

Abstract
Alzheimer’s disease (AD) onset is associated with changes in hypothalamic-pituitary–gonadal (HPG) function. The 54 amino acid kisspeptin (KP) peptide regulates the HPG axis and alters antioxidant enzyme expression. The Alzheimer’s amyloid-β (Aβ) is neurotoxic, and this action can be prevented by the antioxidant enzyme catalase. Here, we examined the effects of KP peptides on the neurotoxicity of Aβ, prion protein (PrP), and amylin (IAPP) peptides. The Aβ, PrP, and IAPP peptides stimulated the release of KP and KP 45–54. The KP peptides inhibited the neurotoxicity of Aβ, PrP, and IAPP peptides, via an action that could not be blocked by kisspeptin-receptor (GPR-54) or neuropeptide FF (NPFF) receptor antagonists. Knockdown of KiSS-1 gene, which encodes the KP peptides, in human neuronal SH-SY5Y cells with siRNA enhanced the toxicity of amyloid peptides, while KiSS-1 overexpression was neuroprotective. A comparison of the catalase and KP sequences identified a similarity between KP residues 42–51 and the region of catalase that binds Aβ. The KP peptides containing residues 45–50 bound Aβ, PrP, and IAPP, inhibited Congo red binding, and were neuroprotective. These results suggest that KP peptides are neuroprotective against Aβ, IAPP, and PrP peptides via a receptor independent action involving direct binding to the amyloid peptides.
Keywords: KiSS-1, kisspeptin, amyloid-β, prion protein, amylin, neuroprotection
Neuroendocrine hormone changes are seen in aging, and there are disease specific changes associated with Alzheimer’s disease (AD).1,2 One of the neuroendocrine systems to show profound changes with aging is the hypothalamic–pituitary–gonadal (HPG) system, and the pathology of AD, Creutzfeldt–Jakob disease (CJD), and type 2 diabetes mellitus (T2DM) is also associated with changes in the hormones of this system.3,4 A major regulator of the HPG-axis is the 54 amino acid kisspeptin (KP) peptide, which acts on gonadotrophin-releasing hormone (GnRH) neurons to activate GnRH release.5
The KP peptide is produced in neurons by processing of the KiSS-1 preproprotein to yield the 54 amino acid KP peptide, which corresponds to residues 68–121 of KiSS-1.6 Shorter derivatives of KP peptide comprising the C-terminal 14 (KP 41–54), 13 (KP 42–54), and 10 (KP 45–54) amino acids have also been found in tissues and corresponding to residues 108–121, 109–121, and 112–121 of KiSS-1.6,7 Biological activity of KP peptides requires the KP 45–54 sequence and is mediated via a specific KP receptor (GPR-54).6,7 Lack of KP signaling via the GPR-54 receptor is associated with reproductive system failure.8 Cleavage of the KP 45–54 peptide between residues 51 and 52 by matrix metalloproteinases abolishes the activation of GPR-54 by the KP 45–54 peptide.9 The KP 42–54 and KP 45–54 peptides also activate NPFF receptors,10,11 and some of the actions of KP peptides could be mediated by NPFF receptor activation.
There are profound changes in KP signaling at menopause12 and notable sex differences in hypothalamic neurodegeneration in the elderly.13 This correlates with a marked elevation of KP peptides in the hypothalamus of postmenopausal women that is not seen in males.12 The onset of AD is normally postmenopausal, and susceptibility may be increased in women, raising the possibility that changes in reproductive function may play a role in the process.14 Changes in estrogen receptors15 in the hypothalamus are linked to AD neuropathology, and these receptors regulate KP levels.16 This raises the possibility that endogenous KP could play a role in AD neurodegeneration. Andropause in males is associated with reduced memory function suggesting that the changes in HPG-axis hormones may influence memory and that the associated hormones may be targets for AD therapy.17 It is possible that the changes in reproductive hormones contribute to the pathology since estrogen is protective in AD, but the levels decline during aging.18
The amyloid-β (Aβ), prion protein (PrP), and amylin (IAPP) peptides play a key role in the pathology of AD, CJD, and T2DM, respectively.19−21 The related amyloid-Bri (A-Bri) and amyloid-Dan (A-Dan) peptides are central to the pathology Familial British and Familial Danish dementias.22 One of the shared features of the Aβ, PrP, A-Bri, A-Dan, and IAPP peptides is their neurotoxicity, which appears to involve activation of similar pathways.23 The accumulation of fibrillar deposits of Aβ and PrP is linked to neurodegeneration in AD24 and CJD,25,26 while the accumulation of fibrillar deposits of IAPP is linked to degeneration of pancreatic islets in T2DM.27 The pathology of T2DM includes neurodegenerative changes, and this appears to be linked to hyperinsulinemia and insulin resistance.28 The mechanisms underlying cell death in these diseases include oxidative stress, apoptosis, impaired mitochondrial function, calcium channel formation cell cycle re-entry, and receptor mediated effects.29 Compounds that specifically bind Aβ have been shown to be neuroprotective.30
Herein we have explored the effects of the amyloid peptides Aβ, PrP, A-Bri, A-Dan, and IAPP on the release of KP and KP 45–54 from human SH-SY5Y neuronal cultures31 and studied the effects of KiSS-1 and KP peptides on the toxicity of amyloid peptides. The effects of KP peptides on Aβ, PrP, A-Bri, A-Dan, and IAPP toxicity in human SH-SY5Y neuronal cultures31 and rat cortical neuron cultures32,33 have been characterized. For comparison, we have also tested KP peptides in a cell model of T2DM using IAPP induced cell death in a pancreatic islet cell line.34 Using both overexpression of KiSS-1 and siRNA knockout of KiSS-1 expression, we have characterized the role of endogenous KiSS-1 the toxicity of amyloid peptides in human SH-SY5Y neuronal cultures.31 The KP receptor antagonist P23435 and the NPFF antagonist RF936 have been used to determine the role of these receptors in the observed responses to KiSS-1 overexpression. The amyloid-binding domain in KiSS-1 has been characterized using specific peptide binding assays,37 and the effects of KP peptides on amyloid peptide interactions with Congo red have also been determined.38,39
Results and Discussion
Effects of Amyloid Peptides on KP 1–54 and KP 45–54 Release from SH-SY5Y Neurons
The KP peptides are neuroendocrine hormones and are normally released in response to stimuli. The SH-SY5Y neuroblastoma cells contain the necessary secretory vesicle machinery for neuroendocrine hormone release40 and also express the KiSS-1 gene.41 We therefore measured KP release in response to the amyloid peptides Aβ, PrP, IAPP, A-Bri, and A-Dan peptides. Cells were exposed to the amyloid peptides at a subtoxic dose (50 nM) for 2 h and the immunoreactive (ir) KP 1–54 and KP 45–54 levels in the media determined by EIA. Results showed that all of the amyloid peptides tested (Aβ 1–42, Aβ 25–35, PrP 106–126, PrP 118–135, IAPP 1–37, IAPP 20–29, A-Bri, and A-Dan) caused a significant increase in both irKP 1–54 (Figure 1A) and irKP 45–54 release (Figure 1B). The irKP 1–54 levels rose 3–5-fold, while the irKP 45–54 levels rose 2–3-fold. The KP 1–54 EIA assay showed no significant cross-reactivity (<0.00001%) with KP 42–54, KP 45–54, or NPFF, while the KP 45–54 EIA assay showed significant cross-reactivity with KP 1–54 (77%), KP 42–54 (98%) and NPFF (54%). This is in agreement with previous studies for KP antibodies that have suggested the cross-reactivity of some with NPFF.42 This raises the possibility of a contribution of NPFF to the levels of irKP 45–54 measured; however, this was not the case for irKP 1–54 measurements and suggests that the results represent specific changes in KP peptides in response to amyloid peptides.
Figure 1.
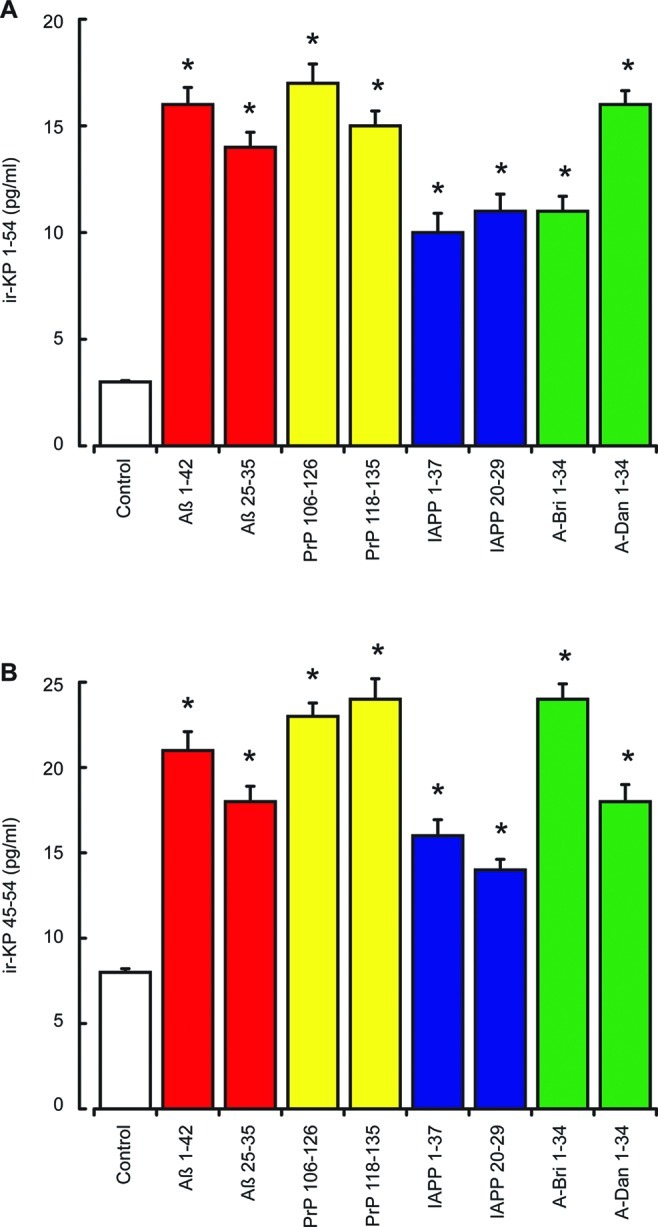
Effects of amyloid peptides on KP 1–54 and KP 45–54 release from SH-SY5Y neurons. Neuronal SHSY-5Y cell cultures were exposed to Aβ 1–42, Aβ 25–35, PrP 106–126, PrP 118–135, IAPP 1–37, IAPP 20–29, A-Bri 1–34, and A-Dan 1–34 peptides (50 nM each) for 2 h. The release of ir-KP 1–54 (A) and ir-KP 45–54 (B) into the cell culture media was determined by EIA. All results are expressed as the mean ± SEM (n = 8). (* = P < 0.05 vs control (media alone); one-way ANOVA.)
To confirm that irKP peptides released into the media were products from the endogenous KiSS-1 gene, we used siRNA knockdown of KiSS-1 expression and measurement of KP levels by EIA. Measurement of KP released into the media showed irKP 1–54 levels of 3.5 ± 0.2 pg/mL (n = 8) and irKP 45–54 levels of 8.3 ± 0.7 pg/mL (n = 8) in media from control siRNA treated cells. In KiSS-1 siRNA treated cells, the irKP 1–54 levels were significantly reduced to 1.3 ± 0.1 pg/mL (n = 8), and the irKP 45–54 levels were significantly reduced to 3.1 ± 0.2 pg/mL (n = 8). This confirms that the basal levels of irKP were derived from the KiSS-1 preproprotein. Stimulation of siRNA treated cells with 50 nM Aβ 25–35 resulted in a significant increase in irKP 1–54 levels to 8.4 ± 0.6 pg/mL (n = 8) and irKP 45–54 levels to 18.7 ± 1.2 pg/mL (n = 8) in control siRNA treated cells. However, in KiSS-1 siRNA treated cells there was no significant change in irKP 1–54 levels, which were 1.6 ± 0.2 pg/mL (n = 8), or irKP 45–54 levels, which were 3.7 ± 0.5 pg/mL (n = 8) in response to 50 nM Aβ 25–35. These results confirm that the stimulation of irKP release by Aβ was indeed KP from translation and processing of the KiSS-1 gene.
Effects of KP Peptides on Aβ, PrP, A-Bri, A-Dan, and IAPP Neurotoxicity in SH-SY5Y and Rat Cortical Neurons
The ability of a range of amyloid peptides to stimulate irKP 1–54 release led us to test the effects of KP 1–54 on the toxicity of Aβ 1–42, Aβ 1–40, Aβ 25–35, Aβ 29–40, Aβ 31–35, PrP 106–126, PrP 118–135, A-Bri 1–34, A-Dan 1–34, IAPP 1–37, IAPP 8–37, and IAPP 20–29 peptides. Results showed KP 1–54 was significantly protective against the Aβ 1–42, Aβ 1–40, Aβ 25–35, Aβ 29–40, PrP 106–126, PrP 118–135, A-Bri 1–34, A-Dan 1–34, IAPP 1–37, IAPP 8–37, and IAPP 20–29 peptides (Figure 2A). KP 1–54 did not prevent the toxicity of Aβ 31–35, A-Bri 1–34, and A-Dan 1–34. The lack of protection against Aβ 31–35 is a feature shared with the endocannabinoids and corticotrophin releasing hormone receptor ligands,43 which protect against Aβ 25–35 and the longer Aβ forms.
Figure 2.

Effects of KP peptides on Aβ, PrP, A-Bri, A-Dan, and IAPP neurotoxicity in SH-SY5Y neurons. The effects of 10 μM KP 1–54 on the toxicity of Aβ 1–42, Aβ 1–40, Aβ 25–35, Aβ 29–40, Aβ 31–35, PrP 106–126, PrP 118–135, IAPP 1–37, IAPP 8–37, IAPP 20–29, A-Bri 1–34, and A-Dan 1–34 peptides (5 μM each) were tested in human SH-SY5Y neuroblastoma cell cultures (A), with cell viability determined by the MTT assay. The effects of KP 1–54, KP 27–54, KP 42–54, KP 45–54, KP 45–50, KP 45–47, KP 47–50, and NPFF peptides (10 μM each) on the toxicity of 5 μM Aβ 1–42 (B), 5 μM PrP 106–126 (C), and 5 μM IAPP 1–37 (D) were tested in human SH-SY5Y neuroblastoma cell cultures, with cell viability determined by the MTT assay. All results are expressed as a % control (SH-SY-5Y cells in media alone) and are expressed as the mean ± SEM (n = 8). (* = P < 0.05 vs control (media alone); † = P < 0.05 vs amyloid fibrils alone; one-way ANOVA.)
To determine the region of KP 1–54 required for neuroprotection against amyloid peptides, we tested the effects of KP 1–54, KP 27–54, KP 42–54, KP 45–54, KP 45–50, KP 45–47, KP 47–50, and NPFF on the toxicity of Aβ 1–42, PrP 106–126, and IAPP 1–37. Results showed that the toxicity of 5 μM Aβ 1–42 (Figure 2B), 5 μM PrP 106–126 (Figure 2C), and 5 μM IAPP 1–37 (Figure 2D) was significantly inhibited in SH-SY5Y cell cultures by addition of 10 μM KP 1–54, 10 μM KP 27–54, 10 μM KP 42–54, 10 μM KP 45–54, and 10 μM KP 45–50. The KP 47–50 and NPFF peptides (10 μM) inhibited 5 μM Aβ 1–42 toxicity but had no effect on 5 μM PrP or 5 μM IAPP toxicity. The KP 45–47 peptide (10 μM) had no effect on 5 μM Aβ 1–42, 5 μM PrP, or 5 μM IAPP toxicity.
Previous studies have questioned the validity of results obtained from neuronal cultures derived from cell lines due to the protein expression patterns in tumor cells and suggested a need for comparison with primary neuronal cultures.32 Since KP peptides are known to modify tumor cell behavior,44,45 we tested the effects of KP 42–54 and KP 45–50 on the toxicity of Aβ 1–42 and PrP 106–126 in rat cortical neuron primary cell cultures.32,33 The results showed that KP 42–54 and KP 45–50 caused a dose-dependent inhibition of 5 μM Aβ 1–42 toxicity, which was significant at concentrations above 2.5 μM (Figure 3A). To determine the mechanism of neuroprotection by KP 42–54 and KP 45–50, the KP receptor antagonist P23435 and the NPFF receptor antagonist RF936 were tested. Results showed that the P234 peptide (10 μM) was neuroprotective against Aβ 1–42 toxicity, while the RF9 had no effect against Aβ 1–42 toxicity (Figure 3B). The P234 increased the protection of KP 42–54 and KP 45–50 against Aβ 1–42 toxicity, while the RF9 had no effect on KP 42–54 and KP 45–50 neuroprotection.
Figure 3.

Effects of KP peptides on Aβ and PrP neurotoxicity in rat cortical neurons. The effects of 10 μM KP 42–54 (closed blue circles) or 10 μM KP 45–50 (closed red squares) on the toxicity of Aβ 1–42 (A) and PrP 106–126 (B) were tested in rat cortical neuron cell cultures, with cell viability determined by the MTT assay. The effects of 10 μM KP receptor antagonist P234 or 10 μM NPFF receptor antagonist RF9 on 2.5 μM KP 42–54 or 2.5 μM KP 45–50 protection against the toxicity of 5 μM Aβ 1–42 (C) and 5 μM PrP 106–126 (D) were tested in rat cortical neuron cell cultures, with cell viability determined by the MTT assay. All results are expressed as % control (rat cortical neurons in media alone) and are expressed as the mean ± SEM (n = 8). (* = P < 0.05 vs Aβ or PrP alone; † = P < 0.05 vs Aβ or PrP plus KP 42–54 or KP 45–50; one way ANOVA.)
The KP 42–54 and KP 45–50 also caused a dose-dependent inhibition of 5 μM PrP 106–126 toxicity, which was significant at concentrations above 0.63 μM (Figure 3C). The KP receptor antagonist P234 peptide (10 μM) was neuroprotective against PrP 106–126 toxicity, while the NPFF antagonist RF9 had no effect against PrP 106–126 toxicity (Figure 3D). The P234 increased the neuroprotection of KP 42–54 and KP 45–50 against PrP 106–126 toxicity, while the RF9 had no effect on KP 42–54 and KP 45–50 neuroprotection.
The ability of KP 45–50 to prevent Aβ 1–42, PrP 106–126, and IAPP 1–37 toxicity suggests that the action is unlikely to be mediated via the KP receptor since when the KP 45–54 is cleaved between residues 51 and 52 by MMP enzymes to give a KP 45–51 sequence it looses its ability to activate the KP receptor.9 The neuroprotective actions of the KP receptor antagonist P234 are additive to the protection observed with KP 42–54 and KP 45–50 and suggest that the neuroprotective mechanism is more complex. Since the P234 peptide is a derivative of KP 45–54 with modifications at residues 1, 5, and 8 and binds the KP receptor, it is possible that it has receptor activity as a partial agonist that is key to the neuroprotection observed but not to GnRH release.35 It can also not be excluded that KP 45–50 has partial agonist activity at the KP receptor that mediates the neuroprotective response. This protective activity may mask actions to block the KP receptor, and we therefore used siRNA to inhibit the expression of the KP receptor (GPR-54) to determine whether loss of the KP receptor signaling would alter the toxicity of the Aβ 1–42, PrP 106–126, and IAPP 1–37 peptides. Results showed that the toxicity of Aβ 1–42, PrP 106–126, and IAPP 1–37 peptides was the same in GPR-54 and control siRNA treated cells (Figure 4). The treatment with GPR-54 siRNA did not have any significant effect on the neuroprotection of KP 45–54 against Aβ 1–42, PrP 106–126, or IAPP 1–37 neurotoxicity.
Figure 4.
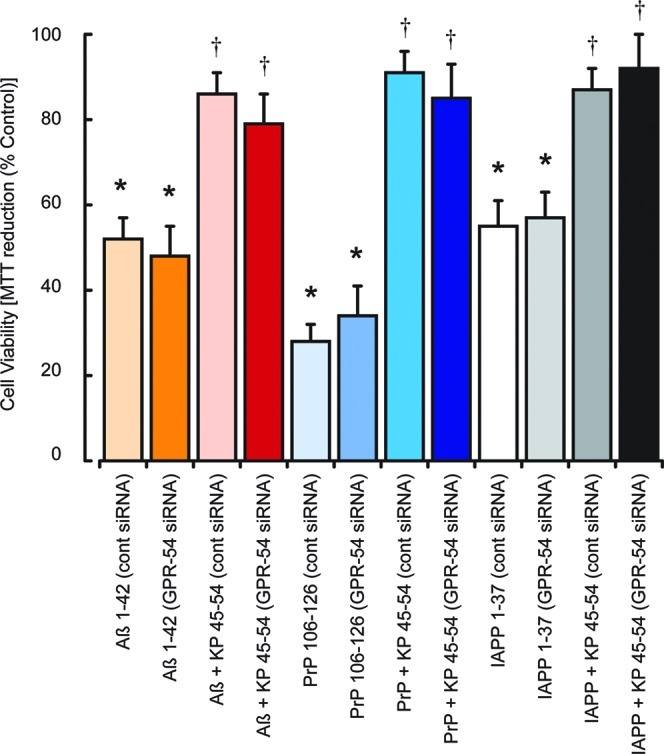
Effects of KP receptor knockdown on KP protection against Aβ, PrP, and IAPP neurotoxicity in SH-SY5Y neurons. Neuronal SHSY-5Y cell cultures were treated with control siRNA or GPR-54 siRNA and allowed to recover for 48 h prior to exposure to Aβ 1–42, PrP 106–126, or IAPP 1–37 (5 μM each) with or without 10 μM KP 45–54. Cell viability was determined by the MTT assay. All results are expressed as % control (siRNA treated SH-SY-5Y cells in media alone) and are expressed as the mean ± SEM (n = 8).
The observation that NPFF protects against Aβ 1–42 toxicity but not PrP 106–126 or IAPP 1–37 toxicity suggests that actions via NPFF are not responsible for KP neuroprotection. The results obtained using the NPFF receptor antagonist RF9 support this conclusion.
KP Peptide Inhibition of IAPP Toxicity in Pancreatic Islet Cultures
The principle site of action of IAPP in T2DM is the pancreas, where IAPP deposits are associated with β-islet degeneration.21 Since IAPP is neurotoxic and KP protects against this action, we tested the effects of KP 45–50 on IAPP 1–37 toxicity in a human pancreatic islet cell line.34 Results showed that KP 45–54 caused a dose-dependent inhibition of 5 μM IAPP 1–37 toxicity, which was significant at concentrations above 0.63 μM (Figure 5A). The KP receptor antagonist P234 peptide (10 μM) was protective against IAPP 1–37 toxicity, while the NPFF antagonist RF9 had no effect (Figure 5B). The P234 increased the protection of KP 45–54 against IAPP 1–37 toxicity, while the RF9 had no effect on KP 45–54 protection. These results indicate that KP peptides can also prevent amyloid peptide toxicity outside a neuronal setting and that KP could also have actions in a T2DM setting where IAPP toxicity in β-islets plays pathological role.21
Figure 5.
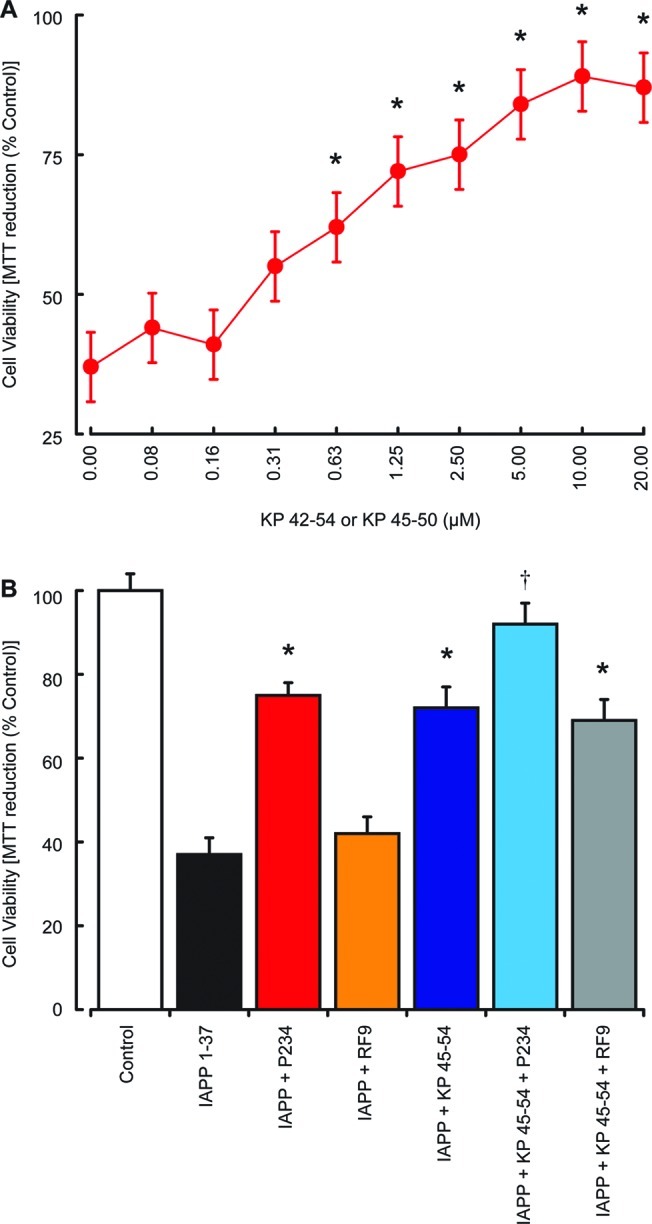
Effects of KP peptides on IAPP toxicity in pancreatic islet cultures. The effects of 10 μM KP 45–54 (closed red circles) on the toxicity of 5 μM IAPP 1–37 was tested in human pancreatic islet cell cultures (A), with cell viability determined by the MTT assay. The effects of 10 μM KP receptor antagonist P234 or 10 μM NPFF receptor antagonist RF9 on 2.5 μM KP 45–54 protection against the toxicity of 5 μM IAPP 1–37 (B) was tested in human pancreatic islet cell cultures, with cell viability determined by the MTT assay. All results are expressed as % control (pancreatic islet cells in media alone) and are expressed as the mean ± SEM (n = 8). (* = P < 0.05 vs IAPP alone; † = P < 0.05 vs IAPP and KP 45–54; one-way ANOVA.)
Effects of Modified KiSS-1 Expression and Anti-KP on Aβ, PrP, and IAPP Neurotoxicity in SH-SY5Y Neurons
The neuroprotective actions of KP peptides against Aβ, PrP, and IAPP toxicity combined with the ability of these peptides to stimulate the release of irKP peptides suggests that endogenous KP could be protective. To assess this, we have used siRNA knockdown of KiSS-1 expression or anti-KP 45–54 antibody treatments to reduce the endogenous KP levels both within cells and released into the culture medium. The expression of KiSS-1 in SH-SY5Y neurons was inhibited using specific siRNAs targeting the human KiSS-1 gene, and results were compared to cells treated with a control siRNA. For siRNA treatments, the cells were incubated for 48 h after treatment of the cells with siRNAs cells before exposure to either Aβ 1–42, Aβ 25–35, PrP 106–126, or IAPP 1–37 peptides for a further 24 h prior to the determination of cell viability by the MTT assay. Results showed that inhibition of KiSS-1 expression significantly enhanced the toxicity of Aβ 1–42, Aβ 25–35, PrP 106–126, and IAPP 1–37 when compared to control siRNA treated neurons (Figure 6A). The ability of KiSS-1 siRNA treatment to abolish Aβ 1–42 induced irKP release suggests that the enhanced toxicity seen could be due to the reduction in KP production.
Figure 6.

Effects of modified KiSS-1 expression and anti-KP on Aβ, PrP, and IAPP neurotoxicity in SH-SY5Y neurons. Neuronal SHSY-5Y cell cultures (A) were treated with control siRNA or KiSS-1 siRNA and allowed to recover for 48 h prior to exposure to Aβ 1–42, Aβ 25–35, PrP 106–126, or IAPP 1–37 (5 μM each). Cell viability was determined by the MTT assay. The effects of 10 μg/mL anti-KP 45–54 antibody, 10 μM KP receptor antagonist P234, or 10 μM NPFF receptor antagonist RF9 on the toxicity of 5 μM Aβ 1–42 (B) were tested in human SH-SY5Y neuroblastoma cell cultures, with cell viability determined by the MTT assay. Neuronal SHSY-5Y cells transfected and stably overexpressing (C) either control expression vector (PCont) or expression vector containing the KiSS-1 gene (PKiSS) were treated with Aβ 1–42, Aβ 25–35, PrP 106–126, or IAPP 1–37 (5 μM each) and cell viability determined by the MTT assay after 24 h. The effects of 10 μg/mL anti-KP 45–54 antibody, 10 μM KP receptor antagonist P234, or 10 μM NPFF receptor antagonist RF9 on the toxicity of 5 μM Aβ 1–42 (D) were tested in KiSS-1 overexpressing SH-SY5Y neuroblastoma cell cultures, with cell viability determined by the MTT assay. All results are expressed as % control (SHSY-5Y neurons, siRNA treated SHSY-5Y neurons, or transfected SHSY-5Y neurons in media alone) and are expressed as the mean ± SEM (n = 8). (* = P < 0.05 vs control (media alone); † = P < 0.05 vs control siRNA (A), Aβ alone (B and D), and PCont vector (C); one-way ANOVA.)
To confirm the role of endogenous KP as the neuroprotective component, we treated naïve SH-SY5Y cells with a KP receptor antagonist (P234),35 an NPFF receptor antagonist (RF9),36 or an anti-KP 45–54 antibody to knockout the effects of endogenous KP-like peptides. The antagonists or antibody were added simultaneously with the Aβ 1–42 peptide and incubated for 24 h prior to measurement of cell viability using the MTT assay. Results showed that the KP receptor antagonist and the NPFF receptor antagonist had no effect on the toxicity of Aβ 1–42, while the anti-KP 45–54 antibody significantly enhanced the toxicity of Aβ 1–42 (Figure 6B).
These results suggest that endogenous KP is neuroprotective against Aβ, PrP, and IAPP. As such elevation of endogenous KP may provide a mechanism for protecting against these toxins, we created a stable SHSY-5Y line overexpressing KiSS-1 to determine if KiSS-1 overexpression was neuroprotective. When compared with a stable SHSY-5Y line containing the expression vector, the KiSS-1 overexpressing cell line was significantly resistant to Aβ 1–42, PrP 106–126, and IAPP 1–37 toxicity (Figure 6C). Incubation of these cells in the presence of the KP receptor antagonist (P234), NPFF receptor antagonist (RF9), and an anti-KP 45–54 antibody was used to determine the mechanism of this neuroprotection. Results showed that the KP receptor antagonist and the NPFF receptor antagonist had no effect on the toxicity of Aβ 1–42 in the KiSS-1 overexpressing cells, while the anti-KP 45–54 antibody significantly enhanced the toxicity of Aβ 1–42 (Figure 6D), confirming that the observed neuroprotection against Aβ was due to an increased presence of KP.
Binding of KP to Aβ, PrP, and IAPP Peptides
The observations with modulation of KiSS-1 expression and anti-KP 45–54 antibodies suggest that endogenous KP peptides are neuroprotective. The ability of amyloid peptides to stimulate the release of irKP peptides (Figure 1) combined with the effects of anti-KP 45–54 antibodies (Figure 6B) suggests that protection against Aβ, PrP, and IAPP peptides involves extracellular KP. However, the failure of the KP receptor antagonist, KP receptor knockdown with siRNA, and the NPFF antagonist to block these responses raises questions about the mechanism of action. The receptor antagonists used in this study target the receptors that KP is known to act on;46 however, it is possible that there are other receptors that can be activated by KP. An alternative mechanism of action could be a direct binding interaction between KP and the Aβ, PrP, or IAPP peptides, and we therefore sought to determine if KP does indeed bind these amyloid peptides.
In previous studies, we have shown that human catalase specifically binds Aβ, IAPP, and PrP.38,39,47 The interactions between Aβ and catalase have been used to identify antiamyloid drugs48 and can also be used to identify proteins with amyloid-binding domains based on sequence similarity.49 We have demonstrated that residues 400–409 of human catalase contain the catalase Aβ binding domain (CAβBD), which is neuroprotective,49 and that a peptide containing these residues can also inhibit interactions between catalase and Aβ fibrils.38
A comparison between the human catalase and human metastasis-suppressor KiSS-1 preproprotein sequences was undertaken using the NCBI BLAST program.50 Alignment of the human metastasis-suppressor KiSS-1 preproprotein sequence (NP_002247.3) with the human catalase sequence (NP_001743.1) reveals that KiSS-1 residues 110–118 show 78% identity with catalase residues 402–410. Similar results were obtained using the UniProt database51 with comparison of human KiSS-1 (Q15726) and human catalase (P04040) sequences, which suggested alignment between KiSS-1 residues 109–118 with catalase 401–410 (Figure 7A). Scheme A (Figure 7A) shows the proposed binding of Aβ, PrP, and IAPP peptides to the CAβBD based on experimental eveidence obtained with both catalase and CAβBD binding to the Aβ peptides.38,39,47,49 The observation that the reverse Aβ 40–1 sequence also binds catalase37 and the palindromic nature of the Aβ and CAβBD sequences raises the possibility of an alternative binding arrangement that is illustrated in scheme B (Figure 7A), which was based on the original experimental results obtained with Aβ and the CAβBD.49 The complete CAβBD-like sequence in KiSS-1 corresponds to residues 42–51 of KP and would be found in KP, KP 41–54, and KP 42–54 forms.
Figure 7.

Binding of KP to Aβ, PrP and IAPP peptides. Alignment of the human metastasis-suppressor KiSS-1 preproprotein sequence (NP_002247.3) with the human catalase sequence (NP_001743.1) is shown in A. The red box highlights the region of KP that prevents Aβ, PrP, and IAPP toxicity; blue and green boxes highlights the Gly-Ala-Ile-Ile region that binds catalase in schemes A and B respectively; the black box highlights Aβ 31–35, which inhibits Aβ 1–42 binding to catalase. Immunoplates were coated with Aβ 1–40, Aβ 1–28, Aβ 29–40, Aβ 25–35, PrP 106–126, PrP 118–135, IAPP 1–37, IAPP 20–29, A-Bri 1–34, or A-Dan 1–34 fibrils. Coated plates were incubated with either biotinylated KP 45–54 (B) alone (open columns) or in the presence of unlabeled KP 45–54 (closed blue columns) and bound material determined by EIA. Plates coated with KP 1–54, KP 27–54, KP 42–54, KP 45–54, KP 45–50, KP 45–47, KP 47–50, or NPFF (C) were incubated with biotinylated Aβ 1–42 (open columns), biotinylated PrP 106–126 (red columns), or biotinylated IAPP 1–37 (black columns) and bound material determined by EIA. All results are expressed as the mean ± SEM (n = 8). (* = P < 0.05 vs control (buffer alone); † = P < 0.05 vs biotinylated KP 45–54; one-way ANOVA.)
The demonstration that KiSS-1 and KP are neuroprotective and the identification of CAβBD-like sequence in the region of the KiSS-1 preproprotein that contains the KP peptides suggest that it is possible that neuroprotection is mediated via a direct binding action. We therefore used EIA assays to characterize interactions between KP and amyloid peptides.37 Biotinylated KP 45–54 (Figure 7B) bound plates coated with Aβ 1–40, Aβ 29–40, Aβ 25–35, PrP 106–126, PrP 118–135, IAPP 1–37, and IAPP 20–29 fibrils. No binding to Aβ 1–28, Aβ 31–35, A-Bri 1–34, or A-Dan 1–34 fibrils was observed. Incubation of amyloid peptide coated plates with unlabeled KP 45–54 inhibited the binding of both biotinylated KP 1–54 and KP 45–54 to Aβ 1–42, Aβ 29–40, Aβ 25–35, PrP 106–126, PrP 118–135, IAPP 1–37, and IAPP 20–29 fibrils.
Previously we have shown that Aβ, PrP, and IAPP fibrils bind catalase and that the binding involves recognition of a region with similarity to the Aβ 29–32 Gly-Ala-Ile-Ile sequence.38,39 The alignment of the CAβBD domain with KP 45–54 suggests that the Aβ 29–32 region aligns with KiSS-1 residues 114–117 (Figure 7A), which correspond to KP 47–50. We therefore obtained KP peptides corresponding to residues 45–50, 47–50, and 45–47 to see if they bound labeled amyloid peptides. The biotinylated Aβ 1–42, biotinylated PrP 106–126, and biotinylated IAPP 1–37 showed significant binding to KP 1–54, KP 27–54, KP 42–54, KP 45–54, and KP 45–50 coated plates, while only biotinylated Aβ 1–42 showed significant binding to KP 47–50 coated plates (Figure 7C). The biotinylated Aβ 1–42 (Figure S1, Supporting Information), biotinylated PrP 106–126 (Figure S3, Supporting Information), and biotinylated IAPP 1–37 (Figure S5, Supporting Information) also bound P234 coated plates. The binding to labeled amyloid peptides to KP 1–54, KP 27–54, KP 42–54, KP 45–54, and KP 45–50 coated plates could be inhibited by the corresponding unlabeled amyloid peptide confirming specificity of binding (Figures S1, S3, and S5, Supporting Information).
Binding of biotinylated Aβ 1–42 to the KP 45–50 peptide was inhibited by 10 μM unlabeled amyloid peptides (Aβ 1–40, Aβ 25–35, Aβ 29–40, IAPP 1–37, IAPP 20–29, PrP 106–126, and PrP 118–135), 10 μM of the antiamyloid nonapeptide R9,38 10 μg/mL of an anti-Aβ 21–32 monoclonal antibody, 10 μg/mL of an anti-KP 45–54 polyclonal antibody, and 10 μg/mL of human catalase47 (Figure S2, Supporting Information). Binding of biotinylated PrP 106–126 to the KP 45–50 was inhibited by 10 μM unlabeled amyloid peptides (PrP 106–126, PrP 118–135, IAPP 1–37, IAPP 20–29, Aβ 1–40, Aβ 25–35, Aβ 29–40, PrP 106–126, and PrP 118–135), 10 μg/mL of an anti-KP 45–54 polyclonal antibody and 10 μg/mL of human catalase (Figure S4, Supporting Information). The anti-Aβ 21–32 monoclonal antibody (10 μg/mL) and 10 μM antiamyloid nonapeptide R9 had no effect on PrP binding to KP 45–50. Binding of biotinylated IAPP 1–37 to the KP 45–50 peptide was inhibited by 10 μM unlabeled amyloid peptides (IAPP 1–37, IAPP 20–29, Aβ 1–40, Aβ 25–35, Aβ 29–40, PrP 106–126, and PrP 118–135), 10 μg/mL of an anti-KP 45–54 polyclonal antibody, and 10 μg/mL of human catalase (Figure S6, Supporting Information). The anti-Aβ monoclonal antibody (10 μg/mL) and 10 μM antiamyloid nonapeptide R9 had no effect of IAPP binding to KP 45–50.
Addition of 10 μM Aβ fragments Aβ 1–28 and Aβ 31–35 had no effect on the binding of biotinylated Aβ 1–42, IAPP 1–37, and PrP 106–126 peptides to KP 45–50 (Figures S2, S4, and S6, Supporting Information), while 10 μM Aβ 25–35 and Aβ 29–40 both strongly inhibited binding suggesting that residues 29 and 30 of Aβ and some of the surrounding residues may be key. The 29–32 region of Aβ comprises a Gly-Ala-Ile-Ile sequence and is known to play a role in catalase binding.38 This region is similar to the Gly-Ala-Val-Val sequence of PrP and the Gly-Ala-Ile-Leu sequence of IAPP, which are thought to play a role in catalase binding to these peptides.39 The binding of Aβ, PrP, and IAPP peptides to KP residues 45–50 and the failure of Aβ 31–35 to inhibit the binding suggest the proposed binding alignment in scheme A (Figure 7A) in which the 29–32 region of Aβ binds to residues 45–48 on KP in an antiparallel alignment.
Affinity constants (KD) for binding to KP 45–50 were 0.62 ± 0.07 nM (n = 5) for biotinylated Aβ 1–42; 0.47 ± 0.04 nM (n = 5) for biotinylated Aβ 1–40; 11.6 ± 1.2 nM (n = 5) for biotinylated IAPP 1–37; and 34.2 ± 3.7 nM (n = 5) for biotinylated PrP 106–126. These constants are similar to those previously determined for biotinylated Aβ 1–42 and biotinylated IAPP 1–37 binding to human catalase and a peptide containing residues 400–409 of catalase.39,47,49
Effects of KP Peptides on Cellular Aβ Phosphorylation and Aβ Inhibition of Catalase Activity
In previous studies, we have shown that amyloid binding compounds prevent the uptake the Aβ peptide and generation of the serine 26 phosphorylated derivative of Aβ (pSAβ).52,53 The KP 45–54 peptide (10 μM) prevented the increase in intracellular ir-Aβ caused by the addition of 10 μM Aβ 17–35 and also prevented the increase in the ir-pSAβ. The changes in the levels (expressed as % level in untreated cells) of ir-Aβ and ir-pSAβ were 315 ± 22% (ir-Aβ; mean ± SEM; n = 8) and 429 ± 34% (ir-pSAβ; mean ± SEM; n = 8) in Aβ 17–35 treated cells. In Aβ 17–35 and KP 45–54 treated cells, the levels were significantly lower with ir-Aβ levels of 181 ± 17% (mean ± SEM; n = 8) and ir-pSAβ levels of 212 ± 15% (mean ± SEM; n = 8). These results suggest that the KP 45–54 is blocking the uptake and subsequent phosphorylation of exogenously added Aβ in a manner similar to that of other Aβ binding molecules rather than by blocking just the intracellular phosphorylation as observed with the cannabinoids, which act via cannabinoid receptors to inhibit phosphorylation.53
Another biological activity of Aβ that can be blocked by amyloid binding molecules is the inhibition of catalase breakdown of hydrogen peroxide.47 Preincubation of 2 μM Aβ 25–35 either alone or with 10 μM KP 45–54 for 2 h prior to the addition to catalase was tested to determine if KP altered Aβ inhibition of catalase activity. The catalase inhibition by Aβ 25–35 alone was 27 ± 2.1% (mean ± SEM; n = 8), and the catalase inhibition in the presence of KP 45–54 was significantly reduced to 2.3 ± 0.1% (mean ± SEM; n = 8). These results confirm that KP 45–54 can block the Aβ inhibition of catalase activity and support the suggestion from the binding studies that KP 45–54 binds to the same region of Aβ as catalase.
Effects of KP on Congo Red Binding to Aβ, PrP, and IAPP
Compounds that bind to amyloid peptides have the potential to modify the formation of aggregates.54 The ability of Congo red to identify amyloid aggregates is well documented, and indeed, the definition of amyloid peptides includes an interaction with Congo red as a specific requirement for a protein being termed an amyloid protein.19 We therefore used an assay of Congo red binding38,39 to determine whether the KP 45–54 peptide would alter Congo red binding to the amyloid peptides.
Results showed that the Aβ 1–42, IAPP 1–37, PrP 106–126, PrP 118–135, Amyloid-Bri, and Amyloid-Dan peptides all showed significant Congo red binding amyloid aggregate formation. The concentrations of Congo red binding amyloid aggregates formed by Aβ 1–42, IAPP 1–37, PrP 106–126, and PrP 118–135 were significantly reduced by treatment with KP 45–54 (Figure 8A). The KP 45–54 peptide had no significant effect on the Congo red binding amyloid aggregate formation by either A-Bri 1–34 or A-Dan 1–34 peptides. Previous studies have shown that rat KP 45–54 can form aggregates but that this requires the addition of enhancers such as heparin;55 in the system used here, KP 45–54 showed no detectable Congo red binding aggregate formation.
Figure 8.
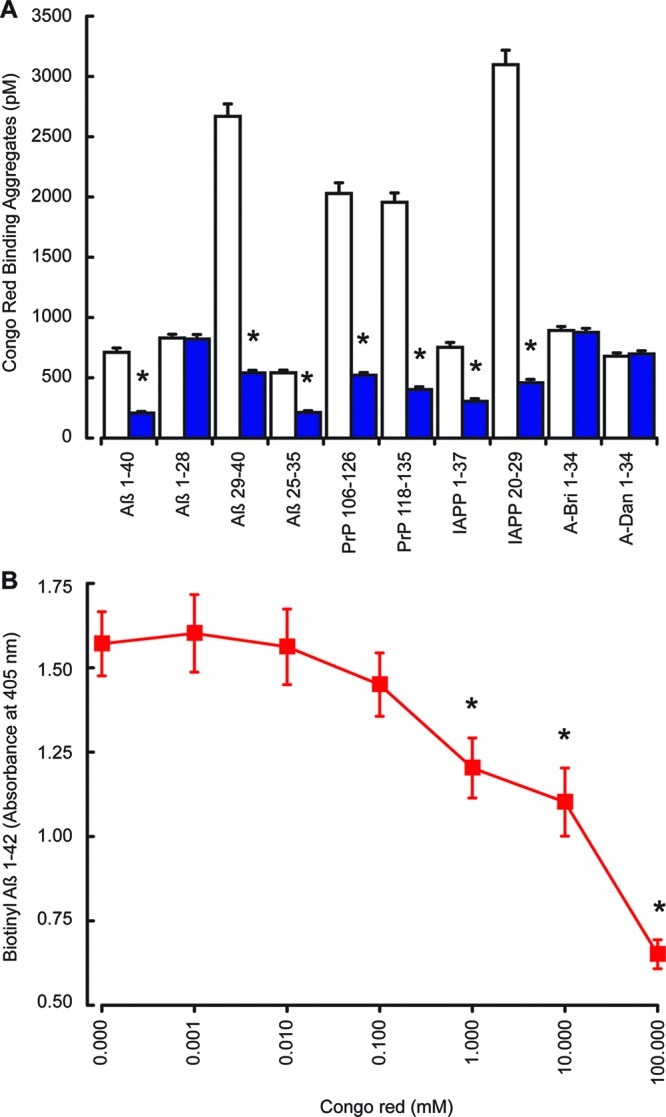
Effects of KP on Congo red binding to Aβ, PrP, and IAPP. The Aβ 1–40, Aβ 1–28, Aβ 25–35, Aβ 29–40, PrP 106–126, PrP 118–135, IAPP 1–37, IAPP 20–29, A-Bri 1–34, and A-Dan 1–34 peptides were dissolved in PBS and incubated at 37 °C for 24 h, with constant oscillation, with or without KP 45–50. Congo red binding assays (A) were performed with amyloid peptides alone (open columns) or in the presence of KP 45–50 (closed blue columns). The effect of Congo red (0–100 mM) on biotinylated Aβ 1–42 binding to KP 45–54 (B) was determined by EIA. All results are expressed as the mean ± SEM (n = 8). (* = P < 0.05 vs amyloid fibrils alone; one-way ANOVA.)
The binding of biotinylated Aβ 1–42 to KP 45–54 was dose dependently inhibited by Congo red (Figure 8B). The concentration of Congo red required to inhibit Aβ binding to KP is 100× higher than that used in the detection of aggregates, raising the possibility that the effects of KP peptides on Congo red binding aggregates could be due to the inhibition of Congo red binding by KP rather than the inhibition of aggregate formation. However, Congo red binding to Aβ has been shown to occur with a range of nonoverlapping regions,38 some of which are not involved in KP binding, suggesting that in the case of Aβ Congo red binding may not be prevented by KP binding and therefore that the reduction in Congo red binding in the presence of KP reflects a reduction in aggregate formation.
Conclusions
The results from this study demonstrate that both endogenous KP and exogenously added KP peptides are neuroprotective. The minimal region of KP to provide protection against Aβ, PrP, and IAPP peptides is the KP 45–50 sequence (Tyr-Asn-Trp-Asn-Ser-Phe), and this is also the minimal region to which these amyloid peptides bind. Catalase binds to Aβ, PrP, and IAPP,38,39,47,48 and this interaction involves a CAβBD49 that shows sequence similarity to KP (Figure 7A). Both catalase and a CAβBD peptide are neuroprotective,38,39,47,48 and the mechanism of neuroprotection involves a direct binding interaction for these compounds. This suggests that the mechanism of neuroprotection involves direct binding of KP to the amyloid peptide and subsequent prevention of toxicity. The ability of KP to reduce the uptake and subsequent phosphorylation of Aβ further supports this mechanism of action. The failure of KP to protect against A-Bri and A-Dan neurotoxicity is matched by the failure of these peptides to bind KP peptides. The A-Bri and A-Dan peptides do not contain a Gly-Ala-Ile-Ile like motif that is found in Aβ, PrP, and IAPP sequences. The ability of an anti-KP 45–54 antibody to block neuroprotection and also KP binding to amyloid peptides further supports this mechanism. The actions of the KP peptides in the neuronal cell systems do not appear to be mediated via activation of either the GPR-54 or NPFF receptor systems that are known targets of KP.6,7,10,11
Compounds that bind to amyloid peptides are also likely to modify Congo red binding,54 and the KP peptides shared this property. Congo red binding is suggestive of fibril formation,19 and the ability of KP peptides to inhibit this suggests that there is potential inhibition of amyloid aggregation. However, the assay we used shows cross-reactivity with Aβ fragments that do not form fibrils visible by transmission electron microscopy (TEM) analysis.38 The ability of Congo red to displace KP binding suggests that there is a shared binding domain; however, in the case of Aβ we have identified multiple Congo red binding domains.38 Further studies using, for example, TEM will be required to determine whether KP peptides can actually disrupt amyloid peptide fibril formation.
The ability of Aβ, PrP, and IAPP peptides to activate irKP release from neuronal cells and the enhancement of Aβ toxicity by an anti-KP 45–54 antibody suggest that the endogenous system plays a role in observed toxicity of these amyloid peptides. The relatively low levels of irKP release combined with the relatively high concentrations of amyloid peptides required to kill cells in culture suggests that under these experimental conditions the endogenous system is insufficient to prevent amyloid peptide toxicity. Contributions of the activated KP release to other effects cannot be excluded. For example, KP inhibits corticotrophin releasing hormone release,56 an effect that would remove the protective actions of endogenous corticotrophin releasing hormone.43 The KP peptide also activates neuropeptide Y expression,57 and neuropeptide Y is neuroprotective against Aβ toxicity.32 The actions of KP inducers may also provide a neuroprotective strategy and could be mediated via KP, for example, leptin activates KP57 and is known to reduce Aβ levels both in vitro and in vivo.58,59 Further in vivo studies will determine whether KP neuroprotection has potential for development as a neuroprotective strategy against Aβ, PrP, and IAPP. Such studies will also help to determine the role of KP neuroprotection in the effects of other protective endogenous neuromodulators.
The observations that KP levels are elevated in the hypothalamus of women postmenopause12 and that in the hypothalamus there is significantly less neurodegeneration in women compared to men13 suggests that the endogenous KP system may play a role in vivo. Menopause is associated with a loss of estrogen production and hence feedback in the regulation of KP,16 a mechanism that would suggest that women should be protected postmenopause rather than more susceptible to AD.14 However, KiSS-1 expression is both positively and negatively regulated by estrogens.60,61 There are roles for the α and β forms of the estrogen receptor in the regulation of KiSS-1 expression,60,61 and the levels of these receptor types are altered in AD.15 Thus, it may be that a loss of this protection in brain regions where neurodegeneration is seen in AD is associated with menopause.
In conclusion, we have identified a neuroprotective action of KP peptides against Aβ, PrP, and IAPP toxicity that is mediated via a KP receptor independent direct binding interaction between the amyloid and KP peptides.
Methods
Test Peptides
N-Terminally biotinylated KP 1–54, KP 45–54, Aβ 1–42, Aβ 1–40, PrP 106–126, and IAPP 1–37 were purchased from Bachem or Alpha Diagnostics. KP peptides (KP 1–54, KP 27–54, KP 42–54, KP 45–54, KP 45–50, KP 45–47, and KP 47–50), NPFF peptides (NPFF and SQA-NPFF), Aβ peptides (Aβ 1–43, Aβ 1–42, Aβ 1–40, Aβ 1–38, Aβ 1–28, Aβ 1–20, Aβ 10–35, Aβ 10–20, Aβ 12–28, Aβ 17–40, Aβ 17–35, Aβ 17–28, Aβ 20–29, Aβ 22–35, Aβ 25–35, Aβ 29–40, Aβ 31–35, and Aβ 33–42), IAPP peptides (IAPP 1–37 and IAPP 20–29), and PrP peptides (PrP 106–126 and PrP 118–135) were purchased from American Peptides, Bachem, and Sigma-Aldrich.
Cell Cultures
Human SH-SY5Y neuroblastoma were routinely grown in a 5% CO2 humidified incubator at 37 °C in a 1:1 mixture of Dulbecco’s modified Eagle’s medium and HAM’s F12 with Glutamax (Invitrogen) supplemented with 10% fetal calf serum (FCS), 1% nonessential amino acids, penicillin (100 units/mL), and streptomycin (100 mg/mL).31 Human pancreatic islet cell line 1.1B4 were maintained in RPMI 1640 medium (Sigma) supplemented with 10% FCS and antibiotics.34
Primary cultures of rat cortical neurons were prepared from cryopreserved rat cortex neurons isolated from day-18 Fisher 344 rat embryos (Invitrogen) and cultured according to the supplier instructions in Neurobasal/B27 medium.33
KiSS-1 Overexpression
The human KiSS-1 cDNA clone (NM_002256) was obtained from Origene and PCR cloned into the pcDNA4/TO/myc–His expression vector using forward (5′-TTAGGATCCATGAACTCACTGGTTTCTTGGCA-3′) and reverse (5′-ATACTCGAGGCCCCGCCCAGCGCTTCT-3′) oligonucleotides to create the PKiSS-1 expression vector. SH-SY5Y cells were transfected with PKiSS-1 or control vector using lipofectamine (Invitrogen), and stably expressing clones were selected by culturing in 100 μg/mL Zeocin (Invitrogen). The presence of KiSS-1 overexpression was confirmed by Western blot analysis.
siRNA Transfection
Using 24 well plates, 5 × 104 SH-SY5Y cells/mL were incubated, after differentiation for 7 days with 10 mM retinoic acid, in normal media without antibiotics for 18 h. Cells were washed with 1 mL oftransfection medium (Santa Cruz; sc-36868). The human KiSS-1 siRNA (Santa Cruz; sc-37443) or GPR-54 siRNA (Santa Cruz; sc-60747), each containing a pool of 3 target-specific 20–25 nucleotide siRNAs designed to knock down gene expression or control siRNA (Santa Cruz; sc-37007), was diluted in transfection medium (Santa Cruz; sc-36868) and added 0.1 mL/well prior to incubation with cells for 6 h at 37 °C in a CO2 incubator. A further 1 mL of normal media containing 20% fetal calf serum, 2% penicillin, and 2% streptomycin was added directly to the cells, which were incubated for a further 18 h. Medium was replaced with normal media for 24 h prior to exposure to test substances and viability assays. For KiSS-1 siRNA transfections, the ir-KP levels were determined by EIA to confirm gene expression knockdown. For GPR-54, Western blots of cell membrane extracts were prepared to confirm reduction in GPR-54 expression.
Cell Viability
Test peptides or drugs were added directly to culture medium prior to incubation for 24 h. Cell viability was determined either by trypan blue dye exclusion with at least 100 cells counted per well or by MTT reduction.31 For MTT reduction determination, after incubation with test substances MTT (10 μL: 12 mM stock) was added and cells incubated for a further 4 h. Cell lysis buffer [100 μL/well; 20% (v/v) SDS and 50% (v/v) N,N-dimethylformamide, pH 4.7] was added, and after repeated pipetting to lyse cells, the MTT formazan product formation was determined by measurement of absorbance change at 570 nm. Control levels in the absence of test substances were taken as 100%, and the absorbance in the presence of cells lysed with Triton X-100 at the start of the incubation period with test substances taken as 0%.
Western-Blot Analysis of KiSS-1 and GPR-54
Cells were lysed on ice in 20 mM HEPES buffer supplemented with 1% Nonindet P-40, 1 mM EDTA (EDTA), 150 mM sodium chloride (NaCl), 0.25% sodium deoxycholate, and protease inhibitors. Cell lysates were incubated for 1 h in lysis buffer and centrifuged at 12,000g for 10 min at 4 °C. Total protein was measured by using the BCA assay. Supernatants were diluted to 1 mg/mL and resuspended in sample buffer before boiling for 5 min and separation of samples using a 15% SDS–PAGE gel. Proteins were then transferred to a nitrocellulose membrane and membranes blocked with 3% nonfat dried milk powder in PBS containing 0.1% Tween 20 (1 h at room temperature). Membranes were incubated overnight at 4 °C with rabbit anti-GPR-54 or rabbit anti-KP 45–54 antibody. Unbound antibody was rinsed from the membranes before incubation with horseradish peroxidase-conjugated goat antirabbit secondary antibody. Immunoreactivity was detected using an enhanced chemiluminescence substrate and UVP BioImaging system.
Kisspeptin EIA
Tissue culture media concentrations of KP 1–54 and KP 45–54 were determined using EIA kits (Bachem) according to the manufacturer's instructions. Samples or standards were mixed with biotinyl-kisspeptin and rabbit anti-KP antiserum in antirabbit IgG coated plates. Binding of biotinyl-KP was detected using a streptavidin-conjugated horseradish peroxidase conjugate and TMB substrate. Absorbance readings were measured at 450 nm after the addition of 2 M HCl to samples to stop the reaction.
Cellular Aβ and pAβ Measurement
Human SH-SY-5Y neurons were incubated either alone or with Aβ 17–35 with or without KP 45–54 for 24 h. Extracts from SH-SY-5Y neurons were prepared in DEA buffer supplemented with 0.1 mM sodium vanadate. Using a polyclonal anti-Aβ 15–30 antiserum plus protein-A agarose, the Aβ was immunoprecipitated. The resultant extracts were further purified using a Sep-Pak C18 extraction step. Columns were prewetted with methanol and 0.5 M acetic acid, samples applied in 20% acetonitrile in 0.1% TFA, and columns washed with 20% acetonitrile in 0.1% TFA prior to elution of bound peptide with 70% acetonitrile. After drying under a stream of nitrogen, samples were resuspended in PBS.62
ELISA plates were coated with either anti-Aβ 15–30 antiserum, for ir-Aβ measurement, or antiphosphoserine antibody, for ir-pSAβ measurement, and blocked with 5% marvel. Samples or synthetic Aβ standards (Aβ 1–40 or pAβ 1–40) were applied in PBS containing 0.1% BSA and 0.05% Tween 20. Monoclonal antibody ALI-01 was added and incubated for 2 h. After washing to remove unbound material, ir-Aβ or ir-pSAβ was detected using an antimouse IgG-HRP conjugate and TMB substrate.62
Aβ Inhibition of Catalase Activity
Catalase (EC 1.11.1.6) from human erythrocytes (Sigma, Dorset, UK) was used for all incubation experiments. Activity of catalase (50 kU/L) incubated with Aβ (2 μM) preincubated for 2 h either alone or with KP 45–54 (10 μM) was determined by spectrophotometric measurement of H2O2 breakdown as previously described.37
Binding Studies
Ninety-six-well immunoplates were coated either with KP peptides (1 μg/mL), NPFF peptides (1 μg/mL), Aβ peptides (1 μg/mL), IAPP peptides (1 μg/mL), PrP peptides (1 μg/mL), Amyloid-Bri (1 μg/mL), Amyloid-Dan (1 μg/mL), or catalase (1 μg/mL) in carbonate buffer, pH 9.6, and unoccupied sites blocked with 0.2% (w/v) marvel. Biotinylated peptides (200 pM) were incubated alone, with control peptides, with KP peptides, or with unlabeled forms of the respective amyloid peptides in 50 mM TRIS (containing 0.1% BSA and 0.1% Triton X-100) at 4 °C for 16 h. After washing to remove unbound material, an alkaline phosphatase polymer–streptavidin conjugate was added and incubated at 24 °C for 2 h. After washing to remove unbound material, p-nitrophenylphosphate substrate was added and absorbance at 405 nm determined. Affinity constants were determined by incubating KP 45–50 coated plates with biotinylated peptides (200 pM) and unlabeled peptides over a range of concentrations (0–100 nM) and detection of bound peptides by EIA.37
Scatchard analysis was performed using the following equation: A0/(A0 – A) = 1 + KD/a0 and plotting v [(A0 – A)/A0] against v/a, where A0 = absorbance in the absence of unlabeled peptide, A = absorbance in the presence of unlabeled peptide, a0 = total concentration of unlabeled peptide, and a = concentration of unlabeled peptide added. The KD was equal to −1/slope of v against v/a.47
Spectrophotometric Analysis Congo Red Binding to Amyloid Peptides
Freshly prepared 50 μM solutions of amyloid peptides (Aβ 1–42, IAPP 1–37, PrP 106–126, PrP 118–135, A-Bri, and A-Dan) were incubated either alone or in the presence of either KP 45–54 or KP 45–50, after dissolving in distilled water, at 37 °C for 24 h with constant oscillation. Following in vitro fibrillogenesis of peptides, an aliquot of each test sample was prepared to give a 50 μM concentration of amyloid peptide in phosphate buffered saline (PBS). Congo red, prepared as a 200 μM stock in PBS containing 10% ethanol, was then added to give final concentration 10 μM Congo red to 9.09 μM amyloid peptide, and 100 μL aliquots were added to 96 well microtiter plates. After 15 min of incubation, the absorbance levels at 405 and 540 nm were determined. The concentration of amyloid aggregates ([Amyloidagg]) was then calculated, with correction for the path length of the reader used, as follows: [Amyloidagg] = 10((540At/4780) – (405At/6830) – (405ACR/8620)), where 540At = absorbance of amyloid peptide + Congo red solution at 540 nm, 405At = absorbance of amyloid peptide + Congo red solution at 405 nm, and 405ACR = absorbance of Congo red solution at 405 nm. Absorbance readings at 405 and 540 nm were taken for each amyloid peptide, and the ratio of 540AAmyloid /405AAmyloid was checked, where 405AAmyloid = absorbance of amyloid peptide solution at 405 nm and 540AAmyloid = absorbance of amyloid peptide solution at 540 nm.38,39
Data Analysis
All data are expressed as means ± SEM. For cytotoxicity experiments. data are expressed as % dead (trypan blue stained) cells or % control cells MTT reduction. The significance of differences between data was evaluated by one-way analysis of variance (ANOVA). A P value of <0.05 was considered statistically significant.
Acknowledgments
We thank Dr. Mark Odell and Marc Rhyan Puno for assistance with cloning and overexpression of KiSS-1.
Glossary
Abbreviations
- KP
kisspeptin
- Aβ
amyloid-β
- PrP
prion protein
- A-Bri
amyloid-Bri
- A-Dan
amyloid-Dan
- IAPP
islet amyloid polypeptide or amylin
- NPFF
neuropeptide FF
- AD
Alzheimer’s disease
- CJD
Creutzfeldt–Jakob disease
- T2DM
type 2 diabetes mellitus
- GPR-54
kisspeptin receptor
- ir
immunoreactive
- EIA
enzyme immunoassay
- HPG
hypothalamic–pituitary–gonadal
- GnRH
gonadotrophin-releasing hormone
Supporting Information Available
Characterization of amyloid peptide binding to KP peptides, results from biotinylated Aβ, PrP, and IAPP binding to KP peptides determined by EIA and inhibition of binding by unlabeled amyloid peptides and other compounds. This material is available free of charge via the Internet at http://pubs.acs.org.
Author Contributions
N.G.N.M. conceived and designed the experiments. N.G.N.M., A.C., E.R.-F., A.N.N., and M.A. performed the experiments. N.G.N.M., A.C., E.R.-F., A.N.N., and M.A. analyzed the data. N.G.N.M. wrote the paper. N.G.N.M., A.C., E.R-F., A.N.N., and M.A. critically reviewed the manuscript.
The work was funded by the Universities of Roehampton and Westminster, and by grants from WestFocus (Park Board, PCF II 104 to N.G.N.M.), the Wellcome Trust (Biomedical Vacation Scholarship to E.R.-F., M.A., and N.G.N.M.), and the Nuffield Foundation (URB/38335 to E.R.-F., M.A., and N.G.N.M.).
The authors declare the following competing financial interest(s): N.G.N.M. is named as the inventor on patent applications held by the University of Roehampton for the use of kissorphin peptides to treat Alzheimer's disease, Creutzfeldt–Jakob disease or diabetes mellitus (PCT/EP2011/058213); under the University's rules, he could benefit financially if the patent is commercially exploited.
Supplementary Material
References
- Bao A.-M.; Meynen G.; Swaab D. F. (2008) The stress system in depression and neurodegeneration: focus on the human hypothalamus. Brain Res. Rev. 57, 531–553. [DOI] [PubMed] [Google Scholar]
- Tortosa-Martínez J.; Clow A. (2012) Does physical activity reduce risk for Alzheimer’s disease through interaction with the stress neuroendocrine system?. Stress 15, 243–261. [DOI] [PubMed] [Google Scholar]
- Verdile G.; Yeap B. B.; Clarnette R. M.; Dhaliwal S.; Burkhardt M. S.; Chubb S. A. P.; de Ruyck K.; Rodrigues M.; Mehta P. D.; Foster J. K.; Bruce D. G.; Martins R. N. (2008) Luteinizing hormone levels are positively correlated with plasma amyloid-beta protein levels in elderly men. J. Alzheimer's Dis. 14, 201–208. [DOI] [PubMed] [Google Scholar]
- George J. T.; Millar R. P.; Anderson R. A. (2010) Hypothesis: kisspeptin mediates male hypogonadism in obesity and type 2 diabetes. Neuroendocrinology 91, 302–307. [DOI] [PubMed] [Google Scholar]
- Navarro V. M.; Tena-Sempere M. (2012) Neuroendocrine control by kisspeptins: role in metabolic regulation of fertility. Nat. Rev. Endocrinol. 8, 40–53. [DOI] [PubMed] [Google Scholar]
- Kotani M.; Detheux M.; Vandenbogaerde A.; Communi D.; Vanderwinden J. M.; Le Poul E.; Brézillon S.; Tyldesley R.; Suarez-Huerta N.; Vandeput F.; Blanpain C.; Schiffmann S. N.; Vassart G.; Parmentier M. (2001) The metastasis suppressor gene KiSS-1 encodes kisspeptins, the natural ligands of the orphan G protein-coupled receptor GPR54. J. Biol. Chem. 276, 34631–34636. [DOI] [PubMed] [Google Scholar]
- Bilban M.; Ghaffari-Tabrizi N.; Hintermann E.; Bauer S.; Molzer S.; Zoratti C.; Malli R.; Sharabi A.; Hiden U.; Graier W.; Knöfler M.; Andreae F.; Wagner O.; Quaranta V.; Desoye G. (2004) Kisspeptin-10, a KiSS-1/metastin-derived decapeptide, is a physiological invasion inhibitor of primary human trophoblasts. J. Cell Sci. 117, 1319–1328. [DOI] [PubMed] [Google Scholar]
- de Roux N.; Genin E.; Carel J.-C.; Matsuda F.; Chaussain J.-L.; Milgrom E. (2003) Hypogonadotropic hypogonadism due to loss of function of the KiSS1-derived peptide receptor GPR54. Proc. Natl. Acad. Sci. U.S.A. 100, 10972–10976. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Takino T.; Koshikawa N.; Miyamori H.; Tanaka M.; Sasaki T.; Okada Y.; Seiki M.; Sato H. (2003) Cleavage of metastasis suppressor gene product KiSS-1 protein/metastin by matrix metalloproteinases. Oncogene 22, 4617–4626. [DOI] [PubMed] [Google Scholar]
- Lyubimov Y.; Engström M.; Wurster S.; Savola J.-M.; Korpi E. R.; Panula P. (2010) Human kisspeptins activate neuropeptide FF2 receptor. Neuroscience 170, 117–122. [DOI] [PubMed] [Google Scholar]
- Oishi S.; Misu R.; Tomita K.; Setsuda S.; Masuda R.; Ohno H.; Naniwa Y.; Ieda N.; Inoue N.; Ohkura S.; Uenoyama Y.; Tsukamura H.; Maeda K.-I.; Hirasawa A.; Tsujimoto G.; Fujii N. (2010) Activation of neuropeptide FF receptors by kisspeptin receptor ligands. ACS Med. Chem. Lett. 2, 53–57. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Rance N. E. (2009) Menopause and the human hypothalamus: evidence for the role of kisspeptin/neurokinin B neurons in the regulation of estrogen negative feedback. Peptides 30, 111–122. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Schultz C.; Braak H.; Braak E. (1996) A sex difference in neurodegeneration of the human hypothalamus. Neurosci. Lett. 212, 103–106. [DOI] [PubMed] [Google Scholar]
- Bonomo S. M.; Rigamonti A. E.; Giunta M.; Galimberti D.; Guaita A.; Gagliano M. G.; Müller E. E.; Cella S. G. (2009) Menopausal transition: a possible risk factor for brain pathologic events. Neurobiol. Aging 30, 71–80. [DOI] [PubMed] [Google Scholar]
- Hestiantoro A.; Swaab D. F. (2004) Changes in estrogen receptor-alpha and -beta in the infundibular nucleus of the human hypothalamus are related to the occurrence of Alzheimer’s disease neuropathology. J. Clin. Endocrinol. Metab. 89, 1912–1925. [DOI] [PubMed] [Google Scholar]
- Patisaul H. B.; Losa-Ward S. M.; Todd K. L.; McCaffrey K. A.; Mickens J. A. (2012) Influence of ERbeta selective agonism during the neonatal period on the sexual differentiation of the rat hypothalamic-pituitary-gonadal (HPG) axis. Biol. Sex Differ. 3, 2. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Fuller S. J.; Tan R. S.; Martins R. N. (2007) Androgens in the etiology of Alzheimer’s disease in aging men and possible therapeutic interventions. J. Alzheimer's Dis. 12, 129–142. [DOI] [PubMed] [Google Scholar]
- Pike C. J.; Carroll J. C.; Rosario E. R.; Barron A. M. (2009) Protective actions of sex steroid hormones in Alzheimer’s disease. Front. Neuroendocrinol. 30, 239–258. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Buxbaum J. N.; Linke R. P. (2012) A Molecular History of the Amyloidoses. J. Mol. Biol. 10.1016/j.jmb.2012.01.024. [DOI] [PubMed] [Google Scholar]
- Norrby E. (2011) Prions and protein-folding diseases. J. Intern. Med. 270, 1–14. [DOI] [PubMed] [Google Scholar]
- Zraika S.; Hull R. L.; Verchere C. B.; Clark A.; Potter K. J.; Fraser P. E.; Raleigh D. P.; Kahn S. E. (2010) Toxic oligomers and islet beta cell death: guilty by association or convicted by circumstantial evidence?. Diabetologia 53, 1046–1056. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Tsachaki M.; Ghiso J.; Rostagno A.; Efthimiopoulos S. (2010) BRI2 homodimerizes with the involvement of intermolecular disulfide bonds. Neurobiol. Aging 31, 88–98. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kawahara M.; Kuroda Y.; Arispe N.; Rojas E. (2000) Alzheimer’s beta-amyloid, human islet amylin, and prion protein fragment evoke intracellular free calcium elevations by a common mechanism in a hypothalamic GnRH neuronal cell line. J. Biol. Chem. 275, 14077–14083. [DOI] [PubMed] [Google Scholar]
- Larson M. E.; Lesné S. E. (2012) Soluble Aβ oligomer production and toxicity. J. Neurochem. 120 Suppl 1, 125–139. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Mallucci G.; Collinge J. (2005) Rational targeting for prion therapeutics. Nat. Rev. Neurosci. 6, 23–34. [DOI] [PubMed] [Google Scholar]
- Linden R.; Martins V. R.; Prado M. A. M.; Cammarota M.; Izquierdo I.; Brentani R. R. (2008) Physiology of the prion protein. Physiol. Rev. 88, 673–728. [DOI] [PubMed] [Google Scholar]
- Gurlo T.; Ryazantsev S.; Huang C.-J.; Yeh M. W.; Reber H. A.; Hines O. J.; O’Brien T. D.; Glabe C. G.; Butler P. C. (2010) Evidence for proteotoxicity in beta cells in type 2 diabetes: toxic islet amyloid polypeptide oligomers form intracellularly in the secretory pathway. Am. J. Pathol. 176, 861–869. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Park S. A. (2011) A common pathogenic mechanism linking type-2 diabetes and Alzheimer’s disease: evidence from animal models. J. Clin. Neurol. 7, 10–18. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bossy-Wetzel E.; Schwarzenbacher R.; Lipton S. A. (2004) Molecular pathways to neurodegeneration. Nat. Med. 10 Suppl, S2–9. [DOI] [PubMed] [Google Scholar]
- Tayeb H. O.; Yang H. D.; Price B. H.; Tarazi F. I. (2012) Pharmacotherapies for Alzheimer’s disease: Beyond cholinesterase inhibitors. Pharmacol. Ther. 134, 8–25. [DOI] [PubMed] [Google Scholar]
- Milton N. G. N. (2008) Homocysteine inhibits hydrogen peroxide breakdown by catalase. Open Enzym. Inhib. J. 1, 34–41. [Google Scholar]
- Croce N.; Ciotti M. T.; Gelfo F.; Cortelli S.; Federici G.; Caltagirone C.; Bernardini S.; Angelucci F. (2012) Neuropeptide Y Protects rat cortical neurons against β-amyloid toxicity and re-establishes synthesis and release of nerve growth factor. ACS Chem. Neurosci. 3, 312–318. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Schock S. C.; Jolin-Dahel K. S.; Schock P. C.; Theiss S.; Arbuthnott G. W.; Garcia-Munoz M.; Staines W. A. (2012) Development of dissociated cryopreserved rat cortical neurons in vitro. J. Neurosci. Methods 205, 324–333. [DOI] [PubMed] [Google Scholar]
- McCluskey J. T.; Hamid M.; Guo-Parke H.; McClenaghan N. H.; Gomis R.; Flatt P. R. (2011) Development and functional characterization of insulin-releasing human pancreatic beta cell lines produced by electrofusion. J. Biol. Chem. 286, 21982–21992. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Roseweir A. K.; Kauffman A. S.; Smith J. T.; Guerriero K. A.; Morgan K.; Pielecka-Fortuna J.; Pineda R.; Gottsch M. L.; Tena-Sempere M.; Moenter S. M.; Terasawa E.; Clarke I. J.; Steiner R. A.; Millar R. P. (2009) Discovery of potent kisspeptin antagonists delineate physiological mechanisms of gonadotropin regulation. J. Neurosci. 29, 3920–3929. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Simonin F.; Schmitt M.; Laulin J.-P.; Laboureyras E.; Jhamandas J. H.; MacTavish D.; Matifas A.; Mollereau C.; Laurent P.; Parmentier M.; Kieffer B. L.; Bourguignon J.-J.; Simonnet G. (2006) RF9, a potent and selective neuropeptide FF receptor antagonist, prevents opioid-induced tolerance associated with hyperalgesia. Proc. Natl. Acad. Sci. U.S.A. 103, 466–471. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Milton N. G. N. (2006) Interactions between Amyloid-β and Enzymes, in Cell Biology Protocols (Harris J. R., Graham J. M., and Rickwood D., Eds.), pp 359–363, John Wiley & Sons Ltd., Chichester, England. [Google Scholar]
- Milton N. G. N.; Harris J. R. (2009) Polymorphism of amyloid-beta fibrils and its effects on human erythrocyte catalase binding. Micron 40, 800–810. [DOI] [PubMed] [Google Scholar]
- Milton N. G. N.; Harris J. R. (2010) Human islet amyloid polypeptide fibril binding to catalase: a transmission electron microscopy and microplate study. TheScientificWorldJournal 10, 879–893. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Goodall A. R.; Danks K.; Walker J. H.; Ball S. G.; Vaughan P. F. (1997) Occurrence of two types of secretory vesicles in the human neuroblastoma SH-SY5Y. J. Neurochem. 68, 1542–1552. [DOI] [PubMed] [Google Scholar]
- Poomthavorn P.; Wong S. H. X.; Higgins S.; Werther G. A.; Russo V. C. (2009) Activation of a prometastatic gene expression program in hypoxic neuroblastoma cells. Endocr. Relat. Cancer 16, 991–1004. [DOI] [PubMed] [Google Scholar]
- Iijima N.; Takumi K.; Sawai N.; Ozawa H. (2010) An immunohistochemical study on the expressional dynamics of kisspeptin neurons relevant to GnRH neurons using a newly developed anti-kisspeptin antibody. J. Mol. Neurosci. 43, 146–154. [DOI] [PubMed] [Google Scholar]
- Milton N. G. N. (2002) Anandamide and noladin ether prevent neurotoxicity of the human amyloid-beta peptide. Neurosci. Lett. 332, 127–130. [DOI] [PubMed] [Google Scholar]
- Martínez-Fuentes A. J.; Molina M.; Vázquez-Martínez R.; Gahete M. D.; Jimenez-Reina L.; Moreno-Fernández J.; Benito-Lopez P.; Quintero A.; La Riva, de A.; Dieguez C.; Soto A.; Leal-Cerro A.; Resmini E.; Webb S. M.; Zatelli M. C.; Degli Uberti E. C.; Malagón M. M.; Luque R. M.; Castaño J. P. (2011) Expression of functional KISS1 and KISS1R system is altered in human pituitary adenomas: evidence for apoptotic action of kisspeptin-10. Eur. J. Endocrinol. 164, 355–362. [DOI] [PubMed] [Google Scholar]
- Harms J. F.; Welch D. R.; Miele M. E. (2003) KISS1 metastasis suppression and emergent pathways. Clin. Exp. Metastasis 20, 11–18. [DOI] [PubMed] [Google Scholar]
- Kirby H. R.; Maguire J. J.; Colledge W. H.; Davenport A. P. (2010) International Union of Basic and Clinical Pharmacology. LXXVII. Kisspeptin receptor nomenclature, distribution, and function. Pharmacol. Rev. 62, 565–578. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Milton N. G. N. (1999) Amyloid-beta binds catalase with high affinity and inhibits hydrogen peroxide breakdown. Biochem. J. 344 Pt 2, 293–296. [PMC free article] [PubMed] [Google Scholar]
- Habib L. K.; Lee M. T. C.; Yang J. (2010) Inhibitors of catalase-amyloid interactions protect cells from beta-amyloid-induced oxidative stress and toxicity. J. Biol. Chem. 285, 38933–38943. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Milton N. G. N.; Mayor N. P.; Rawlinson J. (2001) Identification of amyloid-beta binding sites using an antisense peptide approach. Neuroreport 12, 2561–2566. [DOI] [PubMed] [Google Scholar]
- Altschul S. F.; Madden T. L.; Schäffer A. A.; Zhang J.; Zhang Z.; Miller W.; Lipman D. J. (1997) Gapped BLAST and PSI-BLAST: a new generation of protein database search programs. Nucleic Acids Res. 25, 3389–3402. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Jain E.; Bairoch A.; Duvaud S.; Phan I.; Redaschi N.; Suzek B. E.; Martin M. J.; McGarvey P.; Gasteiger E. (2009) Infrastructure for the life sciences: design and implementation of the UniProt website. BMC Bioinformatics 10, 136. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Milton N. G. N. (2001) Phosphorylation of amyloid-beta at the serine 26 residue by human cdc2 kinase. Neuroreport 12, 3839–3844. [DOI] [PubMed] [Google Scholar]
- Milton N. G. N. (2005) Phosphorylated amyloid-beta: the toxic intermediate in alzheimer’s disease neurodegeneration. Subcell. Biochem. 38, 381–402. [DOI] [PubMed] [Google Scholar]
- Novick P. A.; Lopes D. H.; Branson K. M.; Esteras-Chopo A.; Graef I. A.; Bitan G.; Pande V. S. (2012) Design of β-amyloid aggregation inhibitors from a predicted structural motif. J. Med. Chem. 55, 3002–3010. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Nielsen S. B.; Franzmann M.; Basaiawmoit R. V.; Wimmer R.; Mikkelsen J. D.; Otzen D. E. (2010) beta-Sheet aggregation of kisspeptin-10 is stimulated by heparin but inhibited by amphiphiles. Biopolymers 93, 678–689. [DOI] [PubMed] [Google Scholar]
- Rao Y. S.; Mott N. N.; Pak T. R. (2011) Effects of kisspeptin on parameters of the HPA axis. Endocrine 39, 220–228. [DOI] [PubMed] [Google Scholar]
- Backholer K.; Smith J. T.; Rao A.; Pereira A.; Iqbal J.; Ogawa S.; Li Q.; Clarke I. J. (2010) Kisspeptin cells in the ewe brain respond to leptin and communicate with neuropeptide Y and proopiomelanocortin cells. Endocrinology 151, 2233–2243. [DOI] [PubMed] [Google Scholar]
- Fewlass D. C.; Noboa K.; Pi-Sunyer F. X.; Johnston J. M.; Yan S. D.; Tezapsidis N. (2004) Obesity-related leptin regulates Alzheimer’s Abeta. FASEB J. 18, 1870–1878. [DOI] [PubMed] [Google Scholar]
- Greco S. J.; Sarkar S.; Johnston J. M.; Tezapsidis N. (2009) Leptin regulates tau phosphorylation and amyloid through AMPK in neuronal cells. Biochem. Biophys. Res. Commun. 380, 98–104. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Glidewell-Kenney C.; Hurley L. A.; Pfaff L.; Weiss J.; Levine J. E.; Jameson J. L. (2007) Nonclassical estrogen receptor alpha signaling mediates negative feedback in the female mouse reproductive axis. Proc. Natl. Acad. Sci. U.S.A. 104, 8173–8177. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Gottsch M. L.; Navarro V. M.; Zhao Z.; Glidewell-Kenney C.; Weiss J.; Jameson J. L.; Clifton D. K.; Levine J. E.; Steiner R. A. (2009) Regulation of Kiss1 and dynorphin gene expression in the murine brain by classical and nonclassical estrogen receptor pathways. J. Neurosci. 29, 9390–9395. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Milton N. G. N. (2006) Amyloid-β Phosphorylation, in Cell Biology Protocols (Harris J. R., Graham J. M., and Rickwood D., Eds.), pp 364–368, John Wiley & Sons Ltd., Chichester, England. [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.