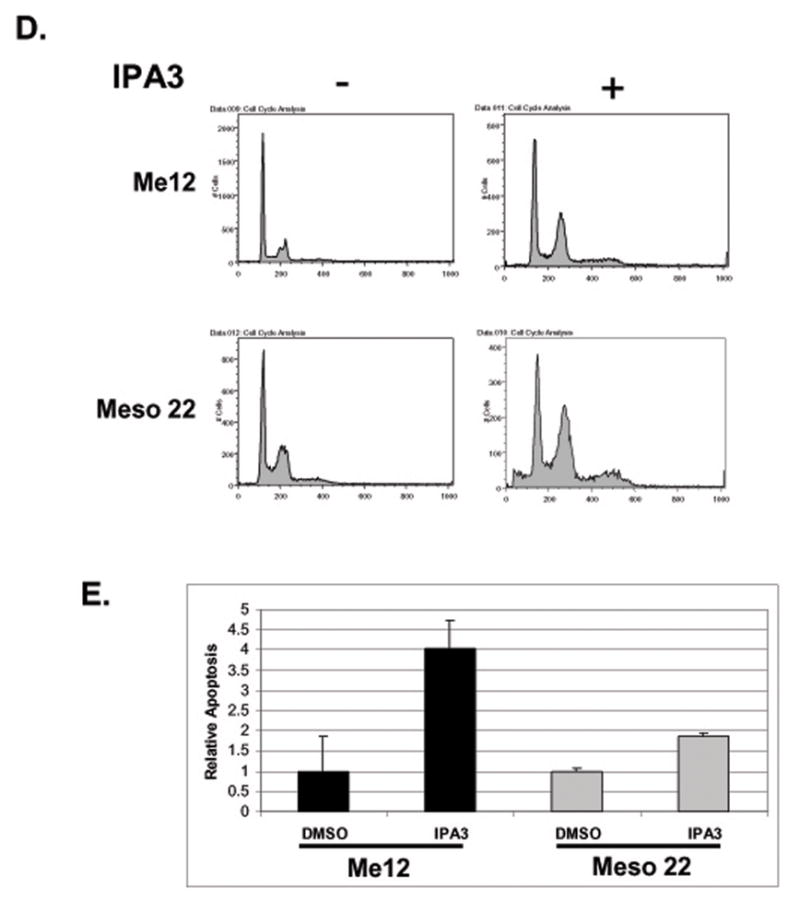
A selective group I PAK inhibitor (IPA-3) inhibits PAK phosphorylation and MM cell viability, proliferation and survival. A) MTS cell viability assay of Me12 and Meso 22 cells 72-hrs post treatment with varying concentrations of IPA-3. B) Immunoblot analysis of P-PAK1/2/3, total PAK and actin in protein lysates of Me12 and Meso 22 cells 72-hrs post-IPA-3 treatment. C) 2-dimensional immunoblot analysis of PAK1 and PAK1/2/3 in Me12 and Meso 22 after treatment for 24 hr with IPA-3. Arrows represent different phosphorylation states of PAKs. D) Cell cycle analysis of propidium iodide-stained Me12 and Meso 22 cells treated with DMSO (−) or IPA-3 (+) for 24 hr. E) Apoptosis as determined by the Cell Death Detection ElisaPLUS Kit (Roche Applied Biosciences) in Me12 and Meso 22 cells treated with DMSO or IPA-3 for 48 hr.
To assess mechanistically how IPA-3 treatment decreases tumor cell viability, we evaluated the effect of PAK inhibition on cell cycle progression/cell proliferation. MM cells were treated with IPA-3 or DMSO control for 48 hr and then fixed and stained with propidium iodide. IPA-3-treated cells were then analyzed by flow cytometry for DNA content. As shown in Figure 4D, IPA-3-treated MM cells showed a marked increase in the percentage of cells in G2/M, suggesting arrest or delay in that stage of the cell cycle. This finding is consistent with other reports implicating a role for PAKs in mitosis (30–32). To address the effect of IPA-3-mediated PAK inhibition on MM cell survival, ME12 and Meso 22 cells were treated with the drug for 48 hr and assessed for apoptosis. As demonstrated in Figure 4E, IPA-3-treated ME12 and Meso 22 showed a significant increase in DNA fragmentation when compared to DMSO-treated control cells, suggesting a role for PAKs in MM cell survival as well. An increase in the sub-G1 cell population was also observed in propidium iodide-stained Meso 22 cells treated with IPA-3, consistent with an increase in apoptotic cells (Fig. 4D, panel 2). These data suggest that inhibiting group I PAKs with the small molecule inhibitor IPA-3 causes a decrease in PAK phosphorylation in association with decreased MM cell viability, proliferation and survival, consistent with the effects observed for dominant-negative expression of PID or knockdown of PAK1 and PAK2 in MM cells.
Group I PAKs promote tumor cell survival and proliferation via the AKT1 and Raf1-MAPK signaling pathways
Our data implicate Group I PAKs in MM cell viability, proliferation and survival. To identify downstream effector pathways that mediate oncogenic PAK signaling, we evaluated the status of the AKT and MAPK signaling pathways. In Figure 5A, IPA-3 treatment reduced the phosphorylation of both AKT and ERK1/2, suggesting that PAKs play a significant role in regulating these signaling molecules in MM cells. A similar effect on AKT and ERK1/2 phosphosphorylation was upon exogenous expression of PID (Supplementary Figure 6). Expression of PID or treatment with IPA-3 also inhibited the phosphorylation of multiple kinases, based on results from reverse-phase antibody arrays, implicating these signaling axes as bona fide targets of PAKs (Supplemental Fig. 7). Some of these kinases, including p70S6 kinase, p90RSK, STAT5a/b, STAT1 and MSK1/2, have not been previously linked to PAK signaling but are implicated in carcinogenesis (33–36).
Figure 5.


Identification of AKT and MAPK signaling as potentially important effector pathways in regulated tumor cell viability. A) Immunoblot analysis of P-PAK1/2/3, total PAK1/2/3, P-AKT, total AKT, P-ERK1/2, and β-actin in Me12 and Meso 22 treated with DMSO or IPA-3 for 24 hr. B) G418-resistent colonies 2-weeks post-nucleofection of pcDNA, pcDNA-PID, pcDNA-PID + pcDNA-HA-myr-AKT1, or pcDNA-PID + myc-Raf1(BXB) in Me12 and Meso 22 cells. C) MTS assay of ME12 or Meso 22 cells 72 hours post-treatment with the following drugs or drug combinations: DMSO (D), 25 uM IPA-3 (I), 15 uM LY294002 (L), 20 uM PD98509 (P) and various combinations labeled with the aforementioned letters. D) Schematic model of PAK involvement in MM cell survival and proliferation through effectors AKT1 and Raf1 and the inhibitors used to target each pathway.
To determine if inhibition of AKT and Raf1 are required for PAK-mediated MM cell viability, constitutively active forms of AKT1 and Raf1 were co-expressed with PID. Both constitutively active AKT1 and Raf1 were able to rescue cell viability in clonogenic assays in the presence of PAK inhibition by PID in two different MM cell lines tested (Fig. 5B). These data implicate AKT1 and Raf1-MAPK signaling as important downstream effectors of PAK for MM cell viability. In a complementary experiment, we inhibited the AKT and MAPK signaling pathways with LY294002 and PD98509, respectively. In Figure 5C, inhibition of both AKT and MAPK with LY294002 and PD98509, respectively (bar 5) was sufficient to phenocopy the effect of PAK inhibition (IPA-3 treatment – bar 2) on tumor cell viability, whereas inhibition of AKT or MAPK signaling alone was not (bars 3 and 4). Interestingly, inhibiting AKT, but not MAPK, sensitized MM cells to PAK inhibition (Fig. 5C – bars 6 and 7), suggesting that the AKT signaling node may be a more dominant effector of PAK signaling than the MAPK pathway for MM cell survival. Collectively, these data implicate the AKT and MAPK signaling pathways as important effector pathways of PAKs in regulating tumor cell viability.
Discussion
In this report, we show that group I PAKs are frequently activated in MM, a tumor type with frequent inactivation of NF2/Merlin. This finding is in agreement with the notion that Merlin acts as a tumor suppressor by restraining PAK signaling. We demonstrate for the first time that inhibition of group I PAKs by genetic or pharmacological means is effective in restraining MM cell viability, proliferation and survival. We also provide proof-of-concept that use of novel small molecule inhibitors of group I PAKs may serve as important therapeutic agents for treating tumor types that show frequent hyperactivation of PAK. Moreover, we demonstrate that inhibition of PAK results in attenuated signaling via AKT and MAPK, two central nodes in tumorigenesis.
Several reports have demonstrated that group I PAKs is important in cellular transformation and tumorigenesis. For example, PAK1 was recently shown to be hyperactivated in schwannomas from patients with neurofibromatosis type 2 syndrome and was required for rat schwannoma cell proliferation (37). Independently, PAK activity was found to be required for tumor cell growth and invasion in a cell-based model of neurofibromatosis type 2 (38). Both groups demonstrated that targeting more than one of the group I PAKs was required to inhibit NIH-3T3-NF2-BBA-transformed cell proliferation and growth in vivo (37, 38). Consistent with these results, we show that both PAK1 and PAK2 are activated in NF2-deficient MM cells and that genetic (shRNA) or pharmacological inhibition of both kinases was sufficient to inhibit tumor cell viability, although our shRNA data demonstrated that targeting either isoform could affect MM cell proliferation (Fig. 3). Additionally, other studies have shown that PAK activity is required for both Ras- and ERBB2-mediated transformation in cell models of breast cancer (12, 39).
To our knowledge, this is the first report demonstrating that pharmacological inhibition of PAKs (with IPA-3) is efficacious in patient-derived MM cells, although PAK kinases have been shown to be effective in other preclinical cancer models (40). IPA-3 was recently shown to inhibit PAK2-mediated cell spreading but not viability of primary human schwannoma cells (41). Although this lack of effect on the viability of these benign tumor cells appears to contradict our findings and those of others who have inhibited PAK by genetic means (37, 38), the schwannoma cells in the aforementioned study were treated with IPA-3 for only 24 hr, whereas in our study MM cells were treated for 72 hr prior to the cell viability analysis. Additionally, schwann cells and mesothelial cells arise from different embryonic lineages; thus, the resulting neoplasms may have different tissue-specific dependency on PAK activity for cell survival and proliferation. Moreover, other investigations have found that dissimilar cell types respond differently to PAK inhibition. For example, Yi et al. (37) and Tange et al. (13) found that although PAK1 perturbation had no effect in NIH3T3 cells, PAK1 ablation/inhibition in the rat schwannoma cell line RT4 resulted in growth arrest.
Although our data suggest group I PAKs are important for tumor cell viability, it is not clear what downstream substrates or effector pathways regulated by PAKs are critical for regulating MM cell proliferation and survival. We found that inhibition of PAKs in MM cells, via dominant-negative or pharmacological means, had a modest but reproducible negative effect on both AKT and ERK signaling, pathways regulated in part by PAKs and that promote tumor cell proliferation and survival. Interestingly, dominant-expression of the PID sequence did not affect AKT or ERK activity in a cell-based model of neurofibromatosis type-2 (38), although these experiments were conducted in vivo, where feedback loops could account for these differences. We demonstrated that AKT1 and Raf-MAPK signaling pathways are potentially important mediators of group PAK’s pro-survival and pro-proliferation signaling in MM cells. A similar result with dual inhibition of AKT and ERK kinases was shown to specifically affect colon cancer cell growth and invasion (42). Additionally, PAK inhibition affected the phosphorylation of multiple kinases involved in various signal transduction pathways, based on results from reverse-phase antibody arrays, implicating these signaling axes as bona fide targets of PAK activity. The kinases that were altered by inhibition of PAK via expression of PID and by treatment with IPA-3 represent novel effectors because both of these methods inhibit PAK activity in a specific-manner and are unlikely to have off-target effects. Phosphorylation of paxillin, a known substrate of PAKs, was also diminished in both PID-expressing and IPA-3-treated MM cells, further supporting the global signaling antibody array approach used herein. Additionally, phosphorylation of p70S6 kinase, p90RSK, STAT5a/b, STAT1 and MSK1/2, proteins previously not considered as direct PAK substrates or effectors, were also decreased in PID-expressing and IPA-3-treated MM cells. These data suggest that many effectors of PAK signaling could be important, either independently or in concert, for the proliferation and survival of tumor cells characterized by hyperactivation of PAK. Importantly, we demonstrate that activation of AKT1 and Raf-MAPK signaling appears to be key downstream effects that contribute to oncogenic PAK signaling in MM.
Together these data implicate group I PAKs as important regulators of tumor cell proliferation and survival in MM and other types of cancer exhibiting hyperactivation of PAK and identifies new potential effector pathways of PAKs in tumor cells. PAK-inhibition was sufficient to attenuate both the AKT and MAPK signaling pathways in MM cells, thus simultaneously inhibiting two important oncogenic effectors critical to tumor cell survival and proliferation, respectively. Thus, in some tumor types, PAK inhibitors could prove to be as effective as single agents as combinatorial AKT and MAPK inhibitors (summarized in Fig. 5D). This report reinforces the importance of developing clinically relevant small molecule inhibitors of PAK as a targeted-therapy for multiple malignancies.
Supplementary Material
Acknowledgments
The authors thank Drs. S. Jhanwar and H. Pass for providing MM cell lines.
Grant Support
This study was supported by National Institutes of Health awards CA-114047 (J.R.T.), CA-148805 (J.C., J.R.T.), GM083025 (J.R.P.), and CA-06927 (Fox Chase Cancer Center), an appropriation from the Commonwealth of Pennsylvania, an award from the Children’s Tumor Foundation (2011-15-012) (J.C.), and by a gift from the Local #14 Mesothelioma Fund of the International Association of Heat and Frost Insulators & Allied Workers in memory of Hank Vaughan and Alice Haas.
Footnotes
Disclosure of Potential Conflicts of Interest
No potential conflicts of interest were disclosed.
References
- 1.Dummler B, Ohshiro K, Kumar R, Field J. Pak protein kinases and their role in cancer. Cancer Metastasis Rev. 2009;28:51–63. doi: 10.1007/s10555-008-9168-1. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2.Molli PR, Li DQ, Murray BW, Rayala SK, Kumar R. PAK signaling in oncogenesis. Oncogene. 2009;28:2545–55. doi: 10.1038/onc.2009.119. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.Xiao GH, Beeser A, Chernoff J, Testa JR. p21-activated kinase links Rac/Cdc42 signaling to merlin. J Biol Chem. 2002;277:883–6. doi: 10.1074/jbc.C100553200. [DOI] [PubMed] [Google Scholar]
- 4.Eswaran J, Soundararajan M, Knapp S. Targeting group II PAKs in cancer and metastasis. Cancer Metastasis Rev. 2009;28:209–17. doi: 10.1007/s10555-008-9181-4. [DOI] [PubMed] [Google Scholar]
- 5.Eswaran J, Soundararajan M, Kumar R, Knapp S. UnPAKing the class differences among p21-activated kinases. Trends Biochem Sci. 2008;33:394–403. doi: 10.1016/j.tibs.2008.06.002. [DOI] [PubMed] [Google Scholar]
- 6.Jaffer ZM, Chernoff J. p21-activated kinases: three more join the Pak. Int J Biochem Cell Biol. 2002;34:713–7. doi: 10.1016/s1357-2725(01)00158-3. [DOI] [PubMed] [Google Scholar]
- 7.Wells CM, Jones GE. The emerging importance of group II PAKs. Biochem J. 2010;425:465–73. doi: 10.1042/BJ20091173. [DOI] [PubMed] [Google Scholar]
- 8.Yi C, Maksimoska J, Marmorstein R, Kissil JL. Development of small-molecule inhibitors of the group I p21-activated kinases, emerging therapeutic targets in cancer. Biochem Pharmacol. 2010;80:683–9. doi: 10.1016/j.bcp.2010.03.012. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Strochlic TI, Viaud J, Rennefahrt UE, Anastassiadis T, Peterson JR. Phosphoinositides are essential coactivators for p21-activated kinase 1. Mol Cell. 2010;40:493–500. doi: 10.1016/j.molcel.2010.10.015. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Eblen ST, Slack JK, Weber MJ, Catling AD. Rac-PAK signaling stimulates extracellular signal-regulated kinase (ERK) activation by regulating formation of MEK1-ERK complexes. Mol Cell Biol. 2002;22:6023–33. doi: 10.1128/MCB.22.17.6023-6033.2002. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Beeser A, Jaffer ZM, Hofmann C, Chernoff J. Role of group A p21-activated kinases in activation of extracellular-regulated kinase by growth factors. J Biol Chem. 2005;280:36609–15. doi: 10.1074/jbc.M502306200. [DOI] [PubMed] [Google Scholar]
- 12.Arias-Romero LE, Villamar-Cruz O, Pacheco A, Kosoff R, Huang M, Muthuswamy SK, et al. A Rac-Pak signaling pathway is essential for ErbB2-mediated transformation of human breast epithelial cancer cells. Oncogene. 2010;29:5839–49. doi: 10.1038/onc.2010.318. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Tang Y, Chen Z, Ambrose D, Liu J, Gibbs JB, Chernoff J, et al. Kinase-deficient Pak1 mutants inhibit Ras transformation of Rat-1 fibroblasts. Mol Cell Biol. 1997;17:4454–64. doi: 10.1128/mcb.17.8.4454. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Kissil JL, Johnson KC, Eckman MS, Jacks T. Merlin phosphorylation by p21-activated kinase 2 and effects of phosphorylation on merlin localization. J Biol Chem. 2002;277:10394–9. doi: 10.1074/jbc.M200083200. [DOI] [PubMed] [Google Scholar]
- 15.Kissil JL, Wilker EW, Johnson KC, Eckman MS, Yaffe MB, Jacks T. Merlin, the product of the Nf2 tumor suppressor gene, is an inhibitor of the p21-activated kinase, Pak1. Mol Cell. 2003;12:841–9. doi: 10.1016/s1097-2765(03)00382-4. [DOI] [PubMed] [Google Scholar]
- 16.Ramos-Nino ME, Testa JR, Altomare DA, Pass HI, Carbone M, Bocchetta M, et al. Cellular and molecular parameters of mesothelioma. J Cell Biochem. 2006;98:723–34. doi: 10.1002/jcb.20828. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Yang H, Testa JR, Carbone M. Mesothelioma epidemiology, carcinogenesis, and pathogenesis. Curr Treat Options Oncol. 2008;9:147–57. doi: 10.1007/s11864-008-0067-z. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Bianchi AB, Mitsunaga SI, Cheng JQ, Klein WM, Jhanwar SC, Seizinger B, et al. High frequency of inactivating mutations in the neurofibromatosis type 2 gene (NF2) in primary malignant mesotheliomas. Proc Natl Acad Sci U S A. 1995;92:10854–8. doi: 10.1073/pnas.92.24.10854. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Altomare DA, Vaslet CA, Skele KL, De Rienzo A, Devarajan K, Jhanwar SC, et al. A mouse model recapitulating molecular features of human mesothelioma. Cancer Res. 2005;65:8090–5. doi: 10.1158/0008-5472.CAN-05-2312. [DOI] [PubMed] [Google Scholar]
- 20.Fleury-Feith J, Lecomte C, Renier A, Matrat M, Kheuang L, Abramowski V, et al. Hemizygosity of Nf2 is associated with increased susceptibility to asbestos-induced peritoneal tumours. Oncogene. 2003;22:3799–805. doi: 10.1038/sj.onc.1206593. [DOI] [PubMed] [Google Scholar]
- 21.Dalgliesh GL, Furge K, Greenman C, Chen L, Bignell G, Butler A, et al. Systematic sequencing of renal carcinoma reveals inactivation of histone modifying genes. Nature. 2010;463:360–3. doi: 10.1038/nature08672. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Sinkre P, Perry A, Cai D, Raghavan R, Watson M, Wilson K, et al. Deletion of the NF2 region in both meningioma and juxtaposed meningioangiomatosis: case report supporting a neoplastic relationship. Pediatr Dev Pathol. 2001;4:568–72. doi: 10.1007/s10024001-0086-2. [DOI] [PubMed] [Google Scholar]
- 23.Sterman DH, Molnar-Kimber K, Iyengar T, Chang M, Lanuti M, Amin KM, et al. A pilot study of systemic corticosteroid administration in conjunction with intrapleural adenoviral vector administration in patients with malignant pleural mesothelioma. Cancer Gene Ther. 2000;7:1511–8. doi: 10.1038/sj.cgt.7700269. [DOI] [PubMed] [Google Scholar]
- 24.Wilson JM. Lessons learned from the gene therapy trial for ornithine transcarbamylase deficiency. Mol Genet Metab. 2009;96:151–7. doi: 10.1016/j.ymgme.2008.12.016. [DOI] [PubMed] [Google Scholar]
- 25.Pass HI, Stevens EJ, Oie H, Tsokos MG, Abati AD, Fetsch PA, et al. Characteristics of nine newly derived mesothelioma cell lines. Ann Thorac Surg. 1995;59:835–44. doi: 10.1016/0003-4975(95)00045-m. [DOI] [PubMed] [Google Scholar]
- 26.Taguchi T, Jhanwar SC, Siegfried JM, Keller SM, Testa JR. Recurrent deletions of specific chromosomal sites in 1p, 3p, 6q, and 9p in human malignant mesothelioma. Cancer Res. 1993;53:4349–55. [PubMed] [Google Scholar]
- 27.Menges CW, Chen Y, Mossman BT, Chernoff J, Yeung AT, Testa JR. A Phosphotyrosine Proteomic Screen Identifies Multiple Tyrosine Kinase Signaling Pathways Aberrantly Activated in Malignant Mesothelioma. Genes Cancer. 2010;1:493–505. doi: 10.1177/1947601910375273. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Deacon SW, Beeser A, Fukui JA, Rennefahrt UE, Myers C, Chernoff J, et al. An isoform-selective, small-molecule inhibitor targets the autoregulatory mechanism of p21-activated kinase. Chem Biol. 2008;15:322–31. doi: 10.1016/j.chembiol.2008.03.005. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Viaud J, Peterson JR. An allosteric kinase inhibitor binds the p21-activated kinase autoregulatory domain covalently. Mol Cancer Ther. 2009;8:2559–65. doi: 10.1158/1535-7163.MCT-09-0102. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Banerjee M, Worth D, Prowse DM, Nikolic M. Pak1 phosphorylation on t212 affects microtubules in cells undergoing mitosis. Curr Biol. 2002;12:1233–9. doi: 10.1016/s0960-9822(02)00956-9. [DOI] [PubMed] [Google Scholar]
- 31.Bompard G, Rabeharivelo G, Frank M, Cau J, Delsert C, Morin N. Subgroup II PAK-mediated phosphorylation regulates Ran activity during mitosis. J Cell Biol. 2010;190:807–22. doi: 10.1083/jcb.200912056. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Thiel DA, Reeder MK, Pfaff A, Coleman TR, Sells MA, Chernoff J. Cell cycle-regulated phosphorylation of p21-activated kinase 1. Curr Biol. 2002;12:1227–32. doi: 10.1016/s0960-9822(02)00931-4. [DOI] [PubMed] [Google Scholar]
- 33.Klampfer L. Signal transducers and activators of transcription (STATs): Novel targets of chemopreventive and chemotherapeutic drugs. Curr Cancer Drug Targets. 2006;6:107–21. doi: 10.2174/156800906776056491. [DOI] [PubMed] [Google Scholar]
- 34.Ulivi P, Arienti C, Amadori D, Fabbri F, Carloni S, Tesei A, et al. Role of RAF/MEK/ERK pathway, p-STAT-3 and Mcl-1 in sorafenib activity in human pancreatic cancer cell lines. J Cell Physiol. 2009;220:214–21. doi: 10.1002/jcp.21753. [DOI] [PubMed] [Google Scholar]
- 35.Pon YL, Zhou HY, Cheung AN, Ngan HY, Wong AS. p70 S6 kinase promotes epithelial to mesenchymal transition through snail induction in ovarian cancer cells. Cancer Res. 2008;68:6524–32. doi: 10.1158/0008-5472.CAN-07-6302. [DOI] [PubMed] [Google Scholar]
- 36.Perez-Cadahia B, Drobic B, Espino PS, He S, Mandal S, Healy S, et al. Role of MSK1 in the malignant phenotype of Ras-transformed mouse fibroblasts. J Biol Chem. 2011;286:42–9. doi: 10.1074/jbc.M110.156687. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37.Yi C, Wilker EW, Yaffe MB, Stemmer-Rachamimov A, Kissil JL. Validation of the p21-activated kinases as targets for inhibition in neurofibromatosis type 2. Cancer Res. 2008;68:7932–7. doi: 10.1158/0008-5472.CAN-08-0866. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38.Chow HY, Stepanova D, Koch J, Chernoff J. p21-Activated kinases are required for transformation in a cell-based model of neurofibromatosis type 2. PLoS One. 2010;5:e13791. doi: 10.1371/journal.pone.0013791. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39.Vadlamudi RK, Adam L, Wang RA, Mandal M, Nguyen D, Sahin A, et al. Regulatable expression of p21-activated kinase-1 promotes anchorage-independent growth and abnormal organization of mitotic spindles in human epithelial breast cancer cells. J Biol Chem. 2000;275:36238–44. doi: 10.1074/jbc.M002138200. [DOI] [PubMed] [Google Scholar]
- 40.Zhu G, Wang Y, Huang B, Liang J, Ding Y, Xu A, et al. A Rac1/PAK1 cascade controls beta-catenin activation in colon cancer cells. Oncogene. 2012;31:1001–12. doi: 10.1038/onc.2011.294. [DOI] [PubMed] [Google Scholar]
- 41.Flaiz C, Chernoff J, Ammoun S, Peterson JR, Hanemann CO. PAK kinase regulates Rac GTPase and is a potential target in human schwannomas. Exp Neurol. 2009;218:137–44. doi: 10.1016/j.expneurol.2009.04.019. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42.Huynh N, Liu KH, Baldwin GS, He H. P21-activated kinase 1 stimulates colon cancer cell growth and migration/invasion via ERK- and AKT-dependent pathways. Biochim Biophys Acta. 2010;1803:1106–13. doi: 10.1016/j.bbamcr.2010.05.007. [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.