

Abstract
We sought to determine direct vascular effects of peroxisome proliferator-activated receptor alpha (PPARα) agonists using isolated mouse aortas and middle cerebral arteries (MCAs). The PPARα agonists GW7647, WY14643, and gemfibrozil acutely relaxed aortas held under isometric tension and dilated pressurized MCAs with the following order of potency: GW7647≫WY14643>gemfibrozil. Responses were endothelium-independent, and the use of PPARα deficient mice demonstrated that responses were also PPARα-independent. Pretreating arteries with high extracellular K+ attenuated PPARα agonist-mediated relaxations in the aorta, but not in the MCA. In the aorta, the ATP sensitive potassium (KATP) channel blocker glibenclamide also impaired relaxations whereas the other K+ channel inhibitors, 4-aminopyridine and Iberiotoxin, had no effect. In aortas, GW7647 and WY14643 elevated cGMP levels by stimulating soluble guanylyl cyclase (sGC), and inhibition of sGC with ODQ blunted relaxations to PPARα agonists. In the MCA, dilations were inhibited by the protein kinase C (PKC) activator, phorbol 12,13-dibutyrate, and also by ODQ. Our results demonstrated acute, nonreceptor-mediated relaxant effects of PPARα agonists on smooth muscle of mouse arteries. Responses to PPARα agonists in the aorta involved KATP channels and sGC, whereas in the MCA the PKC and sGC pathways also appeared to contribute to the response.
1. Introduction
Peroxisome proliferator-activated receptors (PPARs), of which there are three subtypes (α, γ, and β/δ), are clinically important pharmacological targets for the treatment of diabetes, dyslipidemia, and metabolic syndrome [1]. First to be cloned, PPARα is widely expressed in liver, heart, skeletal muscle, brown adipose, endothelium, and vascular smooth muscle [2–8]. Biologic PPARα agonists consist of saturated and unsaturated fatty acids, eicosanoids, and glucocorticoids [9–15]. Synthetic PPARα agonists include herbicides, plasticizers, fibrates, WY14643, and GW7647. Fibrates are in clinical use and have cardioprotective effects including reduced death from coronary heart disease, and prevention of myocardial infarction [16–21]. In addition, fibrates reduce stroke occurrence [22], decrease atherosclerosis [23], suppress inflammatory responses in vascular smooth muscle cells [7, 24, 25], and enhance nitric oxide (NO•) production in endothelial cells [26]. Although not in clinical use, GW7647 prevents atherosclerosis in hyperlipidemic mice [27], and WY14643 suppresses the inflammatory response in human aortic smooth muscle cells [7]. Thus, PPARα agonists appear to protect the cardiovascular system from inflammation and disease.
Although PPARα agonists positively impact cardiovascular outcomes, their effects specifically in the vasculature are less well understood. Treating mice with fenofibrate for ten days enhanced endothelium-dependent dilation of resistance (mesenteric) and large conduit (aorta) arteries, possibly by increasing responsiveness to NO• [28]. Likewise, fourteen days in vivo treatment with fenofibrate modestly improved endothelium-dependent dilation of the mouse middle cerebral artery (MCA) [29]. Feeding low levels of WY14643 to mice over ten days also resulted in reduced systolic pressure [30]. Emerging evidence suggests that PPARα agonists also have acute, possibly nonreceptor-mediated, effects such as visceral analgesia [31], increased insulin-induced glucose uptake [32], and stimulation of mitogen-activated protein kinases [33–35]. In the cardiovascular system, another PPARα agonist, gemfibrozil, acutely lowered systemic arterial pressure, and directly relaxed tail arteries of rats by an undefined smooth muscle-dependent mechanism [36]. The fibrate compounds gemfibrozil, fenofibric acid, and bezafibrate also relaxed the rat thoracic aorta evidently by decreasing intracellular calcium, albeit at relatively high concentrations [37]. Therefore, PPARα agonists appear to have both long-term (genomic and possibly nongenomic), as well as short-term (probably nongenomic and possibly nonreceptor-mediated) beneficial effects for the cardiovascular system. Thus, we aimed to determine direct effects of PPARα agonists on isolated arteries and to delineate the mechanism by which they cause arterial relaxations.
Based on previous results, we predicted that PPARα agonists would promote arterial relaxation. Using isometric tension and isobaric myography we examined the ability of the PPARα agonists gemfibrozil, WY14643, and GW7647 to acutely relax the mouse aorta and to dilate the MCA. In addition, we sought to define the mechanism of action of the observed relaxant effect by using different pharmacological inhibitors and PPARα-deficient (Pparα −/−) mice. Our results demonstrate that PPARα agonists caused relaxation of mouse aorta by activating soluble guanylyl cyclase (sGC) and ATP sensitive potassium (KATP) channels. The dilatory response in the MCA, however, involved activation of sGC as well as inhibition of protein kinase C (PKC).
2. Materials and Methods
2.1. Animals and Reagents
Male C57BL/6J and PPARα-deficient (Pparα −/−) mice (aged 12–16 weeks) were purchased from The Jackson Laboratory (Bar Harbor, ME, USA). Mice were euthanized by CO2 inhalation and decapitated prior to tissue harvesting. Deletion of the PPARα gene was confirmed by using primers available from the Jackson Laboratory database using standard PCR conditions. The Animal Care and Use Committee at the University of Missouri-Kansas City approved all protocols. All reagents were sourced from Sigma (St. Louis, MO, USA) unless otherwise noted. Stock solutions of PPARα agonists were prepared in DMSO and diluted in Krebs buffer prior to use. Concentrations of DMSO in the bath never exceeded 0.01% and controls were always vehicle treated.
2.2. Isobaric Vessel Studies: MCA
Brains were quickly removed and placed in ice-cold Hank's buffered saline solution (HBSS, Invitrogen, Carlsbad, CA, USA). MCAs were studied in a pressurized artery myograph (DMT-USA, Ann Arbor, MI, USA) as previously described [38–41]. Briefly, MCAs were carefully dissected away from the brain, cleared of blood and pia mater, mounted on glass micropipettes, and pressurized to 70 mm Hg with Krebs buffer (in mM: 119 NaCl, 4.7 KCl, 0.24 NaHCO3, 1.18 KH2PO4, 1.19 MgSO4, 5.5 glucose, and 1.6 CaCl2). Elevated external K+ buffers were made isotonic by replacement of NaCl with KCl on an equimolar basis. Prior to beginning experiments smooth muscle function was verified with 60 mM KCl-induced contractions whereas endothelial function in phenylephrine (PE, 10−5 M) preconstricted MCAs was probed by determining dilation to 10−5 M acetylcholine. MCA's were denuded by passage of 1 mL of air, over a period of 8 min at 60 mm Hg as previously described [38]. MCAs were preconstricted with 10−5 M PE prior to determining dilatory responses to PPARα agonists.
2.3. Isometric Tension Myography: Aorta
The thoracic aorta was rapidly excised and placed in ice-cold HBSS where blood, fat, and excess connective tissues were carefully removed. Segments 3-4 mm in length were mounted on pins in chambers of a DMT 610 M wire myograph system (Danish Myo Technology A/S, Aarhus N, Denmark) containing Krebs buffer saturated at 37°C with a gas mixture containing 20% O2/5% CO2/75% N2 (Airgas Mid South Inc., Tulsa, OK, USA). Arterial rings were progressively stretched to 0.75 g equivalent force passive tension in 0.1 g steps and allowed to equilibrate for 45 minutes described previously [41]. Aortic rings were exposed to isotonic KCl (40 and 80 mM), and also to 10−5 M Prostaglandin F2-alpha (PGF2α) followed by 10−6 μM acetylcholine (ACh) to assess the quality of the preparation. Vessels were rinsed once with fresh Krebs every 15 min and several times after concentration-response curves. Dose-response curve to PPARα agonists was determined after preconstriction of aortic segments with 10−5 M PGF2α. In pharmacological inhibition experiments, selective agents were added 15–30 minutes prior to preconstriction with PGF2α, and then relaxation response to PPARα agonists was determined. For denudation, the forceps were inserted into the lumen, and the aortic ring was gently rolled over. This process was repeated 5 times. The effectiveness of endothelium removal was confirmed by the absence of relaxation by 10−6 μM Ach in aortic rings precontracted with 10−5 M PGF2α. Force changes were recorded using an ADinstruments (Colorado Springs, CO, USA) PowerLab 4/30 and associated LabChart Pro software (v6.1) running on a standard Windows XP computer platform.
2.4. Determination of Cyclic Guanosine Monophosphate (cGMP) Content
The thoracic aorta was excised and cleaned of fat and excess connective tissue in ice-cold HBSS. The aorta was cut into rings of 3 mm in length and treated with PDE inhibitor 3-isobutyl-1-methylxanthine (IBMX; 10−1 M) in HBSS for 15 minutes. Aortic rings were then incubated with vehicle (0.1% DMSO), GW7647 (10−5 M), WY14643 (10−4 M), 1H-[1,2,4]oxadiazolo[4,3-a]quinoxalin-1-one (ODQ, 10−5 M), SNP (10−5 M), the combination of GW7647 with ODQ, and WY14643 with ODQ, or SNP and ODQ for 15 minutes. After incubation aortic rings were immediately frozen in liquid nitrogen and stored at −80°C. For cGMP extraction, rings were homogenized in five volumes of ice-cold 5% (w/v) trichloroacetic acid followed by centrifugation at 2000 ×g for 15 minutes at 4°C. The supernatant was recovered, extracted four times with five volumes of water-saturated diethyl ether, and the extracted samples were then evaporated to dryness. cGMP content in the extracted samples was then determined by using a commercially available cGMP EIA kit (Caymen Chemical, Ann Arbor, MI, USA) according to the manufacturer's instructions.
2.5. Statistics
Data are plotted and expressed as means ± SEM and n-values are detailed in the legends to the figures. In myograph experiments, changes in isometric tension are expressed as % relaxation. Changes in the diameter of pressurized and MCAs were calculated as % dilation as previously described [38]. Two-factor ANOVA was used to determine differences between concentration-response curves. One-factor ANOVA with Tukey's multiple-comparison post hoc tests were used cGMP assay data. Where appropriate, data normality was examined with D'Agostino and Pearson tests and homogeneity of variance was determined with Bartlett's test. Data were plotted and statistics computed with Graphpad Prism (v5.01, San Diego, CA, USA). Significance was accepted at P ≤ 0.05.
3. Results
3.1. Responses to PPARα Agonists in Aortas and MCAs
Previous studies demonstrated that relatively nonselective PPARα agonists such as gemfibrozil acutely caused vasodilation of rat tail arteries, and relaxation of rat thoracic aorta [36, 37]. To expand upon and investigate this further we determined the direct and acute effects of three PPARα agonists, gemfibrozil, WY14643, and GW7647, on isolated mouse arteries. In binding assays these agonists range from moderately specific and selective for PPARα (gemfibrozil) to very specific and selective (GW7647) [42]. Cumulative addition of each agonist (10−7 M to 10−4 M) to the bath caused concentration-dependent relaxation of precontracted aortas; however, the effectiveness varied greatly for each agonist (Figure 1(a)). For example, the relaxation caused by 10−4 M GW7647 was 95 ± 1% whereas the same concentration of WY14643 only caused 40 ± 9% relaxation. Even at 10−4 M, gemfibrozil had little to no effect causing just 4 ± 3% relaxation of the aorta. Similar to the aorta, at 10−4 M GW7647 maximally dilated the MCA, whereas dilation to 10−4 M WY14643 and gemfibrozil was 60 ± 11% and 28 ± 5%, respectively, (Figure 1(b)). Vehicle (DMSO) treatment did not have any effect on aortic tension or MCA dilation (Figures 1(a) and 1(b)). Figure 1 also shows that the dilatory potency increased in parallel with selectivity for PPARα such that the highly selective (in specialized assays) PPARα agonist GW7647 was substantially more potent than WY14643 or gemfibrozil.
Figure 1.
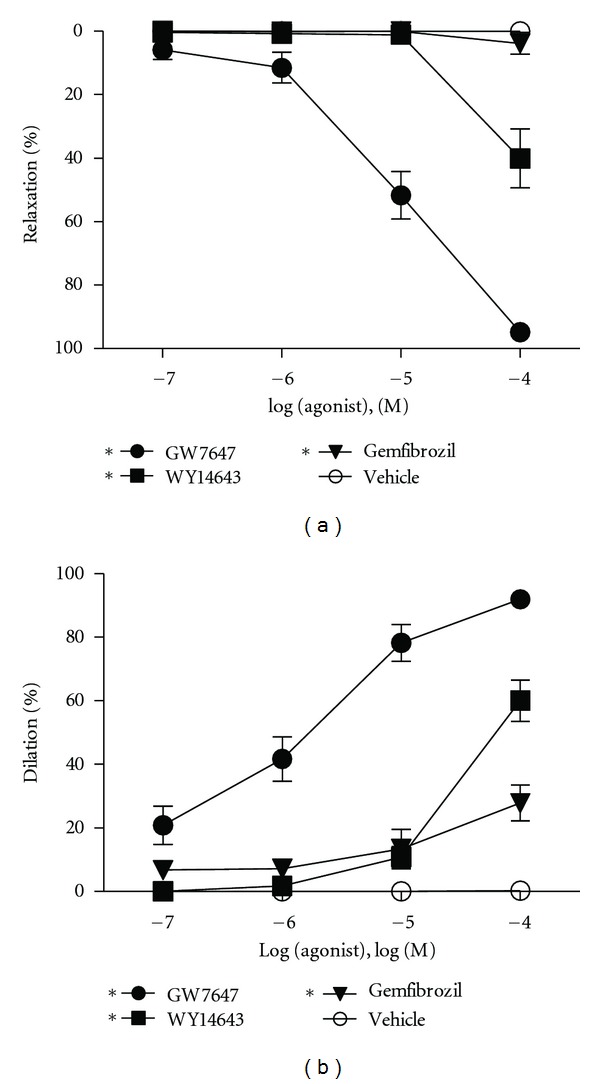
Response to PPARα agonists in aortas and MCAs of WT mice. Concentration-response curves to three different PPARα agonists (gemfibrozil, WY14643, and GW7647) in (a) mouse aortic rings precontracted with 10−5 M PGF2α by using isometric tension myography and (b) MCAs preconstricted with 10−5 M PE by using isobaric myography. All three PPARα agonists relaxed aortic rings (n = 3–5) as well as dilated the MCA (n = 7). Data are means ± SEM. *Indicates P ≤ 0.05.
3.2. Role of PPARα Receptors in Vascular Responses to PPARα Agonists
To determine if the response caused by PPARα agonists was receptor-mediated, we utilized Pparα −/− mice that do not express PPARα. Compared to WT mice, aortic responses were unaltered in Pparα −/− mice for both GW7647 and WY14643 (Figures 2(a) and 2(b)). Likewise, as shown in Figures 2(c) and 2(d) dilation of Pparα −/− MCAs did not differ from WT responses for either GW7647 or WY14643. Thus, these data indicated nonreceptor-mediated effects of PPARα agonists on arteries. Therefore, we sought to determine the mechanism by which GW7647 and WY14643 mediated vasorelaxation.
Figure 2.
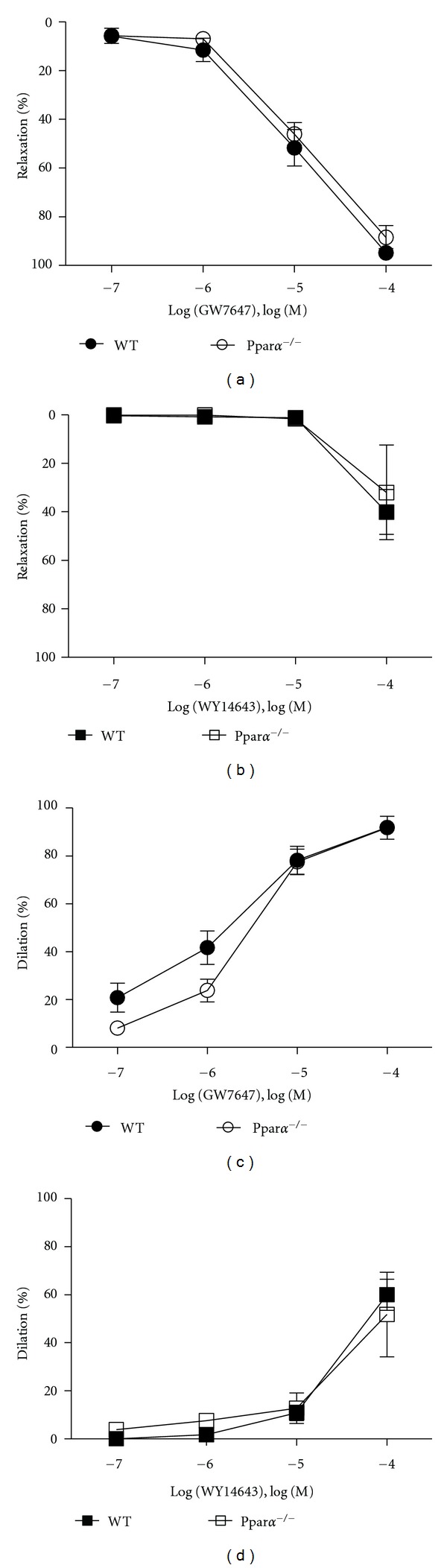
Nonreceptor-mediated responses to PPARα agonists. Concentration responses to GW7647 and WY14643 in aortic rings (a) and (b) and in MCAs (c) and (d) isolated from WT and Pparα −/− mice (n = 3–5). Responses to GW7647 and WY14643 in aortic rings and MCAs of Pparα −/− mice were identical to that of WT (control) mice indicating a receptor-independent effect. Data are means ± SEM. *Indicates P ≤ 0.05.
3.3. Mechanism of Relaxation Caused by PPARα Agonists in the Aorta
Relaxation induced by GW7647 and WY14643 in mouse aortic rings was found to be independent of endothelial derived NO• because preincubation of intact aortic rings with the nitric oxide inhibitor L-NG-nitroarginine methyl ester (L-NAME, 10−4 M) did not impair the response to GW7647 or WY14643. Pretreating aortic rings with indomethacin (10−5 M) also did not change the response to GW7647 or WY14643 suggesting that prostacyclin was not involved (Figures 3(a) and 3(c)). Likewise, endothelial denudation of aortic rings also did not modify the GW7647 or WY14643 responses (Figures 3(a) and 3(c)). Potassium (K+) channel mediated hyperpolarization of smooth muscle is one possible mechanism of smooth muscle relaxation. Elevating the external K+concentration resulted in attenuation of GW7647 (P < 0.0001) and WY14643 (P = 0.0031) mediated relaxations (Figures 3(b) and 3(d)). Figures 3(b) and 3(d) also show that preincubation of intact aortic rings with Iberiotoxin (IbTX) that blocks large-conductance calcium-activated K+ channels (BKca), or 4-aminopyridine (4-AP), which is a blocker of voltage-dependent K+ channels (Kv), did not alter the GW7647 or WY14643 responses. In contrast, glibenclamide, which blocks KATP channels, significantly inhibited GW7647 (P < 0.0001) and WY14643 (P = 0.0473) induced relaxations (Figures 3(b) and 3(d)). Pretreatment of aortic rings with the sGC inhibitor (ODQ, 10−5 M) shifted the GW7647- (P = 0.0003) and WY14643- (P = 0.0085) mediated relaxation curves to the right (Figures 4(a) and 4(c)). Furthermore, by using aortic homogenates, we found that 10−5 M GW7647 elevated cGMP levels by 2.4-fold (P = 0.0013) over baseline (Figure 4(b) left). The NO• donor sodium nitroprusside (SNP, 10−6 M) likewise increased cGMP levels 5.5-fold (P = 0.0006), while 10−5 M ODQ prevented GW7647 (P = 0.0058) and SNP (P = 0.0010) from elevating cGMP levels (Figure 4(b) right). Similar to GW7647, 10−4 M WY14643 also elevated cGMP levels by 2.4-fold (P = 0.0041) while ODQ prevented WY14643 (P = 0.0109) from elevating cGMP (Figure 4(d)). Thus, it appeared that GW7647 and WY14643 possibly activated sGC, which contributed to the relaxant effects of GW7647 and WY14643.
Figure 3.
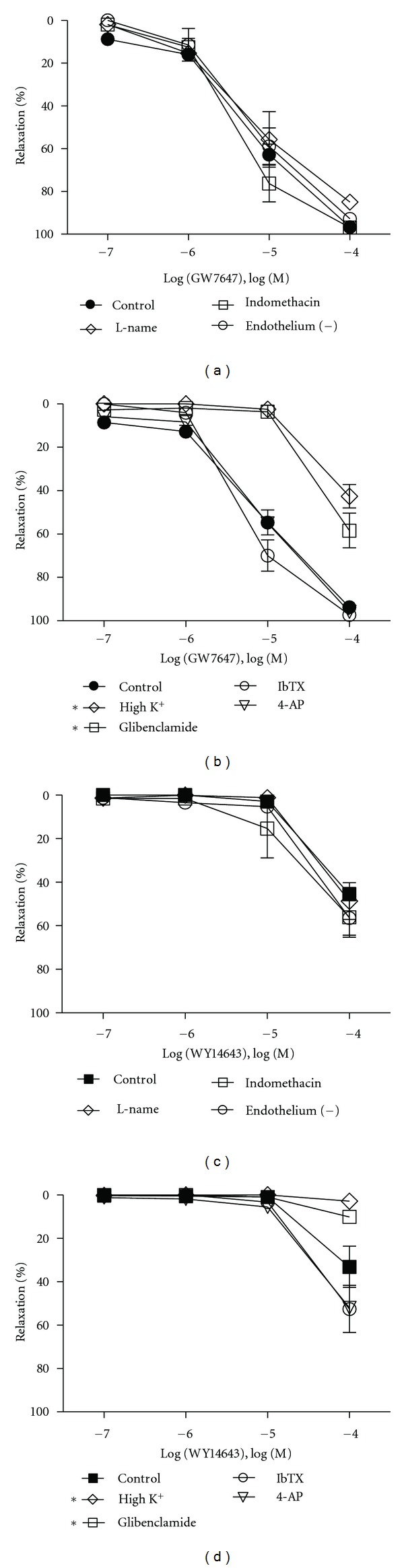
Role of endothelium and K+channels in PPARα agonist-mediated relaxations. The effects of nitric oxide synthase, cyclooxygenase, and endothelial denudation on the relaxant response of GW7647 (a) and WY14643 (c) in WT mice aortic rings (n = 3–5). L-NAME or indomethacin or endothelial denudation had no effect on GW7647- and WY14643-mediated aortic relaxations. Responses to GW7647 (b) and WY14643 (d) after pretreatment of aortic rings with elevated external KCI (80 mM), 10−7 M Iberiotoxin (IbTX), 3 × 10−3 M 4-aminopyridine (4-AP), 10−5 M glibenclamide (n = 3–6). Elevated KCI as well as glibenclamide resulted in attenuation of both GW7647- and WY14643-mediated relaxations. The other K+channel inhibitors did not, however, alter the relaxations to PPARα agonists. Data are means ± SEM. *Indicates P ≤ 0.05.
Figure 4.
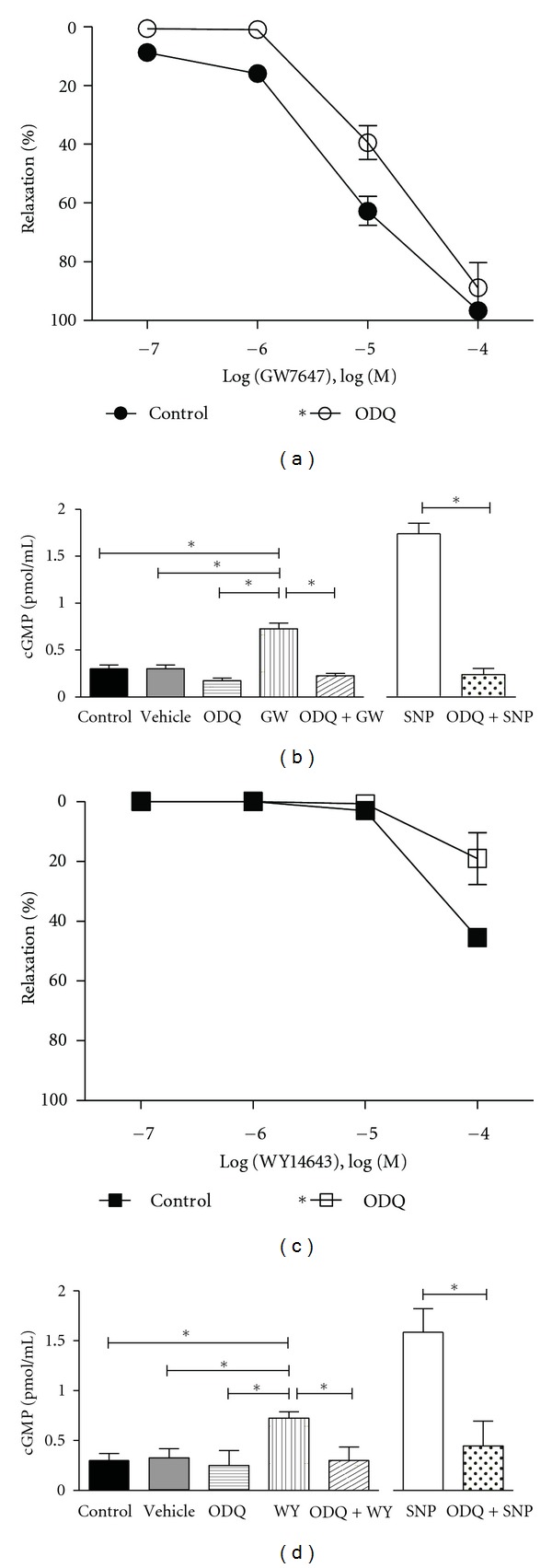
Involvement of sGC in PPARα agonist mediated relaxations. Effect of sGC inhibitor 10−5 M ODQ pretreatment on GW7647 (a) and WY14643 (c) mediated relaxations (n = 3–5). Preincubation with ODQ resulted in a rightward shift in the GW7647 and WY14643 relaxation curves demonstrating the involvement of sGC. Using an EIA kit, cGMP levels were measured in aortic rings freshly isolated from WT mice under basal and stimulated conditions. (b) 10−5 M GW7647 (GW) elevated cGMP content in aortic homogenates 2.4-fold over basal levels, an effect that was inhibitable by pretreatment with the sGC inhibitor 10−5 M ODQ (n = 4). The NO• donor 10−6 M sodium nitroprusside (SNP) elevated cGMP by 5.5-fold in ODQ sensitive manner (n = 4). (d) Similar to GW, 10−4 M WY14643 (WY) also elevated cGMP by 2.4-fold and ODQ was able to attenuate this response to basal levels (n = 4). Compared to untreated control conditions, vehicle treatment (DMSO for GW and WY) did not alter basal cGMP levels. Data are means ± SEM. *Indicates P ≤ 0.05.
3.4. Mechanism of Dilation Caused by PPARα Agonists in the MCA
Since GW7647 and WY14643 relaxant mechanisms were identical in the aorta, and because GW7647 was clearly the more potent dilator, in the demanding MCA studies we focused on the effects of GW7647. In intact MCAs, preincubation with 10−5 M L-NAME and indomethacin did not alter dilation to GW7647 (Figure 5(a)), nor did endothelial denudation (not shown). Elevating extracellular K+ likewise had no effect on the response to GW7647 (Figure 5(a)) suggesting that GW7647 did not activate K+ channels. Preconstricting the MCAs with phorbol 12,13-dibutyrate, which is an activator of PKC, significantly inhibited (P < 0.0001) the dilatory response of GW7647 (Figure 5(b)). Similar to the aorta, ODQ also impaired (P = 0.0028) dilation to GW7647 (Figure 5(b)) in the MCAs especially at lower GW7647 concentrations, suggesting an additional role of sGC.
Figure 5.
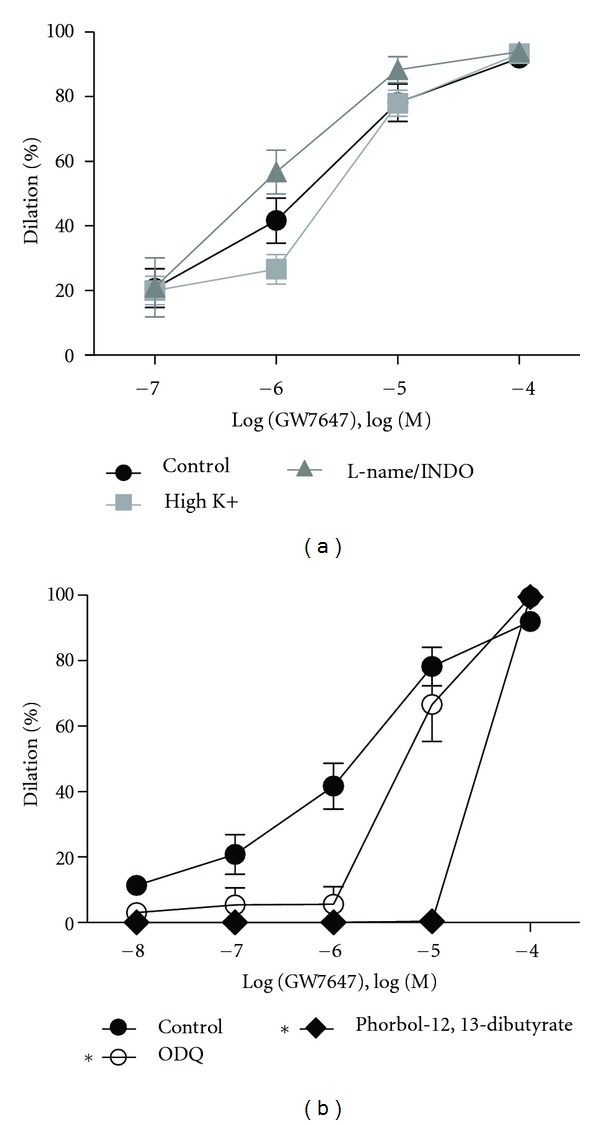
Exploration of the mechanism of PPARα agonist-mediated dilation in MCAs. (a) 10−5 M L-name and indomethacin pretreatment did not alter dilation to GW7647 in PE-preconstricted MCAs (n = 4). Likewise, in MCAs pretreated with high K+, dilation to GW7647 was again unaltered (n = 4). (b) In MCAs, pretreatment with phorbol 12,13-dibutyrate, as well as with 10−5 M ODQ (n = 5) impaired the dilatory response of GW7647 thereby implicating a role for PKC and sGC in the response (n = 3). Data are means ± SEM. *Indicates P ≤ 0.05.
4. Discussion
In the present study, we examined direct vascular effects of three PPARα agonists. The major findings are as follows. First, PPARα agonists acutely and endothelium independently relaxed mouse conduit arteries and dilated resistance arteries. Second, the response to PPARα agonists was nonreceptor-mediated. Third, in the aorta, the relaxant effects of PPARα agonists appeared to involve KATP channels and sGC whereas in the MCA, both the sGC and PKC pathways contributed to the response although K+channels did not. Use of Pparα −/− mice demonstrated that these effects were receptor independent. Although PPARα agonists exert anti-inflammatory and antihyperlipidemic effects in vivo [1, 5, 43], our findings suggest that a direct nongenomic vascular smooth muscle relaxant effect may be a contributor to cardiovascular/cerebrovascular protection by PPARα agonists.
Using arteries isolated from wild-type C57/B6 mice, we found that PPARα agonists elicited relaxation of aortic rings, as well as dilation of MCAs. Similar results using gemfibrozil were found in rat tail arteries by Phelps and Peuler (2010), and in rat thoracic aorta by Liu et al. (2012), although in those studies the mechanism of relaxation was not fully explored [36, 37]. As mentioned above, it was recently found that the fibrates, gemfibrozil, fenofibric acid, and bezafibrate also caused relaxation of rat thoracic aorta [37]. Nevertheless, relaxation of rat aortas to these compounds occurred only at high concentrations, and even so was not complete. Similarly, we found that gemfibrozil was without effect until 10−4 M; however, we did not pursue higher concentrations because in preliminary studies we observed that arteries did not recover from concentrations greater than 10−4 M. In our study, it was interesting to note that the most effective compound, GW7647, also has the greatest pharmacological selectivity for PPARα versus the other PPAR subtypes with an EC50 for receptor activation of mouse PPARα of only 1.0 × 10−9 M [42]. WY14647, which was a less effective relaxant/dilator than GW7647, has a higher EC50 for PPARα activation at 6.3 × 10−7 M [42]. Gemfibrozil, which at 10−4 M essentially did not act on the aorta, and only weakly dilated the MCAs, has the highest EC50 for PPARα receptor activation at 4.5 × 10−5 M [42, 44]. Thus, although the relaxant/dilatory potential paralleled PPARα selectivity, this was apparently coincidental because an identical effect was also observed in Pparα −/− mice.
In our preparation, the relaxant effects of PPARα agonists were rapid occurring within minutes of addition to the bath. In pancreatic beta-cells, PPARα agonists also have rapid nongenomic effects such as decreasing glucose-induced intracellular calcium signals, and insulin secretion [45]. In addition, Jiménez et al.(2010) also reported nongenomic effects of PPARβ agonists on rat aortic relaxations [46]. Agonists for PPARβ/δ/γ were all also found to inhibit platelet activation independent of their genomic effects [47, 48]. Considering that our responses were rapid, and were also observed in Pparα −/− mice, it was likely that effects were nonspecific, acting instead on other sites.
In an attempt to discover if the arterial effects of GW7647 and WY14643 were endothelium-dependent, we found that L-NAME and/or indomethacin did not alter the responses in either the aorta or MCA. Thus, NO• and prostacyclin production in endothelium could not have been responsible for the GW7647 and WY14643 mediated responses. Furthermore, endothelial denudation did not alter the response to GW7647 and WY14643 in both the arteries, thereby showing that no endothelial factor, including endothelium-derived hyperpolarization factor could have accounted for the effect. Thus, the acute vascular effects of PPARα agonists we observed in both conduit and resistance arteries were endothelium-independent, targeting instead the smooth muscle.
To explore the role of smooth muscle in PPARα agonist-mediated relaxations, first we examined K+ channel involvement in general by elevating the external K+concentration. In the aorta, high K+inhibited relaxations to GW7647 and WY14643 whereas no effect was observed in the MCA, indicating that the mechanism of action differs by vascular bed. Inhibiting BKca channels with IbTX, or Kv channels with 4-AP, did not alter aortic responses to GW7647 or WY14643. The KATP channel inhibitor glibenclamide, however, was able to inhibit the response. Thus, in the aorta, relaxations caused by PPARα agonists were partially dependent on activation of KATP channels.
In vascular smooth muscle, one of the pathways that lead to relaxation involves an increase in intracellular cGMP by sGC. Interestingly, blockade of sGC with ODQ also inhibited responses to PPARα agonists in both arteries. But, in aorta, sGC inhibition was not as consistent as KATP channel blockade. Relaxations to GW7647 were only moderately reduced by ODQ, although relaxations to WY14643 were basically eliminated. To further explore this finding, in aortic homogenates we found that both GW7647 and WY14643 raised cGMP concentrations to similar levels by activating sGC, although at different concentrations. Just as greater concentrations of WY14643 than GW7647 were required to relax aortas, a similar elevation of cGMP content was achieved at 10−4 M WY14643, but only 10−5 M GW7647. Thus, our results suggest that in addition to potentially activating KATP channels, PPARα agonists could also activate these channels via sGC [49]. In the MCA, dilation to GW7647 was also blocked by ODQ, but only at low concentrations of GW7647. This suggests that the dilatory response of GW7647 in the MCA was only partially dependent on the activation of sGC. Together, these results indicate that PPARα agonists appear to activate sGC in conduit and resistance arteries, while in the aorta they also activate KATP channels.
Since dilation of MCA to GW7647 was refractory to everything except inhibition of sGC, which was only moderately effective, we sought other targets of GW7647. In cerebral arteries, PKC contributes to myogenic tone [50–52], and we found that pharmacologically activating PKC constricted the mouse MCA. Furthermore, PKC activation largely prevented GW7647 from dilating the MCA suggesting that GW7647 possibly targets this enzyme or an upstream activator of the PKC pathway. While a full exploration of this aspect of the mechanism is beyond the scope of the current investigation, it is unlikely, however, that GW7647 inhibits PKC activation via interactions with other PPAR subtypes. This is because GW7647 is ~200-fold more selective for PPARα than PPARγ or PPARδ. The EC50 of GW7647 for PPARα is 0.006 μM, but is a much greater 1.1 μM for PPARγ, and 6.2 μM for PPARδ [42]. While the in vivo importance of PKC in resistance arteries such as the MCA is incompletely understood, it may be that PKC could even become a target for PPARα agonists in conduit arteries. For example, hypertension can lead to the development of PKC dependent basal tone in the aorta [53]. Thus, in certain disease conditions PPARα agonists may affect both the sGC and PKC pathways in MCAs and aortas.
PPARα agonists also apparently alter vascular function in vivo. For example, the PPARα agonists gemfibrozil [36] and bezafibrate [54] acutely lowered blood pressure in rats. Since PPARα agonists are given to dyslipidemic patients who are also frequently hypertensive [55], direct vasorelaxant effects along with genomic effects may combine to produce cardiovascular protection. Interestingly, chronically administering mice PPARα agonists appears to improve endothelial function. For example, ten days of fenofibrate treatment in mice enhanced endothelium-dependent dilation of mesenteric arterioles as well as relaxation of aortas, possibly by increasing arterial antioxidant capacity, and thus NO• bioavailability [28]. Likewise, fourteen days in vivo treatment with fenofibrate modestly improved endothelium-dependent dilation of the MCA [29]. Although the mechanism of enhanced endothelial function in MCAs was not explored, it was associated with neuroprotection by fenofibrate. Thus, it may be that PPARα agonists evince cardiovascular/cerebrovascular benefits by promoting expression of protective genes via PPARα, and by direct albeit nonreceptor mediated effects on vascular tone.
In summary, we have demonstrated that PPARα agonists directly and acutely relaxed mouse aortas held under isometric tension, and dilated pressurized MCAs. These responses were nonreceptor mediated, and smooth muscle specific. In aorta, the response depended upon elevations of cGMP by activation of sGC and also activation of KATP channels. In the MCA, the responses were dependent upon sGC activation and PKC inhibition. Thus, PPARα agonists may protect the cardiovascular system, in part, by directly promoting relaxation of vascular smooth muscle, which has the potential to affect blood pressure by lowering peripheral vascular resistance. This suggests that continued research into cardioprotective PPARα agonists is warranted. Indeed, a class of drugs that are lipid lowering, anti-inflammatory, and that can genomically activate the NO• signaling pathway without the negative side effects of nitrovasodilators [56], as well as directly promote relaxation of vascular smooth muscle could prove very useful.
5. Conclusions
PPARα agonists are in wide clinical use and protect the cardiovascular system. Our results demonstrated acute, nonreceptor-mediated effects of PPARα agonists on conduit and cerebral resistance arteries. Protection against cardiovascular disease by PPARα agonists may therefore result from long-term genomic, nonspecific acute effects in the cardiovascular system.
Acknowledgments
This work was supported by the American Heart Association under Grants SDG 0735053N (J. Andresen), 11SDG5330016 (M. J. Wacker), and 11POST7650044 (N. Silswal).
References
- 1.Van Raalte DH, Li M, Pritchard PH, Wasan KM. Peroxisome proliferator-activated receptor (PPAR)-α: a pharmacological target with a promising future. Pharmaceutical Research. 2004;21(9):1531–1538. doi: 10.1023/b:pham.0000041444.06122.8d. [DOI] [PubMed] [Google Scholar]
- 2.Issemann I, Green S. Cloning of novel members of the steroid hormone receptor superfamily. Journal of Steroid Biochemistry and Molecular Biology. 1991;40(1–3):263–269. doi: 10.1016/0960-0760(91)90191-7. [DOI] [PubMed] [Google Scholar]
- 3.Issemann I, Green S. Activation of a member of the steroid hormone receptor superfamily by peroxisome proliferators. Nature. 1990;347(6294):645–650. doi: 10.1038/347645a0. [DOI] [PubMed] [Google Scholar]
- 4.Braissant O, Foufelle F, Scotto C, Dauça M, Wahli W. Differential expression of peroxisome proliferator-activated receptors (PPARs): tissue distribution of PPAR-α, -β, and -γ in the adult rat. Endocrinology. 1996;137(1):354–366. doi: 10.1210/endo.137.1.8536636. [DOI] [PubMed] [Google Scholar]
- 5.Lee SST, Pineau T, Drago J, et al. Targeted disruption of the α isoform of the peroxisome proliferator- activated receptor gene in mice results in abolishment of the pleiotropic effects of peroxisome proliferators. Molecular and Cellular Biology. 1995;15(6):3012–3022. doi: 10.1128/mcb.15.6.3012. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Inoue I, Shino K, Noji S, Awata T, Katayama S. Expression of peroxisome proliferator-activated receptor α (PPARα) in primary cultures of human vascular endothelial cells. Biochemical and Biophysical Research Communications. 1998;246(2):370–374. doi: 10.1006/bbrc.1998.8622. [DOI] [PubMed] [Google Scholar]
- 7.Staels B, Koenig W, Habib A, et al. Activation of human aortic smooth-muscle cells is inhibited by PPARα but not by PPARγ activators. Nature. 1998;393(6687):790–793. doi: 10.1038/31701. [DOI] [PubMed] [Google Scholar]
- 8.Auboeuf D, Rieusset J, Fajas L, et al. Tissue distribution and quantification of the expression of mRNAs of peroxisome proliferator-activated receptors and liver X receptor-α in humans: no alteration in adipose tissue of obese and NIDDM patients. Diabetes. 1997;46(8):1319–1327. doi: 10.2337/diab.46.8.1319. [DOI] [PubMed] [Google Scholar]
- 9.Gottlicher M, Widmark E, Li Q, Gustafsson JA. Fatty acids activate a chimera of the clofibric acid-activated receptor and the glucocorticoid receptor. Proceedings of the National Academy of Sciences of the United States of America. 1992;89(10):4653–4657. doi: 10.1073/pnas.89.10.4653. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Yu K, Bayona W, Kallen CB, et al. Differential activation of peroxisome proliferator-activated receptors by eicosanoids. The Journal of Biological Chemistry. 1995;270(41):23975–23983. doi: 10.1074/jbc.270.41.23975. [DOI] [PubMed] [Google Scholar]
- 11.Devchand PR, Keller H, Peters JM, Vazquez M, Gonzalez FJ, Wahli W. The PPARα-leukotriene B4 pathway to inflammation control. Nature. 1996;384(6604):39–43. doi: 10.1038/384039a0. [DOI] [PubMed] [Google Scholar]
- 12.Lehmann JM, Lenhard JM, Oliver BB, Ringold GM, Kliewer SA. Peroxisome proliferator-activated receptors α and γ are activated by indomethacin and other non-steroidal anti-inflammatory drugs. The Journal of Biological Chemistry. 1997;272(6):3406–3410. doi: 10.1074/jbc.272.6.3406. [DOI] [PubMed] [Google Scholar]
- 13.Gonzalez FJ, Peters JM, Cattley RC. Mechanism of action of the nongenotoxic peroxisome proliferators: role of the peroxisome proliferator-activated receptor. Journal of the National Cancer Institute. 1998;90(22):1702–1709. doi: 10.1093/jnci/90.22.1702. [DOI] [PubMed] [Google Scholar]
- 14.Lemberger T, Saladin R, Vázquez M, et al. Expression of the peroxisome proliferator-activated receptor α gene is stimulated by stress and follows a diurnal rhythm. The Journal of Biological Chemistry. 1996;271(3):1764–1769. doi: 10.1074/jbc.271.3.1764. [DOI] [PubMed] [Google Scholar]
- 15.Lemberger T, Staels B, Saladin R, Desvergne B, Auwerx J, Wahli W. Regulation of the peroxisome proliferator-activated receptor α gene by glucocorticoids. The Journal of Biological Chemistry. 1994;269(40):24527–24530. [PubMed] [Google Scholar]
- 16.Rubins HB, Robins SJ, Collins D. The veterans affairs high-density lipoprotein intervention trial: baseline characteristics of normocholesterolemic men with coronary artery disease and low levels of high-density lipoprotein cholesterol. American Journal of Cardiology. 1996;78(5):572–575. doi: 10.1016/s0002-9149(96)00369-4. [DOI] [PubMed] [Google Scholar]
- 17.Rubins HB, Robins SJ, Collins D, et al. Gemfibrozil for the secondary prevention of coronary heart disease in men with low levels of high-density lipoprotein cholesterol. The New England Journal of Medicine. 1999;341(6):410–418. doi: 10.1056/NEJM199908053410604. [DOI] [PubMed] [Google Scholar]
- 18.Rubins HB, Robins SJ, Collins D, et al. Diabetes, plasma insulin, and cardiovascular disease: subgroup analysis from the Department of Veterans Affairs High-density Lipoprotein Intervention Trial (VA-HIT) Archives of Internal Medicine. 2002;162(22):2597–2604. doi: 10.1001/archinte.162.22.2597. [DOI] [PubMed] [Google Scholar]
- 19.Frick MH, Elo O, Haapa K, et al. Helsinki Heart Study: primary-prevention trial with gemfibrozil in middle-aged men with dyslipidemia. Safety of treatment, changes in risk factors, and incidence of coronary heart disease. The New England Journal of Medicine. 1987;317(20):1237–1245. doi: 10.1056/NEJM198711123172001. [DOI] [PubMed] [Google Scholar]
- 20.Manninen V, Elo MO, Frick MH, et al. Lipid alterations and decline in the incidence of coronary heart disease in the Helsinki Heart Study. Journal of the American Medical Association. 1988;260(5):641–651. [PubMed] [Google Scholar]
- 21.Keech A, Simes RJ, Barter P, et al. Effects of long-term fenofibrate therapy on cardiovascular events in 9795 people with type 2 diabetes mellitus (the FIELD study): randomised controlled trial. The Lancet. 2005;366(9500):1849–1861. doi: 10.1016/S0140-6736(05)67667-2. [DOI] [PubMed] [Google Scholar]
- 22.Robinson JG. Update on PPAR agonists: the clinical significance of FIELD and PROACTIVE. Current Atherosclerosis Reports. 2007;9(1):64–71. doi: 10.1007/BF02693930. [DOI] [PubMed] [Google Scholar]
- 23.Calkin AC, Cooper ME, Jandeleit-Dahm KA, Allen TJ. Gemfibrozil decreases atherosclerosis in experimental diabetes in association with a reduction in oxidative stress and inflammation. Diabetologia. 2006;49(4):766–774. doi: 10.1007/s00125-005-0102-6. [DOI] [PubMed] [Google Scholar]
- 24.Collino M, Aragno M, Mastrocola R, et al. Oxidative stress and inflammatory response evoked by transient cerebral ischemia/reperfusion: effects of the PPAR-α agonist WY14643. Free Radical Biology and Medicine. 2006;41(4):579–589. doi: 10.1016/j.freeradbiomed.2006.04.030. [DOI] [PubMed] [Google Scholar]
- 25.Okayasu T, Tomizawa A, Suzuki K, Manaka KI, Hattori Y. PPARα activators upregulate eNOS activity and inhibit cytokine-induced NF-κB activation through AMP-activated protein kinase activation. Life Sciences. 2008;82(15-16):884–891. doi: 10.1016/j.lfs.2008.02.002. [DOI] [PubMed] [Google Scholar]
- 26.Wang Y, Wang Y, Yang Q, et al. Effects of bezafibrate on the expression of endothelial nitric oxide synthase gene and its mechanisms in cultured bovine endothelial cells. Atherosclerosis. 2006;187(2):265–273. doi: 10.1016/j.atherosclerosis.2005.09.008. [DOI] [PubMed] [Google Scholar]
- 27.Li AC, Binder CJ, Gutierrez A, et al. Differential inhibition of macrophage foam-cell formation and atherosclerosis in mice by PPARα, β/δ, and γ . Journal of Clinical Investigation. 2004;114(11):1564–1576. doi: 10.1172/JCI18730. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Tabernero A, Schoonjans K, Jesel L, Carpusca I, Auwerx J, Andriantsitohaina R. Activation of the peroxisome proliferator-activated receptor alpha protects against myocardial ischaemic injury and improves endothelial vasodilatation. BMC Pharmacology. 2002;2(1, article 10) doi: 10.1186/1471-2210-2-10. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Deplanque D, Gelé P, Pétrault O, et al. Peroxisome proliferator-activated receptor-α activation as a mechanism of preventive neuroprotection induced by chronic fenofibrate treatment. Journal of Neuroscience. 2003;23(15):6264–6271. doi: 10.1523/JNEUROSCI.23-15-06264.2003. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Nakagawa K, Holla VR, Wei Y, et al. Salt-sensitive hypertension is associated with dysfunctional Cyp4a10 gene and kidney epithelial sodium channel. Journal of Clinical Investigation. 2006;116(6):1696–1702. doi: 10.1172/JCI27546. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Suardíaz M, Estivill-Torrús G, Goicoechea C, Bilbao A, Rodríguez de Fonseca F. Analgesic properties of oleoylethanolamide (OEA) in visceral and inflammatory pain. Pain. 2007;133(1–3):99–110. doi: 10.1016/j.pain.2007.03.008. [DOI] [PubMed] [Google Scholar]
- 32.González-Yanes C, Serrano A, Bermúdez-Silva FJ, et al. Oleylethanolamide impairs glucose tolerance and inhibits insulin-stimulated glucose uptake in rat adipocytes through p38 and JNK MAPK pathways. American Journal of Physiology. 2005;289(5):E923–E929. doi: 10.1152/ajpendo.00555.2004. [DOI] [PubMed] [Google Scholar]
- 33.Takeda K, Matsuzawa A, Nishitoh H, et al. Involvement of ASK1 in Ca2+—induced p38 MAP kinase activation. EMBO Reports. 2004;5(2):161–166. doi: 10.1038/sj.embor.7400072. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.Gardner OS, Dewar BJ, Earp HS, Samet JM, Graves LM. Dependence of peroxisome proliferator-activated receptor ligand-induced mitogen-activated protein kinase signaling on epidermal growth factor receptor transactivation. The Journal of Biological Chemistry. 2003;278(47):46261–46269. doi: 10.1074/jbc.M307827200. [DOI] [PubMed] [Google Scholar]
- 35.Gardner OS, Dewar BJ, Graves LM. Activation of mitogen-activated protein kinases by peroxisome proliferator-activated receptor ligands: an example of nongenomic signaling. Molecular Pharmacology. 2005;68(4):933–941. doi: 10.1124/mol.105.012260. [DOI] [PubMed] [Google Scholar]
- 36.Phelps LE, Peuler JD. Evidence of direct smooth muscle relaxant effects of the fibrate gemfibrozil. Journal of Smooth Muscle Research. 2010;46(3):125–142. doi: 10.1540/jsmr.46.125. [DOI] [PubMed] [Google Scholar]
- 37.Liu A, Yang J, Huang X, et al. Relaxation of rat thoracic aorta by fibrate drugs correlates with their potency to disturb intracellular calcium of VSMCs. Vascular Pharmacology. 2012;56(3-4):168–175. doi: 10.1016/j.vph.2012.01.003. [DOI] [PubMed] [Google Scholar]
- 38.Andresen JJ, Shafi NI, Durante W, Bryan RM. Effects of carbon monoxide and heme oxygenase inhibitors in cerebral vessels of rats and mice. American Journal of Physiology. 2006;291(1):H223–H230. doi: 10.1152/ajpheart.00058.2006. [DOI] [PubMed] [Google Scholar]
- 39.Bryan RM, Jr., You J, Phillips SC, et al. Evidence for two-pore domain potassium channels in rat cerebral arteries. American Journal of Physiology. 2006;291(2):H770–H780. doi: 10.1152/ajpheart.01377.2005. [DOI] [PubMed] [Google Scholar]
- 40.Parelkar NK, Silswal N, Jansen K, Vaughn J, Bryan RM, Andresen J. 2,2,2-Trichloroethanol activates a nonclassical potassium channel in cerebrovascular smooth muscle and dilates the middle cerebral artery. Journal of Pharmacology and Experimental Therapeutics. 2010;332(3):803–810. doi: 10.1124/jpet.109.162313. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41.Silswa N, Parelkar NK, Wacker MJ, Brotto M, Andresen J. Phosphatidylinositol 3,5-bisphosphate increases intracellular free Ca2+ in arterial smooth muscle cells and elicits vasocontraction. American Journal of Physiology. 2011;300(6):H2016–H2026. doi: 10.1152/ajpheart.01011.2010. [DOI] [PubMed] [Google Scholar]
- 42.Brown PJ, Stuart LW, Hurley KP, et al. Identification of a subtype selective human PPARα agonist through parallel-array synthesis. Bioorganic and Medicinal Chemistry Letters. 2001;11(9):1225–1227. doi: 10.1016/s0960-894x(01)00188-3. [DOI] [PubMed] [Google Scholar]
- 43.Madej A, Okopien B, Kowalski J, et al. Effects of fenofibrate on plasma cytokine concentrations in patients with atherosclerosis and hyperlipoproteinemia IIb. International Journal of Clinical Pharmacology and Therapeutics. 1998;36(6):345–349. [PubMed] [Google Scholar]
- 44.Berger A, Roberts MA. Unraveling Lipid Metabolism with Microarrays. New York, NY, USA: Marcel Dekker; 2005. [Google Scholar]
- 45.Ropero AB, Juan-Picó P, Rafacho A, et al. Rapid non-genomic regulation of Ca2+ signals and insulin secretion by PPARα ligands in mouse pancreatic islets of Langerhans. Journal of Endocrinology. 2009;200(2):127–138. doi: 10.1677/JOE-08-0397. [DOI] [PubMed] [Google Scholar]
- 46.Jiménez R, Sánchez M, Zarzuelo MJ, et al. Endothelium-dependent vasodilator effects of peroxisome proliferator-activated receptor β agonists via the phosphatidyl-inositol-3 kinase-Akt pathway. Journal of Pharmacology and Experimental Therapeutics. 2010;332(2):554–561. doi: 10.1124/jpet.109.159806. [DOI] [PubMed] [Google Scholar]
- 47.Ali FY, Davidson SJ, Moraes LA, et al. Role of nuclear receptor signaling in platelets: antithrombotic effects of PPARβ . FASEB Journal. 2006;20(2):326–328. doi: 10.1096/fj.05-4395fje. [DOI] [PubMed] [Google Scholar]
- 48.Akbiyik F, Ray DM, Gettings KF, Blumberg N, Francis CW, Phipps RP. Human bone marrow megakaryocytes and platelets express PPARγ, and PPARγ agonists blunt platelet release of CD40 ligand and thromboxanes. Blood. 2004;104(5):1361–1368. doi: 10.1182/blood-2004-03-0926. [DOI] [PubMed] [Google Scholar]
- 49.Kubo M, Nakaya Y, Matsuoka S, Saito K, Kuroda Y. Atrial natriuretic factor and isosorbide dinitrate modulate the gating of ATP-sensitive K+ channels in cultured vascular smooth muscle cells. Circulation Research. 1994;74(3):471–476. doi: 10.1161/01.res.74.3.471. [DOI] [PubMed] [Google Scholar]
- 50.Chrissobolis S, Sobey CG. Inhibitory effects of protein kinase C on inwardly rectifying K+- and ATP-sensitive K+ channel-mediated responses of the basilar artery. Stroke. 2002;33(6):1692–1697. doi: 10.1161/01.str.0000016966.89226.67. [DOI] [PubMed] [Google Scholar]
- 51.Goyal R, Mittal A, Chu N, Shi L, Zhang L, Longo LD. Maturation and the role of PKC-mediated contractility in ovine cerebral arteries. American Journal of Physiology. 2009;297(6):H2242–H2252. doi: 10.1152/ajpheart.00681.2009. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 52.Lagaud G, Gaudreault N, Moore EDW, Van Breemen C, Laher I. Pressure-dependent myogenic constriction of cerebral arteries occurs independently of voltage-dependent activation. American Journal of Physiology. 2002;283(6):H2187–H2195. doi: 10.1152/ajpheart.00554.2002. [DOI] [PubMed] [Google Scholar]
- 53.Pucci ML, Tong X, Miller KB, Guan H, Nasjletti A. Calcium- and protein kinase C-dependent basal tone in the aorta of hypertensive rats. Hypertension. 1995;25(4):752–757. doi: 10.1161/01.hyp.25.4.752. [DOI] [PubMed] [Google Scholar]
- 54.Agrawal B, Kopecký J, Kränzlin B, Rohmeiss P, Pill J, Gretz N. Acute effects of bezafibrate on blood pressure and renal haemodynamics in SHR and WKY rats. Nephrology Dialysis Transplantation. 1998;13(2):333–339. doi: 10.1093/oxfordjournals.ndt.a027827. [DOI] [PubMed] [Google Scholar]
- 55.Assmann G, Schulte H. The Prospective Cardiovascular Munster study: prevalence and prognostic significance of hyperlipidemia in men with systemic hypertension. American Journal of Cardiology. 1987;59(14):9G–17G. doi: 10.1016/0002-9149(87)90152-4. [DOI] [PubMed] [Google Scholar]
- 56.Kojda G, Mayer B. Therapeutic Importance of Nitrovasodilators Nitric Oxide. Berlin, Germany: Springer; 2000. [Google Scholar]