

Abstract
With recent advances in high-throughput proteomics and systems biology, there is a growing demand for new instruments that can precisely quantify a wide range of receptor-ligand binding kinetics in a high-throughput fashion. Here we demonstrate a surface plasmon resonance (SPR) imaging spectroscopy instrument capable of extracting binding kinetics and affinities from 50 parallel microfluidic channels simultaneously. The instrument utilizes large-area (~cm2) metallic nanohole arrays as SPR sensing substrates and combines a broadband light source, a high-resolution imaging spectrometer and a low-noise CCD camera to extract spectral information from every channel in real time with a refractive index resolution of 7.7 × 10−6. To demonstrate the utility of our instrument for quantifying a wide range of biomolecular interactions, each parallel microfluidic channel is coated with a biomimetic supported lipid membrane containing ganglioside (GM1) receptors. The binding kinetics of cholera toxin b (CTX-b) to GM1 are then measured in a single experiment from 50 channels. By combining the highly parallel microfluidic device with large-area periodic nanohole array chips, our SPR imaging spectrometer system enables high-throughput, label-free, real-time SPR biosensing, and its full-spectral imaging capability combined with nanohole arrays could enable integration of SPR imaging with concurrent surface-enhanced Raman spectroscopy.
Introduction
The ability to quantify kinetics and affinities of receptor-ligand binding interactions is essential for basic biology, biomarker discovery, proteomics, pharmaceutical development and drug discovery, among others.1 Optical biosensors that utilize surface plasmon waves2 (SPs) – surface electromagnetic waves propagating on the interface between a metallic film and a dielectric medium – have been widely used for this purpose.3–7 Commonly known as surface plasmon resonance (SPR) instruments, they measure refractive index changes near a metal surface caused by the surface binding of biomolecules without fluorescent labels, enabling rapid label-free quantification of association/dissociation rates and equilibrium constants for analytes binding to targets such as proteins, antibodies, nucleic acids, aptamers, or lipids. Owing to rapid developments in proteomics, lipidomics, and systems biology, there is a growing demand to enhance the throughput and sensitivity of conventional SPR instruments.1 High-throughput microarray technologies based on proteins, nucleotides or lipids, in which multiple recognition elements can be arrayed at high spatial densities and investigated directly on a single chip, require detection methods that can quantify their reaction kinetics in an equally high-throughput fashion. Many technological challenges exist, however, since even routine quantification of protein binding kinetics generally requires a series of SPR measurements performed at multiple analyte concentrations, which can take many hours depending on surface modification steps and dissociation rates.
SPR imaging provides an attractive path to expand the throughput of conventional SPR sensors.8–14 Typically an analyte or buffer solution flows over a microarray of receptors on the gold sensing surface, which is illuminated with a light source. The spatial variation in light intensity generated by surface binding is imaged in real time, allowing the extraction of binding kinetics from every sensing spot simultaneously. Target receptors are typically arrayed on the gold sensing surface using robotic spotting tools, microfluidics, or microcontact printing. Among various multiplexing options, intensity-based SPR imaging of two-dimensional (2-D) microarrays has been widely studied11 because many sample spots can be densely packed in this format. High-throughput SPR imaging with a 2-D microarray of proteins, however, is a challenging task, because standard robotic spotting protocols, which work well for nucleic acids, require some process modifications to work well with proteins.15 It is difficult to print small protein spots with a uniform thickness and morphology, and the protein spots must be prevented from drying, which can denature some proteins.16 Also, in a typical SPR imaging experiment with a 2-D microarray, multiple receptor spots are exposed to an analyte solution in a single flow cell and artifacts or errors may be introduced due to cross-contamination, position-dependent depletion of analyte, as well as position-dependent binding rates due to mass transport effects.17 Alternatively, SPR imaging can be performed with parallel microfluidic channels that partition the sensing area on the gold surface. While the multiplexing capacity in space is reduced in this one-dimensional (1-D) array, multiple receptors can be easily patterned on the gold surface via well-established flow-based immobilization protocols, obviating the need for expensive robotic spotting equipment. In fact, many commercial SPR instruments use individual flow cells such that all experimental steps such as receptor immobilization, washing, blocking non-specific binding, analyte binding, and surface regeneration are performed in the same channel using flow-based techniques, rather than spotting.18 Cross-contamination can be eliminated and receptors that are not readily amenable to standard robotic spotting (e.g. lipid membranes) can also be readily reconstituted and confined inside each channel. The width of each microfluidic channel (e.g. 30 µm in this work) can also be smaller than the typical size of a protein spot obtained using standard robotic spotting equipment (100~200 µm).6 However, trade-offs exist on the instrumentation side. Unlike single-channel SPR sensing, wherein a full angular or spectral response of a sensor chip can be recorded in real time, intensity-based 2-D SPR imaging only records the intensity of light at a fixed wavelength, rather than full spectral information, motivating the development of an SPR imaging instrument capable of measuring full spectra in real time from a large array of parallel microfluidic channels.
In this paper, we combine metallic nanohole-based SPR technologies with imaging spectroscopy. Metallic nanohole arrays that can give rise to the extraordinary optical transmission (EOT) effect19 have been widely investigated for SPR sensing because they can simplify optical systems design by exciting SP waves at normal incidence illumination,20–23 while also allowing high-resolution SPR imaging,24–26 flow-through sensing,27,28 and integration of lipid membranes and membrane proteins.29–31 To integrate nanohole SPR sensors with a large array of microfluidic channels, we fabricate large-area periodic Ag nanohole sensor chips with uniform optical properties by a template-stripping technique.32–34 After coating the Ag sensing surface with a thin silica overlayer for biological interfacing, a PDMS microfluidic chip is bonded to partition the sensing region into 50 parallel microfluidic channels. After demonstrating real-time SPR spectral imaging with custom LabVIEW™ software and MATLAB-based image processing, device sensitivity and refractive index resolution are evaluated. The use of partitioned microfluidic channels allows us to readily incorporate membrane-bound receptors in fluidic supported lipid membranes35 without cross-contamination or mixing. Without partitioned microfluidic channels, additional processing steps, such as deposition of metallic grids as lipid diffusion barriers,36 laser ablation,37 or microcontact printing38 would be required to isolate supported membranes with different receptors or receptor concentrations. This is a significant advantage of a 1-D SPR imaging configuration as it facilitates the deposition of many different lipid/receptor compositions and negative controls on a single assay platform with minimal sample processing and a simple, flow-based lipid injection step. To demonstrate the utility of our instrument for high-throughput analysis of protein-lipid binding interactions, full-spectral SPR imaging is performed using cholera toxin b and ganglioside (GM1), and the dissociation rate constant (kd), association rate constant (ka), and equilibrium dissociation constant (KD) are extracted simultaneously from 50 channels.
Results and Discussion
Experimental Set-ups
The concept of full-spectral SPR imaging using metallic nanohole arrays and parallel microfluidic channels is illustrated in Figure 1a. Broadband light illuminates nanohole arrays at normal incidence. Surface plasmons are excited, and resonantly transfer energy through the nanohole substrate. The light is then re-radiated back to free space, exiting the microscope (Nikon Eclipse Inverted) via the side camera port and entering the imaging spectrometer (Newport MS257) through a narrow slit. The multiple channels imaged and aligned along the vertical entrance slit are each spread as a single spectrum by a grating and are recorded on a 1340 × 400 pixel CCD in real time. In this way, the recorded image contains spectrally dispersed information (i.e. an full optical transmission spectrum for each channel) along the microfluidic channel direction, and a spatially resolved 1-D image of multiple channels along the orthogonal direction. The range and resolution of the spectra can be controlled by using diffraction gratings with different line spacing.
Fig_1.
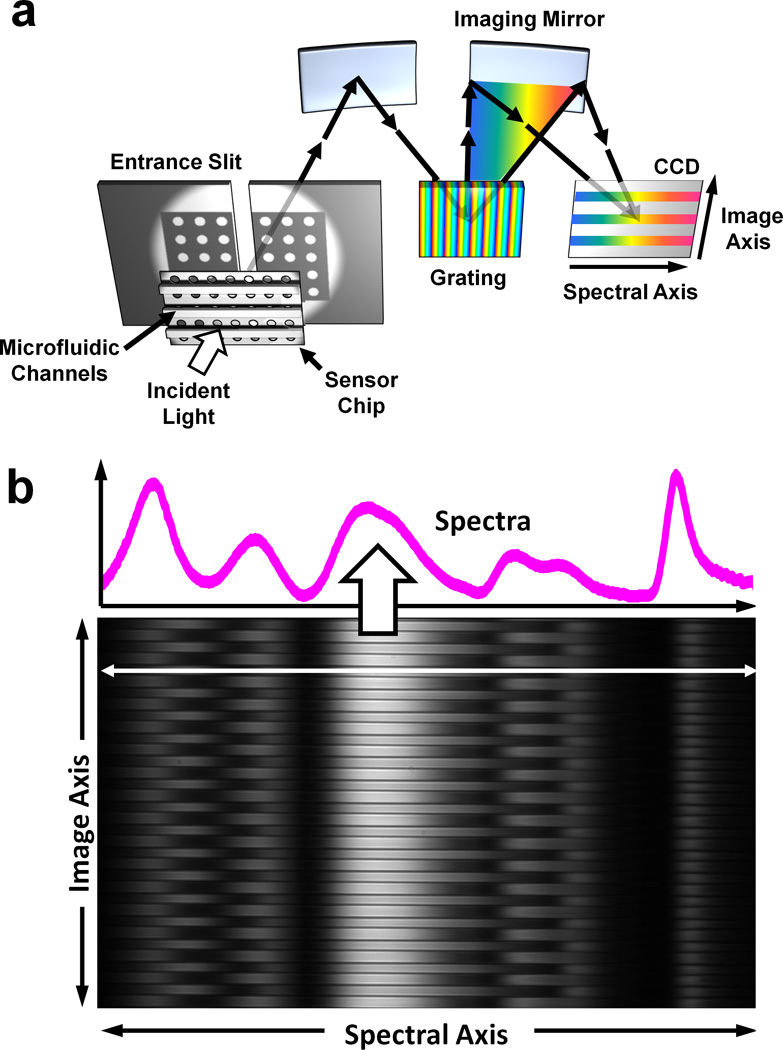
Spectral imaging setup. (a) An imaging spectrometer is used to acquire multiple transmission spectra from a single nanohole array chip with 50 parallel microfluidic channels. An image of the transmitted light is focused onto the entrance slit, with perpendicular microfluidic channels. This light is dispersed along one dimension, forming a spectral image on a CCD chip. The vertical, or imaging, axis corresponds to position along the entrance slit whereas the horizontal axis corresponds to the spectral content of the light entering the slit. (b) Sample data recorded on the CCD. The microfluidic channels are clearly seen spaced vertically. A single horizontal cross section line shows the transmission spectrum from a single channel.
Figure 1b shows a sample spectral image taken from the imaging spectrometer and a deep-cooled, low-noise CCD camera (Princeton PIXIS) with custom LabVIEW™ software. The image recorded by the CCD shows the EOT peaks along the spectral (horizontal) axis. Channels and boundaries can be clearly distinguished along the imaging (vertical) axis due to the difference of refractive index between polydimethylsiloxane (PDMS) and liquid solutions. The data is analyzed using home-made MATLAB image processing scripts to quantify kinetics data from each microfluidic channel in parallel.
Our instrumentation technique is different from conventional SPR imaging that measures a 2-D intensity distribution, since full spectral information from each channel can be acquired without any data loss, leading to an increase in the dynamic range of detection. For example, SPR imaging using a laser (i.e. single-wavelength) illumination requires the excitation wavelength precisely positioned at the position of maximum sensitivity along the SPR curve (i.e. region of maximum slope).39 However, this sets a limitation since further molecular immobilization can shift the SPR response curve to wavelengths not probed by the laser. With full spectral imaging, such restrictions do not exist, since the entire spectrum is recorded. Furthermore, by exploiting the surface-enhanced Raman spectroscopy (SERS) capability of nanohole arrays40 with our imaging spectrometer, it would be possible to use our instrument to perform molecular detection and identification successively in a parallel setup.
Nanofabrication and Microfluidic Integration
The fabrication of large-area nanohole arrays having uniform optical properties is the prerequisite to perform high-throughput SPR imaging spectroscopy of multiple parallel channels. To accomplish this, we use template stripping. Template stripping exploits the poor adhesion between noble metals such as Au or Ag and a Si substrate. With this technique, many high fidelity metallic nanostructures can be replicated from a single patterned silicon master template, as previously demonstrated.32,33,41,34 Figure 2 shows photographs of Si templates and template-stripped nanoholes. The Ag film is stripped off from this Si template after curing UV optical epoxy (Fig. 2a), producing a large area (8 × 8 mm) periodic nanohole array (Fig. 2b). The Si master templates (Fig. 2c) can be reused more than 30 times. The nanohole array substrate is attached to a PDMS microfluidic chip (Fig. 2d), which partitions the nanohole array into 50 individual channels. The channel period is 70 µm and the width of each channel is 30 µm. Optical transmission or reflection spectral from all 50 channels can be monitored simultaneously in our instrument (Figs. 2e–f). While the number of channels could be increased beyond 100, attachment of inlet and outlet tubes and syringe pumps limits the practical throughput in our proof-of-concept experiment. Before assembling microfluidics on the Ag nanohole arrays, a thin silica shell is coated on the surface by atomic layer deposition (ALD). This dense and conformal silica layer will protect Ag from oxidation as well as promote supported lipid bilayer formation.30 In addition, the PDMS chip containing microfluidic channels can be permanently bonded to silica-coated Ag nanohole arrays, which eliminates fluid leakage (Fig. 2d). Prior to multi-channel biosensing, the uniformity of the optical spectra from each channel was evaluated. After the spectral dispersion, 50 channels with a 70 µm period over a 3.5 × 2 mm area were successfully recorded on the CCD (Fig. 2e). Multiple points of enhanced transmission (i.e. bright spots) in each channel in the image represent the EOT peaks from the various grating orders. The peak transmission wavelength of EOT can be approximated by the following equation42:
where a0 is the periodicity of the array, the integers (i, j) represent the Bragg resonance orders, and εm and εd are the dielectric functions of the metal and dielectric, respectively. Figure 2f shows the spectra extracted simultaneously from all 50 channels. Positions 1, 2, and 3 represent the peak transmission wavelength at (1,1) water, (1,1) substrate, (1,0) water, respectively. Highly uniform spectral peak positions over the 50 channels are obtained from the template-stripped Ag nanohole arrays. The resolution of one pixel in the spectral image can be tuned by changing the grating used in the experiment from 0.623 nm/pixel with a 121 lines/mm grating to 0.0623 nm/pixel with a 1200 lines/mm grating. Based on the gratings used, the entire spectral range can cover from 250 nm, which can accommodate major EOT peaks, to 25 nm for monitoring the peaks at a narrow spectral region.
Fig_2.
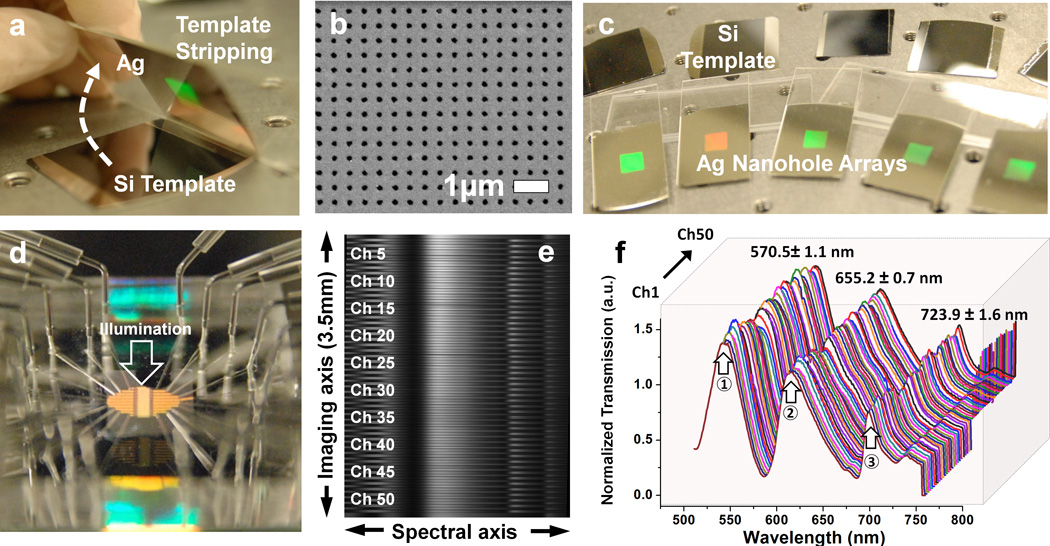
Chip fabrication procedure. (a) Using a silicon template, a large-area nanohole array is replicated by template stripping. (b) Scanning electron micrograph (SEM) of an area of the nanohole chip. (c) Photograph showing multiple chips and their templates. A single metal deposition was used to produce the entire batch and the templates are reusable. (d) Photograph of the complete microfluidic chip with inlet and outlet ports. The direction of the incident light is indicated. The transmitted light is imaged as described in figure 1, giving (e) full spectral content from 50 parallel channels. (f) Due to the high-quality template, the transmission spectra from each channel are highly uniform.
1-D Full-Spectral Imaging
To evaluate the refractive index sensitivity of nanohole device, index-calibrated glycerol-water solutions were serially injected into all channels. When the refractive index increases from 1.333 to 1.353 in steps of 0.05 refractive index units (RIU), the EOT spectrum shifts linearly due to the increase of refractive index near sensing surface (Fig. 3a). The spectral shift is homogenous over all 50 channels, and the measured (1,0) bulk sensitivity of the device is 470 ± 11.7 nm / RIU. (Fig. 3b)
Fig_3.
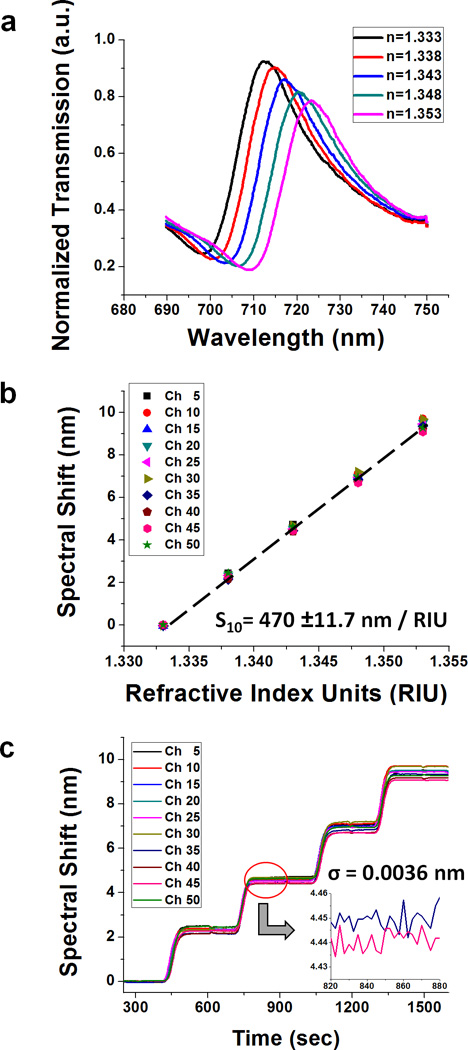
(a) The transmission spectrum shifts after injecting the index-calibrated water-glycerol solutions. Each spectrum is obtained from one of the 50 channels using a 600 lines/mm grating. (b) The refractive index sensitivity measured from spectral shift versus refractive index units (RIU) change. It shows linear increase and is very homogeneous over all 50 channels. The measured sensitivity is 470 ± 11.7 nm / RIU. (c) Spectral resolution based on the refractive index sensitivity and the standard deviation of noise (Ïf) of the experiment above. With 2.5 sec temporal resolution, the spectral noise is Ïf = 3.6×10<sup>−3</sup> nm, giving ~10<sup>−6</sup> RIU spectral resolution.
Spectral and temporal resolutions are also important factors for defining the performance of the system. Spectral resolution (i.e. refractive index resolution) is obtained by dividing the noise level (σ) by the refractive index sensitivity. Figure 3c shows the spectral resolution based on the refractive index sensitivity and the noise (σ) at the 2.5 sec (1 sec exposure with 2 spectra averaged) temporal resolution. The noise is decreased to 3.6×10−3 nm by doing 2 times temporal averaging. Based on the sensitivity and the noise level, 7.7 × 10−6 RIU spectral resolution is obtained from our current instrument configuration. With faster spectral acquisition and more frequent frame averaging, the instrumental resolution can be further improved.43,44
Binding kinetics of CTX-b vs. GM1 using 50-channel SPR imaging spectroscopy
To demonstrate the multi-channel microfluidic SPR spectral imaging capability, cholera toxin b (CTX-b) and ganglioside GM1 are used. CTX-b binding to GM1 is a well-characterized system of a protein binding to a lipid receptor.45–50 Figure 4a shows schematic of a supported lipid bilayer (SLB) on a nanohole sensor and CTX-b binding to GM1 in the SLB. Phospholipid vesicles containing GM1 are injected into microfluidic channels then rupture on the silica-coated nanohole array to form SLBs.51,35 After SLB formation, CTX-b is introduced and binds with the oligosaccharide portion of the GM1 receptor. Prior to SPR imaging, we employed fluorescence imaging to evaluate SLB formation on the silica surface and CTX-b binding to GM1 in multiple channels. Vesicles containing 1 mol % rhodamine-labeled phosphatidylethanolamine (Rho-PE) but with different concentrations of GM1 were injected into each individual flow cell. After the formation of SLBs, 50nM CTX-b was introduced simultaneously into all channels. Figure 4b shows fluorescence images of SLBs with 1 mol % Rho-PE (top) and Alexa 488-conjugated CTX-b (bottom). The Rho-PE intensity is consistent across all channels, indicating little channel-to-channel variability in SLB formation. However, the Alexa 488 intensities increase when the GM1 densities increase, implying more CTX-b binds when the receptor density increases, as expected. Figure 4c shows a line profile of fluorescence intensity across the microfluidic channels.
Fig_4.
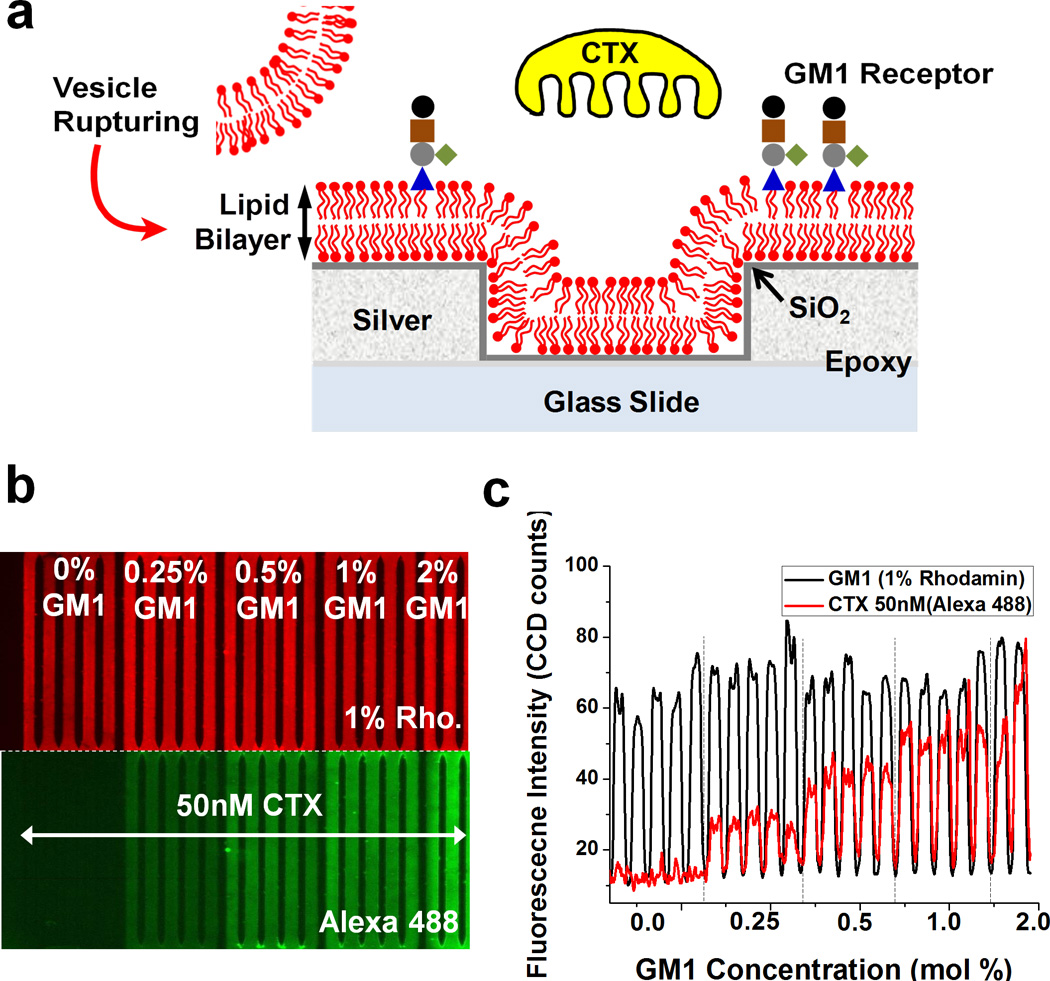
(a) Schematic of cholera toxin b subunits (CTX-b) binding to ganglioside GM1 receptors incorporated in lipid membranes. First, a silica shell is coated on a silver surface to promote vesicle rupture. Then, GM1 containing vesicles are injected into microfluidic channels and rupture to form supported lipid bilayers (SLBs). The CTX-b is introduced and binds with GM1. (b) Fluorescence images showing homogeneous SLB formation and different amount of CTX-b binding to various concentration of GM1 in supported lipid bilayers. (c) A line profile of fluorescence intensity across the microfluidic channels. The fluorescence intensity of the SLBs indicates a homogeneous lipid membrane across the channels, while the intensity of CTX-b bound to GM1 increases due to the increase in receptor density.
The experimental cycle including SLB formation, blocking and CTX-b binding is shown in Figure 5a. Vesicles were injected after 10 min of stable baseline for an hour. After injecting PBS to remove excess vesicles, the resonance peak position stabilized at a 2 nm shift from initial (1,0) maximum position, indicating stable formation of an SLB on the nanohole array. After BSA blocking to reduce nonspecific adsorption, various concentrations of CTX-b solution are simultaneously injected into each channel.
Fig_5.
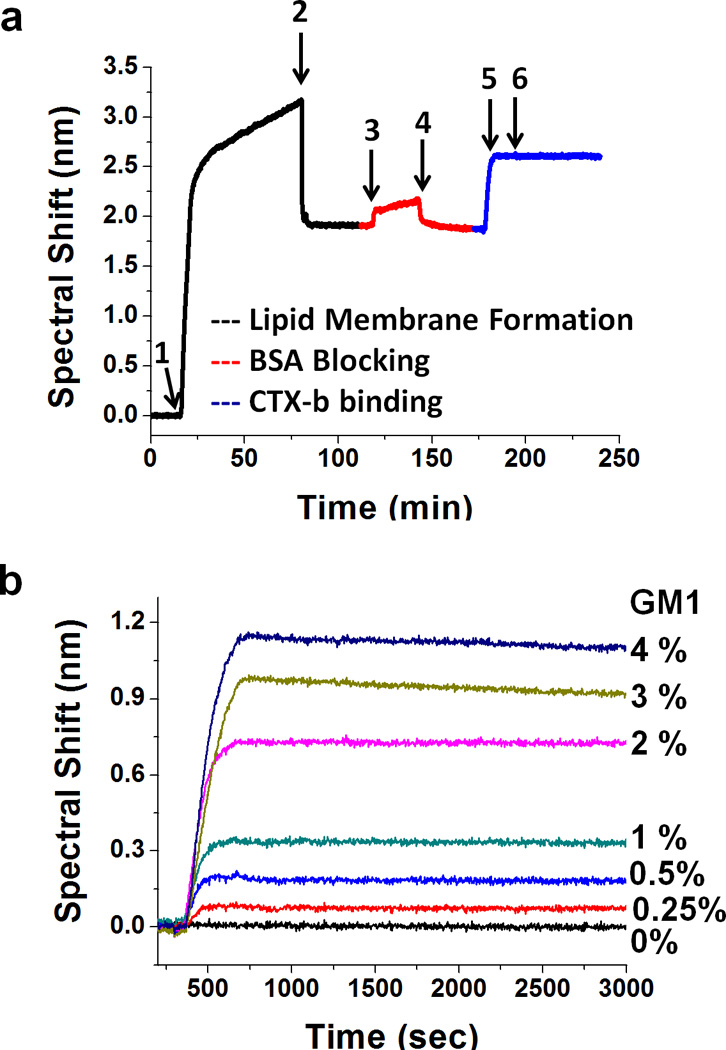
(a) Sensorgram showing SLB formation, BSA blocking and CTX-b binding to GM1. 1) vesicle injection; 2) washing excess vesicles from surface; 3) BSA blocking; 4) wash excess BSA; 5) inject CTX-b; 6) end CTX-b injection and begin dissociation step by washing the channel with PBS. (b) Binding kinetics curves of 50 nM CTX-b with SLBs containing various concentration of GM1. The kinetic curves clearly show fast association and a slow dissociation of CTX-b binding to GM1. The maximum spectral shifts increase proportionally with the amount of GM1 receptor up to 2 mol % concentration.
Though CTX-b is pentavalent, a 1:1 kinetic interaction model can be used for low receptor densities, while more complex models are required for higher receptor densities where polyvalent interactions occur. Therefore, the first set of experiments was to determine the range of GM1 concentrations for which a 1:1 binding model is appropriate. Figure 5b shows kinetic curves for 50 nM CTX-b binding on SLBs with GM1 concentrations ranging from 0 – 4 mol %. During a 5 min CTX-b injection, rapid association between CTX-b and GM1 were observed. Up to 2 mol % GM1, the maximum spectral shift is linearly proportional to the amount of GM1 in the SLB. Thus, in subsequent experiments the GM1 concentration was fixed at 2 mol %. In all kinetic analyses, bulk refractive index change was subtracted. During the 1 hour dissociation phase, all channels showed slow negative peak shifts, confirming the high affinity between CTX-b and GM1.
Figure 6a shows a concentration assay of CTX-b binding to GM1 using multichannel SPR imaging spectroscopy. CTX-b concentration ranged from 3.13 to 400 nM and the GM1 receptor concentration is fixed to 2 mol %. 40 channels were used for simultaneous binding kinetic experiments (5 channels × 8 different concentrations). The other 10 channels are used for the negative control SLBs with no GM1 and for channels that compensate for bulk refractive index shift. 50 nM CTX-b was also injected into the negative control channels. Due to the multichannel microfluidics and SPR spectral imaging setup, 40 different binding interactions can be measured in parallel. The binding kinetic curves are highly stable over all channels and clear kinetics were observed with CTX-b concentrations as low as 6.25 nM. Figure 6b shows representative binding kinetics with CTX-b concentrations ranging from 3.13 to 400 nM. With the increase of analyte concentration, the spectral response increases but approaches a maximum. The kinetic constants calculated using a 1:1 binding analysis method (See analysis of molecular binding kinetics in the Supporting Information) are shown in Table 1. Although the dissociation constant (KD) obtained from each concentration varies from sub-nM to ~ 2 nM, they are comparable to previously reported values.47,52–54
Fig_6.
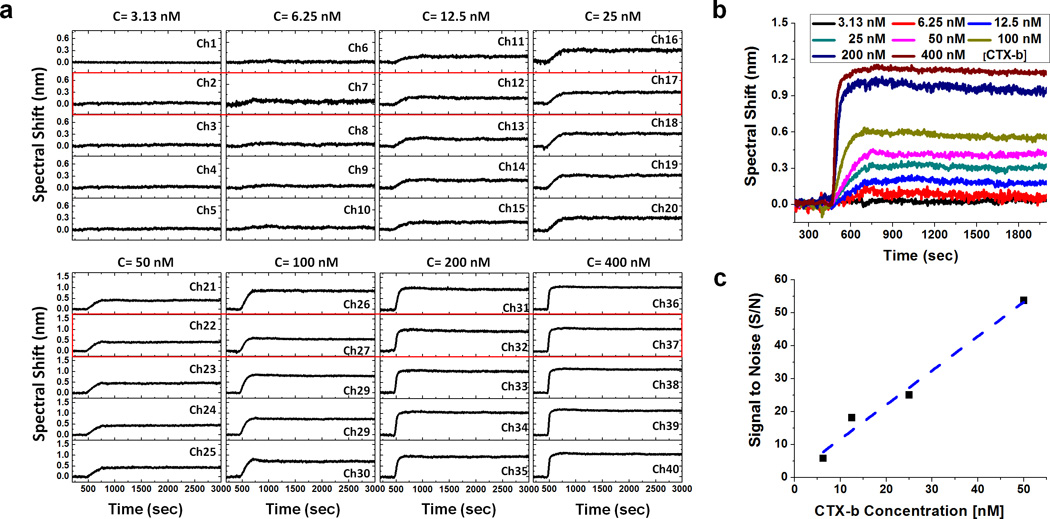
(a) A CTX-b concentration assay with 2 mol % GM1 using multichannel SPR imaging spectroscopy. 40 different binding interactions are measured in one experiment. (b) The representative binding kinetics at different concentrations of CTX-b. The spectral response increases with the analyte concentration but approaches to a maximum. (c) The plot of signal to noise (S/N) versus concentration of CTX-b to estimate the limit of quantification (LOQ). The maximum spectral shifts and the SD of the baseline at the representative kinetic curve are used for the signal and the noise, respectively. LOQ corresponding to S/N = 10 is calculated to 8.4 nM.
Table_1.
Dissociation constants and binding rate constants for CTX-b binding to lipid membranes containing GM1 measured by multichannel SPR imaging spectroscopy using metallic nanohole arrays.
| CTX-b Concentration (nM) |
kd (1/s) | ka (1/M·s) | KD (nM) |
|---|---|---|---|
| 6.25 | 6.46 ± 2.7 × 10−5 | 1.64 ± 0.6 × 105 | 0.39 ± 0.08 |
| 12.5 | 4.27 ± 1.0 × 10−5 | 3.26 ± 1.9 × 105 | 0.19 ± 0.15 |
| 25 | 4.10 ± 2.7 × 10−5 | 1.44 ± 1.2 × 105 | 0.32 ± 0.06 |
| 50 | 3.55 ± 3.5 × 10−5 | 3.29 ± 3.2 × 104 | 1.07 ± 0.03 |
| 100 | 5.13 ± 2.1 × 10−5 | 3.29 ± 0.9 × 104 | 1.53 ± 0.29 |
| 200 | 1.25 ± 0.4 × 10−4 | 6.01 ± 2.0 × 104 | 2.34 ± 1.35 |
| 400 | 9.40 ± 0.7 × 10−5 | 8.20 ± 1.1 × 104 | 1.17 ± 0.24 |
The limit of detection (LOD), commonly defined as a signal-to-noise ratio (S/N) of 3, is generally the smallest detectable concentration for a given instrument. However, when seeking to extract quantitative binding relationships, it is more advantageous to determine the practical limit of quantification (LOQ), which we estimate as S/N = 10. To calculate LOQ of CTX-b binding to GM1 in the SPR spectral imaging setup, the S/N for the maximum spectral shift is plotted vs. CTX-b concentration. (Fig. 6c) Based on the S/N versus analyte concentration plot, LOQ corresponding S/N=10 is computed to 8.4 nM. Thus, we did not attempt to extract binding kinetic constants for CTX-b concentrations below 6.25 nM.
Conclusions
We have demonstrated a 50-channel SPR imaging spectroscopy instrument based on large-area metallic nanohole arrays. The instrument combines an imaging spectrometer, a cooled CCD camera and a PDMS microfluidic chip to produce high throughput full-spectral imaging and accurate quantification of receptor-ligand binding kinetics. Multi-channel microfluidics are assembled on the silica-coated nanohole arrays. Highly uniform optical properties, a prerequisite for SPR spectral imaging, can be acquired through homogeneous, large-area nanohole arrays fabricated by the template-stripping method. Device sensitivity extracted from the multichannel SPR spectral image is highly consistent over all channels. The measured refractive index resolution is around 7.7×10−6 RIU level, which can be further improved via more frequent frame averaging. The parallel microfluidic channels partition the sensing area on the nanohole array into 50 isolated compartments, which is particularly valuable for SPR measurements using lipid or protein receptors incorporated in biomimetic supported lipid membranes. To confirm the utility of our system, various conditions for binding kinetics of cholera toxin b subunit (CTX-b) to ganglioside (GM1) receptors are measured in a single experiment from 50 channels. The dissociation constant (~sub-nM) acquired from nanohole-based multichannel SPR imaging spectroscopy matches very well with the previously reported results. The practical LOQ defining the smallest concentration from which kinetic constants can be reliably extracted is calculated as 8.4 nM, similar to the minimum concentration showing appreciable spectral shift in real-time binding kinetics. Since this general-purpose multi-channel imaging system allows real-time acquisition of full SPR spectra with high-throughput and individual functionalization of each channel with a wide range of receptors, it may benefit a wide range of bioanalytical research and high-throughput proteomics.
Materials and Methods
Nanohole Array Fabrication
Periodic nanohole array patterns were transferred on a Si chip (1 × 1 inch) having a 200 nm-thick thermally grown SiO2 layer by nanoimprint process. First, A NXR-1025 nanoimprint resist (Nanonex Corp., NJ USA) was spin-coated on a Si chip and cured at 150 °C for 1 min. (Heptadecafluoro-1,1,2,2-tetrahydrodecyl)trichlorosilane (Gelest Inc., Morrisville, PA USA) anti-sticking layer was formed on the nanoimprint Si stamp for 12 hours prior to the imprinting. Nanoimprint was performed with a pressure of 300 psi for 2 min at 130 °C. Then the imprinted nanohole pattern was transferred to SiO2 layer by reactive ion etching (RIE, STS model 320). After removing the residual nanoimprint resist by O2 plasma ashing, anisotropic Si etching was performed to make the periodic nanohole Si template by reactive ion etching (Plasma Therm SLR-770) with SiO2 hard mask. The Si template was cleaned in a 1:1 mixture of sulfuric acid and hydrogen peroxide for 10 min and the remaining SiO2 layer was removed by a buffered oxide etchant (BOE) solution. Before an e-beam evaporation to deposit 100 nm-thick Ag films onto the Si template, Si molds were dipped in a 1:1 mixture of sulfuric acid and hydrogen peroxide for 10 min and rinsed with DI water. Metal deposition rate was controlled at 0.2 Å/sec for the initial 20 nm and was increased to 1 Å/sec for the rest of the thickness. After metal deposition, the Ag film was covered by a UV-curable optical epoxy (NOA 61, Norland Products) with a glass slide. After curing the epoxy under UV light, the metallic film was peeled off from the template to reveal the patterned nanohole arrays.
Spectral Imaging Measurement Setup and Data Acquisition
To record multichannel SPR spectral imaging, a spectrometer (Newport MS257), an inverted microscope (Nikon Eclipse), and a deep-cooled CCD (Princeton, PIXIS) with custom LabVIEW™ software were used. A tungsten-halogen lamp illuminated nanohole arrays through a pinhole at normal incidence. The EOT signal from the multichannel microfluidics was collected through a microscopic objective (4×, NA=0.13) and entered to the spectrometer through 50 um width slit. The transmitted light was spectrally dispersed by a grating and two mirrors in a spectrometer. Before the images were transferred to LabVIEW™ software from the CCD, they were binned by 4 × 1 (image axis × spectral axis) to reduce the file size. For 50 channel refractive index sensing or biosensing, the CCD was rotated 90 degrees, assigning the long axis of the CCD to the image axis, and accommodating more channels. The images were taken with a 1 sec exposure and were averaged twice, giving 2.5 sec of temporal resolution. The pixel resolution was set to 0.623 nm / pixel by using a 121 lines / mm grating for testing the uniformity of optical properties. It was improved to 0.128 nm / pixel by using a 600 lines / mm grating for the refractive index sensing and biosensing. The multichannel spectra were extracted from the images by a custom MATLAB image-processing script. The peak position was calculated by fitting the spectra with a second-order polynomial function around the measured peak and calculating the centroid of the peak automatically.
Microfluidics
To make a master mold for microfluidics, SU-8 50 (Microchem) negative photoresist was spin coated on a 4-inch Si wafer and cured at 65 °C for 6 min and 95 °C for 20 min. Soft-cured SU-8 resist was exposed through a Cr-on-glass photomask using a Karl Suss MA6 mask aligner. Post exposure bake was done at 65 °C for 1 min and 95 °C for 5 min. The resist was developed in SU-8 developer. After applying an anti-sticking coating (heptadecafluoro-1,1,2,2-tetrahydrodecyl)trichlorosilane on the SU-8 mold, PDMS was cured at 60 °C overnight.
Refractive Index Sensing
Various index-calibrated water-glycerol solutions were made by adding glycerol to DI water and confirmed by a refractometer (Refracto 30PX, Mettler Toledo). Different index solutions (from n=1.333 to 1.533) were injected into 50 channel microfluidics sequentially by using a four-way valve (Cole-Parmer,USA) which selectively sends the solution to a flow cell and two syringe pumps (Harvard Apparatus). Real-time spectra shifts were measured with a temporal resolution of 1.2 sec (1 sec of exposure time and no averaging) and 2.5 sec (1 sec of exposure time and 2 averaging).
Vesicle Formation
For the majority of the experiments, vesicles were composed of Egg PC (L-alpha-phosphatidylcholine) (chicken eggs) and varying amounts of Ganglioside GM1 (ovine brain-ammonium salt) (0 – 4 mol %). For fluorescence imaging 1 mol % Rhodamine-DPPE (1,2-dipalmitoyl-sn-glycero-3-phosphoethanolamine-N-(lissamine Rhodamine B sulfonyl), ammonium salt) was incorporated. All lipids were obtained from Avanti Polar Lipids (Alabaster, AL USA) and used without further purification. Lipids in chloroform/methanol were mixed to desired amounts and then dried under vacuum in glass vials for at least 6 hours to remove all solvent. The dried lipid film was then hydrated with Tris buffer (100 mM NaCl, 10 mM Tris, 1 mM EDTA, pH = 8) and refrigerated overnight. Next the lipids were vortex-mixed for 10 seconds to disperse the vesicles into solution, followed by bath sonication for 10 min, and lastly by 15 passes through an extruder with a polycarbonate membrane filter having 200 nm diameter holes (Avanti Mini-Extruder), all performed at room temperature. Prior to injecting the vesicles on the chip, an equal amount of Tris buffer with calcium (100mM NaCl, 10mM Tris, 10mM CaCl2, pH = 8) was mixed with the vesicle solution to promote rupture on the chip surface. Cholera toxin b subunits (CTX-b) from recombinant origin, conjugated with Alexa Fluor-488 dye (5 mol dye/mol CTX-b), was purchased from Invitrogen and diluted in PBS.
SPR Kinetic Measurements
For kinetic assays for multichannel SPR spectral imaging, a 50-channel PDMS microfluidic device with 30 µm wide and 50 µm high channels was bonded permanently on the 15 nm-thick silica coated template-stripped nanohole arrays. The channel has an equivalent width of 150 µm since 5 channels are merged with one inlet and outlet. Before SLB formation, all channels were wet with a PBS solution for at least 10 min. Lipid membranes were formed by injecting egg PC vesicles containing GM1 molecules into the channels with a constant flow rate of 20 µl/min for 2 min and 5 µl/hr for the rest of time. Egg PC vesicles without GM1 were injected into one channel for a negative control. After incubation of vesicles for 1 hour to allow vesicle rupture on the silica surface, excess unruptured vesicles were washed by PBS for another 30 min with a flow rate of 20 µl/min. To reduce non-specific adsorption of CTX-b on the lipid membrane, the surface was blocked by injecting a 1 mg/ml BSA solution at 20 µl/min for 5 min and 1 µl/min for 25 min, followed by PBS washing for another 30 min. After 5 min baseline with the PBS solution, different concentrations of CTX-b solution were injected simultaneously for 5 min, followed by PBS washing for another 60 min. All solutions were introduced into channels continuously by using a four-way valve (Cole-Parmer, USA) and two syringe pumps (Harvard Apparatus). The real-time kinetics of CTX-b binding to GM1 receptor were obtained by monitoring the spectral shifts of the maximum transmission peak around 700 nm.
Acknowledgement
This work was supported by grants to S.-H.O. from the National Institutes of Health (R01 GM 095638), the National Science Foundation (NSF DBI 0964137), and the Minnesota Partnership Award for Biotechnology and Medical Genomics. S.-H.O. also acknowledges support from the NSF CAREER Award and the Office of Naval Research (ONR) Young Investigator Award (N00014-11-1-0645). S.H.L. was supported by a Samsung Fellowship. L.R.J. was supported by 3M Science and Technology Fellowship. Device fabrication was performed at the University of Minnesota Nanofabrication Center, which receives support from NSF through the National Nanotechnology Infrastructure Network.
Footnotes
Published as part of a themed issue dedicated to Emerging Investigators
References
- 1.Ramachandran N, Larson DN, Stark PRH, Hainsworth E, LaBaer J. FEBS J. 2005;272:5412–5425. doi: 10.1111/j.1742-4658.2005.04971.x. [DOI] [PubMed] [Google Scholar]
- 2.Barnes WL, Dereux A, Ebbesen TW. Nature. 2003;424:824–830. doi: 10.1038/nature01937. [DOI] [PubMed] [Google Scholar]
- 3.Liedberg B, Nylander C, Lundstrom I. Sens. Actuators. 1983;4:299–304. [Google Scholar]
- 4.Liedberg B, Nylander C, Lundstrom I. Biosens. Bioelectron. 1995;10:R1–R9. doi: 10.1016/0956-5663(95)96965-2. [DOI] [PubMed] [Google Scholar]
- 5.Mrksich M, Sigal GB, Whitesides GM. Langmuir. 1995;11:4383–4385. [Google Scholar]
- 6.Cooper MA. Nature Rev. Drug Discov. 2002;1:515–528. doi: 10.1038/nrd838. [DOI] [PubMed] [Google Scholar]
- 7.Homola J. Chem. Rev. 2008;108:462–493. doi: 10.1021/cr068107d. [DOI] [PubMed] [Google Scholar]
- 8.Rothenhausler B, Knoll W. Nature. 1988;332:615–617. [Google Scholar]
- 9.Yeatman E, Ash EA. Electron Lett. 1987;23:1091–1092. [Google Scholar]
- 10.Brockman JM, Nelson B, Corn RM. Annu. Rev. Phys. Chem. 2000;51:41–63. doi: 10.1146/annurev.physchem.51.1.41. [DOI] [PubMed] [Google Scholar]
- 11.Smith E, Corn RM. Appl. Spectrosc. 2003;57:320A–332A. doi: 10.1366/000370203322554446. [DOI] [PubMed] [Google Scholar]
- 12.Shumaker-Parry JS, Aebersold R, Campbell CT. Anal. Chem. 2004;76:2071–2082. doi: 10.1021/ac035159j. [DOI] [PubMed] [Google Scholar]
- 13.Luo Y, Yu F, Zare RN. Lab Chip. 2008;8:694–700. doi: 10.1039/b800606g. [DOI] [PubMed] [Google Scholar]
- 14.Ouellet E, Lausted C, Lin T, Yang CWT, Hood L, Lagally ET. Lab Chip. 2010;10:581–588. doi: 10.1039/b920589f. [DOI] [PubMed] [Google Scholar]
- 15.Ramachandran N, Hainsworth E, Bhullar B, Eisenstein S, Rosen B, Lau A, Walter J, LaBaer J. Science. 2004;305:86–90. doi: 10.1126/science.1097639. [DOI] [PubMed] [Google Scholar]
- 16.Natarajan S, Katsamba PS, Miles A, Eckman J, Papalia GA, Rich RL, Gale BK, Myszka DG. Anal. Biochem. 2008;373:141–146. doi: 10.1016/j.ab.2007.07.035. [DOI] [PubMed] [Google Scholar]
- 17.Rich RL, Cannon MJ, Jenkins J, Pandian P, Sundaram S, Magyar R, Brockman J, Lambert J, Myszka DG. Anal. Biochem. 2008;373:112–120. doi: 10.1016/j.ab.2007.08.017. [DOI] [PubMed] [Google Scholar]
- 18.Johnsson B, Löfås S, Lindquist G. Anal. Biochem. 1991;198:268–277. doi: 10.1016/0003-2697(91)90424-r. [DOI] [PubMed] [Google Scholar]
- 19.Ebbesen TW, Lezec HJ, Ghaemi HF, Thio T, Wolff P. Nature. 1998;391:667–669. [Google Scholar]
- 20.Brolo AG, Gordon R, Leathem B, Kavanagh KL. Langmuir. 2004;20:4813–4815. doi: 10.1021/la0493621. [DOI] [PubMed] [Google Scholar]
- 21.Stewart ME, Mack NH, Malyarchuk V, Soares JANT, Lee TW, Gray SK, Nuzzo RG, Rogers JA. Proc. Natl. Acad. Sci. USA. 2006;103:17143–17148. doi: 10.1073/pnas.0606216103. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Tetz K, Pang L, Fainman Y. Opt. Lett. 2006;31:1528–1530. doi: 10.1364/ol.31.001528. [DOI] [PubMed] [Google Scholar]
- 23.Lesuffleur A, Im H, Lindquist NC, Oh S-H. Appl. Phys. Lett. 2007;90 243110. [Google Scholar]
- 24.Yang J-C, Ji J, Hogle JM, Larson DN. Nano Lett. 2008;8:2718–2724. doi: 10.1021/nl801043t. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Lindquist NC, Lesuffleur A, Im H, Oh S-H. Lab Chip. 2009;9:382–387. doi: 10.1039/b816735d. [DOI] [PubMed] [Google Scholar]
- 26.Menezes JW, Ferreira J, Santos MJL, Cescato L, Brolo AG. Adv. Funct. Mater. 2010;20:3918–3924. [Google Scholar]
- 27.Eftekhari F, Escobedo C, Ferreira J, Duan X, Girotto EM, Brolo AG, Gordon R, Sinton D. Anal. Chem. 2009;81:4308–4311. doi: 10.1021/ac900221y. [DOI] [PubMed] [Google Scholar]
- 28.Jonsson MP, Dahlin AB, Feuz L, Petronis S, Höök F. Anal. Chem. 2010;82:2087–2094. doi: 10.1021/ac902925e. [DOI] [PubMed] [Google Scholar]
- 29.Dahlin AB, Zäch M, Rindzevicius T, Käll M, Sutherland DS, Höök F. J. Am. Chem. Soc. 2005;127:5043–5048. doi: 10.1021/ja043672o. [DOI] [PubMed] [Google Scholar]
- 30.Im H, Wittenberg NJ, Lesuffleur A, Lindquist NC, Oh S-H. Chem. Sci. 2010;1:688–696. doi: 10.1039/C0SC00365D. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Wittenberg NJ, Im H, Johnson TW, Xu X, Warrington AE, Rodriguez M, Oh S-H. ACS Nano. 2011;5:7555–7564. doi: 10.1021/nn202554t. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Nagpal P, Lindquist NC, Oh S-H, Norris DJ. Science. 2009;325:594–597. doi: 10.1126/science.1174655. [DOI] [PubMed] [Google Scholar]
- 33.Im H, Lee SH, Wittenberg NJ, Johnson TW, Lindquist NC, Nagpal P, Norris DJ, Oh S-H. ACS Nano. 2011;5:6244–6253. doi: 10.1021/nn202013v. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.Lindquist NC, Nagpal P, McPeak KM, Norris DJ, Oh S-H. Rep. Prog. Phys. 2012;75 doi: 10.1088/0034-4885/75/3/036501. 036501. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35.Yang T, Jung S, Mao H, Cremer PS. Anal. Chem. 2001;73:165–169. doi: 10.1021/ac000997o. [DOI] [PubMed] [Google Scholar]
- 36.Groves JT, Ulman N, Boxer SG. Science. 1997;275:651. doi: 10.1126/science.275.5300.651. [DOI] [PubMed] [Google Scholar]
- 37.Smith AM, Huser T, Parikh AN. J. Am. Chem. Soc. 2007;129:2422–2423. doi: 10.1021/ja065626p. [DOI] [PubMed] [Google Scholar]
- 38.Hovis JS, Boxer SG. Langmuir. 2001;17:3400–3405. [Google Scholar]
- 39.Im H, Lesuffleur A, Lindquist NC, Oh S-H. Anal. Chem. 2009;81:2854–2859. doi: 10.1021/ac802276x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40.Brolo AG, Arctander E, Gordon R, Leathem B, Kavanagh KL. Nano Lett. 2004;4:2015–2018. [Google Scholar]
- 41.Lindquist NC, Johnson TW, Norris DJ, Oh S-H. Nano Lett. 2011;11:3526–3530. doi: 10.1021/nl2005737. [DOI] [PubMed] [Google Scholar]
- 42.Ghaemi HF, Thio T, Grupp DE, Ebbesen TW, Lezec HJ. Phys. Rev. B. 1998;58:6779–6782. [Google Scholar]
- 43.Dahlin AB, Tegenfeldt JO, Höök F. Anal. Chem. 2006;78:4416–4423. doi: 10.1021/ac0601967. [DOI] [PubMed] [Google Scholar]
- 44.Im H, Sutherland JN, Maynard JA, Oh S-H. Anal. Chem. 2012;84:1941–1947. doi: 10.1021/ac300070t. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 45.Fukuta S, Magnani JL, Twiddy EM, Holmes RK, Ginsburg V. Infect. Immun. 1988;56:1748–1753. doi: 10.1128/iai.56.7.1748-1753.1988. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 46.Picking WL, Moon H, Wu H, Picking WD. Biochimica et Biophysica Acta (BBA)-Protein Structure and Molecular Enzymology. 1995;1247:65–73. doi: 10.1016/0167-4838(94)00208-x. [DOI] [PubMed] [Google Scholar]
- 47.MacKenzie CR, Hirama T, Lee KK, Altman E, Young NM. J. Biol. Chem. 1997;272:5533–5538. doi: 10.1074/jbc.272.9.5533. [DOI] [PubMed] [Google Scholar]
- 48.Cai X-E, Yang J. Biochemistry. 2003;42:4028–4034. doi: 10.1021/bi027016h. [DOI] [PubMed] [Google Scholar]
- 49.Schoen A, Freire E. Biochemistry. 1989;28:5019–5024. doi: 10.1021/bi00438a017. [DOI] [PubMed] [Google Scholar]
- 50.Merritt EA, Sarfaty S, Akker FVD, L'Hoir C, Martial JA, Hol WGJ. Protein Science. 1994;3:166–175. doi: 10.1002/pro.5560030202. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 51.Keller C, Kasemo B. Biophys. J. 1998;75:1397–1402. doi: 10.1016/S0006-3495(98)74057-3. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 52.Kuziemko GM, Stroh M, Stevens RC. Biochemistry. 1996;35:6375–6384. doi: 10.1021/bi952314i. [DOI] [PubMed] [Google Scholar]
- 53.Cooper MA, Williams DH. Anal. Biochem. 1999;276:36–47. doi: 10.1006/abio.1999.4333. [DOI] [PubMed] [Google Scholar]
- 54.Shi J, Yang T, Kataoka S, Zhang Y, Diaz AJ, Cremer PS. J. Am. Chem. Soc. 2007;129:5954–5961. doi: 10.1021/ja069375w. [DOI] [PMC free article] [PubMed] [Google Scholar]