

Abstract
Morphine and related drugs are widely employed as analgesics despite the side effects associated with their use. Although morphine is thought to mediate analgesia through mu opioid receptors, delta opioid receptors have been implicated in mediating some side effects such as tolerance and dependence. Here we present evidence in rhesus monkeys that morphine, fentanyl, and possibly methadone selectively activate mu-delta heteromers to produce antinociception that is potently antagonized by the delta opioid receptor antagonist, naltrindole (NTI). Studies with HEK293 cells expressing mu-delta heteromeric opioid receptors exhibit a similar antagonism profile of receptor activation in the presence of NTI. In mice, morphine was potently inhibited by naltrindole when administered intrathecally, but not intracerebroventricularly, suggesting the possible involvement of mu-delta heteromers in the spinal cord of rodents. Taken together, these results strongly suggest that, in primates, mu-delta heteromers are allosterically coupled and mediate the antinociceptive effects of three clinically employed opioid analgesics that have been traditionally viewed as mu-selective. Given the known involvement of delta receptors in morphine tolerance and dependence, our results implicate mu-delta heteromers in mediating both antinociception and these side effects in primates. These results open the door for further investigation in humans.
Keywords: Analgesics, Allosterism, GPCR heteromers, opioid antagonism, primates
Pain is a universal phenomenon that can be expressed as an underlying symptom of different diseases or injury. When pain becomes a chronic condition, it can be a disease unto itself. According to the American Association of Pain Medicine, 40% of all Americans experience pain daily and over 100 billion dollars is spent on treatments annually.1 Morphine and other opiates derived from the opium poppy have been the first line therapy for chronic pain for over a hundred years. Synthetic analgesics such as methadone and fentanyl have also been widely employed.2 A feature common to these analgesics is that they produce side effects that include tolerance, physical dependence, and respiratory depression.2
Morphine, methadone, and fentanyl (Figure 1) are believed to bind selectively and produce their effects via the mu opioid receptor,3−7 which is one of three types of opioid receptors that mediate antinociception. The receptors are commonly referred to as mu (MOP), kappa (KOP), and delta (DOP)8−11 and are members of the class-A family of G protein-coupled receptors. Opioid receptor proteins are products of different genes,12 they have dissociable anatomic distributions, and they play distinct roles in physiology and behavior.2,13,14
Figure 1.

Chemical structures of opioid agonists used in the study.
The tripartite classification of opioid receptors has served as a powerful organizing principle in the classification of existing opioids and the development of new analgesics. The origin of this classification is based primarily on binding data and the assumption that opioid receptors exist as monomers or homomers. However, the concept of mu, delta, and kappa receptors as distinct entities has been challenged by the discovery of higher order complexity of opioid receptor organization and function. Thus, mu, delta, and kappa receptors can oligomerize with identical opioid receptors, different opioid receptors, or nonopioid receptors to form larger protein complexes.16 For example, three types of homomeric (mu-mu, delta-delta, kappa-kappa) and heteromeric (mu-delta, mu-kappa, delta-kappa) opioid receptors have been reported in cultured cells.17−19
Studies with delta receptor antisense knockdown,20 delta receptor knockout,21 coadministration of delta antagonists with mu agonists,22 and bivalent ligands with mu agonist and delta antagonist pharmacophores23,24 have all indicated the possible relevance of interaction between mu and delta receptors in development of analgesic tolerance and dependence. The possibility that physically coupled mu-delta heteromeric receptors could be responsible for these effects requires colocalization of these receptors in the same neuron. This was challenged by a recent study using delta-GFP knockin mice which showed that mu and delta receptors do not colocalize in nociceptor neurons in these mice.25 However, Hökfelt and colleagues have recently rebutted this finding as an artifact of modified receptor trafficking due to the GFP fusion protein.26 In other strains of mice with wild-type delta receptors, extensive colocalization was observed using selective antibodies for native mu and delta receptors.26 Also, antibodies selective for mu-delta heteromers have been developed that show mu-delta heteromers to be extensively distributed in the CNS of rodents.27 Moreover, the report that coexpressed mu and delta receptors are transported as mu-delta heteromers from the endoplasmic reticulum (ER) to the surface of cultured cells lends credence to the idea that this may occur in vivo.28
A major deficiency in relating behavioral effects to specific opioid receptors is the lack of information on ligands that are selective for oligomeric receptors. Recent studies of commonly used opioid ligands using both the [35S]GTPγS and intracellular calcium release assays revealed that morphine and DAMGO, both considered to be mu-selective agonists, display greater potency in HEK293 cells coexpressing both mu and delta receptors than in cells expressing only a single receptor type (mu, delta, or kappa) or in cells coexpressing other opioid receptor pairs (mu-kappa, delta-kappa).29 Given that the physical association of mu and delta receptors as heteromers in cultured cells is well established, these results were interpreted to suggest that heteromeric receptors rather than homomeric receptors may contribute to the clinically relevant physiological and behavioral effects of morphine and other opioid analgesics.
In the present study, we show that the clinically employed analgesics, morphine, methadone, and fentanyl, all are selective activators of mu-delta heteromers in HEK293 cells and are antagonized by the selective delta opioid receptor antagonist, naltrindole (NTI).30 Moreover, NTI antagonized morphine when coadministered intrathecally (i.t.), but not intracerebroventricularly (i.c.v.), suggesting that mu-delta heteromers are expressed in the spinal cord of drug-naive rodents. Significantly, antagonism of the antinociceptive effects of these analgesics by NTI in rhesus monkeys implicates the involvement of mu-delta heteromers in mediating antinociception.
Results
Fentanyl and Methadone Activate Mu-Delta Opioid Heteromers
We have previously shown that the mu agonist, morphine, selectively activated mu-delta opioid receptors in HEK-293 cells,29. To determine the functional selectivity of methadone and fentanyl, we utilized the intracellular calcium release method that has been described previously.29,31 HEK-293 cells stably expressing mu or delta receptors alone, mu and delta receptors in pairs, and mu and kappa receptors together were used for this study. These cell lines have been characterized using colocalization and coimmunoprecipitation studies and shown to contain heteromeric opioid receptors.29,31−33 We have also conducted radioligand binding studies to show that mu receptors are similarly expressed in mu, mu-delta, and mu-kappa cell lines (Table S1 in the Supporting Information). Both methadone and fentanyl, while activating mu receptors, produced more potent activation in cells coexpressing mu and delta opioid receptors (Figure 2). Methadone was ∼12-fold more potent at mu-delta (EC50 = 67.7 ± 1.9 nM) than at mu (EC50 = 837.5 ± 23.5 nM) receptors, while fentanyl was ∼6-fold more potent in cells expressing both mu and delta receptors (EC50 = 54.7 ± 18.2 nM) when compared to cells containing only mu receptors (EC50 = 326.6 ± 12.0 nM). Neither of these ligands produced significant activity in cells expressing only delta opioid receptors.
Figure 2.

Methadone and fentanyl selectively activate mu-delta opioid receptor heteromers. Intracellular Ca2+ ion release mediated by increasing concentrations of either (a) fentanyl or (b) methadone, were measured in HEK-293 cells stably expressing opioid receptors and transiently transfected with the chimeric G-protein (Δ6-Gαqi4-myr). We have previously shown that morphine also selectively activates mu-delta opioid receptor heteromers (ref (29)). Response was measured as change in relative fluorescence units (ΔRFU = RFUmax – RFUmin). All points in the panels represent mean ± SEM from triplicate biological replications.
NTI Antagonism in HEK-293 Cells
To relate the contribution of the delta protomer in mu-delta heteromers to the agonist effect of morphine, methadone, and fentanyl in HEK293 cells, we examined the ability of the delta-selective antagonist, NTI,30 to affect the activation at homomeric and heteromeric opioid receptors (Figure 3). NTI did not produce any antagonism in cells expressing mu opioid receptors alone (Figure 3a–c). Significantly, NTI antagonized the activation of all three analgesics at mu-delta heteromers (Figure 3d–f). However, the antagonism of methadone by NTI is surmountable by increasing the concentration of methadone (Figure 3f), unlike morphine and fentanyl (Figure 3d,e).
Figure 3.

Antagonism of morphine, methadone, and fentanyl by naltrindole (NTI) occurs only at mu-delta heteromers. (a–c) NTI (1 μM) did not produce significant antagonism of morphine, methadone, or fentanyl (all 1 μM) in HEK-293 cells expressing only mu opioid receptors. However, NTI (d–f) significantly antagonized all three agonists (0.1 μM and 1 μM) in HEK-293 cells coexpressing both mu and delta opioid receptors. The antagonism of NTI (1 μM) could be surmounted by increasing the concentration of methadone (f), but not morphine (d) or fentanyl (e). The Y-axis represents % change in relative fluorescence units (RFU), and all the data are plotted as mean ± SEM.
NTI Antagonism in Rhesus Monkeys and Mice
To evaluate whether these agonists were producing antinocicpetion via mu-delta heteromers or homomeric mu receptors, NTI was employed to determine if it would antagonize their effects in rhesus monkeys. Indeed, antagonism of the antinociceptive effects of morphine, methadone, and fentanyl was observed upon NTI pretreatment (0.1–3.2 mg/kg) (Figure 4a–c). Naltrindole produced a dose-dependent rightward shift in the morphine and fentanyl dose–effect curves with significant increases in morphine and fentanyl ED50 values at NTI doses of 0.32–3.2 mg/kg. While NTI also shifted the methadone dose–effect curve to the right, the antagonist effect was significant only at the highest dose of NTI (3.2 mg/kg). Figure 4d and Table S2 in the Supporting Information show NTI antagonism quantified as dose ratios. Doses of 1.0 and 3.2 mg/kg of NTI produce greater antagonism of morphine and fentanyl than of methadone.
Figure 4.

Effects of pretreatment with NTI on (a) morphine-, (b) fentanyl-, and (c) methadone-induced antinociception in the assay of thermal nociception using a 50 °C thermal stimulus in rhesus monkeys (n = 3–4). Naltrindole was administered 30 min before the start of the behavioral session. Abscissae for panels (a–c): Dose of test drug in mg/kg (log scale). Ordinates for panels (a–c): Percent maximal possible effect (% MPE). Dashed lines indicate the antinociceptive effects of each mu agonist alone. Panel (d) shows dose ratios (ED50 of mu agonist in the presence of naltrindole ÷ ED50 of the mu agonist alone) for morphine (triangles), fentanyl (circles), and methadone (squares) as a function of the naltrindole dose. Closed symbols show no significance, while open symbols in panel (d) represent points significantly different from methadone within a naltrindole dose as indicated by nonoverlapping confidence limits shown in Table S2 in the Supporting Information. All drugs were administered via intramuscular injections. All points in all panels represent mean ± SEM.
To determine if mu-delta heteromers mediated opioid analgesia in mice, we pretreated mice with NTI and then administered morphine (both i.c.v. and i.t.). The control ED80 dose of morphine (5 nmol/mouse) produced similar antinociception (88% MPE ± 8.5) after NTI (5 nmol/mouse) administration demonstrating that NTI had no effect on morphine when both ligands were administered i.c.v. However, NTI shifted the dose response curve of morphine by almost 10-fold when administered i.t. (Figure 5), suggesting that mu-delta heteromers mediate antinociception in the spinal cord of mice.
Figure 5.
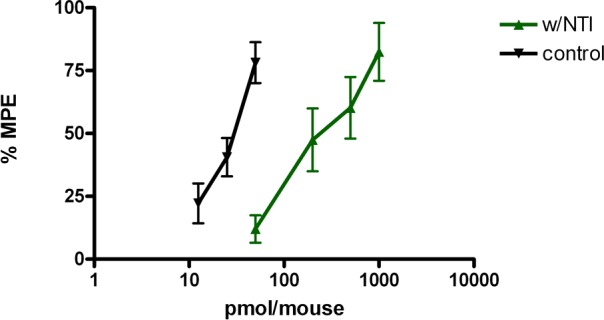
Effect of intrathecal administration of NTI on the antinociceptive activity of morphine in mice. Antinociceptive dose–response curves were established for morphine in the absence and presence of NTI (5 nmol/mouse). NTI and morphine were administered 20 and 10 min, respectively, before the start of the tail-flick experiment. The dose–response curve of morphine (ED50 = 25.6 pmol/mouse; CI, 21.3–35.7) was 9.7-fold right-shifted by NTI (ED50 = 267.0 pmol/mouse; CI, 155.9–457.1).
Discussion
The functional selectivity of methadone and fentanyl was determined using the intracellular calcium release method as described previously (29) in HEK-293 cells. Given that the calcium release and [35S]GTPγS assays give qualitatively similar results with respect to the selectivity of morphine for mu-delta heteromers, calcium release is a reasonable method for such studies. The HEK-293 cellular data was consistent with potent activation in cells coexpressing mu and delta opioid receptors (Figure 2) by both methadone and fentanyl. Methadone and fentanyl were ∼12-fold and ∼6-fold more potent at mu-delta than at mu receptors (Figure 2), respectively. Neither of these ligands produced significant activity in cells expressing only delta opioid receptors. Thus, when taken together with the fact that morphine has been shown to selectively activate mu-delta heteromers,29 all three of these commonly used analgesics selectively activate mu-delta opioid receptor heteromers.
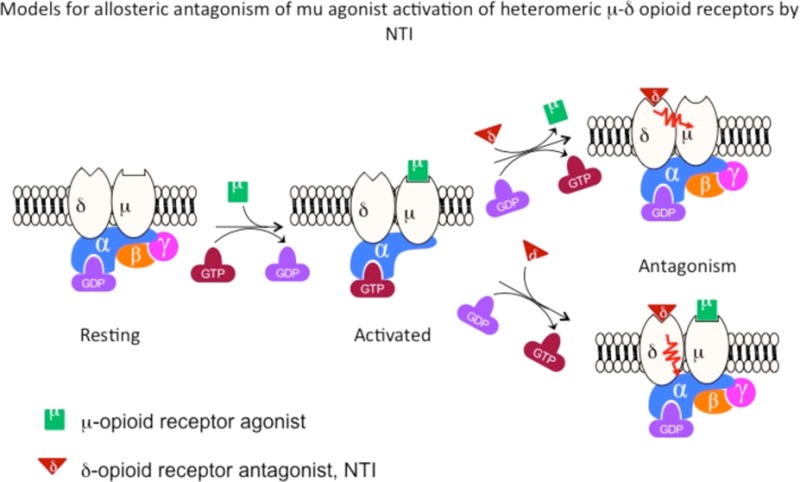
It was then hypothesized that occupying the delta protomer in the mu-delta heteromer with the delta antagonist, NTI,30 would lead to allosteric inhibition of morphine, methadone, and fentanyl. NTI did not produce any antagonism in cells expressing mu opioid receptors alone (Figure 3a–c), indicating that NTI did not inhibit mu opioid receptors. Significantly, NTI antagonized the activation of all three analgesics at mu-delta heteromers (Figure 3d–f). These data indicate that antagonism was mediated via the delta protomer rather than its mu partner, strongly suggesting allosterism between the delta and mu protomers. Another distinction is that the NTI antagonism of methadone is surmountable by increasing the concentration of methadone (Figure 3f), whereas this is not the case with morphine and fentanyl (Figure 3d,e).
In order to investigate the relevance of our cell-based results with respect to the antinociception produced by morphine, methadone, and fentanyl, we conducted analogous studies in rhesus monkeys. All three ligands were observed to produce dose-dependent thermal antinociception,12 as shown by the dose–effect curves for each opioid agonist administered alone (Figure 4a–c; Table 1). Furthermore, NTI at doses of 0.32–3.2 mg/kg significantly antagonized the antinociceptive effects of morphine, fentanyl, and methadone in rhesus monkeys, strongly suggesting the involvement of mu-delta heteromers in the antinociception produced by the agonists. It should also be noted that, overall, NTI was more potent as an antagonist of antinociception induced by morphine and fentanyl than by methadone.
Table 1. Mean ED50 Values in mg/kg (95% CL) for Morphine, Fentanyl, and Methadone Alone and after Pretreatment with the Delta-Opioid Antagonist Naltrindole in Rhesus Monkeys.
| agonist alone | 0.1 mg/kg naltrindole | 0.32 mg/kg naltrindole | 1.0 mg/kg naltrindole | 3.2 mg/kg naltrindole | |
|---|---|---|---|---|---|
| morphine | 2.67 (2.21–3.23) | 2.41 (1.72–3.38) | 4.33*a(3.54–5.29) | 5.84*(5.52–6.18) | 15.48*(10.35–23.16) |
| fentanyl | 0.011 (0.008–0.015) | 0.011 (0.008–0.016) | 0.017*(0.016–0.018) | 0.030*(0.020–0.045) | 0.048*(0.035–0.065) |
| methadone | 1.79 (1.64–1.95) | not determined | 2.19 (1.77–2.72) | 2.22 (1.83–2.69) | 4.35*(3.76–5.05) |
Asterisk indicates that 95% confidence intervals do not overlap compared to morphine, fentanyl, or methadone alone.
The potency of NTI in the antagonism of the antinociceptive effect of morphine and fentanyl bears a striking similarity to the antagonism of the behavioral effects of selective delta agonists in rhesus monkeys, in that the doses of NTI are in a similar range as the present study.34−37 In light of this and our antagonism studies in HEK293 cells, it is conceivable that NTI binds to the delta protomer, thereby allosterically inhibiting signaling by agonist bound to the mu protomer. Indeed, we have observed that morphine and fentanyl did not surmount the antagonistic effects of the highest dose of NTI (3.2 mg/kg) in the dose ranges tested in rhesus monkeys (Figure 4). [Note that higher morphine and fentanyl doses were not tested due to the emergence of severe sedative and/or respiratory depressant effects that were apparently insensitive to naltrindole antagonism. In these cases, monkeys were treated with 0.1 mg/kg IM naltrexone, which effectively reversed morphine- and fentanyl-induced untoward effects.]
That insurmountable antagonism was also observed in HEK-293 cells (Figure 3) suggests the possibility of such an allosteric mechanism. Insofar as NTI has relatively low affinity for isolated mu receptors, these results are consistent with the hypothesis that the thermal antinociceptive effects of morphine and fentanyl are not mediated by homomeric mu receptors, but rather by mu-delta heteromeric opioid receptors in vivo.
Unlike morphine and fentanyl, the antagonism of methadone by NTI appears to be surmountable (Figure 3f, Figure 4c). This supports the idea that the cell-based studies represent a credible model for the role of mu-delta heteromers in rhesus monkeys. The lower sensitivity of methadone to NTI-induced antagonism of antinociception could arise as a consequence of a methadone-induced conformational perturbation of the mu-delta heteromer that is different from that induced by morphine or fentanyl.
Another major question relates to the tissue distribution of mu-delta heteromers in vivo. In this context, we determined the ability of NTI to antagonize morphine in mice when both ligands were administered via either i.c.v. or i.t. Interestingly, morphine was only antagonized when both ligands were administered i.t., but not i.c.v. (Figure 5), suggesting that mu-delta heteromers may be present in spinal cord of mice.
In conclusion, it appears that morphine, methadone, and fentanyl, which have been traditionally viewed as mu-selective ligands, selectively activate heteromeric mu-delta receptors in rhesus monkeys. Data supporting this conclusion were derived from studies involving the antagonism of activation of mu-delta heteromers in HEK293 cells, and antagonism of antinociception in mice and monkeys using the delta opioid receptor antagonist, NTI. Evidence for allosterically mediated antagonism is presented in cell-based, mouse, and monkey studies. Given the high homology between rhesus monkeys and humans, this work opens the possibility of heteromeric mu-delta receptor-mediated analgesia by morphine, fentanyl, and methadone in humans. While the extent of antinociception mediated by mu-delta heteromers in vivo remains unanswered due to paucity of knowledge about their presence and distribution, mu-delta heteromeric opioid receptors also may mediate some of the deleterious side effects as implicated in prior studies.20−24 Because delta-selective antagonists are widely employed as pharmacologic tools for characterization of delta opioid receptors in vivo, the results of the present study reveal possible pitfalls in attributing antagonism of antinociception to homomeric delta receptors alone. Consequently, the literature will require re-examination in light of the present results. Finally, given the side effects associated with analgesics that act through mu-delta heteromers, the present work has implications with respect to development of new analgesics, as it clearly suggests a paradigm shift involving a greater reliance on coexpressed receptors in the screening effort.
Methods
Subjects
Four adult rhesus monkeys (two female and two male) were used in studies of thermal nociception. Subjects weighed 4.5 to 12 kg during the course of these studies. All monkeys had prior exposure to central nervous system acting drugs (primarily opioid compounds) and to the behavioral procedures in which they were tested. The subjects were individually housed, and water was freely available. Their diet consisted of Lab Diet high protein monkey chow (Purina, Framingham, MA). This diet was supplemented with fresh fruit twice daily. A 12 h light/12 h dark cycle was in effect (lights on from 7:00 a.m. to 7:00 p.m.).
Animal maintenance and research were conducted in accordance with the guidelines provided by the National Institutes of Health Committee on Laboratory Animal Resources. The facility was licensed by the United States Department of Agriculture, and accredited by the Association for the Assessment and Accreditation of Laboratory Animal Care. Protocols were approved by the Institutional Animal Care and Use Committee. The health of the monkeys was monitored daily by technical staff and periodically by a veterinarian. Monkeys had visual, auditory, and olfactory contact with other monkeys throughout the study. Monkeys also had access to puzzle feeders, mirrors, and chew toys to provide environmental enrichment. Music was played daily in all housing rooms.
Drugs for Rhesus Monkey Studies
Morphine sulfate, fentanyl HCl, and (±)-methadone HCl were provided by the National Institute on Drug Abuse (Besthesda, MD) and dissolved in sterile water. Naltrindole HCl was generously provided by Dr. Kenner Rice (Chemical Biology Research Branch, National Institute on Drug Abuse and National Institute on Alcohol Abuse and Alcoholism). Morphine, fentanyl, methadone, and naltrindole were dissolved in sterile water. Drugs were administered intramuscularly in the thigh, and doses were determined based on the salt forms listed above.
Assay of Thermal Nociception
Monkeys were seated in acrylic restraint chairs so that their tails hung down freely. The bottom 10 cm of each monkey’s shaved tail was immersed in a thermal container of warm water. If the subject did not remove its tail within 20 s, the tail was removed by the experimenter, and a latency of 20 s was assigned to that measurement. During each cycle of measurements, tail-withdrawal latencies were measured from water heated to 38 and 50 °C. The order in which the temperatures were presented varied from one set of measurements to the next. Experiments were conducted no more than twice a week with at least two days between tests. A stopwatch was used to measure and record time intervals.
Each test session consisted of multiple 15 min cycles. Before the first drug dose was administered, baseline latencies to tail-withdrawal at 38 and 50 °C water were determined. Testing continued only if tail withdrawal from 38 °C water did not occur before the 20 s cutoff, and if tail withdrawal occurred in ≤2 s from 50 °C water. In our experiments, vehicle treatment did not produce any within-session baseline shifts. This criterion was met during every session with every monkey in this study. During cumulative dosing experiments, a single drug dose was administered at the start of each sequential 15 min cycle, and each dose increased the total cumulative dose by one-fourth or one-half log unit. Starting 10 min after each intramuscular injection, tail-withdrawal latencies were recorded as described above. Initially, complete dose–effect curves were determined for morphine (0.1–10 mg/kg), fentanyl (0.001–0.056 mg/kg), and methadone (0.1–5.6 mg/kg) alone. Subsequently, doses of naltrindole (0.1–3.2 mg/kg) were randomly administered 30 min as a pretreatment before redetermining the morphine, fentanyl, or methadone dose–effect curve.
Data analysis
Drug effects were expressed as % maximum possible effect (% MPE) using the following equation:
where test latency was the tail withdrawal latency from 50 °C water obtained after drug administration, and control latency was the latency obtained at the beginning of the session prior to drug administration. ED50 for each drug was defined as the dose that produced 50% MPE, and these values were determined by interpolation when only two data points were available (one below and one above 50% control response rate) or by linear regression when at least three data points were available on the linear portion of the dose–effect curve. Individual ED50 values were averaged to determine mean values and 95% confidence limits, and values were considered to be significantly different if confidence limits did not overlap.
To quantify antagonist effects of naltrindole, dose ratios were calculated for each subject as the ED50 of the mu agonist in the presence of naltrindole ÷ ED50 of the mu agonist alone. Individual dose ratios were averaged to determine mean values and 95% confidence limits, and values were considered to be significantly different if confidence limits did not overlap.
Tail-Flick Assay in Mice
The tail flick assay was performed as described previously.31,38 All ligands were administered in a 5 μL volume in conscious mice according to previously published methods for i.c.v. and i.t. injections.39,40 For the latency of the tail-flick measurement, the mice were held gently in one hand with the tail positioned in the apparatus (Tail Flick Analgesia Meter, Columbus Instruments, Columbus, Ohio) for radiant heat stimulus. The tail-flick response was elicited by applying radiant heat to the dorsal side of the tail. The intensity of the heat was set at setting 8 so that the animal flicked its tail within 2 to 3 s. The test latency was measured once before drug treatment (control) and again after the drug treatment (test) at the peak time of the compound; a 10 s maximum cutoff time was used to prevent damage to the tail. Antinociception was quantified according to a previously published method (41) as the percent maximal possible effect (% MPE), which is calculated as: % MPE = [(test – control)/(10 – control)] × 100. At least three groups of eight to ten mice were used for each dose response curve, and each mouse was used only once. ED50 values with 95% confidence intervals (CI) were computed with GraphPad Prism 4 by using nonlinear regression methods.
Cell Culture and Intracellular Ca2+ Release Assay
HEK-293 cells containing singly expressed opioid receptors were generated using pcDNA3 vectors (Invitrogen, Carlsbad, CA) encoding human kappa, mu, and murine delta opioid receptors. HEK-293 cells stably coexpressing mu-kappa, kappa-delta, and mu-delta receptors were obtained from Dr. Jennifer Whistler, and their construction has been previously described and verified.31−33 The chimeric G-protein, Δ6-Gαqi4-myr, was graciously provided by Dr. Evi Kostenis, and its construction has been previously described.42
HEK-293 cells were cultured at 37 °C in Dulbelcco’s modified Eagle’s medium supplemented with 10% bovine calf serum, Pen/Strep antibiotics. For cells singly expressing opioid receptors, G418 was used as the selection antibiotic; G418 and Zeocin were used for selecting for cells coexpressing two opioid receptors. The protocol for the intracellular calcium release has been previously described in detail.29,31 Briefly, the stable opioid receptor cell lines were transiently transfected with 200 ng/20,000 cells of the chimeric G-protein, Δ6-Gαqi4-myr, using OptiMEM medium (Invitrogen) and Lipofectamine 2000 (Invitrogen, Carlsbad CA) reagent according to manufacturer’s protocol (1:2 wt/vol ratio for DNA:Lipofectamine). The cells were seeded into 96 well plates (half-area; Corning) at 20,000 cells/well after 24 h and assayed 48 h after transfection using the FLIPR calcium kit (Molecular devices) in a Flexstation-III apparatus (Molecular devices). The response was measured as area under the curve (relative fluorescence units × seconds) and plotted using nonlinear regression using Prism 4 (GraphPad Inc.).
Acknowledgments
We thank Dr. Evi Kostenis for providing us with the cDNA for Δ6-Gαqi4-myr chimeric Gα subunit and Dr. Jennifer Whistler for stable dual transfected HEK-293 cells. We also thank Michael Powers for his technical assistance in designing the graphical table of contents, and for invaluable input and discussions.
Glossary
Abbreviations
- NTI
naltrindole
- DAMGO
[d-Ala2, N-MePhe4, Gly-ol]-enkephalin
- i.t.
intrathecal
- i.c.v.
intracerebroventricular
- MPE
maximum possible effect
Supporting Information Available
Tables of Bmax values for [3]HDAMGO and mean dose ratios for morphine, fentanyl, and methadone after pretreatment with naltrindole in rhesus monkeys. This material is available free of charge via the Internet at http://pubs.acs.org.
The authors declare no competing financial interest.
Notes
A portion of this work was presented at the 2010 International Narcotic Research Conference (INRC) held in Malmö, Sweden. Yekkirala, A. S., Banks, M. L., Negus, S. S. and Portoghese, P. S. Clinically active opioid ligands selectively activate heterodimeric opioid receptors in HEK-293 cells and rhesus monkeys. International Narcotics Research Conference, Malmö, Sweden; 2010.
Author Contributions
§ Contributed equally to the work.
This work was supported by NIH Grants R01-DA01533, R01-DA011460 and T32-DA007027 from the National Institute on Drug Abuse (NIDA). A portion of this work was also supported by Intramural Research Programs of NIDA and National Institute on Alcohol Abuse and Alcoholism (NIAAA).
Author Contributions
Participated in research design: A.S.Y., M.L.B., S.S.N., and P.S.P. Conducted experiments: A.S.Y., M.L.B., and M.M.L. New reagents: K.C.R. and P.S.P. Data analysis: A.S.Y., M.L.B., S.S.N., K.C.R., and P.S.P. Wrote the manuscript: A.S.Y., M.L.B., S.S.N., and P.S.P.
Funding Statement
National Institutes of Health, United States
Supplementary Material
References
- Dubois M. Y.; Gallagher R. M.; Lippe P. M. (2009) Pain medicine position paper. Pain Med. 10(6), 972–1000. [DOI] [PubMed] [Google Scholar]
- Gutstein H. B., Akil H. (2006) Opioid analgesics, in Goodman & Gilman's The Pharmacological Basis of Therapeutics (Brunton L. L., Lazo J. S., Parker K. L., Eds.) 11th ed., pp 547–90, McGraw Hill, New York. [Google Scholar]
- Bowen C. A.; Fischer B. D.; Mello N. K.; Negus S. S. (2002) Antagonism of the antinociceptive and discriminative stimulus effects of heroin and morphine by 3-methoxynaltrexone and naltrexone in rhesus monkeys. J. Pharmacol. Exp. Ther. 302(1), 264–73. [DOI] [PubMed] [Google Scholar]
- Emmerson P. J.; Liu M. R.; Woods J. H.; Medzihradsky F. (1994) Binding affinity and selectivity of opioids at mu, delta and kappa receptors in monkey brain membranes. J. Pharmacol. Exp. Ther. 271(3), 1630–7. [PubMed] [Google Scholar]
- Emmerson P. J.; Clark M. J.; Mansour A.; Akil H.; Woods J. H.; Medzihradsky F. (1996) Characterization of opioid agonist efficacy in a C6 glioma cell line expressing the mu opioid receptor. J. Pharmacol. Exp. Ther. 278(3), 1121–7. [PubMed] [Google Scholar]
- Negus S. S.; Burke T. F.; Medzihradsky F.; Woods J. H. (1993) Effects of opioid agonists selective for mu, kappa and delta opioid receptors on schedule-controlled responding in rhesus monkeys: antagonism by quadazocine. J. Pharmacol. Exp. Ther. 267(2), 896–903. [PubMed] [Google Scholar]
- Matthes H. W.; Maldonado R.; Simonin F.; Valverde O.; Slowe S.; Kitchen I.; Befort K.; Dierich A.; Le Meur M.; Dolle P.; Tzavara E.; Hanoune J.; Roques B. P.; Kieffer B. L. (1996) Loss of morphine-induced analgesia, reward effect and withdrawal symptoms in mice lacking the mu-opioid-receptor gene. Nature 383(6603), 819–23. [DOI] [PubMed] [Google Scholar]
- Evans C. J.; Keith D. E. Jr.; Morrison H.; Magendzo K.; Edwards R. H. (1992) Cloning of a delta opioid receptor by functional expression. Science (New York, N.Y.) 258(5090), 1952–5. [DOI] [PubMed] [Google Scholar]
- Kieffer B. L.; Befort K.; Gaveriaux-Ruff C.; Hirth C. G. (1992) The delta-opioid receptor: isolation of a cDNA by expression cloning and pharmacological characterization. Proc. Natl. Acad. Sci. U.S.A. 89(24), 12048–52. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Meng F.; Xie G. X.; Thompson R. C.; Mansour A.; Goldstein A.; Watson S. J.; Akil H. (1993) Cloning and pharmacological characterization of a rat kappa opioid receptor. Proc. Natl. Acad. Sci. U.S.A. 90(21), 9954–8. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Thompson R. C.; Mansour A.; Akil H.; Watson S. J. (1993) Cloning and pharmacological characterization of a rat mu opioid receptor. Neuron 11(5), 903–13. [DOI] [PubMed] [Google Scholar]
- Stevenson G. W.; Folk J. E.; Linsenmayer D. C.; Rice K. C.; Negus S. S. (2003) Opioid interactions in rhesus monkeys: effects of delta + mu and delta + kappa agonists on schedule-controlled responding and thermal nociception. J. Pharmacol. Exp. Ther. 307(3), 1054–64. [DOI] [PubMed] [Google Scholar]
- Kieffer B. L. (1995) Recent advances in molecular recognition and signal transduction of active peptides: receptors for opioid peptides. Cell Mol. Neurobiol. 15(6), 615–35. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Mansour A.; Fox C. A.; Akil H.; Watson S. J. (1995) Opioid-receptor mRNA expression in the rat CNS: anatomical and functional implications. Trends Neurosci. 18(1), 22–9. [DOI] [PubMed] [Google Scholar]
- Jordan B. A.; Cvejic S.; Devi L. A. (2000) Opioids and their complicated receptor complexes. Neuropsychopharmacology 23(4), S5–S18. [DOI] [PubMed] [Google Scholar]
- Wang D.; Sun X.; Bohn L. M.; Sadee W. (2005) Opioid receptor homo- and heterodimerization in living cells by quantitative bioluminescence resonance energy transfer. Mol. Pharmacol. 67(6), 2173–84. [DOI] [PubMed] [Google Scholar]
- Jordan B. A.; Devi L. A. (1999) G-protein-coupled receptor heterodimerization modulates receptor function. Nature 399(6737), 697–700. [DOI] [PMC free article] [PubMed] [Google Scholar]
- George S. R.; Fan T.; Xie Z.; Tse R.; Tam V.; Varghese G.; O’Dowd B. F. (2000) Oligomerization of mu- and delta-opioid receptors. Generation of novel functional properties. J. Biol. Chem. 275(34), 26128–35. [DOI] [PubMed] [Google Scholar]
- Sanchez-Blazquez P.; Garcia-Espana A.; Garzon J. (1997) Antisense oligodeoxynucleotides to opioid mu and delta receptors reduced morphine dependence in mice: role of delta-2 opioid receptors. J. Pharmacol. Exp. Ther. 280(3), 1423–31. [PubMed] [Google Scholar]
- Nitsche J. F.; Schuller A. G.; King M. A.; Zengh M.; Pasternak G. W.; Pintar J. E. (2002) Genetic dissociation of opiate tolerance and physical dependence in delta-opioid receptor-1 and preproenkephalin knock-out mice. J. Neurosci. 22(24), 10906–13. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Abdelhamid E. E.; Sultana M.; Portoghese P. S.; Takemori A. E. (1991) Selective blockage of delta opioid receptors prevents the development of morphine tolerance and dependence in mice. J. Pharmacol. Exp. Ther. 258(1), 299–303. [PubMed] [Google Scholar]
- Daniels D. J.; Lenard N. R.; Etienne C. L.; Law P. Y.; Roerig S. C.; Portoghese P. S. (2005) Opioid-induced tolerance and dependence in mice is modulated by the distance between pharmacophores in a bivalent ligand series. Proc. Natl. Acad. Sci. U.S.A. 102(52), 19208–13. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lenard N. R.; Daniels D. J.; Portoghese P. S.; Roerig S. C. (2007) Absence of conditioned place preference or reinstatement with bivalent ligands containing mu-opioid receptor agonist and delta-opioid receptor antagonist pharmacophores. Eur. J. Pharmacol. 566(1–3), 75–82. [DOI] [PubMed] [Google Scholar]
- Scherrer G.; Imamachi N.; Cao Y. Q.; Contet C.; Mennicken F.; O’Donnell D.; Kieffer B. L.; Basbaum A. I. (2009) Dissociation of the opioid receptor mechanisms that control mechanical and heat pain. Cell 137(6), 1148–59. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wang H. B.; Zhao B.; Zhong Y. Q.; Li K. C.; Li Z. Y.; Wang Q.; Lu Y. J.; Zhang Z. N.; He S. Q.; Zheng H. C.; Wu S. X.; Hokfelt T. G.; Bao L.; Zhang X. (2010) Coexpression of delta- and mu-opioid receptors in nociceptive sensory neurons. Proc. Natl. Acad. Sci. U.S.A. 107(29), 13117–22. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Gupta A.; Mulder J.; Gomes I.; Rozenfeld R.; Bushlin I.; Ong E.; Lim M.; Maillet E.; Junek M.; Cahill C. M.; Harkany T.; Devi L. A. (2010) Increased abundance of opioid receptor heteromers after chronic morphine administration. Sci. Signaling 3(131), ra54. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hasbi A.; Nguyen T.; Fan T.; Cheng R.; Rashid A.; Alijaniaram M.; Rasenick M. M.; O’Dowd B. F.; George S. R. (2007) Trafficking of preassembled opioid mu-delta heterooligomer-Gz signaling complexes to the plasma membrane: coregulation by agonists. Biochemistry 46(45), 12997–3009. [DOI] [PubMed] [Google Scholar]
- Yekkirala A. S.; Kalyuzhny A. E.; Portoghese P. S. (2010) Standard opioid agonists activate heteromeric opioid receptors: evidence for morphine and [D-Ala2-MePhe4-Glyol5]enkephalin as selective μ-∂ agonists. ACS Chem. Neurosci. 1(1), 146–54. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Portoghese P. S.; Sultana M.; Takemori A. E. (1988) Naltrindole, a highly selective and potent non-peptide delta opioid receptor antagonist. Eur. J. Pharmacol. 146(1), 185–6. [DOI] [PubMed] [Google Scholar]
- Yekkirala A. S.; Lunzer M. M.; McCurdy C. R.; Powers M. D.; Kalyuzhny A. E.; Roerig S. C.; Portoghese P. S. (2011) N-naphthoyl-beta-naltrexamine (NNTA), a highly selective and potent activator of mu/kappa-opioid heteromers. Proc. Natl. Acad. Sci. U.S.A. 108(12), 5098–103. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Waldhoer M.; Fong J.; Jones R. M.; Lunzer M. M.; Sharma S. K.; Kostenis E.; Portoghese P. S.; Whistler J. L. (2005) A heterodimer-selective agonist shows in vivo relevance of G protein-coupled receptor dimers. Proc. Natl. Acad. Sci. U.S.A. 102(25), 9050–5. [DOI] [PMC free article] [PubMed] [Google Scholar]
- van Rijn R. M.; Whistler J. L. (2009) The δ1 Opioid Receptor Is a Heterodimer That Opposes the Actions of the δ2 Receptor on Alcohol Intake. Biol. Psychiatry 66(8), 777–84. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Negus S. S.; Butelman E. R.; Chang K. J.; DeCosta B.; Winger G.; Woods J. H. (1994) Behavioral effects of the systemically active delta opioid agonist BW373U86 in rhesus monkeys. J. Pharmacol. Exp. Ther. 270(3), 1025–34. [PubMed] [Google Scholar]
- Negus S. S.; Gatch M. B.; Mello N. K.; Zhang X.; Rice K. (1998) Behavioral effects of the delta-selective opioid agonist SNC80 and related compounds in rhesus monkeys. J. Pharmacol. Exp. Ther. 286(1), 362–75. [PubMed] [Google Scholar]
- Brandt M. R.; Negus S. S.; Mello N. K.; Furness M. S.; Zhang X.; Rice K. C. (1999) Discriminative stimulus effects of the nonpeptidic delta-opioid agonist SNC80 in rhesus monkeys. J. Pharmacol. Exp. Ther. 290(3), 1157–64. [PubMed] [Google Scholar]
- Brandt M. R.; Furness M. S.; Mello N. K.; Rice K. C.; Negus S. S. (2001) Antinociceptive effects of delta-opioid agonists in Rhesus monkeys: effects on chemically induced thermal hypersensitivity. J. Pharmacol. Exp. Ther. 296(3), 939–46. [PubMed] [Google Scholar]
- Dewey W. L.; Harris L. S.; Howes J. F.; Nuite J. A. (1970) The effect of various neurohumoral modulators on the activity of morphine and the narcotic antagonists in the tail-flick and phenylquinone tests. J. Pharmacol. Exp. Ther. 175(2), 435–42. [PubMed] [Google Scholar]
- Hylden J. L.; Wilcox G. L. (1981) Intrathecal substance P elicits a caudally-directed biting and scratching behavior in mice. Brain Res. 217(1), 212–5. [DOI] [PubMed] [Google Scholar]
- Haley T. J.; McCormick W. G. (1957) Pharmacological effects produced by intracerebral injection of drugs in the conscious mouse. Br. J. Pharmacol. Chemother. 12(1), 12–5. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Harris L. S.; Pierson A. K. (1964) Some Narcotic Antagonists in the Benzomorphan Series. J. Pharmacol. Exp. Ther. 143, 141–8. [PubMed] [Google Scholar]
- Kostenis E. (2001) Is Galpha16 the optimal tool for fishing ligands of orphan G-protein-coupled receptors?. Trends Pharmacol. Sci. 22(11), 560–564. [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.