

Abstract
AIM: To investigate the correlation of hyperlipemia (HL) and acute cerebral ischemia/reperfusion (I/R) injury on liver damage and its mechanism.
METHODS: Rats were divided into 4 groups: control, HL, I/R and HL+I/R. After the induction of HL via a high-fat diet for 18 wk, middle cerebral artery occlusion was followed by 24 h of reperfusion to capture I/R. Serum alanine transaminase (ALT) and aspartate aminotransferase (AST) were analyzed as part of liver function tests and liver damage was further assessed by histological examination. Hepatocyte apoptosis was evaluated by terminal deoxynucleotidyl transferase dUTP nick-end labeling (TUNEL) assay. The expression of genes related to apoptosis (caspase-3, bcl-2) was assayed by immunohistochemistry and Western blotting. Serum tumor necrosis factor-α (TNF-α), interleukin-1 (IL-1) and liver mitochondrial superoxide dismutase (SOD), glutathione peroxidase (GSH-Px), malondialdehyde (MDA) and Ca2+ levels were measured to determine inflammatory and oxidative/antioxidative status respectively. Microsomal hydroxylase activity of the cytochrome P450 2E1 (CYP2E1)-containing enzyme was measured with aniline as the substrate, and CYP2E1 expression in the liver tissue and microsome was determined by immunohistochemistry and Western blotting respectively.
RESULTS: HL alone induced by high-fat diet for 18 wk resulted in liver damage, indicated by histopathological analysis, and a considerable increase in serum ALT (25.13 ± 16.90 vs 9.56 ± 1.99, P < 0.01) and AST levels (18.01 ± 10.00 vs 11.33 ± 4.17, P < 0.05) compared with control. Moreover, HL alone induced hepatocyte apoptosis, which was determined by increased TUNEL-positive cells (4.47 ± 0.45 vs 1.5 ± 0.22, P < 0.01), higher caspase-3 and lower bcl-2 expression. Interestingly, compared with those in control, HL or I/R groups, massive increases of serum ALT (93.62 ± 24.00 vs 9.56 ± 1.99, 25.13 ± 16.90 or 12.93 ± 6.14, P < 0.01) and AST (82.32 ± 26.92 vs 11.33 ± 4.17, 18.01 ± 10.00 or 14.00 ± 6.19, P < 0.01) levels in HL+I/R group were observed suggesting severe liver damage, which was confirmed by liver histology. In addition, HL combined with I/R also caused significantly increased hepatocyte apoptosis, as evidenced by increased TUNEL-positive cells (6.20 ± 0.29 vs 1.5 ± 0.22, 4.47 ± 0.45 or 1.97 ± 0.47, P < 0.01), elevated expression of caspase-3 and lower expression of bcl-2. Furthermore, when compared to HL or I/R alone, HL plus I/R enhanced serum TNF-α, IL-1, liver mitochondrial MDA and Ca2+ levels, suppressed SOD and GSH-Px in liver mitochondria, and markedly up-regulated the activity (11.76 ± 2.36 vs 4.77 ± 2.31 or 3.11 ± 1.35, P < 0.01) and expression (3.24 ± 0.38 vs 1.98 ± 0.88 or 1.72 ± 0.58, P < 0.01) of CYP2E1 in liver.
CONCLUSION: The coexistence of HL and acute cerebral I/R induces severe liver damage, suggesting that cerebral ischemic stroke would exaggerate the damage of liver caused by HL. This effect is possibly due to enhanced CYP2E1 induction which further promotes oxidative damage, inflammation and hepatocyte apoptosis.
Keywords: Hyperlipidemia, High-fat diet, Cerebral ischemia/reperfusion, Liver damage, Hepatocyte apoptosis, Cytochrome P450 2E1
INTRODUCTION
The history of hyperlipidemia (HL), an increasing public health problem, is positively associated with acute cerebrovascular diseases (ACD) such as ischemic stroke[1]. Although HL per se is not a direct predictor of ACD, the condition with high plasma levels of inflammatory-sensitive proteins associated with HL links to the increased incidence of stroke[2]. Positive correlations between excess lipid levels and the promotion of inflammation have been documented[2]. Moreover, it has been widely accepted that oxidative stress, an early event in the evolution of HL[3], also plays an essential role in the pathophysiology of ischemic stroke[4]. So it can be speculated that HL, as a highly prevalent risk factor for ACD, frequently accompanies ACD.
In addition, HL is also a major risk factor for liver disease[5]. The spectrum of HL-induced liver disease, also known as non-alcoholic fatty liver disease (NAFLD), ranges from “simple” steatosis to non-alcoholic steatohepatitis, which is characterized by oxidative stress, inflammation, and liver damage as well as fat deposition[6]. Of interest is that one of the main complications of the patient with ACD is the risk of secondary liver dysfunction. Ischemic stroke will further trigger the activation of inflammatory cascades leading to systemic inflammatory response[7]. In the systemic inflammatory environment developed by circulating reactive oxygen species (ROS) and pro-inflammatory cytokines after ACD, nonneurological organs, including liver, would be impaired leading to multiple organ dysfunction syndromes (MODS). The incidence of MODS in patients with ACD was reported as 11.5%, the mortality rate was 40.3%[8]. It is noteworthy that one of the major systemic complications of ACD is liver dysfunction, the presence of which is associated with mortality and overall poor outcome[9]. Clinical studies in China have showed, in patients of ACD with MODS, 26.83%[8] or 15.5%[10] had abnormal liver function. Therefore, HL or ACD is respectively known to be the independent risk factor for liver injury. It is thus reasonable to hypothesize that the coexistence of HL and ACD may contribute to more severe liver dysfunction.
As mentioned above, the pathological changes in liver caused by either HL or cerebral ischemia are associated with inflammation and oxidative stress. Inflammatory response and even apoptosis could be triggered by oxidative stress[11], which may be a key mechanism of liver injury induced by HL or cerebral ischemia. The source of oxidative stress is controversial but may result in part from over-expression of the prooxidant enzyme cytochrome P450 2E1 (CYP2E1), that has been reported to occur in human and experimental NAFLD[12]. As a major microsomal source of ROS, CYP2E1 mainly located in the endoplasmic reticulum and played a significant role in the biotransformation of fatty acid oxidation. ROS overproduction induces oxidative damage to liver DNA and contributes to hepatocellular injury[13]. The expression of CYP2E1 in liver can be enhanced in rats treated with high-fat diet[14]. In addition, brain injury or inflammation has influence on some liver cytochrome P450 enzymes[15]. But the effect of HL together with ACD on CYP2E1 is still unclear.
In this study, we hypothesized that HL or cerebral ischemia/reperfusion (I/R) injury may directly induce a series of changes leading to a pro-oxidant and pro-inflammatory state, which may finally result in the onset of liver injury, and the coexistence of HL and cerebral I/R injury may cause much more severe changes and the exacerbation of liver injury. To verify this assumption, a repeatable experimental hyperlipemic rat model with cerebral I/R injury was established to investigate the correlation of HL and/or acute cerebral I/R injury on liver damage, and further examine the effects of HL and/or I/R on hepatocyte apoptosis, CYP2E1, oxidative/antioxidant system and inflammatory process to understand the mechanism of liver damage.
MATERIALS AND METHODS
Animals
Adult male Sprague-Dawley rats, weighing 200-250 g, were obtained from Hubei Laboratory Animals Center for Medical Science and Research (No. 00006231). The animals were allowed to acclimatize for 1 wk while being maintained at a room temperature of 22 ± 2 °C on a 12 h light/dark cycle with free access to standard rodent chow food and water (Standard sustain feed, from Hubei Laboratory Animals Center for Medical Science and Research, Wuhan, China). Housing facilities and all experimental protocols were carried out according to the National Institutes of Health Guide for the Care and Use of Laboratory Animals, and approved by the Animals Care and Use Committee of Wuhan University of Science and Technology Medical College which adopts the guideline for the care and use of laboratory animals published by the US National Institutes of Health (NIH Publication No. 85-23, revised 1996).
Experiment design
The rats were fed either a standard low-fat rat chow diet (control diet) or a high-fat diet (HFD, 1% cholesterol, 10% lard, 20% sugar, 5% egg yolk, 0.2% bile salts and 63.8% standard chow, finally analyzed by the Laboratory Animal Centre of Hubei). After 18 wk of HFD, blood was withdrawn from the tail vein of overnight fasted rats for confirming HL. Serum triglycerides (TG), total cholesterol (TC), high-density lipoprotein cholesterol (HDL-C) and low-density lipoprotein cholesterol (LDL-C) were detected by Beckman Coulter synchron LX20 autoanalyzer (Beckman Coulter Inc., United States). The animals with serum TC level > 3.87 mmol/L were considered HL and included in the study[16]. Subsequently, the control diet group and HL group were respectively divided into two subgroups treated with or without cerebral I/R (Table 1). After HL was confirmed, the rats in I/R or HL+I/R group were subjected to middle cerebral artery occlusion (MCAO)[17] for 2 h and followed by 24 h reperfusion to induce focal cerebral I/R injury. As the sham control, rats in control or HL groups underwent the same surgical procedure as in I/R or HL+I/R group without inserting the nylon thread.
Table 1.
The examined animal groups
| Group | Administration |
| Control | Rats given normal diet (control) for 18 wk and without I/R |
| HL | HL rats given high-fat diet for 18 wk and without I/R |
| I/R | Rats given normal diet (control) for 18 wk and with I/R |
| HL+I/R | HL rats given high-fat diet for 18 wk and with I/R |
HL: Hyperlipidemia; I/R: Ischemia/reperfusion.
Behavioral score and brain infarct volume were evaluated at 24 h after reperfusion by researchers and another assistant who was not involved in this experiment. The neurological deficits were scored on a 5-point scale based on the report of Longa et al[18]. All animals were sacrificed after the evaluation of behavioral scores. The area of cerebral infarction was quantified with TTC (2, 3, 5-triphenyltetrazolium chloride) staining as previous described[13].
Measurement of liver function and serum pro-inflammatory cytokines
Blood samples were obtained from the common carotid artery to detect serum alanine aminotransferase (ALT), aspartate aminotransferase (AST), tumor necrosis factor-α (TNF-α) and interleukin-1 (IL-1) when rats were sacrificed at 24 h after reperfusion. The levels of serum ALT and AST were measured using corresponding kits (Jiancheng, Nanjing, China) to assess the liver function. Serum TNF-α and IL-1 levels were measured by radioimmunoassay kits (Albert Poole Biotechnology, Beijing, China).
Histological and immunohistochemical examination of liver
Harvested livers were placed into 10% formalin immediately after excision, and immersed for 24 h. Liver specimens were then embedded in paraffin, and sections were cut at 5 μm. Sections were stained for: (1) Hematoxylin and eosin (HE); (2) Apoptosis was detected by the TdT-mediated dUTP-biotin nick end labeling (TUNEL) method using an in situ apoptosis detection kit (Boster Biological Engineering, Wuhan, China) as previously described[19]. The number of cells with TUNEL-positive nuclei was counted from 20 randomly selected fields at × 200 magnification per liver sample. Results were expressed as the mean number of TUNEL-positive apoptotic hepatocytes per microscopic field; and (3) Immunohistological analysis of CYP2E1, caspase-3, bcl-2 expression in paraffin wax-embedded liver sections were performed using a standard peroxidase-antiperoxidase technique as described previously[20], using a CYP2E1 rabbit anti-mouse polyclonal antibody (Abcam Inc, United Kingdom) or mouse monoclonal antibodies against caspase-3 and bcl-2 (Santa Cruz, CA, United States) at a 1:150 dilution, with a biotinylated goat anti-rabbit or goat anti-mouse antibody (Santa Cruz, CA, United States) as the secondary antibody. Brown colour in the cytoplasm of the hepatocytes was evaluated as positive staining.
Determination of antioxidant enzymes activities, MDA and Ca2+ content in liver mitochondria and microsomal CYP2E1-dependent aniline hydroxylase activity
Liver subcellular fractions were prepared by differential centrifugation according to Khemawoot et al[21] with slight modifications. Livers were homogenized (20% w/v) with phosphate buffer (50 mmol/L K2HPO4 containing 0.1 mmol/L EDTA, pH 7.4). The homogenate was centrifuged at 200 g for 10 min at 4 °C, and then the supernatant was recentrifuged at 9000 g for 20 min. The mitochondrial pellet was collected, resuspended in 0.25 mol/L sucrose solution, and stored at -80 °C until detection of superoxide dismutase (SOD), glutathione peroxidase (GSH-Px), malondialdehyde (MDA) and Ca2 + levels using the corresponding kits (Jiancheng, Nanjing, China) respectively.
Then the supernatant was recentrifuged at 105 000 × g for 60 min. The microsomal pellet was washed, resuspended in 0.25 mol/L sucrose solution, and stored at -80 °C until determination of CYP2E1 activity and expression. Microsomal protein concentration was measured according to the method of Lowry et al[22]. Microsomal hydroxylase activity of the CYP2E1-containing enzyme was measured with aniline as the substrate according to the procedure described previously[23].
Western blotting of liver caspase-3, bcl-2 and microsomal CYP2E1
Proteins of liver homogenate or microsome were separated by SDS-PAGE and then transferred onto a polyvinylidene difluoride membrane by electroblotting. Membrane was blocked overnight and then incubated for 2 h with a 1:1000 dilution of a rabbit polyclonal anti-rat CYP2E1 antibody (Abcam Inc, United Kingdom) or a rabbit polyclonal anti-mouse, caspase-3, bcl-2 antibody (Santa Cruz, CA, United States). After incubation with the secondary antibody, proteins were detected with an electrochemiluminescence chemiluminescence detection kit (Amersham Biosciences, Buckinghamshire, United Kingdom), and scanned with a Typhoon 9200 scanner (Amersham Biosciences Europe GmbH, Freiburg, Germany). The amount of protein expression was corrected by the amount of β-actin in the same sample.
Statistical analysis
All data are expressed as mean ± SD. Statistical analysis was performed using Student’s t test. Probability values less than 0.05 (P < 0.05) were considered statistically significant.
RESULTS
Evaluation of animal model
After 18 wk of HFD and prior to ischemia, serum levels of TC, TG and LDL-C in HL or HL+I/R group were significantly higher than those in control or I/R group, whereas HDL-C was lower (P < 0.05 or P < 0.01) (Table 2). There was no significant difference between the HL group and the HL+I/R group.
Table 2.
Blood lipids in different groups after high-fat diet treatment for 18 wk and before middle cerebral artery occlusion (mean ± SD, mmol/L)
| Group | n | TC | TG | HDL-C | LDL-C |
| Control | 10 | 1.69 ± 0.14 | 0.21 ± 0.08 | 1.14 ± 0.05 | 0.40 ± 0.10 |
| HL | 6 | 5.11 ± 0.55be | 0.84 ± 0.20af | 0.68 ± 0.15af | 1.72 ± 0.52ae |
| I/R | 10 | 1.57 ± 0.19 | 0.19 ± 0.02 | 1.21 ± 0.16 | 0.36 ± 0.10 |
| HL+I/R | 10 | 5.33 ± 0.44be | 0.79 ± 0.17af | 0.70 ± 0.19af | 1.64 ± 0.48ae |
HL: Hyperlipidemia; I/R: Ischemia/reperfusion; TC: Total cholesterol; TG: Triglyceride; HDL-C: High-density lipoprotein cholesterol; LDL-C: Low-density lipoprotein cholesterol.
P < 0.05,
P < 0.01 vs control group;
P < 0.05,
P < 0.01 vs I/R group.
At 24 h after reperfusion, rats in the I/R or HL+I/R groups that were successfully operated with MCAO and reperfusion appeared to have a significantly higher neurological deficit score and brain infarct volume than those in the control or HL group (P < 0.01) (Table 3). There was no significant difference between the I/R group and the HL+I/R group.
Table 3.
Brain infarct volume and serum alanine aminotransferase and aspartate aminotransferase in different groups after high-fat diet treatment for 18 wk and/or 2 h transient focal cerebral ischemia followed by 24 h reperfusion (mean ± SD)
| Group | n | Neurological deficit score | Infarct volume (mm3) | ALT (U) | AST (U) |
| Control | 10 | 0 | 0 | 9.56 ± 1.99 | 11.33 ± 4.17 |
| HL | 6 | 0 | 0 | 25.13 ± 16.90b | 18.01 ± 10.00a |
| I/R | 7 | 3.25 ± 0.36bd | 33.4 ± 4.16bd | 12.93 ± 6.14 | 14.00 ± 6.19 |
| HL+I/R | 6 | 3.53 ± 0.45bd | 34.5 ± 6.23bd | 93.62 ± 24.00bdf | 82.32 ± 26.92bdf |
HL: Hyperlipidemia; I/R: Ischemia/reperfusion; ALT: Alanine aminotransferase; AST: Aspartate aminotransferase.
P < 0.05,
P < 0.01 vs control group;
P < 0.01 vs HL group;
P < 0.01 vs I/R group.
These results strongly suggested that the animal model of HL combined with I/R was established successfully.
Liver damage caused by HL plus I/R
Plasma ALT and AST levels are common biomarkers for liver damage[24]. As compared to the control group, plasma activities of ALT and AST were elevated in the HL group (P < 0.05; P < 0.01), but not affected significantly by I/R alone; marked changes in serum ALT and AST activities suggesting hepatic damage were shown in the HL+I/R group (P < 0.01). The most severe liver damage was found in the HL+I/R group compared to HL or I/R alone (P < 0.01) (Table 3).
To analyze the extent of liver injury, liver sections were stained with HE (Figure 1A). No apparent damage was found in liver sections from the control or I/R group. Hepatocytes of the HL group showed fatty infiltration changes, light vacuolar change, scattered inflammatory cells and mild hepatocyte necrosis. However, extensive damage was detected in liver sections from HL+I/R rats. The affected livers displayed remarkable fatty accumulation, infiltration of a mixed population of inflammatory cells, hepatocyte ballooning degeneration and focal necrosis. These changes were more prominent when compared to HL or I/R alone.
Figure 1.
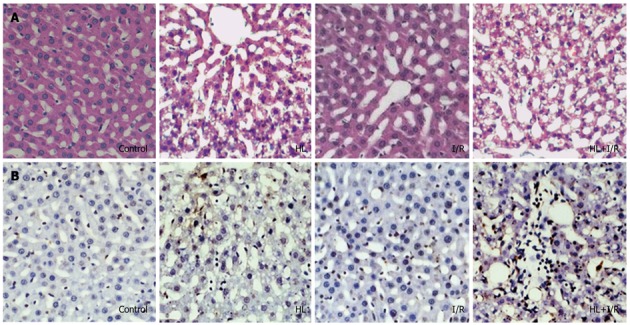
Histology and in situ apoptosis of liver sections from rats after high-fat diet treatment for 18 wk and/or 2 h transient focal cerebral ischemia followed by 24 h reperfusion. A: HE staining showed liver damage in the hyperlipemia (HL) group and especially in the HL+ ischemia/reperfusion (I/R) group (× 200); B: Transferase dUTP nick-end labeling (TUNEL) staining showed TUNEL-positive apoptotic hepatocytes in the HL+I/R group are significantly greater than those in control, HL or I/R groups (× 200). Slides are representative of 6-10 animals per group.
Hepatocyte apoptosis caused by HL plus I/R
We assessed hepatocyte apoptosis using the TUNEL assay. TUNEL-positive cells appeared occasionally in liver sections of control or I/R groups, diffused widely in the HL group, and were most frequently observed in the HL+I/R group (Figure 1B). The number of TUNEL-positive apoptotic hepatocytes (6.2 ± 0.29 apoptotic cells/× 200 field) in the HL+I/R group was significantly greater than those in the control group (1.5 ± 0.22 apoptotic cells/× 200 field), HL group (4.47 ± 0.45 apoptotic cells/× 200 field), and I/R group (1.97 ± 0.47 apoptotic cells/× 200 field) (P < 0.01).
Encouraged by the severe liver damage and hepatocyte apoptosis caused by HL plus I/R, we further investigated whether the CYP2E1, oxidative/antioxidant system and pro-inflammatory cytokines are involved in this pathological condition.
Induction of hepatic CYP2E1 by HL plus I/R
The activities of CYP2E1-dependent aniline hydroxylase activity in the microsome, where CYP2E1 is mainly located in liver, were higher in the HL or the I/R group (P < 0.01; P < 0.05 vs control), and increased considerably especially in the HL+I/R group (P < 0.01 vs control, HL or I/R groups) (Figure 2B). To understand the basis of aniline hydroxylase activity in rat liver microsome, we carried out immunohistochemistry and Western blotting analysis of CYP2E1. Immunohistochemistrical staining for hepatic CYP2E1 was occasionally detectable in the control group, but increased in the HL or I/R groups. In the HL+I/R group, strong and dense CYP2E1 immunoreactivity was observed, which paralleled the improvement in histological severity of liver damage (Figure 2A). Similar effects were observed in Western blotting. The protein expression of microsomal CYP2E1 was induced by HL (P < 0.05 vs control) and I/R (P < 0.05 vs control), and highly enhanced by HL+I/R (P < 0.01 vs control, HL or I/R group) (Figure 2C).
Figure 2.
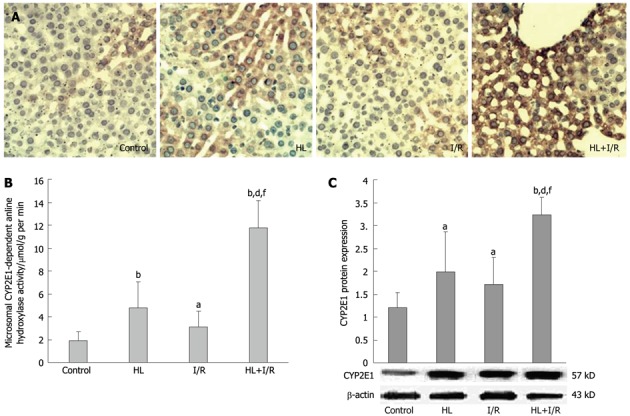
Effect of hyperlipemia plus ischemia/reperfusion on the activity and expression of CYP2E1 in the liver of rats after high-fat diet treatment for 18 wk and/or 2 h transient focal cerebral ischemia followed by 24 h reperfusion. A: Hepatocytes in the hyperlipemia (HL) + ischemia/reperfusion (I/R) group showed increased cytochrome P450 2E1 (CYP2E1) expression examined by immunohistochemical staining (× 200) than those in other groups. Slides are representative of 6-10 animals per group; B: Microsomal hydroxylase activity of the CYP2E1-containing enzyme measured with aniline as the substrate was much higher in the HL+I/R group; C: HL+I/R enhanced the expression level of CYP2E1 protein in liver microsomes, which was confirmed by Western blotting. Values are the mean ± SD. aP < 0.05, bP < 0.01 vs control group; dP < 0.01 vs HL group; fP < 0.01 vs I/R group.
Effect of HL plus I/R on the levels of antioxidant enzymes, MDA and Ca2+ in liver mitochondria
The activities of antioxidant enzymes, including SOD and GSH-Px, fell dramatically in the HL+I/R group (P < 0.01 vs control, P < 0.05 or P < 0.01 vs HL alone, P < 0.01 vs I/R alone). SOD activities of the HL, I/R and HL+I/R groups were, respectively, 41.1%, 70.3% and 24.1% of control level; GSH-Px activities were 43.2%, 81.1% and 22.4% of control level, respectively (Table 4).
Table 4.
Levels of antioxidant enzymes, malondialdehyde and Ca2+ in liver mitochondria of different groups after high-fat diet treatment for 18 wk and/or 2 h transient focal cerebral ischemia followed by 24 h reperfusion (mean ± SD)
| Group | n | SOD (U/μg protein) | GSH-Px (nmol/g protein per min) | MDA (mmol/g protein) | Ca2+ (μmol/g protein) |
| Control | 10 | 244.93 ± 33.70 | 2172.26 ± 241.34 | 7.02 ± 2.58 | 1.329 ± 0.279 |
| HL | 6 | 100.59 ± 40.72b | 937.57 ± 228.68b | 21.84 ± 6.01b | 2.552 ± 0.317b |
| I/R | 7 | 172.15 ± 57.31a | 1762.65 ± 409.18 | 12.58 ± 5.34 | 2.180 ± 0.431b |
| HL+I/R | 6 | 59.01 ± 21.48bcf | 486.41 ± 230.52bdf | 47.27 ± 7.11bcf | 3.136 ± 0.517bcf |
HL: Hyperlipidemia; I/R: Ischemia/reperfusion; SOD: Superoxide dismutase; GSH-Px: Glutathione peroxidase; MDA: Malondialdehyde.
P < 0.05,
P < 0.01 vs control group;
P < 0.05,
P < 0.01 vs HL group;
P < 0.01 vs I/R group.
MDA and Ca2+ levels in liver mitochondria were higher in the HL group than the control group (P < 0.01). Liver mitochondrial Ca2+ levels also increased in the I/R group (P < 0.01 vs control). In the HL+I/R group, MDA and Ca2+ levels were most significantly increased compared with all other groups (P < 0.01 vs control or I/R group, P < 0.05 vs HL group) (Table 4).
Effect of HL plus I/R on the level of serum pro-inflammatory cytokines (TNF-α and IL-1)
The levels of serum TNF-α and IL-1 in HL, I/R or HL+I/R groups were significantly higher compared to those in the control group (P < 0.01; P < 0.05). When compared with HL or I/R alone, the increase of these two pro-inflammatory cytokines in the HL+I/R group also had statistical significance (P < 0.05 vs HL, P < 0.01 vs I/R group) (Figure 3).
Figure 3.
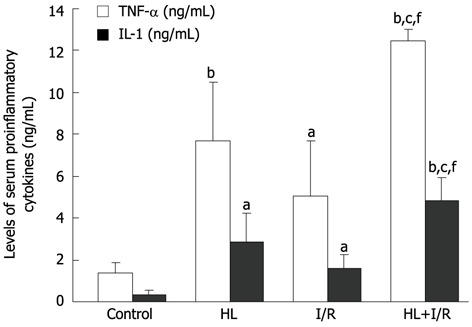
Effect of hyperlipemia + ischemia/reperfusion on the level of serum pro-inflammatory cytokines after high-fat diet treatment for 18 wk and/or 2 h transient focal cerebral ischemia followed by 24 h reperfusion. Serum pro-inflammatory cytokines in the hyperlipemia (HL) + ischemia/reperfusion (I/R) group were significantly higher compared to those in other groups. A: Tumor necrosis factor (TNF)-α; B: Interleukin (IL)-1. Values are the mean ± SD. aP < 0.05, bP < 0.01 vs control group; cP < 0.05 vs HL group; fP < 0.01 vs I/R group.
Effect of HL plus I/R on the expressions of caspase-3 and bcl-2
We further investigated the expression levels of two key apoptosis-related genes- caspase-3 and bcl-2-in liver tissue, which were determined by immunohistochemistry and Western blotting. Immunohistochemistry assay showed protein expression of caspase-3 (Figure 4A) in livers of HL and HL+I/R rats was markedly increased, and the anti-apoptosis gene bcl-2 (Figure 4B) was dramatically decreased. These findings were confirmed by Western blotting, which showed increased expression of caspase-3 (Figure 4C) and decreased expression of bcl-2 (Figure 4D), especially in the HL+I/R group.
Figure 4.
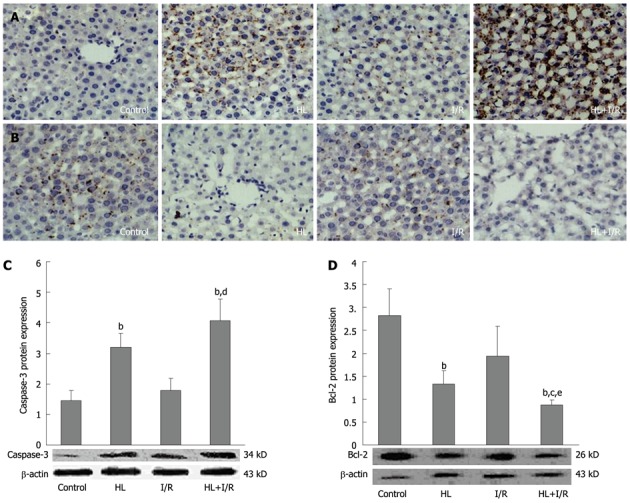
Expressions of bcl-2 and caspase-3 protein in liver after high-fat diet treatment for 18 wk and/or 2 h transient focal cerebral ischemia followed by 24 h reperfusion. Liver sections from different groups were immunohistochemically stained to assay the expression of caspase-3 and bcl-2 (× 200). A: Protein expression of caspase-3 was markedly elevated in the hyperlipemia (HL) + ischemia/reperfusion (I/R) group; B: Protein expression of bcl-2 was dramatically decreased in the HL+I/R group. C, D: Slides are representative of 6-10 animals per group. Liver homogenates were prepared, and then the expression levels of caspase-3 (C) and bcl-2 (D) proteins in liver were confirmed by Western blotting. Values are the mean ± SD. bP < 0.01 vs control group; cP < 0.05, dP < 0.01 vs HL group; eP < 0.05 vs I/R group.
DISCUSSION
Dyslipidemia is usually responsible for liver disease including cirrhosis, hepatocellular carcinoma and liver failure[5]. It has also been reported that “high-fat diet” can induce lipid accumulation in rat liver by nutritional intervention[25]. In agreement with previous work[26-28], our study also demonstrated that HL alone induced by HFD for 18 wk resulted in considerable increases in serum ALT and AST levels, marked pathological changes in the liver, increased TUNEL-positive cells, higher caspase-3 and lower bcl-2 expression, suggesting liver damage and hepatocyte apoptosis. Oxidative stress and subsequent lipid peroxidation, together with the production of proinflammatory cytokines have been shown to play important roles in the pathogenesis of nonalcoholic fatty liver disease induced by HFD[29,30], which were also confirmed in our current experiment.
Besides liver damage caused by long-term HFD, our findings are consistent with the literature stating ischemic stroke per se is possibly associated with liver dysfunction[31]. Although no significant change in serum ALT and AST levels, histologic examination of liver, the number of TUNEL-positive cells, caspase-3 and bcl-2 expression in liver was found, systemic inflammation and the imbalance in oxidant production and antioxidant defense systems, which are strongly associated with the development of MODS due to I/R, were indeed discovered in the I/R group. Actually, circulating pro-inflammatory cytokines, for example, TNF-α and IL-1, and systemic oxidative stress would influence the nonneurological organs including liver[32].
Taken together, HL per se induced systemic inflammation and oxidative stress, and eventually caused hepatocyte apoptosis and liver damage, while I/R per se also induced systemic inflammation and oxidative stress but did not cause obvious hepatocyte apoptosis and liver damage.
Most interestingly, we demonstrated that, as expected, the combined action of HL and I/R brought about an abrupt reduction in hepatic function with enhanced hepatocyte apoptosis, which is much worse than the damage caused by I/R or HL alone. The incidence of ischemic stroke increases dramatically with advancing age. Due to aging, many patients with cerebral ischemia also suffer from HL, which is another common geriatric disease. It has also been reported that HL, as a cardiovascular risk factor, would lead to intracranial artery atherosclerosis or blockage, aggravate cerebral infarction, and exacerbate the deleterious effects of I/R on the brain[33]. In our study, we found the coexistence of HL and cerebral I/R injury would be more dangerous because it induces more severe visceral dysfunction i.e., liver damage than HL alone. This kind of systemic complication may be fatal subsequently.
This liver damage secondary to the coexistence of HL and cerebral I/R injury can be explained readily by the high levels of circulating pro-inflammatory cytokines and oxidative stress, which possibly result from the synergy of systemic inflammatory and oxidative milieu induced by HL per se and those induced by I/R per se. In support of these concepts, we showed that, compared with I/R or HL alone, enhanced CYP2E1 induction, an increase in serum TNF-α, IL-1, liver mitochondrial MDA, and a decrease in SOD and GSH-Px in liver mitochondria were also measured in the HL+I/R group, which indicated that the coexistence of HL with cerebral I/R injury is associated with increased systemic inflammation and oxidative stress.
A significant change in CYP2E1 should be noted in the liver of HL rats with cerebral I/R injury. CYP2E1, sometimes mediating liver damage through oxidative stress[34,35], is a major adaptive system in the CYP super-family. CYP2E1 has adjustability, and is inducible under a variety of physiological or pathophysiological conditions. It has been demonstrated that CYP2E1 levels increased in obese rats by feeding with an energy-dense diet[14] and responded quickly to acute stress, such as hypoxia, cold or hunger[36]. In fact, acute cerebral I/R injury also induces an acute stress condition in animals. In the present study, we have corroborated the induction of CYP2E1 by HL per se or I/R per se, especially I/R combined with HL, as an increase in hepatic CYP2E1 activity and protein expression was found in the HL and I/R groups, and especially in the HL+I/R group. CYP2E1 is known to be a key indicator of lipid peroxidation, and a significant source of ROS[12] contributing to hepatocellular injury. Enhanced CYP2E1 induction oxidizes mitochondrial DNA, proteins and lipids, and triggers hepatic TNF-α formation by activating nuclear factor-κB, which further increases mitochondrial ROS formation. ROS overproduction leads to inflammatory recruitment and apoptosis from oxidative stress[11], and finally exacerbates the disease.
Consistent with the change in MDA, we also revealed that mitochondrial Ca2+ content increased in the HL+I/R group. An imbalance of Ca2+ homeostasis is thought to be the “common pathway” of hepatocyte damage. Mitochondrial and cellular Ca2+ overload facilitates lipid peroxidation, which attenuates the oxidative phosphorylation of the mitochondrion resulting in damage to the mitochondrial structure and function and then the reduction of ATP synthesis. On the other hand, lipid peroxidation will in turn exacerbate the Ca2+ overload by acting on the membrane structure[37]. Accordingly, mitochondrial Ca2+ overload of hepatocytes is closely relevant to lipid peroxidation, and both are likely to be involved in hepatic injury after HL+I/R.
Moreover, we have evaluated, by immunohistochemistry and Western blotting analysis, two key mediators of apoptosis (caspase-3 and bcl-2), which appear to be adjusted by ROS and cytokines, including TNF-α and IL-1[32,38]. Increased protein expression of key pro-apoptotic factor caspase-3 and markedly down-regulated anti-apoptotic bcl-2 detected in livers of HL rats with cerebral I/R injury in our study which resulted in hepatocyte apoptosis and liver damage.
In summary, based on an experimental HL rat model with cerebral I/R, the present study revealed that the coexistence of HL and acute cerebral I/R induced severe liver damage. These findings suggested cerebral ischemic stroke would exaggerate the damage of liver caused by HL. So it can be reckoned that patients of HL combined with cerebral I/R injury have a higher risk of liver damage compared with patients with HL or cerebral I/R injury alone, the mechanism of which is possibly due to enhanced CYP2E1 induction, which can further increase mitochondrial ROS formation, oxidative damage, inflammation and apoptosis with increased protein expression of key pro-apoptotic factor caspase-3, and markedly down-regulated anti-apoptotic bcl-2. Therefore, special precautions against subsequent liver injury are required after the burst of cerebral ischemic stroke in patients with HL. It seems that the therapeutic approaches to inhibit CYP2E1 activation, rebalance the oxidative/antioxidant system, and suppress inflammation or apoptosis might be efficacious in preventing the liver damage induced by HL plus cerebral I/R injury.
COMMENTS
Background
The history of hyperlipidemia (HL), an increasingly public health problem, is positively associated with acute cerebrovascular diseases (ACD) such as ischemic stroke. Moreover, HL is also a major risk factor for liver disease. One of the main complications of the patient with ACD is the risk of secondary liver dysfunction. It is thus reasonable to hypothesize that the coexistence of HL and ACD may contribute to more severe liver dysfunction.
Research frontiers
The pathological changes in liver caused by either HL or cerebral ischemia are associated with inflammation and oxidative stress. An inflammatory response and even apoptosis could be triggered by oxidative stress, which may be a key mechanism of liver injury induced by HL or cerebral ischemia. The source of oxidative stress is controversial but may result in part from over-expression of the prooxidant enzyme cytochrome P450 2E1 (CYP2E1). CYP2E1 has adjustability, and is inducible under a variety of physiological or pathophysiological conditions. It has been demonstrated that CYP2E1 levels are enhanced in obese rats by feeding with an energy-dense diet and respond quickly to acute stress, such as hypoxia, cold or hunger.
Innovations and breakthroughs
HL or ACD have been known to be independent risk factors for liver injury. In this study, the authors developed a repeatable experimental hyperlipemic rat model with acute cerebral ischemia/reperfusion injury, and investigated the correlation of HL and/or acute cerebral ischemia/reperfusion injury on liver damage, and further examine the effects of HL and/or ischemia/reperfusion on hepatocyte apoptosis, CYP2E1, the oxidative/antioxidant system and inflammatory process to understand the mechanism of liver damage.
Applications
The study results suggest that cerebral ischemic stroke would exaggerate the damage of liver caused by HL. So it can be reckoned that patients with HL combined with acute cerebral ischemia/reperfusion injury have a higher risk of liver damage compared with patients with HL or cerebral ischemia/reperfusion injury alone, the mechanism of which is possibly due to enhanced CYP2E1 induction, which can further increase mitochondrial reactive oxygen species formation, oxidative damage, inflammation and apoptosis with increased protein expression of key pro-apoptotic factor caspase-3, and markedly down-regulated anti-apoptotic bcl-2. Therefore, special precautions against subsequent liver injury are required after the burst of cerebral ischemic stroke in patients with HL. It seems that therapeutic approaches to inhibit CYP2E1 activation, rebalance the oxidative/antioxidant system, suppress inflammation or apoptosis might be efficacious in prevention and treatment of liver damage induced by HL plus cerebral ischemia/reperfusion injury.
Peer review
The topic is of great clinical importance as the prevalence and incidence of hyperlipidemia and related cerebrovascular diseases as well as non-alcoholic fatty liver disease are increasing in industrialized countries; hence the associated financial problems is a major point to cope with. The rat model based study is well designed and the results are significant.
Footnotes
Peer reviewer: Ferenc Sipos, MD, PhD, Cell Analysis Laboratory, 2nd Department of Internal Medicine, Semmelweis University, Szentkirályi u. 46., Budapest 1088, Hungary
S- Editor Cheng JX L- Editor O’Neill M E- Editor Zhang DN
References
- 1.Zheng L, Sun Z, Li J, Yu J, Wei Y, Zhang X, Liu S, Li J, Xu C, Hu D, et al. Mean arterial pressure: a better marker of stroke in patients with uncontrolled hypertension in rural areas of China. Intern Med. 2007;46:1495–1500. doi: 10.2169/internalmedicine.46.0178. [DOI] [PubMed] [Google Scholar]
- 2.Engström G, Lind P, Hedblad B, Stavenow L, Janzon L, Lindgärde F. Effects of cholesterol and inflammation-sensitive plasma proteins on incidence of myocardial infarction and stroke in men. Circulation. 2002;105:2632–2637. doi: 10.1161/01.cir.0000017327.69909.ff. [DOI] [PubMed] [Google Scholar]
- 3.Yang RL, Shi YH, Hao G, Li W, Le GW. Increasing Oxidative Stress with Progressive Hyperlipidemia in Human: Relation between Malondialdehyde and Atherogenic Index. J Clin Biochem Nutr. 2008;43:154–158. doi: 10.3164/jcbn.2008044. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Lagowska-Lenard M, Stelmasiak Z, Bartosik-Psujek H. Influence of vitamin C on markers of oxidative stress in the earliest period of ischemic stroke. Pharmacol Rep. 2010;62:751–756. doi: 10.1016/s1734-1140(10)70334-0. [DOI] [PubMed] [Google Scholar]
- 5.Ma X, Li Z. Pathogenesis of nonalcoholic steatohepatitis (NASH) Chin J Dig Dis. 2006;7:7–11. doi: 10.1111/j.1443-9573.2006.00237.x. [DOI] [PubMed] [Google Scholar]
- 6.Brunt EM. Nonalcoholic steatohepatitis. Semin Liver Dis. 2004;24:3–20. doi: 10.1055/s-2004-823098. [DOI] [PubMed] [Google Scholar]
- 7.Palasik W, Fiszer U, Lechowicz W, Czartoryska B, Krzesiewicz M, Lugowska A. Assessment of relations between clinical outcome of ischemic stroke and activity of inflammatory processes in the acute phase based on examination of selected parameters. Eur Neurol. 2005;53:188–193. doi: 10.1159/000086355. [DOI] [PubMed] [Google Scholar]
- 8.Liu HB, Tian J, Zhao JX, Song DB, Tian JK. Study on the clinical epidemiological features of acute cerebral stroke inducing systemic inflammatory response syndrome and multiple organ dysfunction syndrome. Zhonghua Liu Xing Bing Xue Zazhi. 2008;29:294–296. [PubMed] [Google Scholar]
- 9.Molchanova LV, Chernobaeva GN, Shcherbakova LN, Luk’ianova LD. [Effect of occlusive cerebral ischemia on functional status of the internal organs] Anesteziol Reanimatol. 2001;(6):54–56. [PubMed] [Google Scholar]
- 10.Cao MH. Acute cerebrovascular disease with multiple organ dysfunction syndrome in 161 cases. J Nantong University ( Medical Sciences) 2001;21:192–193. [Google Scholar]
- 11.Schreuder TC, Verwer BJ, van Nieuwkerk CM, Mulder CJ. Nonalcoholic fatty liver disease: an overview of current insights in pathogenesis, diagnosis and treatment. World J Gastroenterol. 2008;14:2474–2486. doi: 10.3748/wjg.14.2474. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Bradford BU, Kono H, Isayama F, Kosyk O, Wheeler MD, Akiyama TE, Bleye L, Krausz KW, Gonzalez FJ, Koop DR, et al. Cytochrome P450 CYP2E1, but not nicotinamide adenine dinucleotide phosphate oxidase, is required for ethanol-induced oxidative DNA damage in rodent liver. Hepatology. 2005;41:336–344. doi: 10.1002/hep.20532. [DOI] [PubMed] [Google Scholar]
- 13.Robin MA, Anandatheerthavarada HK, Fang JK, Cudic M, Otvos L, Avadhani NG. Mitochondrial targeted cytochrome P450 2E1 (P450 MT5) contains an intact N terminus and requires mitochondrial specific electron transfer proteins for activity. J Biol Chem. 2001;276:24680–24689. doi: 10.1074/jbc.M100363200. [DOI] [PubMed] [Google Scholar]
- 14.Raucy JL, Lasker JM, Kraner JC, Salazar DE, Lieber CS, Corcoran GB. Induction of cytochrome P450IIE1 in the obese overfed rat. Mol Pharmacol. 1991;39:275–280. [PubMed] [Google Scholar]
- 15.Poloyac SM, Perez A, Scheff S, Blouin RA. Tissue-specific alterations in the 6-hydroxylation of chlorzoxazone following traumatic brain injury in the rat. Drug Metab Dispos. 2001;29:296–298. [PubMed] [Google Scholar]
- 16.Nepal S, Malik S, Sharma AK, Bharti S, Kumar N, Siddiqui KM, Bhatia J, Kumari S, Arya DS. Abresham ameliorates dyslipidemia, hepatic steatosis and hypertension in high-fat diet fed rats by repressing oxidative stress, TNF-α and normalizing NO production. Exp Toxicol Pathol. 2011:Epub ahead of print. doi: 10.1016/j.etp.2011.01.003. [DOI] [PubMed] [Google Scholar]
- 17.Shimamura N, Matchett G, Tsubokawa T, Ohkuma H, Zhang J. Comparison of silicon-coated nylon suture to plain nylon suture in the rat middle cerebral artery occlusion model. J Neurosci Methods. 2006;156:161–165. doi: 10.1016/j.jneumeth.2006.02.017. [DOI] [PubMed] [Google Scholar]
- 18.Longa EZ, Weinstein PR, Carlson S, Cummins R. Reversible middle cerebral artery occlusion without craniectomy in rats. Stroke. 1989;20:84–91. doi: 10.1161/01.str.20.1.84. [DOI] [PubMed] [Google Scholar]
- 19.Feldstein AE, Canbay A, Angulo P, Taniai M, Burgart LJ, Lindor KD, Gores GJ. Hepatocyte apoptosis and fas expression are prominent features of human nonalcoholic steatohepatitis. Gastroenterology. 2003;125:437–443. doi: 10.1016/s0016-5085(03)00907-7. [DOI] [PubMed] [Google Scholar]
- 20.Kapucuoglu N, Coban T, Raunio H, Pelkonen O, Edwards RJ, Boobis AR, Iscan M. Immunohistochemical demonstration of the expression of CYP2E1 in human breast tumour and non-tumour tissues. Cancer Lett. 2003;196:153–159. doi: 10.1016/s0304-3835(03)00277-5. [DOI] [PubMed] [Google Scholar]
- 21.Khemawoot P, Yokogawa K, Shimada T, Miyamoto K. Obesity-induced increase of CYP2E1 activity and its effect on disposition kinetics of chlorzoxazone in Zucker rats. Biochem Pharmacol. 2007;73:155–162. doi: 10.1016/j.bcp.2006.09.006. [DOI] [PubMed] [Google Scholar]
- 22.Lowry OH, Rosebrough NJ, Farr AL, Randall RJ. Protein measurement with the Folin phenol reagent. J Biol Chem. 1951;193:265–275. [PubMed] [Google Scholar]
- 23.Pahan K, Smith BT, Singh AK, Singh I. Cytochrome P-450 2E1 in rat liver peroxisomes: downregulation by ischemia/reperfusion-induced oxidative stress. Free Radic Biol Med. 1997;23:963–971. doi: 10.1016/s0891-5849(97)00017-8. [DOI] [PubMed] [Google Scholar]
- 24.Adams LA, Angulo P, Lindor KD. Nonalcoholic fatty liver disease. CMAJ. 2005;172:899–905. doi: 10.1503/cmaj.045232. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Asai A, Miyazawa T. Dietary curcuminoids prevent high-fat diet-induced lipid accumulation in rat liver and epididymal adipose tissue. J Nutr. 2001;131:2932–2935. doi: 10.1093/jn/131.11.2932. [DOI] [PubMed] [Google Scholar]
- 26.Svegliati-Baroni G, Candelaresi C, Saccomanno S, Ferretti G, Bachetti T, Marzioni M, De Minicis S, Nobili L, Salzano R, Omenetti A, et al. A model of insulin resistance and nonalcoholic steatohepatitis in rats: role of peroxisome proliferator-activated receptor-alpha and n-3 polyunsaturated fatty acid treatment on liver injury. Am J Pathol. 2006;169:846–860. doi: 10.2353/ajpath.2006.050953. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Lieber CS, Leo MA, Mak KM, Xu Y, Cao Q, Ren C, Ponomarenko A, DeCarli LM. Model of nonalcoholic steatohepatitis. Am J Clin Nutr. 2004;79:502–509. doi: 10.1093/ajcn/79.3.502. [DOI] [PubMed] [Google Scholar]
- 28.Zou Y, Li J, Lu C, Wang J, Ge J, Huang Y, Zhang L, Wang Y. High-fat emulsion-induced rat model of nonalcoholic steatohepatitis. Life Sci. 2006;79:1100–1107. doi: 10.1016/j.lfs.2006.03.021. [DOI] [PubMed] [Google Scholar]
- 29.Farrell GC, Larter CZ. Nonalcoholic fatty liver disease: from steatosis to cirrhosis. Hepatology. 2006;43:S99–S112. doi: 10.1002/hep.20973. [DOI] [PubMed] [Google Scholar]
- 30.McCullough AJ. Pathophysiology of nonalcoholic steatohepatitis. J Clin Gastroenterol. 2006;40 Suppl 1:S17–S29. doi: 10.1097/01.mcg.0000168645.86658.22. [DOI] [PubMed] [Google Scholar]
- 31.Fradette C, Du Souich P. Effect of hypoxia on cytochrome P450 activity and expression. Curr Drug Metab. 2004;5:257–271. doi: 10.2174/1389200043335577. [DOI] [PubMed] [Google Scholar]
- 32.Tilg H. Cytokines and liver diseases. Can J Gastroenterol. 2001;15:661–668. doi: 10.1155/2001/746736. [DOI] [PubMed] [Google Scholar]
- 33.Ishikawa M, Stokes KY, Zhang JH, Nanda A, Granger DN. Cerebral microvascular responses to hypercholesterolemia: roles of NADPH oxidase and P-selectin. Circ Res. 2004;94:239–244. doi: 10.1161/01.RES.0000111524.05779.60. [DOI] [PubMed] [Google Scholar]
- 34.Abdelmegeed MA, Moon KH, Chen C, Gonzalez FJ, Song BJ. Role of cytochrome P450 2E1 in protein nitration and ubiquitin-mediated degradation during acetaminophen toxicity. Biochem Pharmacol. 2010;79:57–66. doi: 10.1016/j.bcp.2009.07.016. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35.Lu Y, Cederbaum AI. CYP2E1 and oxidative liver injury by alcohol. Free Radic Biol Med. 2008;44:723–738. doi: 10.1016/j.freeradbiomed.2007.11.004. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36.Bayanov AA, Brunt AR. Role of hypoxia and constitutionally different resistance to hypoxia/stress as the determiners of individual profile of cytochrome P450 isozyme activity. Gen Pharmacol. 1999;33:355–361. doi: 10.1016/s0306-3623(98)00288-2. [DOI] [PubMed] [Google Scholar]
- 37.Lin MS, Miao HL, Gong XG, Bao ST. Effect of L-arginine on calcium in hepatic mitochondrion in rats with obstructive jaundice. Hepatobiliary Pancreat Dis Int. 2006;5:432–435. [PubMed] [Google Scholar]
- 38.Singh R, Czaja MJ. Regulation of hepatocyte apoptosis by oxidative stress. J Gastroenterol Hepatol. 2007;22 Suppl 1:S45–S48. doi: 10.1111/j.1440-1746.2006.04646.x. [DOI] [PubMed] [Google Scholar]