

Abstract
The last 2 decades have seen discoveries in highly excited states of atoms and molecules of phenomena that are qualitatively different from the “planetary” model of the atom, and the near-rigid model of molecules, characteristic of these systems in their low-energy states. A unified view is emerging in terms of approximate dynamical symmetry principles. Highly excited states of two-electron atoms display “molecular” behavior of a nonrigid linear structure undergoing collective rotation and vibration. Highly excited states of molecules described in the “standard molecular model” display normal mode couplings, which induce bifurcations on the route to molecular chaos. New approaches such as rigid–nonrigid correlation, vibrons, and quantum groups suggest a unified view of collective electronic motion in atoms and nuclear motion in molecules.
Chemical systems range from relatively simple atoms to large and complex mesoscopic structures such as proteins. This is the microscopic substructure that explains, at least in a reductive sense, the appearance and behavior of the world of our ordinary experience and its living beings. As such, the chemical world occupies an intermediate size in nature between the very large and very small. There is a rough hierarchy of scales of organization in nature, each admitting a description in terms of relatively independent concepts and theories. Central to most of these are various ideas of “symmetry” that come into play at the different scales.
My purpose is to give an indication of a unified view of novel kinds of symmetry in chemical systems that is emerging from the work of many people over the past 2 decades. This view goes well beyond the traditional notions of chemical symmetry. These have to do on the one hand with orderly arrays of atoms in molecules or crystals, and on the other, with the “planetary” or independent particle model of electron motion in atoms and molecules. This usual kind of symmetry is most pronounced at low energy. The new view has emerged in response to an explosion of interest in highly excited quantum states of atoms and molecules. This is fueled on the one hand by advances in experimental technology, especially laser systems, and on the other, by the importance of highly excited chemical species in many aspects of science and technology carried out in extreme environments, including combustion, atmospheric, and interstellar phenomena.
I will speak of these symmetries of the dynamics of highly excited states as “dynamical symmetries.” After giving an overview of the new dynamical symmetry view in chemistry, I will return to the theme of a hierarchy of symmetries in nature. There are currently powerful attempts in physics to use symmetry as a fundamental principle to build models of the elementary particles, and even more ambitiously a Theory of Everything. Based on lessons that can be learned from experience with chemical systems, I will present some personal, perhaps highly idiosyncratic, views of these efforts.
I have tried to make the presentation as interesting and useful to as broad an audience as possible, minimizing technical details. For the intended audience curious about a new area of symmetry in science, I have tried to give a reasonably coherent overview, with reference to key original sources, in which can be found many other interesting references that must be left out here.
The Need for Dynamical Symmetry in Chemical Systems
Excitation to Chaos in Molecules.
The earliest notions of symmetry in chemistry had to do with the orderly arrays of atoms found in many molecules and crystalline solids. For example, Fig. 1 shows the rigid structure of the water molecule H2O. This structure is symmetric in that it is left unchanged by a rotation of 180° about the vertical axis through the oxygen atom, and by planes parallel and perpendicular to the molecule. This symmetry is described mathematically (1) by the point group C2v.
Figure 1.

Equilibrium rigid structure of water molecule H2O with point group C2v.
This symmetry is very useful for describing the static equilibrium structure of the molecule and many properties of its excited electronic, vibrational, and rotational states. However, there are important aspects of excited states, about which the point group symmetry just doesn’t have much to say. These excited states comprise the quantum mechanical spectrum. Spectra, observed directly in experiments, are our window on the molecular world, through which to obtain information such as the forces between atoms that govern molecular dynamics and reactivity. For example, the spectrum of vibrational excitations is traditionally described in terms of normal modes (2). However, at high energies, the modes can become strongly coupled to each other. As the atoms perform progressively large excursions from the equilibrium rigid structure, the motion can become chaotic, in the sense of nonlinear dynamical systems. Eventually, the atoms may rearrange into a different molecular structure, or the molecule can fragment. The normal modes description, adequate for a rational account of the spectrum at low energies, is no longer physically valid. But it is precisely here that the spectrum may become fearsomely complex. Spectra are a window through which we are allowed to view the molecular world, but as with all visualization, a great deal of sophisticated processing is necessary to extract the information encoded in the raw data of experience. The search for new ways of modelling spectra and organizing them into meaningful patterns, if any such exist, has been a concerted effort of fundamental chemical science of the past 2 decades.
Breakdown of the Planetary Model in Atoms.
And so, at low energy the atoms in molecules are positioned in relatively well-defined geometrical structures, but they undergo sizeable excursions in highly excited states. In contrast, the individual electrons in atoms and molecules do not have fixed positions relative to the atomic nuclei. It is not evident that they have a “structure” in any geometrical sense. In a classical model of the simplest atom, hydrogen, the electron orbits the nucleus in an ellipse, as in Fig. 2. In the quantum mechanical description of this “planetary atom,” the orbit is replaced by an orbital of probability density. (It is often useful to pass freely between classical and quantum mechanical models, as long as one remembers that a classical picture is at best a guide to thinking about the quantum mechanics.)
Figure 2.
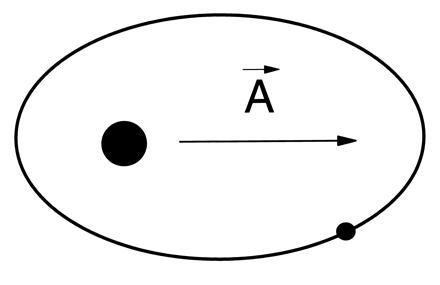
Elliptical Kepler orbit of electron around the nucleus in the hydrogen atom. Runge-Lenz vector is a conserved quantity.
In a multi-electron atom, the Coulomb repulsion between electrons substantially perturbs their elliptical orbits. This mutual perturbation is much greater than between the planets in the solar system, because of the relative magnitudes of the relevant forces (in particular, the huge gravitational force of the sun relative to the planets). This is the problem of electron correlation. The simplest system with correlation is the two-electron atom. This electron correlation problem extends to larger atoms and molecules, and is one of the central problems in a quantum mechanical description of chemical phenomena.
The “Molecular” Atom.
As the simplest prototype of the correlation problem, two-electron atoms continue to elicit intense interest. Conventionally, one would describe an excited state of the atom in terms of a configuration. For example, a state with both electrons in a 2p orbital has a configuration 2p2. This configuration description is the quantum mechanical analog of the independent particle or “planetary” model. From what has been said about correlation, it comes as no surprise that the configuration description is severely inadequate for highly excited states.
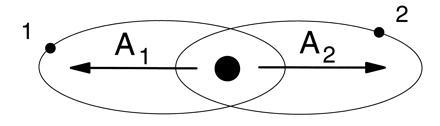
Nonetheless, it came as something of a shock (3–7) to discover that the excited atom had many properties in common with the motion of a triatomic molecule! In this picture, the atom is like a highly nonrigid linear triatomic molecule, with the electrons playing the role of the outer atoms. A crude picture of the rotating molecular atom is shown in Fig. 3a. (The molecule can also vibrate, as described later.) Of course, a rigid structure like Fig. 3a is not to be taken literally as a picture of the molecular atom. A more realistic picture is shown in Fig. 3b, where the structure is more like that of an elongated cloud or blob of charge, with the density of the two electrons concentrated at the ends. This can be rationalized by saying that the repulsion of the electrons tends to correlate them on opposite sides of the nucleus. At the same time, a residual effect of the quantum mechanical configuration is to concentrate the electrons at a most favored distance from the nucleus, but only very loosely.
Figure 3.
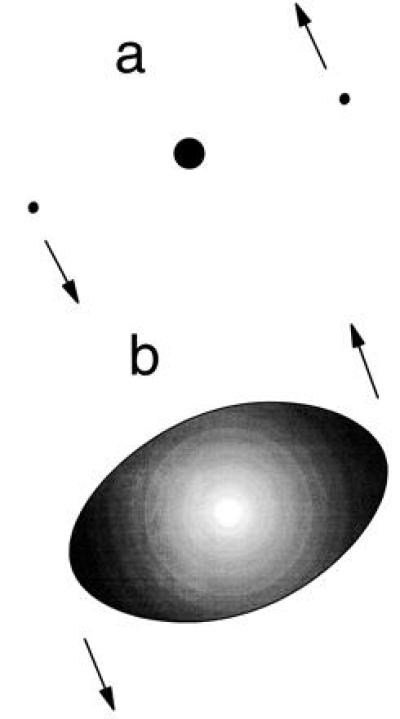
(a) Literal depiction of molecular model of the two-electron atom. (b) More realistic depiction, with the linear molecular structure a “blob” of charge surrounding the nucleus.
A Unified Picture of Dynamical Symmetry.
We have seen that highly excited vibrations and rotations of molecules involve significant departures from the orderly, rigid structures of the ground state, whereas highly excited states of electrons in atoms behave in some ways like highly nonrigid molecular structures. Can there be a unified viewpoint for these two very different types of problems? Strong indications are emerging that this is the case.
I will first describe in more detail the physical and mathematical basis of the molecular atom. Then I will return to molecular vibrations, indicating how their description must change with excitation and development of chaos. I start with new developments in what I call the “standard molecular model” or SMM. Then I discuss two very interesting recent variants of the SMM. One is known as the “vibron” model. The other is based on so-called “quantum groups,” which are having a wide impact on many areas of physics. I then return to the molecular atom, and indicate that there are deep formal connections with the new developments in molecules. Finally, I consider possible lessons for systems at other scales in the hierarchy of symmetries, in particular the “standard model” of elementary particles.
The Molecular Atom: Rotations, Vibrations, and Supermultiplets
The molecular model of the atom developed out of investigations of doubly excited states of two-electron atoms. These are states with both electrons excited from the 1s ground state. I will try to give the briefest cogent outline of the relevant symmetry methods, with an emphasis on the physical ideas. The reader should consult the references for the many technical details.
Fig. 2 shows the elliptical or Kepler planetary orbit of the electron in a one-electron atom. The angular momentum, represented as a vector L⃗ , is an exact constant of motion because of exact invariance of the system under any rotation in space. The set of all possible rotations (1) in three-dimensional space is the group O(3). Unlike the point group of a crystal lattice or symmetrical molecule, this is a continuous, or Lie group. The three components of the angular momentum vector L⃗ are said to be generators of the group O(3).
The Kepler or Coulomb orbit in Fig. 2 actually has far more regularity or “symmetry” than demanded merely by rotational invariance, however. A planetary orbit that conserves angular momentum L⃗ could still have a precessing perihelion, as shown in Fig. 4, just as a gyroscope can precess. The elliptical Kepler orbit of Fig. 2 must have an additional constant of the motion (8) in addition to the angular momentum L⃗. This additional constant is the Runge-Lenz vector A⃗, which points along the semi-major axis of the ellipse. It is defined in terms of the instantaneous position and momentum at each point in the orbit, but as a constant of motion, is invariant in size and direction. The existence of the constant A⃗ in addition to the angular momentum L⃗ suggests that the Coulomb or Kepler problem has a higher symmetry group than the O(3) rotation group responsible for the conservation of L⃗. This higher symmetry group (8) is O(4), the group of rotations in four dimensions. This is an abstract 4-dimensional space, which includes our physical 3-dimensional space as a subspace. The abstract O(4) group of the Coulomb problem is generated by the components of L⃗ and A⃗.
Figure 4.
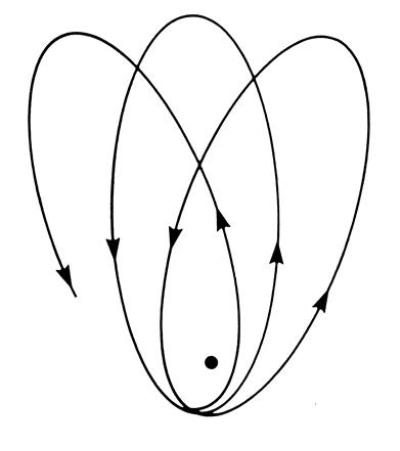
Precessing perihelion of an orbit that conserves angular momentum but not the Runge-Lenz vector.
We now turn to the two-electron atom. If it behaved like a planetary atom, each electron would move in an independent elliptical orbit. In fact, because the system is invariant under rotations in space, the total angular momentum
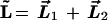 |
1 |
is an exact constant of motion. The total angular momentum L⃗ itself generates a group denoted O(3)12 of rotations of the total system of two electrons.
By analogy with the one-electron atom, one might very reasonably think to combine L⃗ with the total Runge-Lenz vector
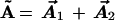 |
2 |
to try to form a two-electron O(4)12 group. In fact, the O(4)12 generated in this way is mathematically an O(4) group. However, it is not an invariance group, with A⃗ = A⃗1 + A⃗2 a conserved quantity, because A⃗ is badly “broken” as a constant of motion by the strong Coulomb repulsion between electrons. This means the electrons do not move in independent elliptical orbits, and it makes the two-electron problem impossible to solve exactly, either in classical or quantum mechanics.
Even though the O(4)12 group is not an exact symmetry, it is reasonable to try to use it as an approximate symmetry. One might hope this would be of use in ordering the spectrum, but in a less complete way than if O(4)12 were exact (in which case the spectrum would be trivially simple). This would be very much in the spirit of the use of approximate symmetries earlier in nuclear and elementary particle physics, where these kinds of ideas led to developments such as the quark model. Unfortunately, when this was tried for the two-electron atom, it was found that O(4)12 was not of much use even in this limited sense of an approximate symmetry.
A key advance came when Wulfman (9), and independently Herrick and Sinanoglu (10), tried instead of A⃗ (2), the operator
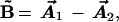 |
3 |
where the crucial difference is the minus sign. A heuristic physical motivation for this is shown in Fig. 5. The idea is that overall, maximizing the difference between the Runge-Lenz vectors A⃗1, A⃗2 minimizes the Coulomb repulsion between the electrons. It was found that B⃗ is an approximate constant of motion, unlike the experience with A⃗. Furthermore, it can easily be shown that L⃗ and B⃗ together generate a distinct O(4) group, which therefore is an approximate symmetry group! We call this O(4)12′ to distinguish it from the earlier unsuccessful O(4)12. [The two O(4) groups are distinct even though they are isomorphic, i.e., their structures are in one-to-one correspondence.]
Figure 5.
Heuristic picture of electron correlation in which the most favorable configuration maximizes the difference B⃗ = A⃗1 − A⃗2 of Runge-Lenz vectors.
The approximate O(4)12 symmetry has many uses that were further explored by Herrick and Sinanoglu (11). A new development, the beginning of the molecular model, came when Kellman and Herrick (3) used the O(4)12′ structure to define “rotor series,” that is O(4)12′ multiplets or sets of states that behaved like a nonrigid rotor, something like Fig. 3. Subsequently (4–6), the O(4)12′ multiplets were combined into “supermultiplets.” This gave the classification of states of the atom with both electrons in the n = 2, 3, 4, and 5 shells; n = 3 is shown in Fig. 6. Evidently, this classification reveals a great deal of order in a spectrum that does not at first show much order at all. The spectral pattern of Fig. 6 is similar to that of a linear, nonrigid triatomic molecule, which undergoes bending vibrations as well as rotations.
Figure 6.
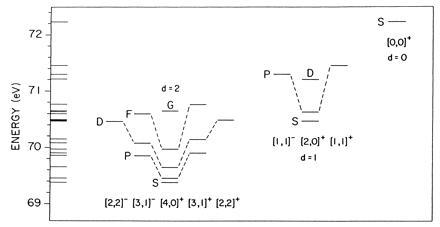
Supermultiplet rotor-vibrator molecular classification of doubly excited states of He with both electrons excited to n = 3 shell, from ref. 4.
There have been significant developments since the work of Herrick and Kellman. Some highlights: I showed (7) that the molecular picture could be applied to atoms with two electrons outside a closed shell, i.e., alkaline earth atoms. Hunter and Berry (12) analyzed the atomic wave functions in terms of a basis with bending vibrations and stretching modes. In the original model of Kellman and Herrick, the molecule was taken to have rotations and bending vibrations, but not stretching excitations, inasmuch as the radial degrees of freedom seemed quite reasonably to be encompassed in the n = 2, 3, … shell structure. Ezra et al. (13) developed a semi-classical treatment of the radial motion of a collinear atom, and found evidence for radial motions that can be described as an anti-symmetric stretch. Rost et al. (14, 15) gave a unified “molecular orbital” treatment of the radial, bending, and rotational motion. They found an intricate interweaving of bend and stretch excitations. These investigations strongly support the idea of the atom as being like a highly nonrigid triatomic molecule, but now understood to have a systematic involvement of radial motion in the classification. Finally, I will describe later some connections between the vibron model of molecules and the atomic supermultiplet classification.
Order in Chaos in Highly Excited Molecules?
We have seen that the spectrum of a highly excited two-electron atom resembles that of a linear molecular structure undergoing rotation and vibration. This molecular atom differs from an ordinary molecule in that its structure is far less rigid. This can be turned around to ask, what happens to a real molecule when it is excited high above the ground state? How does one describe the changes that must occur from the low-energy picture of small-amplitude oscillations about a near-rigid structure? The standard approach to low-energy spectra starts with the notion of a set of N normal vibrational modes (2). (For simplicity, I will neglect rotation. Most of the statements carry over when rotation is included.) At all but the lowest energies, it is necessary to take into account anharmonicity of the normal modes. In addition, there are important couplings: in general, the vibrations are not truly separable into independent modes. At low energy, the combination of anharmonicity and coupling for the most part can be “swept under the rug.” But this becomes untenable at higher energy, where anharmonicity and coupling become inexorably larger.
This leads to three aspects of the problem of complex spectra of polyatomics: (i) When the most important couplings are taken into account are there any remaining approximate constants of motions? If so, these intact portions of the molecular phase space act as “channels for energy flow” to energy flow within the molecule. This is important for efforts to control molecular dynamics with intense laser pulses. (ii) What happens to the dimensions of phase space that are not left intact? This is the problem of the branching of the normal modes in bifurcations on the route to molecular chaos. (iii) New techniques are allowing probes that give an unprecedented “global” view of the molecular phase space. As the number of atoms in a molecule grows, there is explosive growth of the number of parameters needed to model the spectrum. This parameter explosion has catastrophic implications for highly desirable probes of important molecules. Can ways be found to get around this?
I will outline some promising approaches to these problems, developed through the intense efforts of many workers.
The Standard Molecular Model.
I begin by describing a very widely used effective Hamiltonian or energy function for molecular vibrations. I will refer to this as the standard molecular model, or SMM—not to be confused with the “standard elementary particle model,” (SEPM) about which more will be said in the final section. I develop the model through specific examples such as H2O, working through the aspects of anharmonicity and coupling to arrive at the SMM Hamiltonian in Eq. 6.
The SMM is a quantum mechanical model of vibrational normal modes and their couplings. The SMM contains a number of variable parameters. These are optimized to give the best fit to experimental data. The SMM is in no sense a fundamental description of the molecule. That involves treating it as a collection of nuclei and electrons, and essentially solving for the exact quantum mechanics. When feasible, as for a few small molecules, that leads to a molecular potential energy surface, with vibrations emerging as quantum mechanical motion of the atoms in this potential. This is very difficult to carry out in practice. The SMM is intended as an “effective Hamiltonian” to bridge the gap between experimental spectral data and the fundamental description of the true molecular quantum mechanics. A natural question is how the SMM is related to the true Hamiltonian. Fried and Ezra (16) and, independently, Sibert (17) have shown how to obtain the SMM Hamiltonian by beginning with a potential surface and transforming approximately to SMM form by means of perturbation theory.
The starting point of the SMM is a set of elementary normal modes described by a Hamiltonian function H0, which depends on the number of quanta ni in each of the modes i. The simplest model is just a set of N harmonic normal modes. However, anharmonicity is evident from spectra even at low energy. It is therefore standard to use an H0 with harmonic terms proportional to the number of quanta and frequency ωi in each mode, but also anharmonic terms proportional to higher powers of the quantum numbers, including cross-terms:
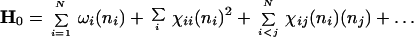 |
4 |
Coupling of the Normal Modes.
One can try to fit an
experimental spectrum by optimizing the parameters
ωi, etc. in H0. For
many systems, especially at low energy, this may suffice to give a good
match with experiment. However, a Hamiltonian of the form
H0 often is simply not adequate. In such cases,
coupling between the normal modes must be introduced beyond the
cross-anharmonic terms
χij(ni)(nj).
For example, Darling and Dennison (18) found that an interaction must
be added to H0, which couples the stretch normal
modes in a molecule such as H2O. This is called
“Darling–Dennison resonance,” because of the near-resonant
frequencies of the symmetric and antisymmetric stretches s
and a. Addition of the Darling–Dennison coupling
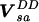 gives a Hamiltonian H = H0 +
gives a Hamiltonian H = H0 +
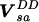 ,
which is no longer separable into modes s and a.
,
which is no longer separable into modes s and a.
The Darling–Dennison coupling is for two modes s and a, which have approximately the same frequency. This is sometimes called a 1:1 resonance coupling. Other couplings can arise between modes with frequency ratio n:m. These are referred to as Fermi resonances (19). For example, there is a prominent 2:1 Fermi resonance in H2O between the symmetric stretch and bend. A global fit of the spectrum, including states with prominent excitation of all three modes, requires a multiresonant Hamiltonian
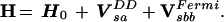 |
5 |
In general, the SMM Hamiltonian consists of a zero-order normal mode part H0 and a sum of resonance couplings Vα:
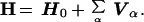 |
6 |
The multiresonance character of spectra has an important connection with identifying approximate constants of motion of the molecule. These act to channel energy flow within the molecular phase space. Furthermore, within the energy flow channels or pathways defined by the constants, the multiple resonances can produce bifurcations of the normal modes and classical chaos. I now consider these in turn.
Polyad Symmetry, Approximate Constants, and Channels for Molecular Energy Flow.
As stated before, excited state spectra are a window through which to view molecular phase space, but considerable processing is necessary to turn the raw data of experience into a meaningful picture. The SMM Hamiltonian can be viewed as an intermediate stage. Further sophisticated analysis is necessary to abstract images and concepts such as energy flow channels, branching of normal modes in bifurcations, and potential energy surfaces.
The theory for finding approximate constants and energy channels fortunately is simple to apply, using a “resonance vector” analysis of the SMM (16, 20). The quantum numbers that are preserved are not necessarily any of the zero-order quantum numbers. Rather, they are “polyad” numbers, which are special combinations of the zero-order quantum numbers. These approximate constants are very important because they channel energy flow in the molecular phase space when energy is deposited at some specific location in the molecule. This could be crucial for efforts to control molecules with intense beams of carefully sculpted light pulses.
The resonance vector method was first applied for experimental data (20) to spectra of acetylene (C2H2) of Smith and Winn (21). These absorption spectra probe the flow of energy initially deposited in the C-H stretch motions. They found a multiplicity of resonance couplings at high energy. Nonetheless, two good quantum numbers are found to remain from the resonance vector analysis (20). The polyad analysis has also been applied by Jonas et al. (22) to spectra that probe the bends of acetylene, with a bearing on observation of hierarchies of time scales for the energy flow. Temsamani and Herman (23) have carefully analyzed the polyads in constructing an effective SMM Hamiltonian. Most recently, Solina et al. (24) have directly “unzipped” the C2H2 spectrum into polyads. When viewed as individual polyads, the spectra reveal a great deal of order that is not otherwise evident. An analogy can be made to the classification of the two-electron spectrum into supermultiplets. An interesting question is whether the polyads can be further unzipped into sub-polyads, revealing still more hidden order in the spectrum; current work is mentioned briefly below.
Branching of the Normal Modes and the Structure of Molecular Chaos.
As shown by the need to use multiresonance Hamiltonians, it is evident that couplings will prove fatal for using the normal modes by themselves in all but the coarsest description of spectra. From the viewpoint of classical mechanics, the destruction of the normal modes occurs when they undergo bifurcations (25). When a normal mode bifurcates, new types of anharmonic or nonlinear motions are formed in place of, or in addition to, the original modes. The goal of recent work (25–28) has been to characterize the bifurcations associated with the SMM applied to data of specific molecules, in order to identify the new nonlinear modes and related spectral patterns that result at high energy.
Multidimensional Systems.
A difficult aspect of using bifurcation theory in spectral analysis is the high number of degrees of freedom in a polyatomic molecule. One confronts the not-well-understood problem of bifurcations in a high-dimensional classical phase space. In addition, the classical dynamics in general show a mixture of chaotic and regular motion, so one also faces the problem that the connection between chaotic classical dynamics and quantum states in the “mixed” regime is notorious for not being well understood.
Completely solving for the bifurcations of a chaotic Hamiltonian is presently impossible, and probably not very useful. Instead, for the three-mode Hamiltonian (5) for H2O we have focused on (27) obtaining the large-scale bifurcation structure. By this is meant the principal branchings of the original normal modes as the system becomes chaotic. This gives the large-scale structure around which the chaotic phase space is organized. This has been used to classify the spectrum in two ways. The first uses a Husimi phase space method (27) to examine the quantum mechanical wave functions. The second is more phenomenological and uses diabatic correlation diagrams (28). These are intended to be useful for larger molecules, where the bifurcation analysis becomes increasingly difficult. Essentially, the correlation diagram technique is a way to “unzip” and assign the polyads of the spectrum further into sub-polyads, while bypassing the bifurcation analysis as much as possible. Successfully carried out for H2O, this is largely uncharted territory for larger molecules, with ongoing investigations for C2H2.
The Parameter Explosion in Larger Molecules.
Techniques which “target” one or more bursts of laser light are giving unprecedented freedom to explore previously hidden portions of the phase space of a molecule. This freedom will allow global probes of increasingly large molecules and access to important information, such as knowledge of the potentials in the chains that make up large biological molecules. The starting point in using spectra to get this information is a fit with a Hamiltonian of the form of Eq. 6.
Unfortunately, inspection of the zero-order Hamiltonian of Eq. 4 reveals a horrendous problem in fitting global spectra of molecules with more than a few atoms. The problem comes from the cross-anharmonicity terms like χij(ni)(nj). The number of normal modes for a system with n atoms is N = (3n − 6), subtracting six degrees of freedom for rotation and translation. The number of independent parameters for the cross terms is N(N − 1)/2 = (3n − 6)(3n − 5)/2, which scales approximately as n2. For a molecule with just 12 atoms, there are 495 parameters altogether, including the χij, ωi, and χii. This is already an unreasonably large number of parameters to fit. For larger molecules, the problem becomes catastrophic.
A possible step toward a remedy can be found in two new approaches to spectra, the vibron model and methods based on quantum groups. These appear at first to be quite different from the SMM Hamiltonian (6), but closer analysis shows all three methods to be related. I will describe the vibron and quantum group approaches and how they provide hints of a remedy for the parameter explosion, and then indicate how this remedy might be extended to the more familiar SMM of Eq. 6.
The vibron model also turns out to have a relation to the supermultiplet description of two-electron atoms. I will return to this briefly following the discussion of molecules.
The Vibron Model.
The vibron model (29, 30) is related to early work on rigid and nonrigid molecular systems in terms of correlation diagrams (31, 32). The basic idea is to represent the rotation–vibration spectrum of each bonded pair of atoms in a molecule in terms of representations of a continuous group. The group is the unitary group U(4), which is related to the O(4) rotation group described above for atoms. The unitary group can be thought of as the generalization of rotations to spaces of complex numbers. In the vibron model, a set of n bonded pairs is described by first coupling a separate U(4) for each bond: U(4)1 × U(4)2 × … × U(4)n.
Then, subgroup chains are used to describe operators that resemble terms in the standard zero-order Hamiltonian (4). Couplings analogous but not identical to the Darling–Dennison or Fermi resonance couplings can also be defined in terms of the group operators. Although this procedure might appear far different from the SMM, there is a term-by-term correspondence of a vibron Hamiltonian to a similar Hamiltonian built in the standard way. It is therefore not surprising that sufficiently elaborate versions of the vibron model (with enough independent terms in the Hamiltonian) can give spectral fits of comparable accuracy to the SMM.
An interesting feature of the vibron approach is that it may offer at least a partial remedy to the problem of parameter explosion. This economy comes about because mathematical Lie group constraints lead to a Hamiltonian with automatic relations among the parameters. This greatly reduces the number of independent parameters. The reduced parameter fits appear to be surprisingly accurate (33). Of course, they cannot be as accurate as a model, whether of standard or vibron form, in which all possible parameters are allowed to vary freely. Nonetheless, this facet of the vibron model may be an important step toward a way around the parameter explosion.
Quantum Groups.
Another new approach uses quantum groups. These are currently of great interest in many branches of physics, as well as pure mathematics. They are not actually groups, but rather Hopf algebras, a generalization of the mathematical structure of continuous, or Lie groups. They arise in molecular problems (34, 35) as a kind of anharmonic generalization of the Lie group structure of the harmonic oscillator. In the quantum group approach, a Hamiltonian is obtained which is analogous to the SMM, but not identical. Moreover, as in the vibron model, the quantum group structure imposes relations between the parameters, potentially alleviating the problem of parameter explosion. To test this, detailed fits of experimental data are needed.
Can the Parameter Explosion Be Contained Within the Standard Molecular Model?
The problem of parameter explosion has catastrophic implications for important goals of spectral analysis of molecules with more than a few atoms. The vibron and quantum group approaches point toward the possibility of at least a partial remedy. But these hints raise questions that need further exploration. Is the success of the reduced parameter sets simply fortuitous? To my knowledge, the way that the vibron model simplifies the cross-anharmonicities is based mainly on a mathematical construct, without evident connection to a physical principle governing molecules. It is possible that practically any reasonable operator construction could give a reduced parameter fit of comparable accuracy, but this needs to be tested. If there is a physical basis for the success of the reduced parameter vibron Hamiltonian, it merits further exploration.
It is possible that the reduced parameter fits in the vibron model are not fortuitous, but also not entirely dependent on the elaborate group theoretical apparatus. I am suggesting that it may be possible to construct operators within the SMM that incorporate reduced parameter sets, without resort to Lie groups. Taking into account the polyad structure and intuition about sets of physically related modes might give guidance in constructing such operators. Methods such as genetic algorithms might be useful to explore optimized operators with reduced parameter sets. It may well be possible to obtain far better results with reduced parameter sets than achieved so far. The severity of the difficulty posed by the parameter explosion warrants exploration of every reasonable possibility.
Atomic Supermultiplets and the Vibron Model.
I now describe work that shows connections between the supermultiplet “molecular” model of atoms, the vibron model of molecules, and work on nonrigid systems. These connections are an indication that a unified symmetry viewpoint is emerging for atoms and molecules.
Despite the compelling physical motivation and the spectral patterns revealed by the supermultiplets, one fundamental aspect of the supermultiplets was mysterious from the beginning. This was the formal group theoretical basis of the supermultiplet classification. The supermultiplets were constructed by collecting together the approximate two-electron O(4)12 multiplets into larger entities—the supermultiplets. The mystery was that despite the beauty of their structure and molecular interpretation, the supermultiplets did not appear to correspond to any larger group structure encompassing the two-electron O(4)12′. In a recent paper (36), I was able to shed some light on this by embedding the O(4) group structure in a larger group, U(4). The U(4) lends its structure to a larger organization scheme, the supermultiplets, than can be obtained from O(4) alone.
The use of U(4) in the two-electron problem is related to the vibron molecular model. The embedding of the atomic problem in a U(4) vibron-type structure is based on the observation that the pattern of correlation from nonrigid to rigid states in linear molecules (32) results in groupings analogous to the atomic supermultiplets.
The U(4) embedding procedure could be very useful if it could be used to classify collective molecule-like behavior in the electronic motions of atoms with three or more electrons in the outer shell. At present, classification schemes for molecular atoms with three or more valence electrons, analogous to the two-electron supermultiplets, are not known.
A Hierarchy of Symmetries
I have tried to indicate how a new view of chemical systems is emerging in terms of dynamical symmetries and methods of nonlinear systems, impelled by recent discoveries of phenomena of highly excited states. I would like to put this in larger perspective by returning to the theme of a hierarchy of scales in nature, each governed by a different aspect of regularity or symmetry in its concepts and laws. I will try to draw possible lessons for other areas of science from the appearance of dynamical symmetry principles in chemical systems. I will present some views of attempts to found theories of elementary particles and even a Theory of Everything (37) on the idea that all the fundamental laws of nature are based on exact symmetry principles. These views are personal and idiosyncratic; the reader can judge whether they are misguided.
At the largest scale, the structure of the universe is dominated by gravitational forces, described by Einstein’s general relativity. The scale of day-to-day life is characterized by very large systems such as biological organisms. The chemical level is described by quantum mechanics and the laws of electricity and magnetism, from which there emerge atomic and molecular behavior. Smaller than the chemical systems are the levels of atomic nuclei, the even smaller “elementary” particles, and their quark constituents. At the smallest scale, the Planck length 10−33 cm, gravitation and quantum mechanics are believed to be intertwined in a fundamental way.
Theories of Everything take as their touchstone a description of matter at the very largest and smallest scales in terms of a few unifying principles. At the largest scale, described by Einstein’s general relativity, nature is governed by space-time symmetry. This is invariance under local coordinate transformations, with the local invariance a matter of profound importance. Skipping to the scale of elementary particles, the SEPM gives an account of the electromagnetic, weak, and strong interactions in terms of quantum field theory (38, 39). These theories are formally related in some measure (40) to general relativity in that one of their basic principles is a type of local symmetry known as gauge invariance. This is well established as a fundamental symmetry for quantum electrodynamics.
Because of this surprising unifying feature of theories devised for such different scales and phenomena, and also because of the unsolved problem of uniting general relativity and quantum mechanics, there are attempts to bring together general relativity, the elementary particles, and the Planck scale in superstring theory as a Theory of Everything, with local symmetry as one of its overarching principles. In both general relativity and quantum electrodynamics, the local symmetries practically define the structure of the theory. It is no wonder that one is tempted to base a unified theory of all forces entirely on principles of local symmetry.
One of the intriguing things about this program is that it leaves most of the world as we encounter it, our chemical and biological world, as almost an accidental by-product of the “fundamental” levels of matter, in particular, of the electromagnetic particles and fields. This raises interesting questions. What, if anything, do gauge principles have to tell us about the molecular world? If they are really so basic, is it possible to construct the molecular world essentially “from scratch” out of gauge principles that are independent of those that describe the “fundamental” scales? What would this tell us about the fundamental status of gauge theories of other scales, such as the SEPM? What is the status of the dynamical symmetries discussed here, and their connection with gauge principles? As a scale that is believed to be essentially completely understandable from first principles, do molecules have anything unique to tell us about the fundamental status of theories of other scales? The remainder of this paper is devoted to these questions.
It is not evident that the fundamental gauge symmetries are themselves of much help for the chemical world. Formal analogies to gauge ideas exist for some molecular phenomena (41–43), such as coupled rotations and vibrations. However, they hardly give a complete picture of the most salient features of molecules even at low energy, nor is it clear how much they have to tell us about the dynamical regularities of highly excited states I have been discussing here. The dynamical symmetries of chemistry are phenomenological, without pretense to being fundamental aspects of nature. The SMM, the vibron model, and the quantum group models are all viable descriptions, and none has a clear claim to any absolute superiority or significance of its concepts or formal basis. That we can make such a statement is due to the very fortunate circumstance that chemical systems are in principle completely understood; in terms of the nuclei and electrons as constituents, Maxwell’s theory of electromagnetism as dynamical law, and quantum laws in place of Newton’s kinematics. In fact, what in principle is possible for all of chemistry can as yet be carried out only for very small systems. But we are far enough along that we can calculate a molecular potential surface for small molecules from first principles. From the molecular potential we can calculate vibrational spectra directly if we wish, or by perturbation methods (16, 17) infer a phenomenological theory such as the SMM of (6), and perhaps such approaches as vibrons or quantum groups. Consider what might have happened if this “first principles” view were not available. We could easily have fallen into the trap of mistaking the phenomenological models and dynamical symmetries as fundamental aspects of nature at the chemical scale, with anharmonic oscillators, vibrons, or quantum groups as basic features of molecules!
Therefore, small chemical systems comprise a gap, as regards both scale and the status of symmetry as a fundamental principle, between the domains of general relativity and the elementary particles. The connection of gauge ideas seems even further removed from the principles, themselves currently rather murky, which govern the key physical behavior at the high end of the size scale in chemistry in biomolecules such as proteins. In many proteins there is manifestly a great deal of regularity that can be regarded as a kind of symmetry. However, as yet, this has little relation even to the symmetry of point group theory, though lattice models of proteins (44) suggest at least hints (45) of a connection. It seems to me that to build a Theory of Everything with an overarching principle such as gauge symmetry should give pause when this principle may not tell us all that much about some of the most interesting phenomena between the scales of the very large and small.
Furthermore, it is not evident that the gauge symmetries of the more restricted SEPM model of elementary particles have any more of a claim to fundamental status than the dynamical symmetries of chemistry, apart from the fact that they describe smaller constituents of matter. The SEPM attempts to describe phenomena that are not manifestly simpler or less “messy” than molecules. It is a theory with many parameters (38) that must be determined by fitting data, much like the molecular models. There is considerable uncertainty both in calculating properties such as the particle spectrum from first principles and in fitting data, certainly much greater than that in molecular spectra. As we saw with the reduced parameter versions of molecular models, the attempt to fit everything into a straitjacket of exact symmetry, or at least a jacket with too tight a fit, may give a pleasing appearance that may or not be completely based on reality. And anyhow, in high symmetry versions or otherwise, the molecular models are simply phenomenological representations that emerge from deeper first principles. Much the same may turn out to be true of current attempts to fit the elementary particles into theories based on exact gauge symmetry.
Inasmuch as molecules are a type of complex system for which we nevertheless can calculate many properties from fundamental principles, they occupy an unusual, if not unique, position in science. They may constitute an immensely valuable arena for investigating both the power and conceptual status of general principles of symmetry. As mentioned before, interesting relations have been discovered (41–43) between gauge ideas and molecular principles such as Born–Oppenheimer separability. However, to my knowledge, no attempt has been made to relate to molecules the full panoply of ideas (39) that appear in gauge theories, such as spontaneous symmetry breaking, Nambu–Goldstone bosons, and the Higgs mechanism. It would be fascinating, and perhaps enlightening, to see what kind of molecular world one could build starting from gauge principles. Would this “virtual molecular world” encompass the known phenomenological molecular laws, including the emerging dynamical symmetries? If so, the known “first principles” foundation of chemistry would suggest the likelihood of something similar for the elementary particles, with the gauge models such as SEPM a phenomenological description of something deeper, based perhaps on superstrings, or perhaps with gauge principles not fundamental at all. On the other hand, if the essentials of the molecular world could not be built from gauge principles, this would challenge directly the view of gauge symmetry as the basis for building a Theory of Everything that is found at all the scales in nature.
Summary
Approximate dynamical symmetries are proving very useful for describing very complex aspects of highly excited chemical systems. Some future challenges are global rotation–vibration spectral analysis of systems with more than a few atoms, and collective molecular modes of many electron atoms. Chemical phenomena are in principle completely derivable from first principles. Yet even if a first principles derivation of these phenomena were possible with foreseeable computational abilities, this would not in itself reveal or give insight into the new regularities being discovered at high energy. Moreover, as a scale of nature whose first principles are complete, yet which still is yielding new discoveries, fundamental chemistry provides a unique laboratory for exploring the relation of symmetry principles and fundamental theory.
Acknowledgments
Support of the Department of Energy under Basic Energy Sciences Grant DE-FG06-92ER14236 is gratefully acknowledged.
Footnotes
Abbreviations: SMM, standard molecular model; SEPM, standard elementary particle model.
References
- 1.Hamermesh M. Group Theory and Its Application to Physical Problems. Reading, MA: Addison–Wesley; 1962. [Google Scholar]
- 2.Herzberg G. Infrared and Raman Spectra. New York: Van Nostrand; 1950. [Google Scholar]
- 3.Kellman M E, Herrick D R. J Phys B. 1978;24:L755–L759. [Google Scholar]
- 4.Herrick D R, Kellman M E. Phys Rev A. 1980;21:418–425. [Google Scholar]
- 5.Herrick D R, Kellman M E, Poliak R D. Phys Rev A. 1980;22:1517–1535. [Google Scholar]
- 6.Kellman M E, Herrick D R. Phys Rev A. 1980;22:1536–1551. [Google Scholar]
- 7.Kellman M E. Phys Rev Lett. 1985;55:1738–1741. doi: 10.1103/PhysRevLett.55.1738. [DOI] [PubMed] [Google Scholar]
- 8.Wybourne B G. Classical Groups for Physicists. New York: Wiley; 1974. [Google Scholar]
- 9.Wulfman C E. Chem Phys Lett. 1973;23:370–372. [Google Scholar]
- 10.Herrick D R, Sinanoglu O. J Chem Phys. 1975;62:886–892. [Google Scholar]
- 11.Herrick D R, Sinanoglu O. Phys Rev A. 1975;11:97–110. [Google Scholar]
- 12.Hunter J E, III, Berry R S. Phys Rev A. 1987;36:3042–3053. doi: 10.1103/physreva.36.3042. [DOI] [PubMed] [Google Scholar]
- 13.Ezra G S, Richter K, Tanner G, Wintgen D. J Phys B. 1991;24:L413–L420. [Google Scholar]
- 14.Rost J M, Gersbacher R, Richter K, Briggs J S, Wintgen D. J Phys B. 1991;24:2455–2466. [Google Scholar]
- 15.Rost J M, Briggs J S. J Phys B. 1991;24:4293–4322. [Google Scholar]
- 16.Fried L E, Ezra G S. J Chem Phys. 1987;86:6270–6282. [Google Scholar]
- 17.Sibert E L., III J Chem Phys. 1989;90:2672–2683. [Google Scholar]
- 18.Darling B T, Dennison D M. Phys Rev. 1940;57:128. [Google Scholar]
- 19.Fermi E. Z Phyzik. 1931;71:250. [Google Scholar]
- 20.Kellman M E, Chen G. J Chem Phys. 1991;95:8671–8672. [Google Scholar]
- 21.Smith B C, Winn J S. J Chem Phys. 1991;94:4120–4130. [Google Scholar]
- 22.Jonas D M, Solina S A B, Rajaram B, Silbey R J, Field R W, Yamanouchi K, Tsuchiya S. J Chem Phys. 1993;99:7350–7370. [Google Scholar]
- 23.Temsamani A, Herman M. J Chem Phys. 1995;102:6371–6384. [Google Scholar]
- 24.Solina S A B, O’Brien J P, Field R W, Polik W F. Ber Bunsenges Phys Chem. 1995;99:555–560. [Google Scholar]
- 25.Li Z, Xiao L, Kellman M E. J Chem Phys. 1990;92:2251–2268. [Google Scholar]
- 26.Kellman M E. Annu Rev Phys Chem. 1995;46:395–412. doi: 10.1146/annurev.pc.46.100195.002143. [DOI] [PubMed] [Google Scholar]
- 27.Lu Z, Kellman M E. Chem Phys Lett. 1995;247:195–203. [Google Scholar]
- 28.Rose, J. P. & Kellman, M. E. (1996) J. Chem. Phys., in press.
- 29.Levine R D, Wulfman C. Chem Phys Lett. 1979;60:372–376. [Google Scholar]
- 30.Iachello F, Levine R D. Algebraic Theory of Molecules. New York: Oxford Univ. Press; 1994. [Google Scholar]
- 31.Kellman M E, Amar F, Berry R S. J Chem Phys. 1980;73:2387–2404. [Google Scholar]
- 32.Kellman M E. Chem Phys. 1988;48:89–95. [Google Scholar]
- 33.Iachello F, Oss S, Lemus R. J Mol Spectrosc. 1991;149:132–151. [Google Scholar]
- 34.Bonatsos D, Daskaloyannis C. Phys Rev A. 1993;48:3611–3616. doi: 10.1103/physreva.48.3611. [DOI] [PubMed] [Google Scholar]
- 35.Bonatsos, D., Daskaloyannis, C. & Kolokotronis, P. (1996) J. Chem. Phys., in press.
- 36.Kellman M E. Phys Rev Lett. 1994;73:2543–2546. doi: 10.1103/PhysRevLett.73.2543. [DOI] [PubMed] [Google Scholar]
- 37.Witten E. Phys Today. 1996;49:24–30. [Google Scholar]
- 38.Donoghue J F, Golowich E, Holstein B R. Dynamics of the Standard Model. Cambridge, U.K.: Cambridge Univ. Press; 1992. [Google Scholar]
- 39.Marshak R E. Conceptual Foundations of Modern Particle Physics. Singapore: World Scientific; 1993. [Google Scholar]
- 40.Eguchi T, Gilkey P B, Hanson A J. Phys Rep. 1980;66:213–393. [Google Scholar]
- 41.Shapere, A. & Wilczek, F. (1989) Geometric Phases in Physics, Advanced Series in Mathematical Physics, Vol. 5 (World Scientific, Singapore).
- 42.Jackiw R. Int J Mod Phys A. 1988;3:285–297. [Google Scholar]
- 43.Littlejohn R G, Reinsch M. Phys Rev A. 1995;52:2035–2051. doi: 10.1103/physreva.52.2035. [DOI] [PubMed] [Google Scholar]
- 44.Yue K, Dill K A. Proc Natl Acad Sci USA. 1995;92:146–150. doi: 10.1073/pnas.92.1.146. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 45.Kellman M E. J Chem Phys. 1996;105:2500–2508. [Google Scholar]