

Abstract
Exocytosis of intracellular vesicles, such as insulin granules, is carried out by soluble N-ethylmaleimide-sensitive factor attachment protein receptor (SNARE) and Sec1/Munc18 (SM) proteins. An additional regulatory protein, Doc2b (double C2 domain), has recently been implicated in exocytosis from clonal β-cells and 3T3-L1 adipocytes. Here, we investigated the role of Doc2b in insulin secretion, insulin sensitivity, and the maintenance of whole-body glucose homeostasis. Doc2b heterozygous (Doc2b+/−) and homozygous (Doc2b−/−) knockout mice exhibited significant whole-body glucose intolerance and peripheral insulin resistance, compared with wild-type littermates. Correspondingly, Doc2b+/− and Doc2b−/− mice exhibited decreased responsiveness of pancreatic islets to glucose in vivo, with significant attenuation of both phases of insulin secretion ex vivo. Peripheral insulin resistance correlated with ablated insulin-stimulated glucose uptake and GLUT4 vesicle translocation in skeletal muscle from Doc2b-deficient mice, which was coupled to impairments in Munc18c-syntaxin 4 dissociation and in SNARE complex assembly. Hence, Doc2b is a key positive regulator of Munc18c-syntaxin 4–mediated insulin secretion as well as of insulin responsiveness in skeletal muscle, and thus a key effector for glucose homeostasis in vivo. Doc2b’s actions in glucose homeostasis may be related to its ability to bind Munc18c and/or directly promote fusion of insulin granules and GLUT4 vesicles in a stimulus-dependent manner.
Glucose homeostasis is maintained through the coordinated actions of insulin secretion and insulin action; dysfunction of insulin action yields insulin resistance and, when coupled with dysfunctional insulin secretion, results in type 2 diabetes. Common to both insulin action and insulin secretion mechanisms is the requirement for SNARE protein–regulated vesicle/granule exocytosis. Two target membrane SNAREs (t-SNAREs) and one vesicle/granule membrane SNARE (v-SNARE) combine to form a heterotrimeric SNARE core complex that drives vesicle fusion in the exocytosis process. Insulin-containing secretory granules within the islet β-cell contain the v-SNARE vesicle-associated membrane protein 2 (VAMP2), which assembles at the plasma membrane (PM) with either the t-SNARE combination syntaxin 1A and synaptosome-associated protein of 25 kDa (SNAP25), syntaxin 4 and SNAP25, or syntaxin 4 and SNAP23 to release insulin (1,2). In a similar fashion, insulin promotes glucose uptake into peripheral skeletal muscle and adipose tissues by translocation and fusion of the GLUT4 glucose transporter vesicles to the cell surface (3,4). Insulin action requires only the t-SNAREs syntaxin 4 and SNAP23 and the v-SNARE VAMP2 (5), all of which are also required for insulin secretion from the β-cell. Defects and/or deficiencies of these SNARE proteins are associated with insulin resistance and/or insulin insufficiency in type 2 diabetes (6–9).
Insulin secretion and GLUT4 recruitment events are highly regulated, maintained at very low levels in the absence of appropriate stimuli, and rapidly and robustly activated in response to stimuli. Maintenance and activation of exocytotic processes are regulated by the Sec1/Munc18 (SM) proteins, which bind and facilitate the accessibility of syntaxin to interact with its cognate SNARE partners (10,11). SM-syntaxin proteins pair in a 1:1 stoichiometry and in multiple modes. In one state, syntaxin is kept in a “closed” conformation, preventing its participation in the SNARE core complex (12). Upon stimulation, the SM protein is presumed to dissociate or reposition syntaxin so that it “opens” and is accessible to SNARE complex assembly. In homogenates of β-cells, adipocytes, and skeletal muscle, Munc18c binds to syntaxin 4 in the absence of stimuli, dissociating in response to stimuli while undergoing tyrosine phosphorylation (13–15). We recently identified the insulin receptor to serve as a Munc18c tyrosine kinase in 3T3-L1 adipocytes and skeletal muscle (13).
Concurrent with its dissociation from syntaxin 4, phosphorylated Munc18c switches its affinity toward binding to the protein Doc2b (double C2 domain) in MIN6 clonal β-cells (16). Elevation of calcium is reported to yield complexation of Doc2b with syntaxin 4 as well (17). Doc2b is a member of the double C2 domain protein family, containing two calcium and phospholipid binding domains in its COOH terminus. In vitro, Doc2b selectively binds to Munc18-1 via Doc2b domain C2A and binds to Munc18c via Doc2b domain C2B (18,19). In MIN6 β-cell and 3T3-L1 adipocyte clonal cell studies, the inhibitory RNA–mediated reduction of Doc2b attenuates stimulus-induced insulin exocytosis and GLUT4 exocytosis events, respectively (17,18,20). By contrast, Doc2b overexpression in these cell types enhances stimulus-induced exocytosis but not basal exocytosis (17,18,20). Unlike Munc18c and syntaxin 4, the relevance of Doc2b function for whole-body glucose homeostasis remains untested. Moreover, although Doc2b mRNA abundance in islets of obese mice was significantly reduced (8), Doc2b deficiency has yet to be correlated with type 2 diabetes, such that its promise as a novel therapeutic target remains in question.
In this study, we used classic Doc2b knockout mice to investigate the role of Doc2b in insulin granule exocytosis and insulin-stimulated GLUT4 vesicle translocation, culminating in a new in vivo model of glucose intolerance and insulin resistance. Mechanistically, islet perifusion studies revealed Doc2b to function in both phases of glucose-stimulated insulin secretion, implicating Doc2b to act on both types of SNARE complexes. Furthermore, skeletal muscle fractionation studies demonstrated a requirement for Doc2b in insulin-stimulated GLUT4 accumulation to mediate glucose uptake; SM-SNARE interactions in muscle fractions were found to be altered by Doc2b deletion.
RESEARCH DESIGN AND METHODS
Materials.
Antibodies to Munc18c and syntaxin 4 were generated as previously described (21,22). All other antibodies were purchased: syntaxin 4 (for immunoprecipitation, Chemicon, Temecula, CA); SNAP23 (Affinity BioReagents, Golden, CO); Doc2b (Abcam, Cambridge, MA); clathrin (BD Biosciences, San Jose, CA); Munc18-1 and VAMP2 (Synaptic Systems, Gottingen, Germany); syntaxin 1A (Sigma-Aldrich, St. Louis, MO); phosphotyrosine-specific antibody (PY20), AKT, and phosphorylated protein kinase B (pAKTS473) (Cell Signaling, Beverley, MA); and goat anti–rabbit-horseradish peroxidase and anti–mouse-horseradish peroxidase secondary antibodies (Bio-Rad, Hercules, CA). Anti-GLUT4 antibody and protein G+ agarose beads were acquired from Santa Cruz (Santa Cruz, CA). Enhanced chemiluminescence and SuperSignal West Femto chemiluminescent reagents were purchased from Amersham Biosciences (Pittsburgh, PA) and Thermo, respectively.
Doc2b knockout mice.
Doc2b knockout mice were initially characterized for neuronal alterations (23) with breeders obtained from VU University to establish a colony at Indiana University School of Medicine. The colony stock at VU University consisted of mice back-crossed for >15 generations on the C57BL/6J background at the VU University and VU Medical Center in Amsterdam, and through an additional two generations at the Indiana University School of Medicine Laboratory Animal Resources Center, all according to animal care guidelines. Paired littermates from heterozygous crossings were used, with genotypes determined by PCR as previously described (23).
Tissue homogenization, immunoblotting, and immunoprecipitation.
For immunoblot analysis of protein content, tissues were homogenized in a 1% NP40 detergent buffer (25 mmol/L Tris [pH 7.4], 1% NP40, 10% glycerol, 50 mmol/L sodium fluoride, 10 mmol/L sodium pyrophosphate, 137 mmol/L sodium chloride, 1 mmol/L sodium vanadate, 1 mmol/L phenylmethylsulfonyl fluoride, 10 μg/mL aprotinin, 1 μg/mL pepstatin, and 5 μg/mL leupeptin) and microcentrifuged, and proteins were resolved on 10–12% SDS-PAGE for immunoblotting. Skeletal muscle lysates used for immunoprecipitation, 4 mg per reaction as previously described (13), were prepared from mice fasted for 4 h (0800–1200 h) and injected intraperitoneally with 10 units/kg insulin or saline control for 5 min. Lysates were prepared in buffer supplemented with 1 mmol/L CaCl2 or 2 mmol/L EDTA as specified in the text.
Intraperitoneal glucose tolerance test and insulin tolerance test.
Male Doc2b+/+, Doc2b+/−, and Doc2b−/− mice (4–6 months old) were fasted for either 6 h (0800–1400 h) or 18 h (1800–1200 h) before intraperitoneal glucose tolerance test (IPGTT), as specified in the figure legend. After sample collection of fasted blood, animals were injected intraperitoneally with glucose (2 g/kg body weight) and blood glucose was sampled from the tail vein every 30 min using the Hemocue glucometer (Mission Viejo, CA). For the insulin tolerance test (ITT), the same male mice were fasted for 6 h (0800–1400 h) and, after collection of fasted blood, were injected intraperitoneally with Humulin R (0.75 units/kg body weight; Eli Lilly and Company).
Skeletal muscle subcellular fractionation.
Littermate male mice (4–6 months old) were fasted for 16 h, injected intraperitoneally with Humulin R or vehicle at 21 units/kg body weight for 40 min, and killed for removal of the hindquarter muscles for differential centrifugation to yield the P2 fractions (contain transverse tubule and sarcolemmal membranes) and intracellular membrane components, as previously described (24,25); each immunoprecipitation reaction required 1 mg P2 fraction protein.
In vitro skeletal muscle glucose uptake.
Extensor digitorum longus (EDL) muscles were excised and immediately placed into prewarmed incubation buffer for subsequent use in 3H-2-deoxyglucose uptake assays in vitro in a 20-min insulin stimulation (micromoles per gram of muscle, divided by the water content per gram of muscle), as previously described (26).
Isolation, culture, and perifusion of mouse islets.
Pancreatic mouse islets were isolated, as previously described (27), from 10–14-week-old male mice. Islets cultured overnight were handpicked onto cytodex bead columns, preincubated in Krebs-Ringer bicarbonate buffer (10 mmol/L HEPES [pH 7.4], 134 mmol/L NaCl, 5 mmol/L NaHCO3, 4.8 mmol/L KCl, 1 mmol/L CaCl2, 1.2 mmol/L MgSO4, and 1.2 mmol/L KH2PO4) containing 2.8 mmol/L glucose and 0.1% BSA for 30 min, and perifused at a rate of 0.3 mL/min during stimulations with 20 mmol/L glucose and 35 mmol/L KCl, with fractions collected every 1–3 min for quantitation by radioimmunoassay (Millipore).
Islet RNA isolation and quantitative PCR.
Total RNA from mouse islets was obtained using the RNeasy mini kit (Qiagen). RNA (2 μg) was reverse transcribed with the SuperScript First Strand cDNA Synthesis Kit (Invitrogen), and 1% of the product was used for quantitative PCR (Q-PCR). The primers used were as follows: Doc2b primers, forward 5′-ccagcaaggcaaataagctc and reverse 5′-attgggcttcagcttcttca; and GAPDH primers, forward 5′-atggtgaaggtcggtgtgaacg and reverse 5′-gttgtcatggatgaccttggcc. The Q-PCR conditions were as follows: 50°C for 2-min hold (UDG incubation), 95°C for 2-min hold, 40 cycles of 95°C for 15 s, and 60°C for 30 s.
Statistical analysis.
All data were evaluated for statistical significance using Student t test for pairwise comparison of two groups (i.e., Doc2b+/− or Doc2b−/−, vs. Doc2b+/+). Data are expressed as the mean ± SE.
RESULTS
Doc2b knockout mice are glucose intolerant.
Although the Doc2b−/− mice have been characterized for alterations in neuronal protein expression and synaptic vesicle trafficking function, such studies are lacking in evaluation of Doc2b function in tissues relevant to glucose homeostasis. Gene ablation in the heterozygous and homozygous knockout mice was confirmed by comparing Doc2b mRNA levels in Doc2b+/+ (wild type [WT]) littermates in brain, skeletal muscle (whole hindlimb), liver, and fat (epididymal) by Q-PCR (Fig. 1A). Protein levels of Doc2b were reduced, in the absence of significant differences in other SNARE or SM proteins implicated in insulin exocytosis, in islets isolated from Doc2b+/− and Doc2b−/− knockout mice, compared with the Doc2b+/+ islets (Fig. 1B and Supplementary Fig. 1). The Doc2b antibody showed nonspecific background, consistent with previous work (23). Doc2b levels were reduced in heart, skeletal muscle, liver, and fat tissues of Doc2b+/− and Doc2b−/− mice, without alterations in abundance of syntaxin 4, SNAP23, VAMP2, or Munc18c (Fig. 1C). Abundance of the glucose transporter GLUT4 protein was also unchanged in heart, skeletal muscle, and fat from WT or Doc2b-deficient mice (Supplementary Fig. 1).
FIG. 1.

Protein and mRNA expression in glucose homeostatic tissues from Doc2b+/− and Doc2b−/− knockout mice. A: Brain, skeletal muscle (whole hindlimb), liver, and fat (epididymal) were isolated from Doc2b+/+, Doc2b+/−, and littermate Doc2b−/− mice for use in Q-PCR analysis (quantified relative to GAPDH from three sets of tissues). B: Islets were isolated from Doc2b+/+, Doc2b+/−, and Doc2b−/− knockout mice for analysis of SNARE protein expression; blots for Munc18c and syntaxin 4 were stripped and reblotted for isoforms Munc18-1 and syntaxin 1A, using clathrin as a loading control for those gel lanes. Doc2b immunoblotting was performed using identical/matched islet lysates run in duplicate lanes on the same gel in an effort to reduce nonspecific background, paired with the corresponding clathrin loading control. C: Assessments of GLUT4, SNARE, and SNARE accessory protein abundances were made by immunoblotting for heart, skeletal muscle, liver, and epididymal fat. Doc2b antibody cross-reactivity yielded substantial background in the Doc2b−/− lysate lanes. Data represent three to five matched sets of tissues. Vertical lines indicate splicing of identical lysates resolved on parallel gels. IB, immunoblot.
To determine the effects of Doc2b deficiency upon whole-body glucose tolerance, 4–6-month-old Doc2b+/+, Doc2b+/−, and Doc2b−/− mice were subjected to IPGTTs. Glucose tolerance after either 18 or 6 h fasting in Doc2b+/− and Doc2b−/− male mice was significantly impaired in comparison with WT mice (Fig. 2A and Supplementary Fig. 2). Doc2b−/− and Doc2b+/− mice showed similar fasting glucose levels compared with WT mice (Table 1). Area under the curve (AUC) analysis confirmed Doc2b+/− and Doc2b−/− mice to be significantly less tolerant than WT (Fig. 2B). These data suggest that full or partial depletion of Doc2b exerted a negative effect upon glucose tolerance in vivo.
FIG. 2.

Doc2b+/− and Doc2b−/− knockout mice are glucose intolerant with reduced serum insulin concentration post–glucose injection. A: IPGTT of Doc2b+/−, Doc2b−/−, and littermate Doc2b+/+ mice was performed by intraperitoneal injection of d-glucose (2 g/kg body weight) into 4–6-month-old male mice fasted for 18 h. Blood glucose was monitored over 2 h postinjection as described in research design and methods. B: AUC data shown as the average ± SE from seven sets of mice. *P < 0.05, WT vs. Doc2b−/−; #P < 0.05, WT vs. Doc2b+/−. C: Insulin concentration present in serum taken prior to and 10 min after injection of glucose during the IPGTT was measured by insulin radioimmunoassay analysis. Data shown as the average ± SE from six sets of mice. *P < 0.05 vs. preinjected WT; **P < 0.05, stimulated Doc2b−/− vs. WT.
TABLE 1.
Fasting serum analytes of Doc2b+/+, Doc2b+/−, and Doc2b−/− mice
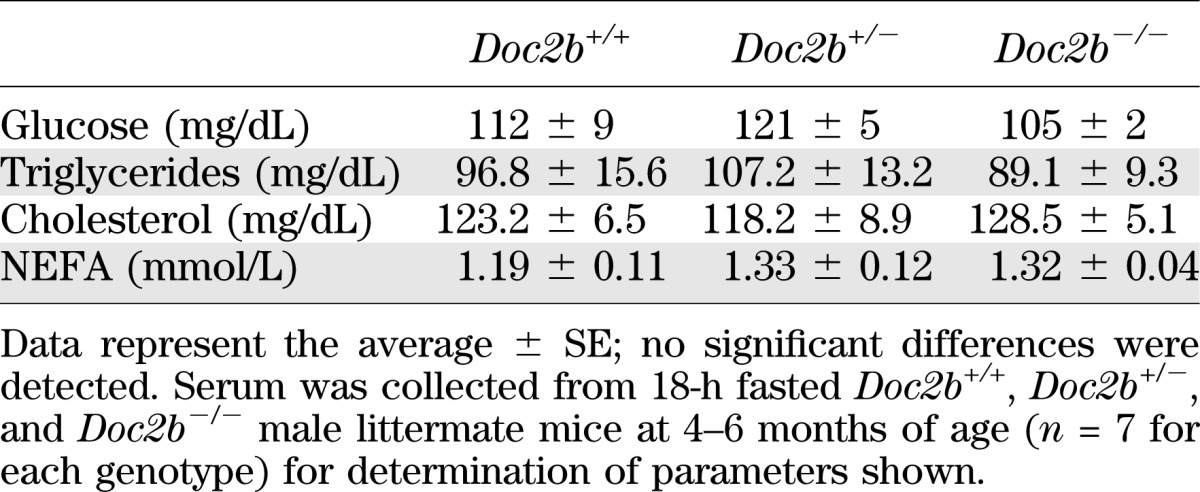
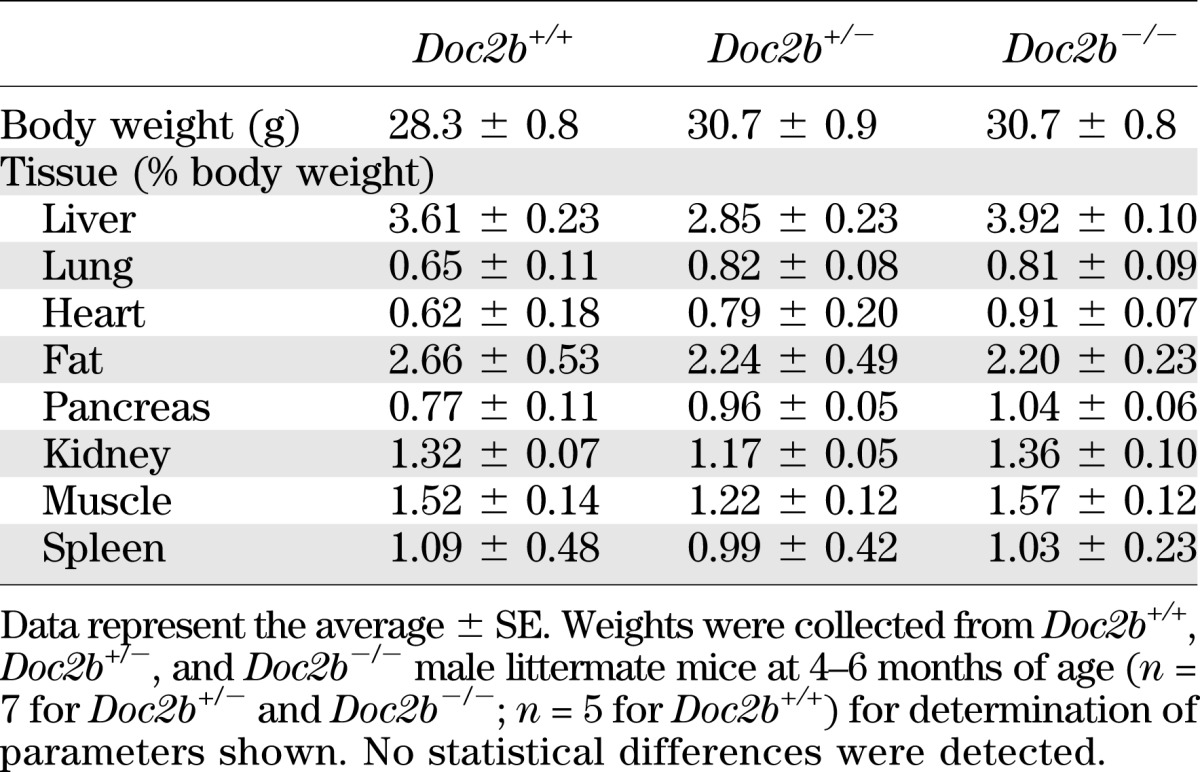
We next determined whether glucose intolerance was related to alterations in body weight or fasted serum metabolites commonly linked to aberrations in glucose metabolism. Table 1 shows serum triglycerides, cholesterol, and nonesterified fatty acids (NEFAs) to be similar in WT versus Doc2b+/− and Doc2b−/− mice. Table 2 shows body weights of Doc2b+/− and Doc2b−/− mice to be equivalent to that of WT littermate mice. Tissue and organ weights were also equivalent, both when normalized to body weight of the whole animal and to absolute tissue weight (Table 1 and Supplementary Table 1). By contrast, serum insulin levels taken 10 min postinjection during the IPGTT were significantly reduced in the Doc2b−/− mice, but not in the Doc2b+/− or WT mice (Fig. 2C). These data indicate an insulin secretory defect.
TABLE 2.
Tissue weights normalized to body weight of Doc2b+/+, Doc2b+/−, and Doc2b−/− mice
Impaired biphasic insulin secretion in islets isolated from Doc2b knockout mice.
To investigate the potential defect in islet β-cell glucose-stimulated insulin release, we isolated islets from male Doc2b+/+, Doc2b+/−, and Doc2b−/− mice for perifusion analyses. Ex vivo, insulin secretion under basal conditions was similar among all three islet groups (Fig. 3A), similar to our findings of insulin content in fasted serum. Glucose stimulation (20 mmol/L) elicited a 12-fold peak increase in insulin release from WT islets during the initial phase, whereas Doc2b+/− and Doc2b−/− islets showed less response. During the second phase, Doc2b+/− and Doc2b−/− islets secreted substantially less insulin (Fig. 3B). Consistent with impaired first-phase glucose-stimulated insulin secretion, KCl-stimulated insulin release was precipitously decreased as Doc2b expression decreased (Fig. 3C). Insulin content in Doc2b+/− and Doc2b−/− islets was comparable to that in WT islets (Fig. 3D). These data indicated that Doc2b-depleted islets lacked function during both phases of glucose-stimulated insulin secretion, corroborating the deficient serum insulin content observed during the IPGTT in the Doc2b−/− mice. This is the first demonstration of a Doc2b requirement in both phases of insulin secretion from islets.
FIG. 3.

Doc2b+/− and Doc2b−/− knockout mouse islets show reduced biphasic insulin release. A: Islets freshly isolated from Doc2b+/−, Doc2b−/−, and littermate Doc2b+/+ mice were cultured overnight, handpicked under a fluorescence microscope into groups of 40, and layered onto cytodex bead columns for perifusion. Islets were first preincubated for 30 min in low glucose (2.8 mmol/L), followed by basal sample collection (1–10 min) at low glucose to establish a baseline. Glucose was then elevated to 20 mmol/L for 35 min and then returned to low glucose for 20 min. Eluted fractions were collected at 1–3-min intervals at a flow rate of 0.3 mL/min, and insulin secretion was determined by radioimmunoassay, as depicted in a representative experiment. B: Quantitation of the AUC for first- (11–17 min) and second-phase (18–45 min) insulin secretion from islets, normalized to baseline; data are presented as average ± SE of at least three independent sets of perifused islets. *P < 0.05 vs. Doc2b+/+. C: Representative traces of perifused islets from A after a 25-min rest under basal conditions and then stimulation with 35 mmol/L KCl for 10 min. D: Average insulin content per 10 islets from Doc2b+/+, Doc2b+/−, and Doc2b−/− littermate male mice used in perifusion studies in panels A\x{2013}C.
Impaired insulin sensitivity, skeletal muscle glucose uptake, and GLUT4 translocation in Doc2b knockout mice.
Whole-body glucose intolerance could also be attributable to defects in insulin sensitivity, causing insulin resistance. To investigate this, 4–6-month-old Doc2b+/+, Doc2b+/−, and Doc2b−/− male mice were subjected to an ITT. As expected of WT mice of this age and strain, insulin injection resulted in a sharp ∼45% decline in blood glucose within 60 min (Fig. 4A). By contrast, neither Doc2b+/− nor Doc2b−/− mice dropped below 70% of starting glucose levels, with levels back on the rise by 60 min postinjection. Analysis of AUC revealed a substantial difference in glucose levels during the ITT ([in arbitrary units] WT = 5,971 ± 238, Doc2b+/− = 7,019 ± 420, and Doc2b−/− = 7,210 ± 420), implicating a defect in the peripheral glucose uptake resulting from Doc2b depletion.
FIG. 4.

Impaired insulin sensitivity in Doc2b-deficient mice is coupled to impaired insulin-stimulated GLUT4 translocation in skeletal muscle. A: ITT of Doc2b+/−, Doc2b−/−, and littermate Doc2b+/+ male mice (seven sets of mice) was performed by intraperitoneal injection of insulin (0.75 units/kg of body weight) into 4–6-month-old male mice fasted for 6 h. Blood glucose was monitored before and at 15, 30, 60, and 90 min after injection as described in research design and methods. Data shown are presented as mean percent of basal blood glucose concentration ± SE. *P < 0.05 vs. WT mice. B: Littermate sets of male WT or Doc2b−/− mice were fasted for 16 h and either left untreated or were injected with 21 units/kg body weight of insulin as described in research design and methods. Hindquarter muscles were homogenized and centrifuged to partition muscle into sarcolemmal/transverse tubule membrane and intracellular vesicular fractions. Proteins were resolved using SDS-PAGE for immunoblotting for GLUT4 (Ponceau S staining shows protein loading). Optical density quantitation of GLUT4 bands in three independent translocation assays is shown in the bar graph. *P < 0.05 compared with basal WT; **P < 0.05 compared with insulin-stimulated WT. C: In vitro 3H-2-deoxyglucose (2DG) uptake assay from EDL muscle of six pairs of WT and Doc2b−/− male mice (for each mouse, one muscle was left in the basal state and one was treated with insulin). *P < 0.05 compared with basal WT; **P < 0.05 compared with insulin-stimulated WT. D: Skeletal muscle and liver homogenates were prepared from mice stimulated with or without insulin and proteins were resolved on 10% SDS-PAGE for immunoblot analysis of AKT activation assessed by anti–phospho-AKTS473 immunoblotting. Blots were stripped and reprobed for total AKT content. Data are representative of three independent sets of tissue homogenates. IB, immunoblot.
Skeletal muscle GLUT4-mediated glucose uptake accounts for ∼80% of whole-body glucose clearance, and so largely controls the response in the ITT (28). To assess insulin-stimulated GLUT4 translocation in skeletal muscle, sarcolemma/transverse tubule–enriched fractions (referred to as P2 fractions) were prepared from insulin- or saline-injected mice as described previously (24,25,29,30). A statistically significant, nearly twofold increase in GLUT4 protein accumulation into the P2 membrane fraction was detected from insulin-stimulated WT mouse muscle (Fig. 4B). Remarkably, no insulin-stimulated increase in GLUT4 accumulation was observed in Doc2b−/− mice. P2 fractions prepared from unstimulated WT and Doc2b−/− mice showed similarly low levels of GLUT4 protein. Consistent with this, EDL muscle from Doc2b−/− mice showed a lack of insulin-stimulated 3H-2-deoxyglucose uptake, in contrast to the nearly twofold increase seen in WT EDL muscle (Fig. 4C). Proximal insulin signaling in skeletal muscle and liver was unaffected, as determined by insulin-stimulated AKTS473 phosphorylation and equivalent AKT expression (Fig. 4D). Taken together, these data demonstrate that insulin-stimulated GLUT4 externalization and glucose uptake is significantly impaired in skeletal muscle tissue of Doc2b−/− mice.
Altered SM and SNARE complex formations in skeletal muscle of the Doc2b knockout mice.
To date, all studies regarding the mechanistic role of Doc2b are from in vitro and cell culture model systems, and results are controversial due to methodological differences (17,18,20). To resolve these issues, we tested previously described Doc2b interactions using skeletal muscle of insulin-injected mice as a more physiologically relevant model system. Because calcium has been shown to trigger Doc2b association with syntaxin 4 in vitro (17), we examined binding under calcium-deficient (2 mmol/L EDTA) and calcium-supplemented (1 mmol/L CaCl2) conditions. In WT muscle lysates, Doc2b binding to Munc18c increased by ∼60% in response to insulin stimulation; calcium addition to the lysis buffer failed to significantly alter either basal or insulin-stimulated binding events (Fig. 5A). Similar results were obtained using basal or glucose-stimulated MIN6 cell lysates supplemented with calcium in the lysis buffer (Supplementary Fig. 3). In skeletal muscle lysates, anti-Munc18c coprecipitated syntaxin 4 regardless of calcium supplementation, whereas neither VAMP2 nor SNAP23 coprecipitated with Munc18c under any conditions (Fig. 5B). Reciprocal anti-VAMP2 immunoprecipitation reactions showed no binding of Munc18c. Calcium supplementation did not impact SNARE complex formation; ratios of SNAP23/VAMP2 and syntaxin 4/VAMP2, normalized to 1.0 in the absence of calcium (2 mmol/L EDTA), were measured to be 0.8 ± 0.2 and 0.8 ± 0.2, respectively, in the presence of supplemental calcium (n = 3 paired experiments, P > 0.05). Moreover, syntaxin 4 failed to coimmunoprecipitate Doc2b, even under calcium-containing and insulin-stimulated conditions from skeletal muscle (Fig. 5C); Doc2b−/− muscle served as control for nonspecific binding. Syntaxin 4 coprecipitated SNAP23 equivalently under all conditions, consistent with SNAP23 participation in binary and ternary SNARE complexes. Like syntaxin 4, which was constitutively present in the P2 fraction (Fig. 5D), Doc2b abundance was unchanged by insulin in P2 fractions prepared 5 min post–insulin injection, the time of peak tyrosine phosphorylation of Munc18c and its association with Doc2b, and Doc2b translocation was not detected within 30 min post–insulin injection (data not shown). These data suggest that in skeletal muscle lysate, Doc2b binds to Munc18c in an insulin-sensitive manner and fails to bind to syntaxin 4 in response to insulin and/or added calcium.
FIG. 5.

Insulin-dependent, but calcium-independent, Doc2b-Munc18c association in mouse skeletal muscle. The impact of insulin stimulation and/or calcium addition to lysis buffer upon association of Munc18c with Doc2b (A) or VAMP2, SNAP23, and syntaxin 4 (B) was assessed by reciprocal coimmunoprecipitation reactions using hindlimb skeletal muscle extracts. Reactions were processed in parallel from the same starting hindlimb muscle extracts from WT mice injected with vehicle (saline) or insulin (10 units/kg body weight) for 5 min in lysis buffers supplemented with either 2 mmol/L EDTA or 1 mmol/L CaCl2. Immunoprecipitated proteins were resolved on 10–12% SDS-PAGE for immunodetection of Munc18c, Doc2b, syntaxin 4, SNAP23, and VAMP2. Equivalent abundance of proteins in the corresponding starting lysates was confirmed by immunoblot (Lysate). *P < 0.05. C: Calcium addition to lysis buffer does not facilitate Doc2b coimmunoprecipitation with anti–syntaxin 4 from skeletal muscle extracts. Muscle extracts used in A and B were subjected to anti–syntaxin 4 immunoprecipitation for immunodetection of Doc2b. SNAP23 binding to syntaxin 4 validated the immunoprecipitation reactions. Control IgG and lysates from Doc2b−/− mice were used in separate reactions to control for nonspecific banding occurring with the Doc2b antibody. D: Evaluation of Doc2b protein recruitment to the PM fraction in response to insulin. P2 fraction extracts prepared from saline or insulin-stimulated WT mice were subjected to SDS-PAGE as described in Fig. 4B for immunodetection of Doc2b and syntaxin 4 (Syn4). Data are representative of three independent sets of homogenates or fractions. IB, immunoblot.
We next sought to determine why GLUT4 accumulation in the target membranes of skeletal muscle was impaired in the Doc2b−/− mice by examining effects upon SM and SNARE protein complexation. In WT extracts, insulin induced phosphorylation of Munc18c, as described previously (13), and was fully recapitulated in reactions using Doc2b−/− extracts (Fig. 6A), suggesting that Doc2b was not required for Munc18c to undergo insulin-stimulated tyrosine phosphorylation. Anti–syntaxin 4 immunoprecipitation reactions using the same extracts revealed 40–50% more Munc18c binding to syntaxin 4 in Doc2b−/− muscle (Fig. 6B), as compared with WT muscle. Input lysates validated the absence of Doc2b in Doc2b−/− lysates and the otherwise comparable expression of Munc18c. Using sarcolemmal/transverse tubule (P2) membrane fractions from hindlimb muscles of insulin-stimulated WT and Doc2b−/− mice, significant reductions of VAMP2 and SNAP23 binding to syntaxin 4 in Doc2b−/− fractions were revealed (Fig. 6C). Coordinately, Munc18c binding to syntaxin 4 was elevated by 46% in Doc2b−/− fractions. Thus, our cumulative data suggest that ablation of insulin-stimulated GLUT4 vesicle translocation in Doc2b−/− muscles is underpinned by increased abundance of Munc18c–syntaxin 4 complexes coordinated with diminished abundance of SNARE complexes.
FIG. 6.

Munc18c–syntaxin 4 binding is increased in Doc2b−/− mouse skeletal muscle. Male 4–6-month-old Doc2b+/+ and Doc2b−/− littermate mice were injected with vehicle (saline) or insulin (10 units/kg body weight) for 5 min, hindlimb muscles were excised, and detergent extracts were prepared for use in anti-Munc18c (A) or anti–syntaxin 4 (Syn4) (B) immunoprecipitation reactions. Immunoprecipitated proteins were resolved on 10% SDS-PAGE for immunodetection of Doc2b and tyrosine-phosphorylated Munc18c (using PY20 antibody), which was stripped and reblotted for total Munc18c and syntaxin 4. Optical density scanning was used to determine the average ratio of phosphotyrosine-Munc18c/Munc18c (pTyr/Munc18c: vs. basal WT = 1.00, insulin-stimulated WT = 1.30 ± 0.07, basal Doc2b−/− = 1.01 ± 0.10, and insulin-stimulated Doc2b−/− = 1.30 ± 0.15) and Munc18c/syntaxin (Munc18c/Syn4: vs. basal WT = 1.00, insulin-stimulated WT = 1.00 ± 0.28, basal Doc2b−/− = 1.40 ± 0.10, and insulin-stimulated Doc2b−/− = 1.52 ± 0.15) as indicated below the blots, as determined using three independent sets of muscle. C: Sarcolemmal/transverse tubule membrane fractions (P2) were used in anti–syntaxin 4 immunoprecipitation reactions to capture binary and ternary SNARE complexes, composed of VAMP2 and SNAP23, and syntaxin 4–Munc18c complexes, all resolved on 12% SDS-PAGE for immunoblotting. Optical density quantitation of three independent pairs of Doc2b+/+ and Doc2b−/− muscle fractions is shown in the bar graphs. *P < 0.05 compared with insulin-stimulated WT. IB, immunoblot; IP, immunoprecipitation.
DISCUSSION
In this study, we present the Doc2b+/− and Doc2b−/− mice as new in vivo models of metabolic dysregulation. The data reveal for the first time that Doc2b is a key effector for insulin-stimulated GLUT4 vesicle translocation in skeletal muscle, and for both phases of glucose-stimulated insulin secretion from pancreatic islets. Doc2b associates with Munc18c in an insulin-dependent manner, but Doc2b binding to syntaxin 4 was not detected. Notably, Munc18c–syntaxin 4 association was increased in the absence of Doc2b, suggesting that this increased association is inhibitory for the insulin-stimulated, syntaxin 4–mediated docking/fusion of GLUT4 vesicles. Strikingly, the disease phenotype of the Doc2b+/− knockout mice was almost as severe as that of the Doc2b−/−mice, suggesting that Doc2b haploinsufficiency is worthy of future investigation in diabetes susceptibility.
Mechanism(s) of Doc2b-dependent insulin granule and GLUT4 vesicle fusion events.
Unlike other secretory cell types, islet β-cells require multiple Munc18 and syntaxin isoforms, otherwise sharing SNAP25/SNAP23 and VAMP2, for two distinct phases of glucose-stimulated insulin secretion. Syntaxin 1A−/− null islets lack first-phase insulin release, whereas Munc18c and syntaxin 4 are imperative for second-phase insulin release from islets (27,31,32); Munc18-1 null islet perifusion has yet to be reported, although Munc18-1 and Munc18-2 were recently implicated in fast calcium–dependent exocytosis in electrophysiological studies (33). Demonstrating here that Doc2b is required for both phases of insulin release from primary islets, we speculate that Doc2b regulates both Munc18-1-syntaxin 1A– as well as Munc18c-syntaxin 4–dependent secretion mechanisms. The role of Doc2b in the first phase went undetected in static incubation studies using stable Doc2b short hairpin RNA clonal β-cells (20) but is consistent with its role in Munc18-1-syntaxin 1–driven exocytosis mechanisms in brain (23). The partial reduction of Doc2b in clonal β-cells may not have been sufficient to uncover the requirement for Doc2b in the first phase. Doc2b+/− islets retained >60% of first-phase function (while losing ∼75% of second phase), and total ablation of Doc2b was required to detect a >50% loss of first-phase function. Our data does confirm the late-phase deficit reported in stable Doc2b short hairpin RNA clonal β-cells (20). Strikingly, second-phase secretion was nearly abolished in Doc2b−/− islets. Although our MIN6 β-cell studies support a mechanistic regulation of Munc18c–syntaxin 4 and SNARE complexes analogous to our studies with these proteins in skeletal muscle, future β-cell studies assessing the impact of Doc2b upon Munc18-1 or -2 with syntaxin 1A are required, as well as assessment of all isoform-binding interactions in primary β-cells.
Doc2b is present in skeletal muscle transverse tubule/sarcolemmal-enriched subcellular fractions under basal conditions, and does not translocate, in contrast to GLUT4, in response to insulin stimulation. This finding is consistent with similar observations in glucose-stimulated MIN6 β-cells, yet counter to calcium-stimulated translocation seen in other cell types (17,34). Doc2b is known to require very little calcium to translocate in neurons (35). Skeletal muscle may have baseline calcium already high enough to translocate Doc2b under resting conditions. Under such conditions, Doc2b can be considered constitutively active (35), which can explain the strong effects in the Doc2b−/− mice observed here, relative to effects previously observed in brain (23). In 3T3-L1 adipocytes, Doc2b is reported to bind to syntaxin 4 only under high calcium buffer conditions (17). Therefore, we simulated those calcium conditions to investigate the physiological occurrence/relevance of this putative Doc2b–syntaxin 4 complex in primary skeletal muscle. However, regardless of calcium levels in skeletal muscle extracts, Doc2b failed to coprecipitate in anti–syntaxin 4 immunoprecipitation reactions, suggesting that such an interaction might not be a dominant factor in primary cells.
Concerning the mechanism of Doc2b actions in both insulin granule and GLUT4 vesicle exocytosis, several possibilities might be considered. One possibility is that Doc2b serves as a platform for transient interactions with Munc18 and syntaxin. According to this “switch hypothesis” model, derived from β-cell studies (16,18), Munc18c becomes tyrosine phosphorylated in response to a stimulus, dissociates from syntaxin 4, and switches its binding preference to Doc2b. Doc2b’s sequestration of Munc18c would facilitate syntaxin 4’s participation in SNARE complexes to promote vesicle fusion. Such a model is consistent with 1) the insulin-stimulated association of Doc2b with Munc18c in skeletal muscle and 2) the strong increase in Munc18c binding to syntaxin 4, concurrent with the reduced binding of VAMP2 and SNAP23 to syntaxin 4 in sarcolemmal/transverse tubule muscle membrane fractions, indicative of attenuated SNARE complex formation in the absence of Doc2b (Fig. 6C). Alternatively, Doc2b may facilitate fusion via a different or additional mechanism, by partially inserting into the PM upon calcium binding, and induce membrane deformations that assist merging vesicle and PM. This property contributes to the exceptional in vitro fusogenic properties of Doc2b relative to all other C2-domain proteins studied (23).
Although the disease phenotype of the Doc2b+/− knockout mice was almost as severe as that of the Doc2b−/− mice, interpreting the relative contribution of insulin secretory defects versus insulin resistance is complex. For example, insulin content in the serum after the acute glucose challenge trended toward a decrease (P = 0.08, n = 6), intermediate between that of the WT and Doc2b−/− mice, but did not reach statistical significance. However, since serum insulin content is not an absolute readout of insulin secretion but rather a net readout of pancreatic insulin release, hepatic insulin clearance, and insulin utilization by other tissues, use of the hyperglycemic clamp approach will be required for full assessment. Also noteworthy was that the initial drop (15–30 min) in blood glucose in the ITT in the Doc2b−/− mice was similar to that of WT mice, seemingly counter to the blunted glucose uptake into the EDL of the Doc2b−/− mice. However, the glucose uptake assay was performed in vitro using excised muscle, whereas the ITT is performed in vivo. In vivo, the insulin bolus will initiate a decrease in hepatic glucose output. Given that hepatic insulin signaling in the Doc2b−/− mice was normal, it would seem to be a likely contributor to the initial blood glucose drop.
Conclusions.
The data presented here demonstrate a key role for Doc2b in multiple exocytotic processes relevant to the maintenance of whole-body glucose homeostasis, including insulin secretion and peripheral glucose clearance. We propose that Doc2b engages in stimulus-dependent association with Munc18c in skeletal muscle similar to that in β-cells; this implicates the mechanisms to be highly conserved, albeit the stimuli are cell-type specific. Furthermore, our data demonstrating the need for Doc2b in first-phase insulin release suggest that it may also participate as a scaffolding platform for Munc18-1 binding in the β-cell. Novel reagents based upon Doc2b may carry promise as dual insulin-sensitizing/insulin secretion enhancement approaches to combating a combinatorial disease like type 2 diabetes.
ACKNOWLEDGMENTS
This study was supported by grants from the National Institutes of Health (DK-067912 and DK-076614 to D.C.T. and F32-DK-094488 to S.M.Y.), the American Heart Association (10PRE3040010 to M.A.K.), and the Indiana University School of Medicine Showalter Foundation (to E.O.).
J.T.B. is an employee of Eli Lilly and Company. No other potential conflicts of interest relevant to this article were reported.
L.R., E.O., S.M.Y., and M.A.K. researched data, contributed to discussion, and reviewed and edited the manuscript. J.T.B. performed the glucose transport assay and contributed to discussion. A.J.G. and M.V. provided the Doc2b−/− mice, reagents, and protocols and edited the manuscript. D.C.T. contributed to discussion and wrote, reviewed, and edited the manuscript. D.C.T. is the guarantor of this work and, as such, had full access to all the data in the study and takes responsibility for the integrity of the data and the accuracy of the data analysis.
Parts of this study were presented at the 71st Scientific Sessions of the American Diabetes Association, San Diego, California, 24–28 June 2011.
The Vanderbilt Mouse Phenotyping Core Facility quantified measurements of serum NEFA.
Footnotes
This article contains Supplementary Data online at http://diabetes.diabetesjournals.org/lookup/suppl/doi:10.2337/db11-1525/-/DC1.
REFERENCES
- 1.Seino S, Shibasaki T, Minami K. Dynamics of insulin secretion and the clinical implications for obesity and diabetes. J Clin Invest 2011;121:2118–2125 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2.Jewell JL, Oh E, Thurmond DC. Exocytosis mechanisms underlying insulin release and glucose uptake: conserved roles for Munc18c and syntaxin 4. Am J Physiol Regul Integr Comp Physiol 2010;298:R517–R531 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.Hoffman NJ, Elmendorf JS. Signaling, cytoskeletal and membrane mechanisms regulating GLUT4 exocytosis. Trends Endocrinol Metab 2011;22:110–116 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Foley K, Boguslavsky S, Klip A. Endocytosis, recycling, and regulated exocytosis of glucose transporter 4. Biochemistry 2011;50:3048–3061 [DOI] [PubMed] [Google Scholar]
- 5.Watson RT, Pessin JE. GLUT4 translocation: the last 200 nanometers. Cell Signal 2007;19:2209–2217 [DOI] [PubMed] [Google Scholar]
- 6.Ostenson CG, Gaisano H, Sheu L, Tibell A, Bartfai T. Impaired gene and protein expression of exocytotic soluble N-ethylmaleimide attachment protein receptor complex proteins in pancreatic islets of type 2 diabetic patients. Diabetes 2006;55:435–440 [DOI] [PubMed] [Google Scholar]
- 7.Bergman BC, Cornier MA, Horton TJ, Bessesen DH, Eckel RH. Skeletal muscle munc18c and syntaxin 4 in human obesity. Nutr Metab (Lond) 2008;5:21. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Keller MP, Choi Y, Wang P, et al. A gene expression network model of type 2 diabetes links cell cycle regulation in islets with diabetes susceptibility. Genome Res 2008;18:706–716 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Yechoor VK, Patti ME, Ueki K, et al. Distinct pathways of insulin-regulated versus diabetes-regulated gene expression: an in vivo analysis in MIRKO mice. Proc Natl Acad Sci USA 2004;101:16525–16530 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Toonen RF, Verhage M. Munc18-1 in secretion: lonely Munc joins SNARE team and takes control. Trends Neurosci 2007;30:564–572 [DOI] [PubMed] [Google Scholar]
- 11.Lang T, Jahn R. Core proteins of the secretory machinery. In Handbook of Experimental Pharmacology 184: Pharmacology of Neurotransmitter Release. Südhof TC, Starke K, Eds. Heidelberg, Germany, Springer-Verlag, 2008, p. 107–127 [DOI] [PubMed] [Google Scholar]
- 12.Misura KM, Scheller RH, Weis WI. Three-dimensional structure of the neuronal-Sec1-syntaxin 1a complex. Nature 2000;404:355–362 [DOI] [PubMed] [Google Scholar]
- 13.Jewell JL, Oh E, Ramalingam L, et al. Munc18c phosphorylation by the insulin receptor links cell signaling directly to SNARE exocytosis. J Cell Biol 2011;193:185–199 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Oh E, Thurmond DC. The stimulus-induced tyrosine phosphorylation of Munc18c facilitates vesicle exocytosis. J Biol Chem 2006;281:17624–17634 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Umahara M, Okada S, Yamada E, et al. Tyrosine phosphorylation of Munc18c regulates platelet-derived growth factor-stimulated glucose transporter 4 translocation in 3T3L1 adipocytes. Endocrinology 2008;149:40–49 [DOI] [PubMed] [Google Scholar]
- 16.Jewell JL, Oh E, Bennett SM, Meroueh SO, Thurmond DC. The tyrosine phosphorylation of Munc18c induces a switch in binding specificity from syntaxin 4 to Doc2beta. J Biol Chem 2008;283:21734–21746 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Fukuda N, Emoto M, Nakamori Y, et al. DOC2B: a novel syntaxin-4 binding protein mediating insulin-regulated GLUT4 vesicle fusion in adipocytes. Diabetes 2009;58:377–384 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Ke B, Oh E, Thurmond DC. Doc2beta is a novel Munc18c-interacting partner and positive effector of syntaxin 4-mediated exocytosis. J Biol Chem 2007;282:21786–21797 [DOI] [PubMed] [Google Scholar]
- 19.Verhage M, de Vries KJ, Røshol H, Burbach JP, Gispen WH, Südhof TC. DOC2 proteins in rat brain: complementary distribution and proposed function as vesicular adapter proteins in early stages of secretion. Neuron 1997;18:453–461 [DOI] [PubMed] [Google Scholar]
- 20.Miyazaki M, Emoto M, Fukuda N, et al. DOC2b is a SNARE regulator of glucose-stimulated delayed insulin secretion. Biochem Biophys Res Commun 2009;384:461–465 [DOI] [PubMed] [Google Scholar]
- 21.Thurmond DC, Ceresa BP, Okada S, Elmendorf JS, Coker K, Pessin JE. Regulation of insulin-stimulated GLUT4 translocation by Munc18c in 3T3L1 adipocytes. J Biol Chem 1998;273:33876–33883 [DOI] [PubMed] [Google Scholar]
- 22.Wiseman DA, Kalwat MA, Thurmond DC. Stimulus-induced S-nitrosylation of syntaxin 4 impacts insulin granule exocytosis. J Biol Chem 2011;286:16344–16354 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Groffen AJ, Martens S, Díez Arazola R, et al. Doc2b is a high-affinity Ca2+ sensor for spontaneous neurotransmitter release. Science 2010;327:1614–1618 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Spurlin BA, Park SY, Nevins AK, Kim JK, Thurmond DC. Syntaxin 4 transgenic mice exhibit enhanced insulin-mediated glucose uptake in skeletal muscle. Diabetes 2004;53:2223–2231 [DOI] [PubMed] [Google Scholar]
- 25.Zhou M, Sevilla L, Vallega G, et al. Insulin-dependent protein trafficking in skeletal muscle cells. Am J Physiol 1998;275:E187–E196 [DOI] [PubMed] [Google Scholar]
- 26.Brozinick JT, Jr, McCoid SC, Reynolds TH, et al. GLUT4 overexpression in db/db mice dose-dependently ameliorates diabetes but is not a lifelong cure. Diabetes 2001;50:593–600 [DOI] [PubMed] [Google Scholar]
- 27.Oh E, Thurmond DC. Munc18c depletion selectively impairs the sustained phase of insulin release. Diabetes 2009;58:1165–1174 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Foster LJ, Klip A. Mechanism and regulation of GLUT-4 vesicle fusion in muscle and fat cells. Am J Physiol Cell Physiol 2000;279:C877–C890 [DOI] [PubMed] [Google Scholar]
- 29.Yang C, Coker KJ, Kim JK, et al. Syntaxin 4 heterozygous knockout mice develop muscle insulin resistance. J Clin Invest 2001;107:1311–1318 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Oh E, Spurlin BA, Pessin JE, Thurmond DC. Munc18c heterozygous knockout mice display increased susceptibility for severe glucose intolerance. Diabetes 2005;54:638–647 [DOI] [PubMed] [Google Scholar]
- 31.Spurlin BA, Thurmond DC. Syntaxin 4 facilitates biphasic glucose-stimulated insulin secretion from pancreatic beta-cells. Mol Endocrinol 2006;20:183–193 [DOI] [PubMed] [Google Scholar]
- 32.Ohara-Imaizumi M, Fujiwara T, Nakamichi Y, et al. Imaging analysis reveals mechanistic differences between first- and second-phase insulin exocytosis. J Cell Biol 2007;177:695–705 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.Mandic SA, Skelin M, Johansson JU, Rupnik MS, Berggren PO, Bark C. Munc18-1 and Munc18-2 proteins modulate beta-cell Ca2+ sensitivity and kinetics of insulin exocytosis differently. J Biol Chem 2011;286:28026–28040 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.Groffen AJ, Brian EC, Dudok JJ, Kampmeijer J, Toonen RF, Verhage M. Ca(2+)-induced recruitment of the secretory vesicle protein DOC2B to the target membrane. J Biol Chem 2004;279:23740–23747 [DOI] [PubMed] [Google Scholar]
- 35.Groffen AJ, Friedrich R, Brian EC, Ashery U, Verhage M. DOC2A and DOC2B are sensors for neuronal activity with unique calcium-dependent and kinetic properties. J Neurochem 2006;97:818–833 [DOI] [PubMed] [Google Scholar]