

Abstract
Muscle fibres are multinucleated post-mitotic cells that can change dramatically in size during adulthood. It has been debated whether muscle fibre hypertrophy requires activation and fusion of muscle stem cells, the satellite cells. Myostatin (MSTN) is a negative regulator of skeletal muscle growth during development and in the adult, and MSTN inhibition is therefore a potential therapy for muscle wasting diseases, some of which are associated with a depletion of satellite cells. Conflicting results have been obtained in previous analyses of the role of MSTN on satellite cell quiescence. Here, we inhibited MSTN in adult mice with a soluble activin receptor type IIB and analysed the incorporation of new nuclei using 5′-bromo-2′-deoxyuridine (BrdU) labelling by isolating individual myofibres. We found that satellite cells are activated by MSTN inhibition. By varying the dose and time course for MSTN inhibition, however, we found that myofibre hypertrophy precedes the incorporation of new nuclei, and that the overall number of new nuclei is relatively low compared to the number of total myonuclei. These results reconcile some of the previous work obtained by other methods. In contrast with previous reports, we also found that Mstn null mice do not have increased satellite cell numbers during adulthood and are not resistant to sarcopaenia. Our results support a previously proposed model of hypertrophy in which hypertrophy can precede satellite cell activation. Studies of the metabolic and functional effects of postnatal MSTN inhibition are needed to determine the consequences of increasing the cytoplasm/myonuclear ratio after MSTN inhibition.
Key points
There is disagreement about whether muscle hypertrophy requires the activation and fusion of satellite cells, the quiescent muscle stem cells, to the multinucleated post-mitotic muscle fibre.
Although the growth factor myostatin is clearly a negative regulator of muscle size, previous studies regarding its role in maintaining satellite cell quiescence have yielded conflicting results.
We injected mice with a myostatin inhibitor and the DNA labelling agent bromodeoxyuridine to label proliferating cells and found that a small number of satellite cells are activated after the onset of hypertrophy.
We also found that myostatin null mice are not resistant to age-related muscle mass or satellite cell loss.
Our results suggest that myostatin inhibition in adult mice causes hypertrophy mainly by acting on myofibres rather than satellite cells, which results in an increase in the cytoplasmic volume to DNA ratio.
Introduction
Skeletal muscle is the largest organ in the body and can dramatically change in size even in adulthood. A decrease in skeletal muscle mass and strength occurs in a wide range of disorders such as the myopathies, disuse atrophy, age-related muscle loss (sarcopaenia), and cachexia. Additionally, recent human studies demonstrate an inverse association of skeletal muscle mass with insulin resistance (Atlantis et al. 2009; Srikanthan & Karlamangla, 2011). The maintenance of normal muscle mass and function would therefore be of great clinical utility to treat or prevent many different disorders.
A muscle fibre is a single post-mitotic cell formed by fusion of differentiated myoblasts containing hundreds of nuclei called myonuclei. Skeletal muscle also contains stem cells which are called satellite cells because of their location adjacent to the muscle myofibre plasma membrane beneath the basal lamina (Mauro, 1961). Stimuli such as stretch or injury activate the normally quiescent satellite cells to proliferate, differentiate, and then fuse to a myofibre to cause hypertrophy or fuse to each other to generate a new myofibre (Shadrach & Wagers, 2011). Satellite cells are required for postnatal growth, repair and regeneration of muscle. Satellite cells are often depleted in myopathies after repeated rounds of activation and decline in number and function with age (Jejurikar & Kuzon, 2003; Shadrach & Wagers, 2011). Therefore, increasing the regenerative potential of satellite cells is thought to be a promising strategy for improving muscle function in patients with muscle degenerative diseases or sarcopaenia. An unresolved question, however, is whether satellite cell activation is always required for hypertrophy of non-damaged myofibres in otherwise normal adults. Satellite cell activation has usually accompanied hypertrophy, but there are some exceptions (McCarthy & Esser, 2007; O’Connor & Pavlath, 2007; O’Connor et al. 2007; Blaauw et al. 2009; Sartori et al. 2009).
Myostatin (MSTN), a member of the transforming growth factor β (TGFβ) superfamily of secreted growth factors, is a regulator of skeletal muscle mass that is expressed predominantly in skeletal muscle (Lee, 2010). Loss of function mutations in the Mstn gene cause an increase in skeletal muscle mass in mice, cattle, dogs, sheep and humans (Grobet et al. 1997; Kambadur et al. 1997; McPherron et al. 1997; McPherron & Lee, 1997; Schuelke et al. 2004; Clop et al. 2006). MSTN is synthesized from a longer pro-protein and is proteolytically cleaved to generate an amino terminal pro-peptide and a mature, receptor-binding region. The MSTN pro-peptide also functions as a MSTN inhibitor by non-covalently binding the mature MSTN after processing to prevent receptor binding (Lee & McPherron, 2001; Thies et al. 2001). This latent complex is activated by further cleavage of the pro-peptide allowing the mature MSTN to bind to the receptor (Wolfman et al. 2003; Lee, 2008). Of the receptors for TGFβ family ligands, the highest affinity receptor for MSTN is the activin receptor type IIB (ACVR2B, also known as ACTRIIB) (Lee & McPherron, 2001; Rebbapragada et al. 2003). Like MSTN, activin is also a ligand for this receptor and inhibits muscle growth (Amthor et al. 2002, 2006; Lee et al. 2010).
The increased muscle mass in Mstn KO mice is due to an increase in both the number and size of myofibres (McPherron et al. 1997; Elashry et al. 2009; McPherron et al. 2009). In mice, the adult number of muscle myofibres is established soon after birth (Ontell & Kozeka, 1984), and Mstn KO muscles have an increase in the number of progenitor cells during the developmental period when myofibres are normally forming (Matsakas et al. 2010; K. J. Savage & A. C. McPherron, unpublished results). MSTN regulates muscle mass during adulthood as well. Generalized increased muscling by MSTN inhibition in adult mice has been achieved by several methods that prevent receptor binding including injection with a neutralizing monoclonal antibody, a mutant pro-peptide that cannot be activated by proteolysis, or a soluble ACVR2B (Lee, 2010). These methods to antagonize MSTN in adulthood, however, result in myofibre hypertrophy rather than hyperplasia, which demonstrates that MSTN regulates hyperplasia during development.
One plausible hypothesis for how MSTN regulates muscle size is by maintaining satellite cell quiescence. Several laboratories have therefore investigated the role of MSTN in the regulation of satellite cell proliferation and differentiation. In support of this hypothesis, one report found that Mstn KO mice have more satellite cells than WT mice (McCroskery et al. 2003). Consistent with this result, satellite cells from Mstn KO mice have been shown to have an increased rate of proliferation in vitro compared to WT mice (McCroskery et al. 2003; Wagner et al. 2005). These results have been challenged, however, by another analysis of the Mstn KO mice which found that KO muscles had a slightly reduced number of satellite cells and no difference in the rate of satellite cell proliferation compared to WT mice (Amthor et al. 2009).
Postnatal treatments with MSTN inhibitors that cause myofibre hypertrophy have also yielded conflicting results. Soluble ACVR2B treatment was shown to increase satellite cell numbers 1 week after treatment (Zhou et al. 2010). Consistent with this result, the hypertrophic effect of follistatin, an antagonist of MSTN and activin, is partially prevented by treating mice with γ-irradiation to inhibit cell proliferation (Gilson et al. 2009). Together, these results suggest that satellite cells are activated by MSTN and/or activin inhibition and are required for the full hypertrophic response to MSTN inhibitors. However, radiation is not specific for satellite cells and has been shown to affect translation (Lu et al. 2006), although there was no decrease in irradiated muscle mass in controls (Gilson et al. 2009). In contrast, an AAV vector encoding a secreted MSTN pro-peptide did not affect satellite cell number in vivo after 1 month or 8 weeks of treatment (Amthor et al. 2009; Foster et al. 2009; Matsakas et al. 2009).
To attempt to reconcile these contradictory results, we employed bromo-deoxyuridine (BrdU) labelling and single myofibre analysis in adult mice treated pharmacologically with a soluble ACVR2B:Fc fusion protein to block MSTN and activin signalling. We varied the time course and dose of ACVR2B:Fc to attempt to separate satellite cell activation from hypertrophy. This approach allowed us to detect BrdU labelling in the entire myofibre and associated satellite cells that have been isolated from non-muscle cells such as fibroblasts or endothelial cells. Satellite cells that were activated but have not yet fused to a myofibre will be detected by this method as well as myonuclei derived from satellite cells that were activated and fused to the myofibre during the labelling period. Because myonuclei are post-mitotic (Stockdale & Holtzer, 1961; Moss & Leblond, 1971; Schiaffino et al. 1972), any BrdU incorporation into myofibres must be due to satellite cell activation and fusion (Shadrach & Wagers, 2011). We also analysed satellite cell number and muscle mass in Mstn KO mice during ageing to see if prior conflicting results were due to differences in the ageing process between genotypes.
Methods
Ethical approval
All animal procedures were approved by Animal Care and Use Committee of the NIDDK, NIH in accordance with NIH guidelines.
Animals
Male Mstn WT and KO mice (McPherron et al. 1997) produced from matings of heterozygotes on a C57BL/6NCr genetic background were genotyped as described (Manceau et al. 2008). For soluble receptor injection experiments, female C57BL/6NCr mice were purchased from the National Cancer Institute Animal Production Program at age 5 weeks and given 1 week of acclimation before the start of experiments. Animals were kept on a 12 h light–dark schedule with free access to food and water. Mice were killed by CO2 inhalation and muscles were dissected, weighed and prepared as described below.
ACVR2B:Fc preparation
A Chinese hamster ovary cell line stably expressing the extracellular domain of mouse ACVR2B fused to a mouse Fc domain for stability were a gift from Se-Jin Lee. ACVR2B:Fc was purified from conditioned medium using protein A/G sepharose (Pierce) and dialysed against phosphate-buffered saline (PBS).
Injections
At age 6 weeks, female C57BL/6NCr mice were randomly assigned to control or treatment groups and i.p. injected daily with 50 mg (kg body weight)−1 sterile BrdU (Sigma) dissolved in PBS. The first day of BrdU injections was considered day 0. No BrdU injections were given on the day of killing.
For 14 days of ACVR2B:Fc exposure, mice were also injected i.p. with 10 mg (kg body weight)−1 ACVR2B:Fc or equivalent volume of PBS on days 0 and 7.
For the dose–response experiment, mice were injected i.p. with ACVR2B:Fc at 5, 10, or 20 mg (kg body weight)−1 or equivalent volume of PBS once on day 0 and killed on day 7.
For a 48 h labelling period, 10 mg (kg body weight)−1 ACVR2B:Fc or equivalent volume of PBS was injected i.p and mice were killed 48 h later.
For the time course experiment, 10 mg (kg body weight)−1 ACVR2B:Fc or equivalent volume of PBS was injected i.p. starting on day 0 (6 days exposure), day 2 (4 days exposure), or day 4 (2 days exposure). PBS was injected in all mice on days when ACVR2B:Fc was not injected so that every mouse received an injection on days 0, 2 and 4. Mice were killed on day 6.
Myofibre isolation
The extensor digitorum longus (EDL) was carefully dissected so that it was not damaged or stretched and digested with 0.2% weight per volume type I collagenase (Sigma) in Dulbecco's modified Eagle's medium (DMEM) at 37°C for 2 h. After washing with warm DMEM twice, single myofibres were separated from the rest of the muscle by trituration using wide mouth pipets. Isolated myofibres were then immediately fixed in 4% paraformaldehyde in PBS for 1 h at room temperature, collected by gravity sedimentation, and washed three times with PBS.
Histology
The tibialis anterior (TA) and EDL muscles were partially embedded together in 10% gum tragacanth (Sigma), mounted onto cork slices, frozen in liquid nitrogen-cooled 2-methylbutane, and stored at −80°C. Twelve-micrometre serial transverse sections were cut with a cryostat and stored at −20oC. Sections from the midbelly were used for analysis of myofibre cross-sectional area.
Immunofluorescence
For anti-BrdU immunostaining, myofibres were permeabilised with 0.5% Triton X-100 for 10 min at room temperature, and treated with DNase I (Roche) in DMEM at 37°C for 30 min. After two washes in PBS, myofibres were blocked with 10% goat serum/1% bovine serum albumin in PBS. Mouse monoclonal anti-BrdU IgG1 (Sigma) was diluted 1:500 in blocking solution and incubated overnight at 4°C. Myofibres were incubated with the secondary antibody, anti-mouse IgG1 (Molecular Probes), diluted 1:200 for 1 h at room temperature. Individual myofibres were then transferred to slides, aligned using forceps, and mounted with mounting medium containing 4′,6-diamidino-2-phenylindole (DAPI) (Vector Laboratories, Burlingame, CA, USA). The same procedure was used to detect satellite cells except DNAse I treatment was omitted, and the primary antibody was conditioned medium from a hybridoma secreting an anti-Pax7 monoclonoal antibody (Developmental Studies Hybridoma Bank; University of Iowa, Iowa City, IA, USA) diluted 1:5 in blocking solution.
Cross sections of TA and EDL muscles were incubated with polyclonal rabbit anti-laminin (Sigma) diluted 1:1000 and Pax7-conditioned medium diluted 1:10 in blocking solution overnight at 4°C, and detected with anti-rabbit IgG and anti-mouse IgG1 (Molecular Probes) secondary antibodies. Sections were mounted as above.
For all Pax7 or BrdU analysis, only staining that was also positive for DAPI was considered to be a BrdU+ or Pax7+ nucleus.
Imaging and analysis
Three-dimensional stacked pictures were taken on a 5 Live laser scanning confocal microscope (Zeiss) to count DAPI-positive nuclei. BrdU+ nuclei and Pax7+ nuclei were counted from confocal images or from images taken using an upright 80i fluorescence microscope (Nikon).
For analysing satellite cell and central nuclei from cross sections of 2-year-old muscles, the number of satellite cells and myofibres with centrally located myonuclei were counted manually on Pax7, laminin and DAPI-stained slides using an upright 80i fluorescence microscope (Nikon).
Myofibre cross-sectional area (CSA) was calculated from laminin-stained slides from randomly chosen fields using NIS Elements software (Nikon). Myofibres were traced automatically using a specially designed macro for this software for the ACVR2B:Fc dose experiment. The traces were visually examined and those that were not myofibres were removed from the data set. For the time course experiment, myofibres were traced by hand.
Statistical analysis
Muscle weights obtained from ACVR2B:Fc injection experiments were normalized to starting body weight. For myofibre analysis, the average value for each mouse was used to determine the mean, standard deviation and standard error of the mean. Data with unequal variance between groups were log transformed to restore equal variance. Data were analysed by Student's t test for comparison between two groups or by single or two factor analysis of variance as appropriate with Bonferroni correction for multiple comparisons (SPSS v. 19). P < 0.05 was considered significant.
Results
Satellite cell activation after ACVR2B:Fc treatment
To ascertain whether satellite cells were activated after MSTN inhibition in normal mice, we treated mice with weekly i.p. injections of a soluble ACVR2B:Fc and daily i.p. injections of BrdU. A 2 week labelling period was chosen because the maximum muscle mass obtained by soluble receptor treatment is known to be reached within this time frame (Lee et al. 2005; Zhou et al. 2010).
Eight muscles, including the extensor digitorum longus (EDL), were increased significantly in mass in response to ACVR2B:Fc by 19–47% compared to control muscles (Fig. 1A). The number of BrdU+ nuclei in control EDL myofibres was very low at ∼1 per myofibre reflecting the low rate of satellite cell turnover (Fig. 1C and D). The number of BrdU+ nuclei increased significantly in ACVR2B:Fc-treated myofibres by ∼8.5 per myofibre (Fig. 1C and D). The total number of myonuclei per myofibre was similarly increased by ∼10 validating our BrdU labelling technique (Fig. 1B; 257.8 ± 11.7, control; 268.4 ± 8.5, ACVR2B:Fc). This increase in myonuclei, however, was not statistically significant given that the number of myonuclei per myofibre is substantially greater.
Figure 1. Satellite cells are activated in vivo by ACVR2B:Fc treatment.
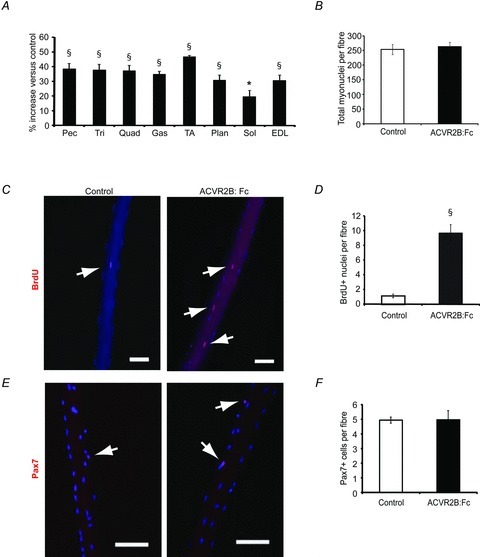
EDL fibre analysis from mice injected with ACVR2B:Fc on days 0 and 7 and analysed on day 14. A, percentage change in muscle weights of ACVR2B:Fc-injected mice expressed relative to vehicle control (n = 7 mice per group). B, number of total myonuclei per myofibre counted from compressed confocal images of DAPI-stained myofibres (39–65 fibres per group, n = 3–4 mice per group). C, immunofluorescence images of BrdU+ nuclei (red) and myonuclei (blue) on single EDL fibres. D, the average number of BrdU+ nuclei per fibre (507–508 fibres per group, n = 7 mice per group). E, compressed confocal images of isolated myofibres taken from EDL muscles showing satellite cells (Pax7, red) and nuclei (DAPI, blue). F, number of Pax7+ cells per myofibre (39–65 fibres per group, n = 3–4 mice per group). Arrows point to BrdU+ or Pax7+ nuclei in C and E, respectively. Pec, pectoralis; Tri, triceps; Quad, quadriceps; Gas, gastrocnemius; TA, tibialis anterior; Plan, plantaris; Sol, soleus; EDL, extensor digitorum longus. *P < 0.05, §P < 0.001 compared to control. Values are means ± SEM. Scale bar = 100 μm.
To determine whether the satellite cell pool is increased by ACVR2B:Fc treatment, we measured the number of satellite cells per myofibre in the EDL from the same animals. The number of nuclei expressing Pax7, a paired box transcription factor that labels the vast majority of satellite cells, was not increased by 2 weeks of ACVR2B:Fc treatment (Fig. 1E and F). This indicates that the BrdU+ nuclei were myonuclei rather than satellite cells.
Dose response effects of ACVR2B:Fc treatment
We treated normal adult mice with different doses of ACVR2B:Fc to determine whether we could separate satellite cell activation from hypertrophy. One week after a single dose of ACVR2B:Fc, muscle weights tended to be increased in all treatment groups (Fig. 2A). Essentially no increase in BrdU labelling in EDL myofibres was seen at a dose of 5 mg (kg body weight)−1 compared to control myofibres, although EDL mass increased non-significantly by 11% (Fig. 2B). At the previously used dose of 10 mg kg−1, there were ∼2 more BrdU-labelled nuclei per myofibre than in control myofibres, while EDL mass increased non-significantly by 15% (Fig. 2B; P = 0.097). At the highest dose of 20 mg kg−1, there were 6 times the number of BrdU-labelled nuclei than in control myofibres while EDL mass increased by 19% (Fig. 2B). This corresponds to an increase of ∼4 more BrdU+ nuclei in treated myofibres on average because control myofibres had on average fewer than one labelled nucleus each. Put another way, we calculate that there were fewer than 10 labelled nuclei in 86% of myofibres at this 20 mg kg−1 dose.
Figure 2. Dose response after a single injection of ACVR2B:Fc.
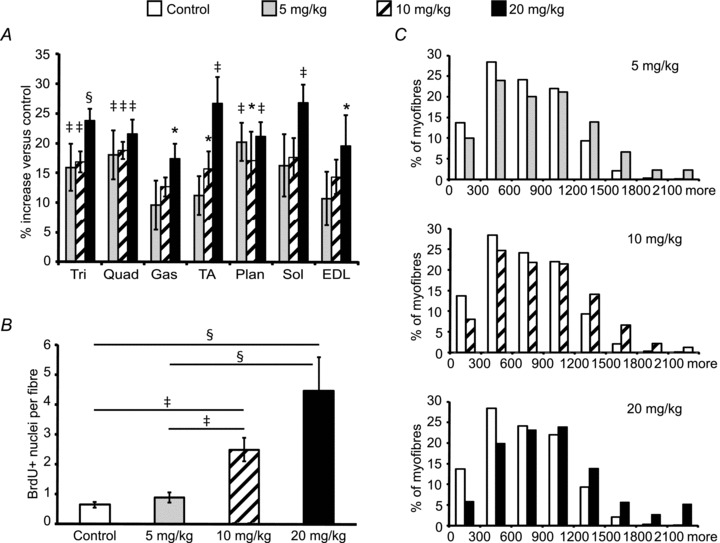
EDL myofibre analysis from mice injected with ACVR2B:Fc at 0, 5, 10 or 20 mg (kg body weight)−1 on day 0 and analysed on day 7. A, percentage change in muscle weights expressed relative to vehicle control (n = 5 mice per group). B, the average number of BrdU+ nuclei per fibre from isolated myofibres (365–498 fibres per group, n = 5 mice per group). C, histograms of EDL fibre cross-sectional area of ACVR2B:Fc injection compared to control at increasing doses (>1000 fibres per cross-section, n = 3–5 mice per group). *P < 0.05, ‡P < 0.01, §P < 0.001 compared to control (A) or as marked (B). Values in A and B are means ± SEM.
To make sure that the increases in muscle mass were due to hypertrophy of myofibres and not proliferation or infiltration of other cell types, we analysed myofibre CSA to compare BrdU labelling with the degree of myofibre hypertrophy. Histogram plots of myofibre CSA were shifted to higher values in all of the ACVR2B:Fc treatment groups including that of the lowest dose that had no increase in BrdU-labelled nuclei (Fig. 2C). The increase in average CSA from treated mice shows that ACVR2B:Fc can induce at least some hypertrophy without activation of satellite cells (739.0 ± 40.6 μm2, control; 886.1 ± 44.4 μm2, 5 mg kg−1; 878.9 ± 38.9 μm2, 10 mg kg−1; 1018.7 ± 171 μm2, 20 mg kg−1).
Time course of ACVR2B:Fc-induced satellite cell activation
To determine when DNA synthesis began relative to myofibre hypertrophy, we gave mice a single injection of 10 mg kg−1 ACVR2B:Fc and measured muscle mass and BrdU incorporation after 48 h. Muscle masses tended to be increased in this short time period (Fig. 3A). ACVR2B:Fc significantly increased EDL mass by 11% (P = 0.045), but there was no increase in BrdU+ nuclei in EDL myofibres from treated mice compared to control mice (Fig. 3A and B).
Figure 3. Muscle hypertrophy occurs without satellite cell activation.
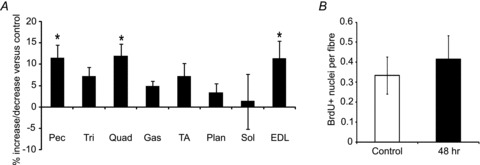
EDL fibre analysis from mice 48 h after injection with ACVR2B:Fc at 10 mg (kg body weight)−1. A, percentage change of muscle weights expressed relative to vehicle control (n = 5 mice per group). B, the average number of BrdU+ nuclei per fibre from isolated fibres (475 fibres, control; 520 fibres, ACVR2B:Fc, n = 5 mice per group). *P < 0.05. Values are means ± SEM.
To see when satellite cells began to divide, we conducted a further time course experiment by injecting the soluble receptor 2, 4, or 6 days prior to analysis (Fig. 4A). The EDL muscle mass seemed to be increased in ACVR2B:Fc-treated animals compared to control EDL although it was only statistically significant with 6 days of treatment in this experiment (Fig. 4B). An increase in myofibre average CSA, however, was detectable after 2, 4 or 6 days of ACVR2B:Fc treatment (Fig. 4D; 890 ± 27 μm2, control; 988 ± 20 μm2, 2 days ACVR2B:Fc; 973 ± 38 μm2, 4 days ACVR2B:Fc; 1135 ± 114 μm2, 6 days ACVR2B:Fc). BrdU labelling was increased by ∼0.7 nuclei per myofibre in ACVR2B:Fc-treated EDL myofibres by 4 days after injection compared to controls, but this increase was not statistically significant (Fig. 4C). By 6 days after treatment, BrdU incorporation was significantly increased compared to control animals to 3.7 times control (Fig. 4C). Given that control labelling was less than one nucleus per myofibre, this corresponds to an increase of only ∼1.7 BrdU+ nuclei per myofibre. These results suggest that satellite cell activation occurs after the onset of hypertrophy caused by ACVR2B:Fc.
Figure 4. Time course of BrdU incorporation after ACVR2B:Fc treatment.
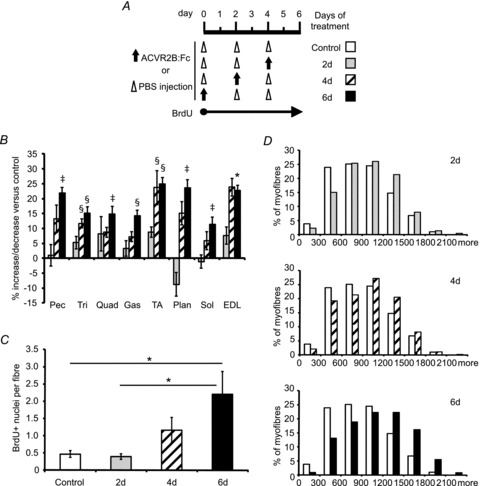
EDL fibre analysis from mice 2, 4 or 6 days after injection with ACVR2B:Fc at 10 mg (kg body weight)−1 or PBS control. A, treatment time course of vehicle, ACVR2B:Fc, and BrdU injections. Open triangles, vehicle injection; filled arrows, ACVR2B:Fc injection. B, percentage change of muscle weights expressed relative to vehicle control (n = 8–9 mice per group). C, the average number of BrdU+ nuclei per fibre from isolated fibres (697–906 fibres per group, n = 9–11 mice per group). D, histograms of EDL myofibre cross-sectional area of ACVR2B:Fc injection compared to control (∼300 fibres per cross-section, n = 3–4 mice per group). D, days of exposure to treatment. *P < 0.05, ‡P < 0.01, §P < 0.001 compared to control (A) or as marked (B). Values in B and C are means ± SEM.
Satellite cell number in Mstn KO mice
To try to reconcile the conflicting results of satellite cell frequency previously obtained in the Mstn KO mice, we counted the number of satellite cells on individual myofibres isolated from the EDL muscle in WT and KO mice. If Pax7+ cells in adult KO mice change in frequency at a rate different from WT mice, a difference in age of the animals analysed may account for the previous conflicting results. We therefore counted the number of Pax7+ cells on EDL myofibres isolated from 2- and 5-month-old mice. Both WT and KO EDL satellite cells declined significantly in number between 2 months and 5 months of age (Fig. 5A). The Pax7+ cell number per myofibre was identical between WT and KO EDL at both ages demonstrating that satellite cells are depleted at the same rate between 2 and 5 months (Fig. 5A). The average total number of myonuclei per myofibre was slightly but non-significantly lower in KO compared to WT myofibres at 2 months of age (Fig. 5B).
Figure 5. Comparision of satellite cell and total myonuclei in isolated EDL myofibres from male WT and Mstn KO mice.
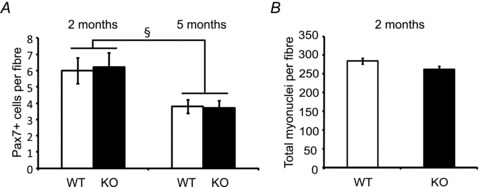
A, the average number of Pax7+ cells per fibre at age 2 months (left) and 5 months (right) (46–58 fibres per group, n = 3 mice per group, 2 month; 642–647 fibres per group, n = 6 mice per group, 5 months old). B, the average number of total myonuclei per fibre from isolated fibres at age 2 months (46–58 fibres per group, n = 3 mice per group). §P < 0.001 compared to the same genotype. Values are means ± SEM.
In sarcopaenia, both satellite cell number and muscle mass are reduced, so we examined the muscle phenotype in aged WT and Mstn KO mice to determine whether a difference in satellite cell depletion or muscle wasting would emerge. The mass of eight different muscles declined in both WT and KO mice by 24 months of age compared to younger ages (Supplemental Table 1). Overall, this decline seemed to be proportional between WT and KO mice because most KO muscles had roughly similar increases in mass compared to age-matched WT muscles at ages 6, 12, 18 and 24 months (Fig. 6A).
Figure 6. The effects of ageing on Mstn KO muscle.
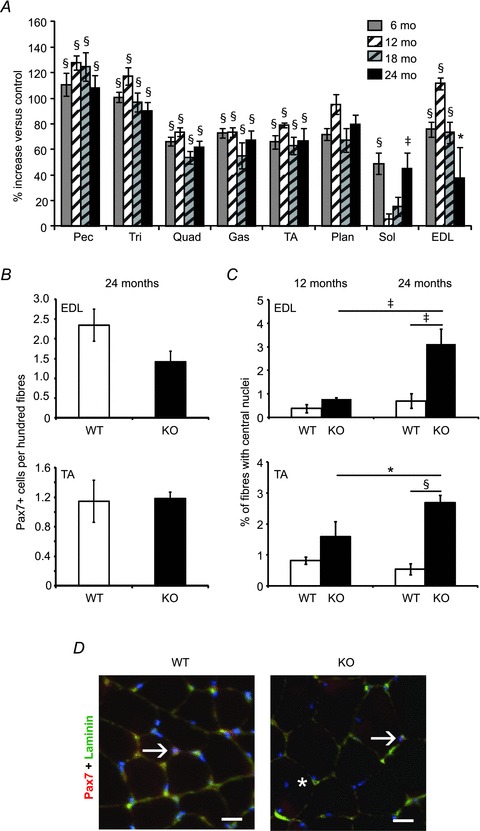
A, percentage change in Mstn KO muscle weights at 6, 12, 18, and 24 months of age expressed relative to age-matched WT muscles (n = 5–10 per group). B, the average number of Pax7+ cells per 100 fibres from cross-sections of EDL (top) and TA (bottom) muscles at age 24 months (>700 fibres per mouse, 5 mice per group). C, percentage of fibres with central nuclei from the EDL (top) or TA (bottom) muscles at age 12 months (left) or 24 months (right) (an average of >700 fibres per mouse, n = 4–5 mice per group). D, cross sections of fibres with Pax7+ satellite cells or central nuclei from the EDL muscles at age 24 months. Arrows point to Pax7+, DAPI+ nuclei and the asterisk marks a fibre with a central nucleus. Pax7, red; DAPI-stained nuclei, blue; laminin, green. *P < 0.05, ‡P < 0.01, §P < 0.001 compared to control (A) or as marked (B). Values in A–C are means ± SEM. Scale bar = 20 μm.
We next examined the frequency of satellite cells in aged mice by counting Pax7+ cells in cross sections of the EDL and TA muscles. The frequency of myofibres with associated satellite cells was non-significantly reduced in Mstn KO compared to WT EDL muscle from 24-month-old mice (Fig. 6B and D). In the TA muscle, the Pax7+ cell frequency was similar between WT and KO myofibres (Fig. 6B).
During this analysis we noticed an increase in the number of centrally located myonuclei in Mstn KO EDL and TA muscles which are evidence of prior satellite cell activation. At 12 months of age, ∼0.4% and 0.8% of myofibres had central nuclei in the WT and KO EDL, respectively (Fig. 6C). By 24 months, the number of myofibres with central nuclei doubled in WT EDL muscle. In the KO EDL, this number was increased 348% between 12 and 24 months (Fig. 6C and D). There was also an increase in central nuclei in the TA muscle with ageing in the KO (Fig. 6C).
Discussion
Whether or not satellite cells are activated by potential therapeutic means to increase muscle mass is an important question because satellite cells are depleted in some myopathies (Jejurikar & Kuzon, 2003). Additionally, it has been hypothesized that if there is an increase in protein synthesis for muscle contractile proteins without a corresponding increase in other proteins, there may be metabolic consequences (Edgerton & Roy, 1991). For example, if there was not a proportional increase in synthesis of nuclear-encoded mitochondrial proteins after hypertrophy, mitochondrial function could be compromised leading to reduced lipid oxidation or increased susceptibility to fatigue.
We have shown here that satellite cells are indeed activated by blocking MSTN and activin signalling in adult muscle. The technique of isolating myofibres to measure BrdU incorporation allowed us to detect rare labelled nuclei in or on myotubes that would be missed by analysing cross sections of muscle tissue while avoiding other non-muscle cell types. One limitation of our method is that we were not able to clearly label BrdU and Pax7 simultaneously, so we cannot tell whether BrdU-labelled nuclei are satellite cells or myonuclei. However, at 14 days after ACVR2B:Fc treatment, there was no increase in Pax7+ nuclei although there were clearly more BrdU+ nuclei. This suggests that BrdU+ nuclei are myonuclei that originated as activated satellite cells that have already fused to the myofibre. Regardless, BrdU labelling of nuclei in or on myofibres by this method is evidence of satellite cell activation because there is no evidence for myonuclear division in mammalian myofibres (Stockdale & Holtzer, 1961; Moss & Leblond, 1971; Schiaffino et al. 1972). While DAPI staining for nuclei is reliable, the interpretation of our results depends on the sensitivity of our BrdU detection. Because the number of total nuclei increased by the same amount as the number of BrdU+ labelled nuclei (Fig. 1), we believe we were able to detect nearly all satellite cell divisions that occurred during the labelling period.
Another limitation is that we did not simultaneously determine myofibre CSA and analyse the same myofibre for the presence of BrdU+ nuclei. Mounting isolated myofibres on slides seemed to distort the shape of the myofibre so we do not believe diameter measurements would be accurate. However, we found an increase in CSA with 5 mg kg−1 ACVR2B:Fc after 7 days and with 10 mg kg−1 ACVR2B:Fc after just 2 days without an increase in BrdU+ nuclei compared to controls (Figs 2 and 4). These results are evidence that CSA can increase after ACVR2B:Fc without any incorporation of satellite cells. In addition, histogram plots of the myofibre CSA of treated mice shows that the entire curves are shifted to greater CSA values. This suggests that the hypertrophy is not due to hypertrophy of a small subset of fibres that might have incorporated new DNA. A third limitation is that our analysis was mainly performed on the EDL muscle because of the ease of isolating fibres from this small muscle. It is possible that other muscles may respond differently depending on such characteristics as contraction frequency, force generation and fibre type composition.
Hypertrophy induced by MSTN inhibition
Although we found clear evidence that satellite cells are activated after MSTN and activin inhibition in adult mice, the number of nuclei that became incorporated into myofibres in all of our experiments was low. For example, with two injections of the soluble receptor over 14 days, a time period when maximal hypertrophy is normally achieved (Lee et al. 2005; Zhou et al. 2010), EDL muscle mass was increased by ∼30%. The number additional BrdU-labelled nuclei, however, was on average only ∼3.4% of the number of total myonuclei in control myofibres. Moreover, after 4 days of ACVR2B:Fc treatment, there was an increase of ∼25% in EDL mass but an increase of less than one labelled nucleus per myofibre compared to control. We estimate that this represents an increase of less than 0.4% of total myonuclei in each myofibre. By comparison, stretch-induced hypertrophy of quail anterior latissimus dorsi muscle caused a 15% increase in mass and an ∼32-fold increase in [3H]thymidine-labelled nuclei on or within myofibres compared to controls within only 1 day (Winchester et al. 1991). Thus, our results suggest that while activation of satellite cells occurs after ACVR2B:Fc treatment, it mainly occurs after the onset of hypertrophy.
In this context, although our results are consistent with recent reports that hypertrophy caused by MSTN inhibition does result in some satellite cell activation (Gilson et al. 2009), satellite cell fusion to the myofibre cannot be the precipitating event that causes the increase in muscle mass. The low number of BrdU-labelled nuclei in myofibres from mice treated with ACVR2B:Fc is likely to explain why an increase in satellite cell numbers using various markers was not detectable after treatment with the soluble receptor (here) or with the MSTN pro-peptide for up to 8 weeks (Amthor et al. 2009; Foster et al. 2009; Matsakas et al. 2009) to induce hypertrophy. In contrast, a massive (4935%) increase in Pax7 immunostaining in cross sections was found after soluble receptor treatment at a higher dose than we used here, although many of these cells were not positive for BrdU (Zhou et al. 2010). The great differences in our results and those of Zhou et al. (2010) are due either to differences in detection technique or in inhibitor dose. Regardless, our results using 5 or 10 mg kg−1 ACVR2B:Fc demonstrate that MSTN inhibition can cause hypertrophy with little to no satellite cell activation.
Recent work suggests mechanisms that are regulated by MSTN signalling to control muscle growth other than satellite cell quiescence. The Akt/mammalian target of rapamycin (mTOR)/p70S6K protein synthesis pathway is inhibited by MSTN in muscle and in differentiating human myoblasts (Amirouche et al. 2009; Trendelenburg et al. 2009). This pathway is upregulated in Mstn KO muscle and in myotubes formed from Mstn null satellite cells in vitro (Lipina et al. 2010; Rodriguez et al. 2011). These effects on protein synthesis are evident within 1 to 2 days in vitro (Trendelenburg et al. 2009; Rodriguez et al. 2011). Furthermore, inhibition of mTOR partially reduces the hypertrophy induced by transfection of a dominant negative kinase dead ACVR2B into myofibres (Sartori et al. 2009). A direct interaction between mediators of the MSTN signalling pathway and protein synthesis pathways, however, has not yet been shown. A recent study of the effects of MSTN on human myoblasts in vitro suggests another potential mechanism. MSTN inhibits the myogenic regulatory factors MyoD and myogenin and the overall muscle differentiation programme not only in myoblasts but also in differentiated myotubes (Trendelenburg et al. 2009). This process is distinct from that induced during atrophy when ubiquitin ligases MurF1 and MAFbx are upregulated (Glass, 2010). Regardless of the exact mechanisms, the majority of data now suggest that MSTN inhibits myofibre growth in adults by signalling directly on the myofibres rather than satellite cells.
Satellite cells and hypertrophy in Mstn KO mice
Our analysis of the Mstn KO phenotype also does not support the conclusion that MSTN regulates satellite cell quiescence in adults. If the function of MSTN is to maintain satellite cell quiescence, one might expect that KO mice should have an increase in satellite cell number. In contrast to other reports describing a higher (McCroskery et al. 2003) or slightly lower (Amthor et al. 2009) satellite cell number in KO mice, we did not find any significant difference compared with WT mice at age 2 or 5 months in the EDL or at age 24 months in the TA. In contrast to the adult Mstn KO, there is a clear increase in Pax7+ progenitor cells during development in KO muscle (Matsakas et al. 2010; K. J. Savage & A. C. McPherron, unpublished results), which might be due to the direct loss of MSTN signalling on embryonic Pax7+ cells. Consistent with this age difference in efficacy of MSTN, the expression of the activin receptors on satellite cells is much lower than that of embryonic myoblasts (Amthor et al. 2009).
It is unclear why different laboratories are finding different numbers of satellite cells in the same mutants. One possible explanation is that the analysis that showed increased satellite cell numbers used CD34 as a satellite cell marker while we and another laboratory that did not find an increase used Pax7 or Myf5 (McCroskery et al. 2003; Amthor et al. 2009). CD34 can label fibroblasts and endothelial cells as well as satellite cells while the only cells in muscle that Pax7 labels are satellite cells. Another explanation for the differences could be that the mouse colonies have been separated for many years and there may be some acquired genetic differences between colonies. Nutritional differences between animal facilities might also contribute to subtle differences in satellite cell development. The lack of a strong effect on satellite cell numbers in our ACVR2B:Fc injection experiments, however, is generally in agreement with our analysis of the Mstn KO mice and that of Amthor et al. (2009).
Alterations in satellite cell activation may become more evident with ageing and affect age-related muscle loss. Siriett et al. (2006), based on measurements of the CSA of type IIB and type IIX myofibres in the TA muscle, found that Mstn KO mice are resistant to sarcopaenia in support of their claim that MSTN regulates satellite cell quiescence. In contrast, we found that most WT and KO muscles decline in mass at similar rates. Our results are consistent with a previous measurement of the quadriceps mass at age 27–30 months (Morissette et al. 2009).
Not only are Mstn KO muscles not resistant to sarcopaenia, the KO mice may even be more susceptible to sarcopaenia than WT mice. In 24-month-old KO mice, we found that the EDL in two mice weighed approximately half that of the other KO mice. The contralateral EDL from these mice appeared to be smaller by cross section, so this reduced weight is unlikely to be due to an error in dissection. We have also analysed muscle weights in 24-month-old mice produced from homozygous matings that were not included here and found that two KO EDL muscles from in this group were half the weight of three others of the same genotype (data not shown). This great a variation in weight was not seen in any of the WT EDL muscles from mice produced from heterozygous or homozygous matings or in any other muscle from either genotype. We also found a non-significant trend toward lower satellite cell numbers in aged Mstn KO mice in the EDL. This was due to low satellite cell numbers in the two mice with very low EDL muscle mass as well, suggesting a correlation. This muscle is one of the smallest we analysed and the functional effect of a reduction in its mass to the animal may be minimal. Multiple muscles will have to be analysed in even older animals to ascertain whether this phenotype was unique to the EDL or is a sign of increased rate of muscle atrophy during ageing.
We also found that the number of myofibres with central nuclei, an indication of prior satellite cell activation and fusion, increases with age in Mstn KO muscle compared to WT muscle. We cannot say whether this is due to a subtle increase satellite cell turnover or repair of myofibres. Ageing KO muscle has an increase in tubular aggregates which are evident on cross sections as small areas of the cytoplasm without myofibrillar structural proteins (Amthor et al. 2007). Although tubular aggregates are not specific to any disease and their importance is unclear, they do indicate some abnormality (Pavlovičováet al. 2003). For example, Mstn KO muscles have a reduced number of mitochondria which could affect fibre function (Amthor et al. 2007). However, developmental deletion of the Mstn gene causes an increase in fibre number and change in fibre type which are not found after treatment with any of the various MSTN inhibitors in adult mice (McPherron, 2010). Some characteristics of the KO muscle may therefore not be relevant to therapeutic use of MSTN inhibitors postnatally, although this needs to be examined in preclinical models.
Hypertrophy in adult mice
Two conceptual models of myofibre hypertrophy have been described based on the multinuclear organization of muscle myofibres (Cheek et al. 1971; Hall & Ralston, 1989; Edgerton & Roy, 1991; Allen et al. 1999). In one model, the rate of transcription could increase in each myonucleus to maintain an appropriate level of transcription for protein synthesis to accommodate the increased cytoplasmic volume. An alternative model is based on the concept of nuclear domain first described by Cheek and colleagues (Cheek, 1985). In this model, the addition of new nuclei to the myofibre maintains the cytoplasmic volume/DNA ratio, the ‘myonuclear domain’, so that each nucleus carries out transcription at the same rate regardless of myofibre size. Because myonuclei themselves are postmitotic, this latter model necessitates cell fusion to maintain a constant cytoplasm/myonuclear ratio. Most evidence from experimental hypertrophy demonstrates an increase in the number of myonuclei and a stable myonuclear domain (Hall & Ralston, 1989; Edgerton & Roy, 1991; Allen et al. 1999). Satellite cells are activated in a variety of hypertrophic conditions, and, in some cases, this activation clearly occurs within a few days, prior to when most of the hypertrophy has been achieved (Schiaffino et al. 1972; Winchester et al. 1991; O’Connor & Pavlath, 2007; Bruusgaard et al. 2010). There has been ongoing controversy, however, about whether satellite cells must be activated to achieve myofibre hypertrophy in all cases (McCarthy & Esser, 2007; O’Connor & Pavlath, 2007; O’Connor et al. 2007). For instance, some hypertrophy relative to myonuclear number has been found in humans during resistance training (Kadi et al. 2004). Myofibre hypertrophy caused by activation of a constitutively active Akt transgene for 3 weeks does not increase BrdU incorporation into myofibres examined by cross sections (Blaauw et al. 2009). Although previous work analysing satellite cell activation after MSTN inhibition in adult rodents is conflicting, different means of MSTN inhibition have consistently produced an increase in the myonuclear domain regardless of whether they also inhibit activin. This is true for treatment with secreted inhibitors that could act on both satellite cells and myofibres (here and Amthor et al. 2009; Foster et al. 2009; Gilson et al. 2009; Matsakas et al. 2009) and for a membrane-bound inhibitor expressed only in myofibres (Sartori et al. 2009).
Some researchers have proposed that there are multiple phases of hypertrophy, essentially a combination of the increased nuclear transcription and myonuclear domain models. In this model, myofibres could have the ability to increase protein synthesis initially until a myofibre size threshold for satellite cell activation is reached after which there is an adaptation phase that requires satellite cells (Rosenblatt et al. 1994; Roy et al. 1999; Kadi et al. 2004; O’Connor & Pavlath, 2007). Our work here could be consistent with multiple phases of hypertrophy because satellite cell activation was low and did not precede hypertrophy. Mechanical disruption of the satellite cell niche by stretch or exercise increases secretion of satellite cell activators (Tatsumi, 2010), and hypertrophy could cause a milder disruption in the satellite cell niche or alter the levels of satellite cell activators or inhibitors secreted from myofibres. This time course might be rather long because treatment with the MSTN pro-peptide resulted in an increase in Pax7+ cells in the EDL after 17 (but not 8) weeks although the CSA of fast fibres was similar at both time points (Matsakas et al. 2009). In this experiment, the number of myonuclei was non-significantly increased by ∼10 at 17 weeks so these extra satellite cells don't seem to have fused in great numbers by this time. Despite finding some activation of satellite cells, this study and our work here do not specifically address whether satellite cell activation and fusion are necessary for long-term maintenance of myofibre function, metabolism, or hypertrophy itself after MSTN inhibition.
Satellite cells are not the only cells to divide during hypertrophy. Myofibre hypertrophy caused by ACVR2B:Fc treatment or activation of a constitutively active Akt transgene is accompanied by proliferation of non-muscle cells (Blaauw et al. 2009; Zhou et al. 2010). Because the constitutively active Akt is cell-autonomous rather than secreted, this result suggests that myofibre hypertrophy changes the local environment to activate proliferation of non-muscle cells. Many of these cells were identified as fibroblasts, endothelial cells and pericytes (Blaauw et al. 2009) suggesting that they may contribute to angiogenesis and to support the hypertrophied muscle. It is speculation, but perhaps some of the reduction in hypertrophic response to follistatin after irradiation (Gilson et al. 2009) could be due to the loss of proliferation of these non-muscle cells rather than satellite cells.
Conclusions
In summary, we have shown that satellite cells are activated after MSTN inhibition but that this activation level is low, does not precede myofibre hypertrophy, and therefore does not account for the resulting hypertrophy. We have also shown that satellite cells are depleted in Mstn KO mice at the same rate as in WT mice, and that Mstn KO mice are not resistant to sarcopaenia. Our results support those previous studies that suggest that MSTN signalling does not directly regulate satellite cell quiescence. The mode of hypertrophy induced by MSTN inhibition is consistent with a multiple phase model of increased myofibre protein synthesis followed by satellite cell activation.
Acknowledgments
This work was supported by the Intramural Research Program of the NIH, NIDDK. We thank Kathleen Savage for helpful discussions, Tingqing Guo for help purifying ACVR2B:Fc, Onur Kanisicak and Shahragim Tajbakhsh for technical advice, Bryan Millis for creating the macro for the imaging analysis, Yunping Wu for help with confocal microscopy, and Kathryn Wagner for comments on the manuscript. The Pax7 hybridoma developed by A. Kawakami was obtained from the Developmental Studies Hybridoma Bank developed under the auspices of the NICHD and maintained by The University of Iowa, Department of Biology, Iowa City, IA 52242, USA.
Glossary
Abbreviations
- ACVR2B
activin receptor type IIB
- BrdU
5′-bromo-2′-deoxyuridine
- EDL
extensor digitorum longus
- Gas
gastrocnemius
- MSTN
myostatin
- Pec
pectoralis
- Plan
plantaris
- Quad
quadriceps
- TA
tibialis anterior
- Tri
triceps
- Sol
soleus
Author contributions
Q.W. and A.C.M. designed the study, grew the CHO cells, collected conditioned medium, and wrote and approved the final version of the manuscript. A.C.M. weighed muscles, and Q.W. carried out all experiments and all other measurements and performed statistical analysis. All authors approved the final version for publication. All work was done at NIDDK.
Disclosures
Under a licensing agreement between Pfizer and the Johns Hopkins University, A.C.M. is entitled to a share of royalty received by the University on sales of myostatin. The terms of these arrangements are being managed by the University in accordance with its conflict of interest policies.
References
- Allen DL, Roy RR, Edgerton VR. Myonuclear domains in muscle adaptation and disease. Muscle Nerve. 1999;22:1350–1360. doi: 10.1002/(sici)1097-4598(199910)22:10<1350::aid-mus3>3.0.co;2-8. [DOI] [PubMed] [Google Scholar]
- Amirouche A, Durieux AC, Banzet S, Koulmann N, Bonnefoy R, Mouret C, Bigard X, Peinnequin A, Freyssenet D. Down-regulation of Akt/mammalian target of rapamycin signaling pathway in response to myostatin overexpression in skeletal muscle. Endocrinology. 2009;150:286–294. doi: 10.1210/en.2008-0959. [DOI] [PubMed] [Google Scholar]
- Amthor H, Huang R, McKinnell I, Christ B, Kambadur R, Sharma M, Patel K. The regulation and action of myostatin as a negative regulator of muscle development during avian embryogenesis. Dev Biol. 2002;251:241–257. doi: 10.1006/dbio.2002.0812. [DOI] [PubMed] [Google Scholar]
- Amthor H, Macharia R, Navarrete R, Schuelke M, Brown SC, Otto A, Voit T, Muntoni F, Vrbova G, Partridge T, Zammit P, Bunger L, Patel K. Lack of myostatin results in excessive muscle growth but impaired force generation. Proc Natl Acad Sci USA. 2007;104:1835–1840. doi: 10.1073/pnas.0604893104. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Amthor H, Otto A, Macharia R, McKinnell I, Patel K. Myostatin imposes reversible quiescence on embryonic muscle precursors. Dev Dyn. 2006;235:672–680. doi: 10.1002/dvdy.20680. [DOI] [PubMed] [Google Scholar]
- Amthor H, Otto A, Vulin A, Rochat A, Dumonceaux J, Garcia L, Mouisel E, Hourde C, Macharia R, Friedrichs M, Relaix F, Zammit PS, Matsakas A, Patel K, Partridge T. Muscle hypertrophy driven by myostatin blockade does not require stem/precursor-cell activity. Proc Natl Acad Sci USA. 2009;106:7479–7484. doi: 10.1073/pnas.0811129106. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Atlantis E, Martin SA, Haren MT, Taylor AW, Wittert GA. Inverse associations between muscle mass, strength, and the metabolic syndrome. Metabolism. 2009;58:1013–1022. doi: 10.1016/j.metabol.2009.02.027. [DOI] [PubMed] [Google Scholar]
- Blaauw B, Canato M, Agatea L, Toniolo L, Mammucari C, Masiero E, Abraham R, Sandri M, Schiaffino S, Reggiani C. Inducible activation of Akt increases skeletal muscle mass and force without satellite cell activation. FASEB J. 2009;23:3896–3905. doi: 10.1096/fj.09-131870. [DOI] [PubMed] [Google Scholar]
- Bruusgaard JC, Johansen IB, Egner IM, Rana ZA, Gundersen K. Myonuclei acquired by overload exercise precede hypertrophy and are not lost on detraining. Proc Natl Acad Sci USA. 2010;107:15111–15116. doi: 10.1073/pnas.0913935107. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Cheek D, Holt A, Hill D, Talbert J. Skeletal muscle cell mass and growth: The concept of the deoxyribonucleic acid unit. Pediatr Res. 1971;5:312–328. [Google Scholar]
- Cheek DB. The control of cell mass and replication. The DNA unit – a personal 20-year study. Early Hum Dev. 1985;12:211–239. doi: 10.1016/0378-3782(85)90144-6. [DOI] [PubMed] [Google Scholar]
- Clop A, Marcq F, Takeda H, Pirottin D, Tordoir X, Bibe B, Bouix J, Caiment F, Elsen JM, Eychenne F, Larzul C, Laville E, Meish F, Milenkovic D, Tobin J, Charlier C, Georges M. A mutation creating a potential illegitimate microRNA target site in the myostatin gene affects muscularity in sheep. Nat Genet. 2006;38:813–818. doi: 10.1038/ng1810. [DOI] [PubMed] [Google Scholar]
- Edgerton VR, Roy RR. Regulation of skeletal muscle fiber size, shape and function. J Biomech. 1991;24(Suppl 1):123–133. doi: 10.1016/0021-9290(91)90383-x. [DOI] [PubMed] [Google Scholar]
- Elashry MI, Otto A, Matsakas A, El-Morsy SE, Patel K. Morphology and myofiber composition of skeletal musculature of the forelimb in young and aged wild type and myostatin null mice. Rejuvenation Res. 2009;12:269–281. doi: 10.1089/rej.2009.0870. [DOI] [PubMed] [Google Scholar]
- Foster K, Graham IR, Otto A, Foster H, Trollet C, Yaworsky PJ, Walsh FS, Bickham D, Curtin NA, Kawar SL, Patel K, Dickson G. Adeno-associated virus-8-mediated intravenous transfer of myostatin propeptide leads to systemic functional improvements of slow but not fast muscle. Rejuvenation Res. 2009;12:85–94. doi: 10.1089/rej.2008.0815. [DOI] [PubMed] [Google Scholar]
- Gilson H, Schakman O, Kalista S, Lause P, Tsuchida K, Thissen JP. Follistatin induces muscle hypertrophy through satellite cell proliferation and inhibition of both myostatin and activin. Am J Physiol Endocrinol Metab. 2009;297:E157–164. doi: 10.1152/ajpendo.00193.2009. [DOI] [PubMed] [Google Scholar]
- Glass DJ. PI3 kinase regulation of skeletal muscle hypertrophy and atrophy. Curr Top Microbiol Immunol. 2010;346:267–278. doi: 10.1007/82_2010_78. [DOI] [PubMed] [Google Scholar]
- Grobet L, Martin LJ, Poncelet D, Pirottin D, Brouwers B, Riquet J, Schoeberlein A, Dunner S, Menissier F, Massabanda J, Fries R, Hanset R, Georges M. A deletion in the bovine myostatin gene causes the double-muscled phenotype in cattle. Nat Genet. 1997;17:71–74. doi: 10.1038/ng0997-71. [DOI] [PubMed] [Google Scholar]
- Hall ZW, Ralston E. Nuclear domains in muscle cells. Cell. 1989;59:771–772. doi: 10.1016/0092-8674(89)90597-7. [DOI] [PubMed] [Google Scholar]
- Jejurikar SS, Kuzon WM., Jr Satellite cell depletion in degenerative skeletal muscle. Apoptosis. 2003;8:573–578. doi: 10.1023/A:1026127307457. [DOI] [PubMed] [Google Scholar]
- Kadi F, Schjerling P, Andersen LL, Charifi N, Madsen JL, Christensen LR, Andersen JL. The effects of heavy resistance training and detraining on satellite cells in human skeletal muscles. J Physiol. 2004;558:1005–1012. doi: 10.1113/jphysiol.2004.065904. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kambadur R, Sharma M, Smith TP, Bass JJ. Mutations in myostatin (GDF8) in double-muscled Belgian Blue and Piedmontese cattle. Genome Res. 1997;7:910–916. doi: 10.1101/gr.7.9.910. [DOI] [PubMed] [Google Scholar]
- Lee S-J, McPherron AC. Regulation of myostatin activity and muscle growth. Proc Natl Acad Sci USA. 2001;98:9306–9311. doi: 10.1073/pnas.151270098. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lee SJ. Genetic analysis of the role of proteolysis in the activation of latent myostatin. PLoS ONE. 2008;3:e1628. doi: 10.1371/journal.pone.0001628. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lee SJ. Extracellular regulation of myostatin: A molecular rheostat for muscle mass. Immunol Endocr Metab Agents Med Chem. 2010;10:183–194. doi: 10.2174/187152210793663748. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lee SJ, Lee YS, Zimmers TA, Soleimani A, Matzuk MM, Tsuchida K, Cohn RD, Barton ER. Regulation of muscle mass by follistatin and activins. Mol Endocrinol. 2010;24:1998–2008. doi: 10.1210/me.2010-0127. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lee SJ, Reed LA, Davies MV, Girgenrath S, Goad ME, Tomkinson KN, Wright JF, Barker C, Ehrmantraut G, Holmstrom J, Trowell B, Gertz B, Jiang MS, Sebald SM, Matzuk M, Li E, Liang LF, Quattlebaum E, Stotish RL, Wolfman NM. Regulation of muscle growth by multiple ligands signaling through activin type II receptors. Proc Natl Acad Sci USA. 2005;102:18117–18122. doi: 10.1073/pnas.0505996102. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lipina C, Kendall H, McPherron AC, Taylor PM, Hundal HS. Mechanisms involved in the enhancement of mammalian target of rapamycin signalling and hypertrophy in skeletal muscle of myostatin-deficient mice. FEBS Lett. 2010;584:2403–2408. doi: 10.1016/j.febslet.2010.04.039. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lu X, de la Pena L, Barker C, Camphausen K, Tofilon PJ. Radiation-induced changes in gene expression involve recruitment of existing messenger RNAs to and away from polysomes. Cancer Res. 2006;66:1052–1061. doi: 10.1158/0008-5472.CAN-05-3459. [DOI] [PubMed] [Google Scholar]
- Manceau M, Gros J, Savage K, Thome V, McPherron A, Paterson B, Marcelle C. Myostatin promotes the terminal differentiation of embryonic muscle progenitors. Genes Dev. 2008;22:668–681. doi: 10.1101/gad.454408. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Matsakas A, Foster K, Otto A, Macharia R, Elashry MI, Feist S, Graham I, Foster H, Yaworsky P, Walsh F, Dickson G, Patel K. Molecular, cellular and physiological investigation of myostatin propeptide-mediated muscle growth in adult mice. Neuromuscul Disord. 2009;19:489–499. doi: 10.1016/j.nmd.2009.06.367. [DOI] [PubMed] [Google Scholar]
- Matsakas A, Otto A, Elashry MI, Brown SC, Patel K. Altered primary and secondary myogenesis in the myostatin-null mouse. Rejuvenation Res. 2010;13:717–727. doi: 10.1089/rej.2010.1065. [DOI] [PubMed] [Google Scholar]
- Mauro A( Satellite cell of skeletal muscle fibers. J Biophys Biochem Cytol. 1961;9:493–495. doi: 10.1083/jcb.9.2.493. [DOI] [PMC free article] [PubMed] [Google Scholar]
- McCarthy JJ, Esser KA. Counterpoint: Satellite cell addition is not obligatory for skeletal muscle hypertrophy. J Appl Physiol. 2007;103:1100–1102. doi: 10.1152/japplphysiol.00101.2007a. discussion 1102–1103. [DOI] [PubMed] [Google Scholar]
- McCroskery S, Thomas M, Maxwell L, Sharma M, Kambadur R. Myostatin negatively regulates satellite cell activation and self-renewal. J Cell Biol. 2003;162:1135–1147. doi: 10.1083/jcb.200207056. [DOI] [PMC free article] [PubMed] [Google Scholar]
- McPherron AC. Metabolic functions of myostatin and Gdf11. Immunol Endocr Metab Agents Med Chem. 2010;10:217–231. doi: 10.2174/187152210793663810. [DOI] [PMC free article] [PubMed] [Google Scholar]
- McPherron AC, Huynh TV, Lee SJ. Redundancy of myostatin and growth/differentiation factor 11 function. BMC Dev Biol. 2009;9:24. doi: 10.1186/1471-213X-9-24. [DOI] [PMC free article] [PubMed] [Google Scholar]
- McPherron AC, Lawler AM, Lee SJ. Regulation of skeletal muscle mass in mice by a new TGF-b superfamily member. Nature. 1997;387:83–90. doi: 10.1038/387083a0. [DOI] [PubMed] [Google Scholar]
- McPherron AC, Lee SJ. Double muscling in cattle due to mutations in the myostatin gene. Proc Natl Acad Sci USA. 1997;94:12457–12461. doi: 10.1073/pnas.94.23.12457. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Morissette MR, Stricker JC, Rosenberg MA, Buranasombati C, Levitan EB, Mittleman MA, Rosenzweig A. Effects of myostatin deletion in aging mice. Aging Cell. 2009;8:573–583. doi: 10.1111/j.1474-9726.2009.00508.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Moss FP, Leblond CP. Satellite cells as the source of nuclei in muscles of growing rats. Anat Rec. 1971;170:421–435. doi: 10.1002/ar.1091700405. [DOI] [PubMed] [Google Scholar]
- O’Connor RS, Pavlath GK. Point:Counterpoint: Satellite cell addition is/is not obligatory for skeletal muscle hypertrophy. J Appl Physiol. 2007;103:1099–1100. doi: 10.1152/japplphysiol.00101.2007. [DOI] [PubMed] [Google Scholar]
- O’Connor RS, Pavlath GK, McCarthy JJ, Esser KA. Last Word on Point:Counterpoint: Satellite cell addition is/is not obligatory for skeletal muscle hypertrophy. J Appl Physiol. 2007;103:1107. doi: 10.1152/japplphysiol.00502.2007. [DOI] [PubMed] [Google Scholar]
- Ontell M, Kozeka K. Organogenesis of the mouse extensor digitorum logus muscle: a quantitative study. Am J Anat. 1984;171:149–161. doi: 10.1002/aja.1001710203. [DOI] [PubMed] [Google Scholar]
- Pavlovičová M, Novotová M, Zahradník I. Structure and composition of tubular aggregates of skeletal muscle fibres. Gen Physiol Biophys. 2003;22:425–440. [PubMed] [Google Scholar]
- Rebbapragada A, Benchabane H, Wrana JL, Celeste AJ, Attisano L. Myostatin signals through a transforming growth factor β-like signaling pathway to block adipogenesis. Mol Cell Biol. 2003;23:7230–7242. doi: 10.1128/MCB.23.20.7230-7242.2003. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Rodriguez J, Vernus B, Toubiana M, Jublanc E, Tintignac L, Leibovitch S, Bonnieu A. Myostatin inactivation increases myotube size through regulation of translational initiation machinery. J Cell Biochem. 2011;112:3531–3542. doi: 10.1002/jcb.23280. [DOI] [PubMed] [Google Scholar]
- Rosenblatt JD, Yong D, Parry DJ. Satellite cell activity is required for hypertrophy of overloaded adult rat muscle. Muscle Nerve. 1994;17:608–613. doi: 10.1002/mus.880170607. [DOI] [PubMed] [Google Scholar]
- Roy RR, Monke SR, Allen DL, Edgerton VR. Modulation of myonuclear number in functionally overloaded and exercised rat plantaris fibers. J Appl Physiol. 1999;87:634–642. doi: 10.1152/jappl.1999.87.2.634. [DOI] [PubMed] [Google Scholar]
- Sartori R, Milan G, Patron M, Mammucari C, Blaauw B, Abraham R, Sandri M. Smad2 and 3 transcription factors control muscle mass in adulthood. Am J Physiol Cell Physiol. 2009;296:C1248–1257. doi: 10.1152/ajpcell.00104.2009. [DOI] [PubMed] [Google Scholar]
- Schiaffino S, Bormioli SP, Aloisi M. Cell proliferation in rat skeletal muscle during early stages of compensatory hypertrophy. Virchows Arch B Cell Pathol. 1972;11:268–273. doi: 10.1007/BF02889406. [DOI] [PubMed] [Google Scholar]
- Schuelke M, Wagner KR, Stolz LE, Hübner C, Riebel T, Komen W, Braun T, Tobin JF, Lee SJ. Myostatin mutation associated with gross muscle hypertrophy in a child. N Engl J Med. 2004;350:2682–2688. doi: 10.1056/NEJMoa040933. [DOI] [PubMed] [Google Scholar]
- Shadrach JL, Wagers AJ. Stem cells for skeletal muscle repair. Philos Trans R Soc Lond B Biol Sci. 2011;366:2297–2306. doi: 10.1098/rstb.2011.0027. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Siriett V, Platt L, Salerno MS, Ling N, Kambadur R, Sharma M. Prolonged absence of myostatin reduces sarcopenia. J Cell Physiol. 2006;209:866–873. doi: 10.1002/jcp.20778. [DOI] [PubMed] [Google Scholar]
- Srikanthan P, Karlamangla AS. Relative muscle mass is inversely associated with insulin resistance and prediabetes. Findings from the third National Health and Nutrition Examination Survey. J Clin Endocrinol Metab. 2011;96:2898–2903. doi: 10.1210/jc.2011-0435. [DOI] [PubMed] [Google Scholar]
- Stockdale FE, Holtzer H. DNA synthesis and myogenesis. Exp Cell Res. 1961;24:508–520. doi: 10.1016/0014-4827(61)90450-5. [DOI] [PubMed] [Google Scholar]
- Tatsumi R. Mechano-biology of skeletal muscle hypertrophy and regeneration: possible mechanism of stretch-induced activation of resident myogenic stem cells. Anim Sci J. 2010;81:11–20. doi: 10.1111/j.1740-0929.2009.00712.x. [DOI] [PubMed] [Google Scholar]
- Thies RS, Chen T, Davies MV, Tomkinson KN, Pearson AA, Shakey QA, Wolfman NM. GDF-8 propeptide binds to GDF-8 and antagonizes biological activity by inhibiting GDF-8 receptor binding. Growth Factors. 2001;18:251–259. doi: 10.3109/08977190109029114. [DOI] [PubMed] [Google Scholar]
- Trendelenburg AU, Meyer A, Rohner D, Boyle J, Hatakeyama S, Glass DJ. Myostatin reduces Akt/TORC1/p70S6K signaling, inhibiting myoblast differentiation and myotube size. Am J Physiol Cell Physiol. 2009;296:C1258–C1270. doi: 10.1152/ajpcell.00105.2009. [DOI] [PubMed] [Google Scholar]
- Wagner KR, Liu X, Chang X, Allen RE. Muscle regeneration in the prolonged absence of myostatin. Proc Natl Acad Sci USA. 2005;102:2519–2524. doi: 10.1073/pnas.0408729102. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Winchester PK, Davis ME, Alway SE, Gonyea WJ. Satellite cell activation in the stretch-enlarged anterior latissimus dorsi muscle of the adult quail. Am J Physiol Cell Physiol. 1991;260:C206–C212. doi: 10.1152/ajpcell.1991.260.2.C206. [DOI] [PubMed] [Google Scholar]
- Wolfman NM, McPherron AC, Pappano WN, Davies MV, Song K, Tomkinson KN, Wright JF, Zhao L, Sebald SM, Greenspan DS, Lee SJ. Activation of latent myostatin by the BMP-1/tolloid family of metalloproteinases. Proc Natl Acad Sci USA. 2003;100:15842–15846. doi: 10.1073/pnas.2534946100. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zhou X, Wang JL, Lu J, Song Y, Kwak KS, Jiao Q, Rosenfeld R, Chen Q, Boone T, Simonet WS, Lacey DL, Goldberg AL, Han HQ. Reversal of cancer cachexia and muscle wasting by ActRIIB antagonism leads to prolonged survival. Cell. 2010;142:531–543. doi: 10.1016/j.cell.2010.07.011. [DOI] [PubMed] [Google Scholar]