

Abstract
Single-cell transcriptome contains reliable gene-gene relationships because gene-gene interaction only happens within a mammalian cell. While the study of gene-gene interaction enables us to understand the molecular mechanism of cellular events and evaluate molecular characteristics of a mammalian cell population, its complexity requires an analysis of a large number of single-cells at various stages. However, many existing microfluidic platforms cannot process single-cells effectively for routine molecular analysis. To address these challenges, we develop an integrated system with individual controller for effective single-cell transcriptome analysis. In this paper, we report an integrated microfluidic approach to rapidly measure gene expression in individual cells for genetic stability assessment of a cell population. Inside this integrated microfluidic device, the cells are individually manipulated and isolated in an array using micro sieve structures, then transferred into different nanoliter reaction chambers for parallel processing of single-cell transcriptome analysis. This device enables us to manipulate individual single-cell into nanoliter reactor with high recovery rate. We have performed gene expression analysis for a large number of HeLa cells and 293T cells expanded from a single-cell. Our data shows that even the house-keeping genes are expressed at heterogeneous levels within a clone of cells. The heterogeneity of Actin expression reflects the genetic stability, and the expression distribution is different between cancer cells (HeLa) and immortalized 293T cells. The result demonstrates that this platform has the potential for assessment of genetic stability in cancer diagnosis.
Introduction
Over recent years there have been significant interests on high-throughput single-cell molecular analysis technologies. Conventional molecular biology techniques require cell lysate (a physical average of thousands to millions of cells) and provide averaged data on a cell population while masking rare and stochastic events. Single cell transcriptome analysis can overcome this limitation and is particularly important in stem cell and cancer research1-2. The proper characterization of the gene expression heterogeneity found in a cell population is the key factor to understand the development, progression, and treatment of cancers3, and has tremendous potential for assessment of genetic stability, identification of biomarkers and development of targeted therapy for personalized medicine4.
One of the hurdles of single-cell molecular analysis is material lost in conventional biochemical reactions5. Conventional biochemical reactions are performed in microliter scale and result in a more than 106 fold dilution of the content in a mammalian cell which has a volume at picoliters scales. Microfluidic systems provide a new set of molecular biology tools for performing biochemical reactions at the nanoliter scale. As demonstrated in our previous work, the small volume of reaction improved reverse transcription (RT) efficiency6. Unlike polymerase chain reaction (PCR) which is a repetitive reaction, RT is a single biochemical event. The concentration of RNA (~20-40 pg of total RNA in a single mammalian cell7) in RT is important for effective biochemical reactions. With these microfluidic devices to minimize the reaction volume to the picoliter and nanoliter scale, we are able to increase the RNA concentration to a level compatible with bulk assay for effective single-cell complementary DNA (cDNA) synthesis. However, for obtaining statistically significant data, a large number of single-cells must be analyzed. It is worth noting that data from these single-cells is different from those from cell lysate which is a physical average and heterogeneity is erased.
The ability of individually addressing and manipulating a cell is essential for the development of high efficiency single cell analysis platforms. Many efforts have been made to develop integrated genetic analysis on chip for large scale single-cell analysis. The basic functionalities for microfluidic single-cell gene expression analysis, including cell manipulation8-9, RNA purification and cDNA synthesis10-12, microfluidic PCR13 has been demonstrated. Recently, White et al. reported a large scale integrated system with on chip RT-qPCR capabilites14. Another novel parallel single cell analysis platform has been demonstrated by van den Brink et al. using individual side channel extraction method15. However, many platforms (including our first generation devices) trap single-cells with stochastic fashion and have little control of individual reactors12-14. We have previously developed microfluidic devices to perform mRNA-to-cDNA conversion within 10-nanoliter reactors, although the capture ability and precise manipulation of target single-cells to individual reactors are still limited. Therefore, the processing efficiency of the microfluidic network has not reached its full potential. Base on our previous devices, we constructed a microfluidic device with individual addressable reactors and hydrodynamic cell capture structures for gene expression studies. The integrated system can capture a large amount of single cells and extract the genetic information in a parallel manner. The device is fully compatible with the commonly used RT-PCR equipments for quantitative PCR analysis. In the second generation devices, we employed a specific cell loading method to ensure only one cell is captured in each reaction chamber. Adding cell trapping units such as mechanical hurdles16 and U-shape structure17 in the fluid channels has been reported to achieve fast immobilization and uniform distribution of cells. In our device, the cell trapping structure with the designed gap ensures the capture of only one single cell and assists their fast movement to the reactors. The individual addressing multiplexer is also implemented in the cell loading portion. With the individual addressing capacity, we can selectively exclude the units with two cells or no cell, hence increase the processing efficiency of a microfluidic chip. With the new integrated microfluidic devices, we measured the gene expression levels in HeLa and 293T cells, and demonstrated that gene expression in individual cells can be used to assess genetic stability of a mammalian cell population. The reported platform has the potential for cancer diagnosis by assessing genetic stability of a cell population.
Materials and methods
Fabrication of the microfluidic chips
Microfluidic single cell analysis chips were manufactured using polydimethylsiloxane (PDMS, RTV 615, GE) by multilayer soft lithography18. Two separated master molds, one for the fluidic layer and the other for the control layer, were fabricated by photolithography. The control mold was fabricated using SU8-25 photoresist to deposit valve features of 20 um in height. The mold of the fluid layer had a combination of cell trap features, sieving structures, and 15 um flow channels. The cell trap features and sieving structures were defined by SU8-25 negative photoresist (Microchem, USA), while the flow channels were fabricated using positive photoresist AZ 50XT (Microchem, USA). To facilitate valve closure in the assembled devices, the flow channel profiles were rounded on a hotplate at 130 °C for 10 minutes to reflow the photoresist. After the hardbake, the molds were silanized with trichlorotrimethylsilane to promote mold release during the PDMS casting process.
The device employs push-up valve geometry and consists of a three-layer elastomeric structure. For flow layer replication, PDMS base and catalyst were mixed in a 5:1 weight ratio, degassed in a vacuum chamber for 30 minutes and then poured on the mold and cured in a 80 °C oven for 45 minutes. The control layer was made by spin-coating a mixture of PDMS (20:1, elastomer to crosslinker) and baked for 30 minutes at 80 °C. The flow layer was carefully aligned with the control layer after holes were punched, and then bake at 80 °C for 45 minutes. The two-layer structure was peeled off the mold, with fluid inlet and outlet punched for tube connections, and bonded to a thin, cured PDMS layer, and incubated in oven again. A homemade COC (Cyclic olefin copolymer) plate was chosen as the chip supporting substrate, and 32 PCR reaction wells were embossed on the plate. This COC substrate is suitable for PCR reaction for its high optical clarity and low water vapour permeability to prevent evaporation19. Finally, the three-layer PDMS chip was bonded to the plastic plate and baked overnight at 80 °C.
Device automation
A homemade pneumatic control system composed of a single-chip microprocessor and multiple solenoid valves was used to actuate the individual valves. A program was written to the single-chip microprocessor. Therefore a combination of multiple valves actuation can be executed for each reaction step and multiplexing. Control channels were filled with water by pressurization before the experiments. The reagents were loaded into pipette tips and driven through the chip with pneumatic pressure. The air pressure for actuating integrated valves was 15 psi, and the pressure for driving sample was 1 psi.
Cell culture and single-cell gene expression profiling
Human cell lines of 293T cells (CRL-11268) and HeLa cells (CCL-2, adenocarcinoma from a 31 year old female patient) were purchased from American Type Culture Collection (ATCC) and cultured as instructed. Briefly, cells were cultured in Dulbecco’s Modified Eagle’s Medium supplemented with 10% v/v fetal bovine serum at 37.0°C with 5% carbon dioxide (CO2). After recovered in cultured, cells were harvested using trypsin solutions (Invitrogen). Serial dilutions were performed to obtain a single cell clone in 96-well plate. Five single-cell clones were chosen from each cell line for expansion. One clone of 293T and one clone of HeLa are used for single-cell gene expression analysis with the microfluidic device. After on-chip reverse transcription (RT), the cDNA of individual cells were subject to real-time PCR analysis. Using known copy number of cDNA of respective genes, standard cures were generated for calculation of copy numbers of mRNA in each cell with the assumption that the RT efficacy is ~50% as described previously12.
Results and discussion
Single-cell processing
The basic components of the microfluidic processor are shown in Fig. 1. A variety of components including cell trapping, manipulation, mRNA purification, cDNA synthesis and PCR amplification units are integrated in the device. This platform has high throughput microfluidic single cell handling capacity, and it can perform transcriptome analysis of dozens of single cells with high efficiency. A composite microscope image in Fig. 1 (Inset 1) demonstrated that a large number of single-cells can be processed at the same time. We designed two sets of multiplexers to ensure the individual addressability of each cell reaction chamber and bead column for high throughput single cell analysis. A homemade device control system with a programmed single-chip microprocessor and multiple solenoid valves was used to manipulate the control valves and the individual addressable multiplexers in the microfluidic device, as shown in Fig. 2. The chip has multiple independent pneumatic valves that can be digitally switched on and off, pushing the liquid into the chambers with great accuracy and consistency. These features significantly improve the simultaneous processing capacity for a large number of cells.
Fig. 1.
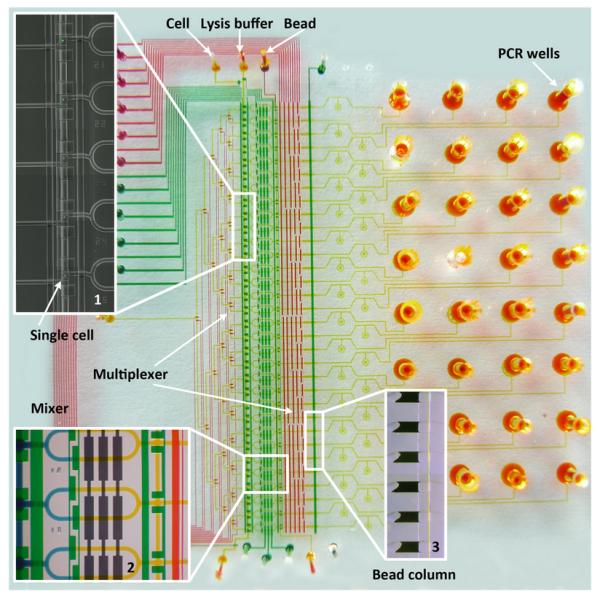
A microphotograph of a multilayer microfluidic chip designed for single-cell analysis. All the microfluidic channels were filled with different food dyes, indicating flow channels (yellow), control valves (green), and multiplexer control channels (red), respectively. Inset 1 is a composite image indicating five single-cells individually captured in the cell loading units. Inset 2 is the cell lysis module. Cell lysis is performed by opening the portion valve and pumping to mix lysis buffer (yellow) with the captured cell (blue). Two sets of multiplexers make each cell manipulation unit and bead column individually addressable for high throughput analysis. Inset 3 shows six stacked oligo-dT bead columns next to the sieve valve. The cell lysate was pushed through oligo-dT bead columns for mRNA capture and cDNA synthesis. Finally, the beads with the synthesized cDNA were collected in the PCR wells in the chip substrate for PCR analysis.
Fig. 2.

The pneumatic control system for microfluidic device operation. The homemade system consists of a single-chip microprocessor and multiple solenoid valves. A control program is written in the single-chip microprocessor for chip automation. A typical microfluidic chip is connected to the pneumatic control system through tygon tubings.
To perform the single cell analysis, we loaded live single-cell suspensions through one of the top inlets in the microfluidic chip. Single cells are trapped in the U-shaped sieve structure in the cell loading channel, as shown in Fig. 3A. The inset in Fig. 3 showed the U-shaped sieve structure with one single cell captured inside. Then the cells were washed by flushing the line with PBS solution, which removed un-trapped cells, after that the cell sample inlet valve was closed. With properly controlled cell density of the suspension and the assistance of the sieve structure, we can achieve uniform distribution of single-cells among all chambers in less than one minute.
Fig. 3.
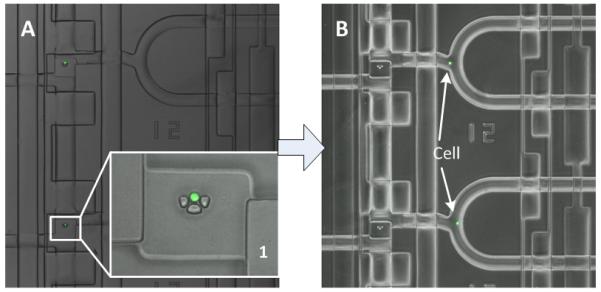
Microscope images of single cells captured in the microfluidic reaction chambers. (A) The cells were captured individually by the U-shape sieve structure in the cell loading area. Inset 1 shows the enlarged cell capture element, the single cell was stained with green fluorescent dye. (B) The captured single cells with green fluorescence were pushed from the cell trapping areas into different reaction chambers individually, followed by mixing with lysis buffer in the reactors.
Next, the cells were pushed in one side of the reaction chamber, while the lysis buffer was loaded from the other side of the reaction chamber, as shown in Fig. 3B. In a series of individual confined micro-mixer, the separation valves were then open and the cells were exposed to lysis buffer through peristaltic pumping in the mixer. After cell lysis, we reconfigured the control valves and flushed the cell lysate towards the bead column. Cell lysis is a crucial step in the gene expression profiling. Therefore, we calculated the volume of the cell and the lysis buffer and utilized peristaltic pumps to mix the two solutions. Mixing times can be reduced to a matter of seconds compared to several hours for passive diffusion.
Purification of mRNA was then carried out after single-cell lysis was performed using solid-phase extraction method in the adjacent microfluidic channels as described previously12. We used Dynabeads functionalized with oligo-dTs (Invitrogen, USA) to capture mRNA. The oligo (dT)25 sequence in Dynabeads acted as both mRNA capture sequence and primer for cDNA synthesis. After washing beads with buffers, reverse transcriptase and dNTPs were flushed over the bead columns to synthesize cDNA from the captured mRNA at 40 °C. The microfluidic device was placed on a thermal plate to maintain the temperature of RT reaction.
After the reverse transcription was completed, the sieve valves were relieved, and the beads with the synthesized cDNA were flushed to the beam-chambers. The beads collected in 32 bead-chambers in the PDMS chip were then pushed directly into the corresponding 32 wells fabricated in the COC plate. After peeling off the PDMS device, the 32 wells were loaded with PCR mixture and sealed with an adhesive PCR sealant film. The COC plate was put on a homemade fixture to be inserted to the RT-PCR equipment. To minimize device expense and complexity, the temperature control and fluorescence detection were performed outside of the chip using commercial thermocycler (Roche LightCycler 480II).
Microfluidic device efficiency
To achieve fast and reproducible loading of single-cells, we designed hydrodynamic single-cell trap within each capture chamber. The U-shaped sieve structure served as the cell blocking structure, as shown in inset1 in Fig. 3. The sieve-like openings in the U-shaped structure are too narrow for the cells to pass, but they add flow components directed through the middle of the capturing elements. Therefore, the small gaps in the capture elements could suppress the effect that the continuity of streamlines dragging the cells around the capturing elements. Using these structures we were able to achieve high single-cell capture rate in the device. With properly controlled cell density of the suspension and the assistance of the sieve structure, we can complete single-cell loading among all capture units in less than one minute. Over 10 separate experiments, we observed high single-cell occupancy of array locations (30 single cells among 32 loading unit on average), therefore the cell capture efficiency is close to 95%. The cell loading concentrations were kept between 5 × 105 and 1.5 × 106 cells/mL, resulting in 95% occupancy of single cells in 1 min at a flow rate of approximately 1μL/min. Lower concentrations were found to require longer time to achieve high occupancy of trapped single cells. Concentrations higher than 1.5 × 106 cells/mL would lead to occasionally clogging the inlet port or the cell trap structures.
To further improve device capture efficiency, we adopted the effective multiplexing addressing method20 in order to individually address each reaction chamber. In the multiplexer units, the control lines are grouped into sets of complementary (binary) valve pairs, and n pairs of control valves can be utilized to control 2^n flow channels. During the single cell loading process, there may be no cell or more than one cell loaded in the capture elements. With the individual addressing ability, we can selectively reload the units with two cells or no cell, hence increase the processing efficiency of one single microfluidic chip. In theory, the overall processing efficiency of the reaction chamber can reach 100% by repeating two or three times of cell loading procedure, making this method applicable to the analysis of limited quantity samples such as rare stem cells or clinical cancer samples.
Gene expression heterogeneity
With this integrated device, we studied the expression of housekeeping genes in two different cell lines, 293T and HeLa. A single-cell was isolated by serial dilution from each cell line and expanded into a colony in Petri dish. Then individual cells from a colony are isolated and profiled for the expression of housekeeping genes, beta-Actin. This experiment setting minimizes the heterogeneity of cell population, however, because of the cell cycle as a fundamental heterogeneous factor among individual cells, the expression of housekeeping genes are expected to be heterogeneous among cells. This has been demonstrated in previous studies by us and other investigators10, 12, 14. Here, we investigate whether the degree of heterogeneity is different among various cell types, specifically cancer and transformed cells which are not cancer cells but are immortalized by DNA mutations. We compared the copy number of Actin in individual cells from these two cell lines, as demonstrated in Fig. 4. The transformed cells, 293T, have a more uniform mRNA copy numbers among cells and the distribution is close to normal distribution. On the other hand, the cancer cells, HeLa (CCL-2 is adenocarcinoma from a 31 year old female patient) have a wider variation of Actin mRNA copy number in each cell. Also the distribution of Actin mRNA is not in a normal distribution pattern as that in 293T cells.
Fig. 4.

Different distribution of Actin expression between HeLa and 293T cells. The expression distribution and degree of heterogeneity are different between two cell lines, HeLa and 293T. The HeLa cells, a cancer cell line, have an irregular distribution of Actin mRNA copy numbers (A) and the heterogeneity of expression is much higher than that of 293 T cells as shown by the wider distribution of amplification plots (B). The transformed cell, 293T, has a normal distribution of Actin mRNA copy number among cells (C) and less heterogeneity of expression as indicated by the tight amplification plots (D). Distribution histograms are constructed with data from 18 and 19 individual cells respectively, and distribution are visualized with best-fitted mathematical patterns.
Housekeeping genes are genes that provide fundamental functions to a cell. Therefore, their expressions are at similar levels in all cells, and are used as control in a lot of gene expression studies. At the single-cell level, mRNAs of house-keeping genes among cells have a random distribution pattern due to random sampling and the continuous synthesis/degradation of mRNA. Our results indicated that the immortalized 293T cells (not cancer cells) have such distribution of Actin mRNA and express Actin at a similar level. The cancer cells (HeLa), however, have an irregular pattern of Actin mRNA copies among cells which indicate that Actin is not expressed at similar level in all cells. The deviation from a normal distribution pattern reflects the genetic stability of HeLa cells. The unstable genome and abnormal regulation of gene expression in cancer cells leads to vary different expression levels of housekeeping genes among individual cells.
Conclusions
In conclusion, we have demonstrated a new integrated single cell analysis platform that combines a high-affinity cell capture assay based on micro sieve capture structure, and individual addressable manipulation. Many devices for single-cell isolation and analysis have been reported12-15. However, the approach of using individual addressable reactor to improve processing efficiency has not been fully explored. Here, we report the implemented individual addressing units to improve single cell analysis efficiency (close to 100%). Different from other systems, the reported system integrates individual addressable units with a variety of components for single-cell gene expression analysis, including cell trapping, manipulation, mRNA purification, cDNA synthesis, and prepares samples for PCR analysis. The device is directly compatible with commercial PCR equipment for rapid quantitative analysis. These new features significantly improve the simultaneous processing capacity for a large amount of cells. The microfluidic system demonstrated here offers a new approach to perform well-controlled high-throughput micro-scale biochemical reactions. The microchip can be employed for a number of related applications for systematic single cell analysis, and the system has great potential for applications in cancer research.
As an example, we measured the expression of a housekeeping gene in two different cell lines: an immortalized cell line (293T) and a cancer cell line (HeLa). An immortalised cell line is a population of cells which normally cannot proliferate indefinitely, but has the ability of undergoing division indefinitely due to mutation. These cells can therefore be grown for prolonged periods in laboratories as a cell culture. However, these cells are not cancer cells which grow and divide at an unregulated and faster cycle with an instable genome. By profiling a larger number of single-cells of a house keeping genes, we demonstrated that the pattern of gene expression can distinguish cancer from immortalized cells. Cancer cells with instable genome (DNA) and abnormal gene regulation express housekeeping genes at very different levels. Under random sampling, the expression pattern deviates from normal distribution. Since immortalized cells can divide indefinitely and have a high potential for developing into cancer cells, our result suggests that our platform can be developed into a potential novel method of evaluating the genetic stability of a cell population and distinguishing immortalized cells from cancer cells. Because the genetic instability is a common feature of many types of cancer, our device has the potential for molecular diagnosis of cancer using small number of cells from solid tumor needle biopsy and from isolated circulating tumor cells.
Gene analysis is a complicated process requiring multiple steps of biochemical reactions. A fully integrated microsystem that is capable of providing sample-to-answer analysis would typically require multiple microfluidic components such as micromixers and multiplexers to carry out the reaction, buffer exchange and material transfer. The compact and programmed pneumatic control system we developed here enables the fast and accurate manipulation of each microfluidic component, providing a useful tool for portable microfluidic genetic analysis device. Such devices have the potential to be developed into a tool for measuring genetic stability of a cell population with the expression pattern of housekeeping genes. With further development, our platform can be used for cancer diagnosis and evaluation of genetic stability of cell populations.
Acknowledgements
This work is supported by the National Natural Science Foundation of China (Grant 61106128), the Knowledge Innovation Project of the Chinese Academy of Science (Grant KGCX2-YW-904), and the Guangdong Innovation Research Team Fund for Low-cost Healthcare Technologies. The authors also thank the support from Grant R21CA134391 from the U.S. National Institutes of Health, and Grant AW 0852720 from the U.S. National Science Foundation.
Footnotes
Published as part of a themed issue dedicated to Emerging Investigators
References
- 1.Dalerba P, Kalisky T, Sahoo D, Rajendran PS, Rothenberg ME, Leyrat AA, Sim S, Okamoto J, Johnston DM, Qian DL, Zabala M, Bueno J, Neff NF, Wang JB, Shelton AA, Visser B, Hisamori S, Shimono Y, van de Wetering M, Clevers H, Clarke MF, Quake SR. Nature Biotechnology. 2011;29:1120–U1111. doi: 10.1038/nbt.2038. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2.Lecault V, VanInsberghe M, Sekulovic S, Knapp D, Wohrer S, Bowden W, Viel F, McLaughlin T, Jarandehei A, Miller M, Falconnet D, White AK, Kent DG, Copley MR, Taghipour F, Eaves CJ, Humphries RK, Piret JM, Hansen CL. Nature Methods. 2011;8:581–U593. doi: 10.1038/nmeth.1614. [DOI] [PubMed] [Google Scholar]
- 3.Klein CA, Seidl S, Petat-Dutter K, Offner S, Geigl JB, Schmidt-Kittler O, Wendler N, Passlick B, Huber RM, Schlimok G, Baeuerle PA, Riethmuller G. Nature Biotechnology. 2002;20:387–392. doi: 10.1038/nbt0402-387. [DOI] [PubMed] [Google Scholar]
- 4.Riethdorf S, Wikman H, Pantel K. International Journal of Cancer. 2008;123:1991–2006. doi: 10.1002/ijc.23825. [DOI] [PubMed] [Google Scholar]
- 5.Bengtsson M, Stahlberg A, Rorsman P, Kubista M. Genome Res. 2005;15:1388–1392. doi: 10.1101/gr.3820805. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Chen Y, Zhong JF. Methods in Molecular Biology. 2008;438:293–303. doi: 10.1007/978-1-59745-133-8_22. [DOI] [PubMed] [Google Scholar]
- 7.Uemura E. Brain Research Bulletin. 1980;5:117–119. doi: 10.1016/0361-9230(80)90182-3. [DOI] [PubMed] [Google Scholar]
- 8.Gong YA, Ogunniyi AO, Love JC. Lab Chip. 2010;10:2334–2337. doi: 10.1039/c004847j. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Skelley AM, Kirak O, Suh H, Jaenisch R, Voldman J. Nature Methods. 2009;6:147–152. doi: 10.1038/nmeth.1290. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Marcus JS, Anderson WF, Quake SR. Analytical Chemistry. 2006;78:3084–3089. doi: 10.1021/ac0519460. [DOI] [PubMed] [Google Scholar]
- 11.Bontoux N, Dauphinot L, Vitalis T, Studer V, Chen Y, Rossier J, Potier MC. Lab Chip. 2008;8:443–450. doi: 10.1039/b716543a. [DOI] [PubMed] [Google Scholar]
- 12.Zhong JF, Chen Y, Marcus JS, Scherer A, Quake SR, Taylor CR, Weiner LP. Lab Chip. 2008;8:68–74. doi: 10.1039/b712116d. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Toriello NM, Douglas ES, Thaitrong N, Hsiao SC, Francis MB, Bertozzi CR, Mathies RA. Proc. Natl. Acad. Sci. U. S. A. 2008;105:20173–20178. doi: 10.1073/pnas.0806355106. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.White AK, VanInsberghe M, Petriv OI, Hamidi M, Sikorski D, Marra MA, Piret J, Aparicio S, Hansen CL. Proc. Natl. Acad. Sci. U. S. A. 2011;108:13999–14004. doi: 10.1073/pnas.1019446108. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.van den Brink FTG, Gool E, Frimat JP, Bomer J, van den Berg A, Le Gac S. Electrophoresis. 2011;32:3094–3100. doi: 10.1002/elps.201100413. [DOI] [PubMed] [Google Scholar]
- 16.Di Carlo D, Wu LY, Lee LP. Lab Chip. 2006;6:1445–1449. doi: 10.1039/b605937f. [DOI] [PubMed] [Google Scholar]
- 17.Wang ZH, Kim MC, Marquez M, Thorsen T. Lab Chip. 2007;7:740–745. doi: 10.1039/b618734j. [DOI] [PubMed] [Google Scholar]
- 18.Unger MA, Chou HP, Thorsen T, Scherer A, Quake SR. Science. 2000;288:113–116. doi: 10.1126/science.288.5463.113. [DOI] [PubMed] [Google Scholar]
- 19.Nunes PS, Ohlsson PD, Ordeig O, Kutter JP. Microfluidics and Nanofluidics. 2010;9:145–161. [Google Scholar]
- 20.Thorsen T, Maerkl SJ, Quake SR. Science. 2002;298:580–584. doi: 10.1126/science.1076996. [DOI] [PubMed] [Google Scholar]