

Abstract
Neuroadrenergic function in type 2 diabetic (T2D) patients without neuropathy is poorly characterized. We therefore compared sympathetic nervous system activity at rest and during an oral glucose tolerance test in obese metabolic syndrome (MetS) subjects classified as glucose intolerant (impaired glucose tolerance [IGT]; n = 17) or treatment-naive T2D (n = 17). Untreated subjects, matched for age (mean 59 ± 1 year), sex, BMI (32.4 ± 0.6 kg/m2), and family history of diabetes were studied. We measured resting muscle sympathetic nerve activity (MSNA) by microneurography, whole-body norepinephrine kinetics by isotope dilution, insulin sensitivity by euglycemic-hyperinsulinemic clamp (steady-state glucose utilization adjusted for fat-free mass and steady-state insulin concentration [M/I]), and MetS components. T2D subjects had higher resting MSNA burst incidence (67 ± 4 versus 55 ± 3 bursts per 100 heartbeats; P = 0.05) and arterial norepinephrine levels (264 ± 33 versus 167 ± 16 pg/mL; P = 0.02), lower plasma norepinephrine clearance (by 17%; P = 0.03), and reduced neuronal reuptake compared with IGT subjects (by 46%; P = 0.04). Moreover, norepinephrine spillover responses to glucose ingestion were blunted in T2D subjects. The M/I value independently predicted whole-body norepinephrine spillover (r = −0.47; P = 0.008), whereas fasting insulin level related to neuronal norepinephrine reuptake (r = −0.35, P = 0.047). These findings demonstrate that progression to T2D is associated with increased central sympathetic drive, blunted sympathetic responsiveness, and altered norepinephrine disposition.
Population aging, urbanization, and the effects of modernization on lifestyle factors such as physical activity and dietary consumption have fueled the escalating prevalence rates of obesity, type 2 diabetes (T2D), and related comorbidities worldwide (1,2). In Australia alone, 60% of the adult population is overweight or obese, and 7.6% has diabetes mellitus, which is projected to rise to 11.4% by 2025 (3,4). The close association of T2D with cardiovascular disease led to the hypothesis that the two arise from a common antecedent, now codified as the metabolic syndrome (MetS) (5,6). An understanding of the pathophysiological mechanisms by which obesity interacts with other factors to produce MetS and diabetes is imperative for tailoring of preventive and therapeutic strategies.
Autonomic nervous system dysfunction, comprising elevated sympathetic neural activity and baroreflex impairment, is a hallmark of MetS obesity (7,8) and relates to insulin resistance via a positive-feedback mechanism (7,9,10). The notion that sympathetic nerve hyperactivity precedes insulin resistance and the onset of diabetes is gaining support from long-term prospective cohort studies (11–14) and investigations in offspring of patients with T2D (15,16). Baseline plasma norepinephrine levels and the norepinephrine response to cold pressor test are both positively associated with future development of insulin resistance in healthy young men (11,12). The Atherosclerosis Risk in Communities study showed significant positive associations between cardiac indices of autonomic function (high resting heart rate, reduced low frequency power heart rate variability) and incident diabetes, even after adjustment for physical activity and BMI (13). Neural vasoconstriction and enhanced adipose tissue lipolysis are two potential mechanisms whereby sympathetic overactivity may predispose to reduced skeletal muscle glucose utilization and insulin resistance (17–19). Reciprocally, there is ample evidence that hyperinsulinemia stimulates central sympathetic outflow (20), albeit the sympathetic response to exogenous and endogenous insulin is blunted in obese insulin resistant subjects (21,22). This may be a consequence of adrenoceptor downregulation and/or impairments in central insulin action (brain insulin uptake, signaling, or neuronal responsiveness) or the peripheral vasodilatory response to insulin (21–23). In the postprandial state, this translates to an impaired ability of insulin to stimulate diet-induced thermogenesis (23,24).
There is however a paucity of clinical data concerning sympathetic nervous system (SNS) activity along the diabetes continuum, particularly in the early stages of the disease, before the onset of peripheral neuropathy. Using the technique of microneurography, Huggett et al. (25) demonstrated elevated muscle sympathetic nerve activity (MSNA) in T2D patients with and without hypertension compared with hypertensive and normotensive nondiabetic control subjects. One limitation of this study was that participants were on a number of antihypertensive and oral hypoglycemic treatments that may have modified sympathetic measurements. Tack et al. (26) found no difference in basal norepinephrine kinetics in lean T2D versus matched control subjects. The primary aim of the present investigation was to compare SNS function at rest and during a standard oral glucose tolerance test (OGTT) in a group of untreated obese MetS subjects classified using World Health Organization criteria (27) as having impaired glucose tolerance (IGT) or treatment-naive T2D and matched for sex, age, BMI, and family history of diabetes. The secondary aim was to examine interrelationships among sympathetic activity, metabolic (including euglycemic clamp–derived measures of insulin sensitivity), anthropometric, and cardiovascular parameters. We hypothesized that the progression from IGT to T2D would be characterized by elevated central sympathetic neural outflow and blunted nutritional sympathetic responsiveness.
RESEARCH DESIGN AND METHODS
Study participants.
The data presented in this study constitute baseline measurements obtained in Caucasian subjects (n = 34), mean age 59 ± 1 years, and BMI 32.4 kg/m2 participating in an ongoing, randomized, controlled trial of pharmacological management of MetS (NCT00408850, clinicaltrials.gov). They were recruited from the general population by newspaper advertisement. To be eligible, they had to fulfill MetS diagnostic criteria, having ≥3 abnormal findings as per the recently harmonized definition (6). Elevated waist circumference was defined as ≥102 cm in men and ≥88 cm in women. Subjects were classified as having IGT (fasting plasma glucose <7.0 mmol/L and 2-h plasma glucose >7.8 and <11.1 mmol/L) or T2D (fasting plasma glucose >7.0 mmol/L or 2-h glucose >11.1 mmol/L) following a 75-g OGTT (Glucaid; Fronine) (27). All participants were nonsmokers and not taking any medications or dietary supplements. They were screened by history and physical and laboratory examinations. Exclusion criteria included secondary hypertension, peripheral or autonomic neuropathy, cardiovascular, cerebrovascular, renal, liver, or thyroid disease. Supine resting blood pressure was recorded by Dinamap monitor (Model 1846SX; Critikon, Tampa, FL) as the average of five readings after 5-min supine rest. Dietary intake was evaluated by 4-day prospective diet records and analyzed using Australian Food Composition Tables (FoodWorks Professional Version 3.02; Xyris Software, Highgate Hill, Australia). Physical activity was quantified by prospective exercise records and 7-day pedometry. The study protocol was approved by the Alfred Hospital Ethics Committee, and all participants provided informed written consent.
Clinical investigations.
Subjects attended at 0800 h on two separate mornings within a 7-day period, having fasted for 12 h and abstained from caffeine and alcohol for 18 and 36 h, respectively. They were instructed not to exercise on the day prior to investigations to eliminate acute effects of exercise. Anthropometric measurements comprised body weight, BMI, waist circumference, and waist/hip ratio. Body composition was measured by dual-energy X-ray absorptiometry (GE-LUNAR Prodigy Advance PA+130510; GE Medical Systems, Lunar, Madison, WI) scans and included a post hoc assessment of abdominal fat mass in the region from L1–L4 (28). Experiments were performed in a quiet room (ambient temperature 22°C) with subjects lying in a supine position. They voided prior to commencement.
Euglycemic clamp.
Insulin sensitivity was evaluated by the euglycemichyperinsulinemic technique (29). The clamp was initiated by an intravenous bolus injection of insulin (9 mU/kg; Actrapid 100 IU/mL; Novo Nordisk, Gentofte, Denmark), followed by a constant infusion rate of 40 mU ⋅ m2 ⋅ min−1. Blood glucose was clamped at 5.0 mmol/L by the infusion of 25% glucose (Baxter, Toongabbie, Australia) at varying rates according to blood glucose concentrations, measured every 5-min using an ABL8XX glucose autoanalyzer (Radiometer, Copenhagen, Denmark). The mean glucose infusion rate for the period 90–120 min was used to calculate whole-body glucose uptake (steady-state glucose utilization adjusted for fat-free mass [M value], mg/kg fat-free mass/min), after space correction for fluctuation in glucose concentration (29). Steady-state plasma insulin was measured at 90, 100, 110, and 120 min and averaged to calculate the steady-state glucose utilization adjusted for fat-free mass and steady-state insulin concentration (M/I) value. Fasting and steady-state plasma C-peptide were measured to ascertain suppression of endogenous insulin secretion.
Sympathetic nervous system activity
Norepinephrine kinetics.
Whole-body SNS activity was assessed using the radioisotope dilution method (30). This technique involves the intravenous infusion of [3H]norepinephrine and measurement of norepinephrine-specific activity and endogenous norepinephrine in arterial blood (sampled from the brachial artery) under steady-state conditions. The method provides a simultaneous estimate of the rate at which norepinephrine released from sympathetic nerve endings enters the plasma compartment (norepinephrine spillover) and norepinephrine plasma clearance. After a priming bolus of 1.94 μCi of 1-[ring-2,5,6-3H]norepinephrine (PerkinElmer, Waltham, MA; specific activity, 10–30 μCi/mmol), an infusion was commenced at 0.094 μCi ⋅ m2 ⋅ min−1. Plasma norepinephrine clearance and spillover rates were calculated as:
 |
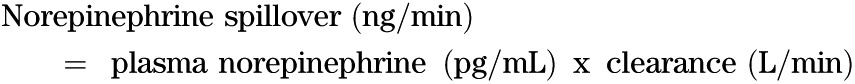 |
We estimated the rate of neuronal uptake of norepinephrine by measurement of its primary intraneuronal metabolite, 3,4-dihydroxyphenylglycol (DHPG). Endogenous DHPG to norepinephrine and steady-state [3H]DHPG to [3H]norepinephrine ratios were calculated as an index of norepinephrine transporter (NET) function (30,31). All measurements were performed under fasting conditions and during a standard OGTT. Fasting blood was obtained for measurement of nonesterified fatty acids (NEFA), cortisol, high-sensitivity C-reactive protein (hs-CRP), leptin, and plasma renin activity (PRA). Catecholamines, glucose, and insulin concentrations were measured at 0, 30, 60, 90, and 120 min during the OGTT. Pancreatic β-cell function was evaluated by homeostasis model assessment of β-cell function (HOMA-β) and from areas under the plasma concentration versus area under the curve of insulin to glucose between time 0 and 30 min during OGTT (insulin AUC0–30/glucose AUC0–30) (32). Subjects provided a 24-h urine specimen on the day of attendance to quantify sodium, creatinine, and albumin excretion.
Microneurography.
Resting multiunit MSNA activity was measured via a tungsten microelectrode inserted into a muscle nerve fascicle of the right peroneal nerve at the fibular head. Once an acceptable nerve-recording site was obtained via visual and acoustic identification of spontaneous sympathetic bursts, resting measurements were recorded over 15 min. The nerve signal was amplified (×50,000), filtered (bandpass, 700–2000 Hz), and integrated. Continuous electrocardiographic and MSNA recordings were digitized with a sampling frequency of 1,000 Hz (PowerLab recording system, model ML785/8SP; AD Instruments). MSNA was manually analyzed and expressed as burst frequency (bursts per minute) and burst incidence (bursts per 100 heartbeats), averaged over 15 min. Burst strength was analyzed by defining the amplitude of the largest burst during the analyzed period as 100, and all other bursts as a percentage of the largest one. Median burst amplitude (percentage) was then calculated for each subject. Total MSNA was calculated by multiplying the mean burst amplitude per min by burst rate, expressed as units per minute and units per 100 heartbeats.
Calf blood flow.
In order to quantify the vasodilatory response to endogenous insulin during the OGTT, calf blood flow measurements were obtained in the left leg by venous occlusion plethysmography (D.E. Hokansen, Bellevue, WA) as previously described (22). Strain gauges were attached around the thickest part of the calf, and the measured leg was supported 10 cm above heart level to empty the venous system. Twelve consecutive readings were averaged at each time point and expressed as milliliters per minute per 100 g of tissue. The blood pressure response to glucose ingestion was monitored by Dinamap and direct intra-arterial recordings.
Spontaneous cardiac baroreflex function.
Baroreflex sensitivity at rest and during OGTT was assessed by the sequence method of Parati and colleagues (22,33). The slope between cardiac interval and systolic blood pressure was calculated for each validated sequence and averaged over 15 min for the resting recording and over 5-min for each time-point during OGTT.
Laboratory analyses.
Plasma glucose and lipid profile were quantified by automated enzymatic methods (Architect C18000 analyzer; Abbott Laboratories), hs-CRP by immunoturbidimetric assay, insulin, leptin, and PRA by radioimmuno assay (Linco Research; REN-CT2, Cis Bio International), cortisol by an in-house extracted radioimmunoassay, and NEFA by enzymatic colorimetry. Plasma norepinephrine and DHPG were determined by high-performance liquid chromatography with electrochemical detection, following extraction from plasma samples using alumina adsorption. [3H]Norepinephrine and [3H]DHPG in the effluent were assayed by liquid scintillation chromatography, and the concentrations were corrected for loss during extraction using recoveries of internal standard. Intra-assay coefficients of variation in our laboratory are 1.3% for norepinephrine and 2.3% for [3H]norepinephrine; interassay coefficients of variation are 3.8 and 4.5%, respectively. Corresponding values for DHPG and [3H]DHPG are 2.0, 6, 8, and 14%.
Statistical analyses.
Data are presented as mean ± SEM. Statistical analysis was performed using SigmaStat Version 3.5 (Systat Software, Point Richmond, CA). Study parameters were compared between groups by unpaired t test and χ2 test for proportions. Metabolic and cardiovascular variables during OGTT were compared by two-way repeat-measures ANOVA and the Holm-Sidak test for multiple post hoc comparisons. Areas under the plasma concentration time curve (AUC) during OGTT were calculated for glucose and insulin by the trapezoidal rule. For the purposes of subgroup analyses, T2D subjects were stratified as insulin hyposecretors (insulin AUC0–120 ≤8,000 mU/L ∙ min−1) or hypersecretors (insulin AUC0–120 >8,000 mU/L ∙ min−1). The distribution of variables was examined by the Kolmogorov-Smirnov test, and nonparametric data were log-transformed. Associations between SNS activity and anthropometric, metabolic, and cardiovascular variables were assessed using linear regression analysis (Pearson and Spearman rank correlations). Age-adjusted forward stepwise regression analyses were carried out with those univariate correlations in which P < 0.05. A two-tailed P value <0.05 was regarded as statistically significant.
RESULTS
Clinical characteristics of the study groups.
Clinical characteristics of the study groups were comparable with regards to sex distribution, age, BMI, waist circumference, lipid profile, and 24-h urinary sodium excretion (Table 1). Dietary parameters and exercise habits did not differ between groups. Energy intake averaged 2,220 kcal/day overall and comprised 35% fat (14% saturated), 41% carbohydrate, 19% protein, and 4% alcohol. Median level of physical activity was 2 h per week (interquartile range 0–4 h) based on pedometry and exercise records. Consistent with the process of group stratification, T2D subjects had higher fasting glucose, 2-h glucose (Fig. 1A), and glucose AUC0–120 during the OGTT (all P < 0.001; Table 2). Indices of pancreatic β-cell function were lower in the T2D versus the IGT group (both P < 0.05). Clamp-derived measures of glucose utilization (M and M/I values) were on average 13 and 12% lower in the T2D subjects, respectively, but these differences were not statistically significant.
TABLE 1.
Demographic, anthropometric, metabolic, and cardiovascular variables of study participants
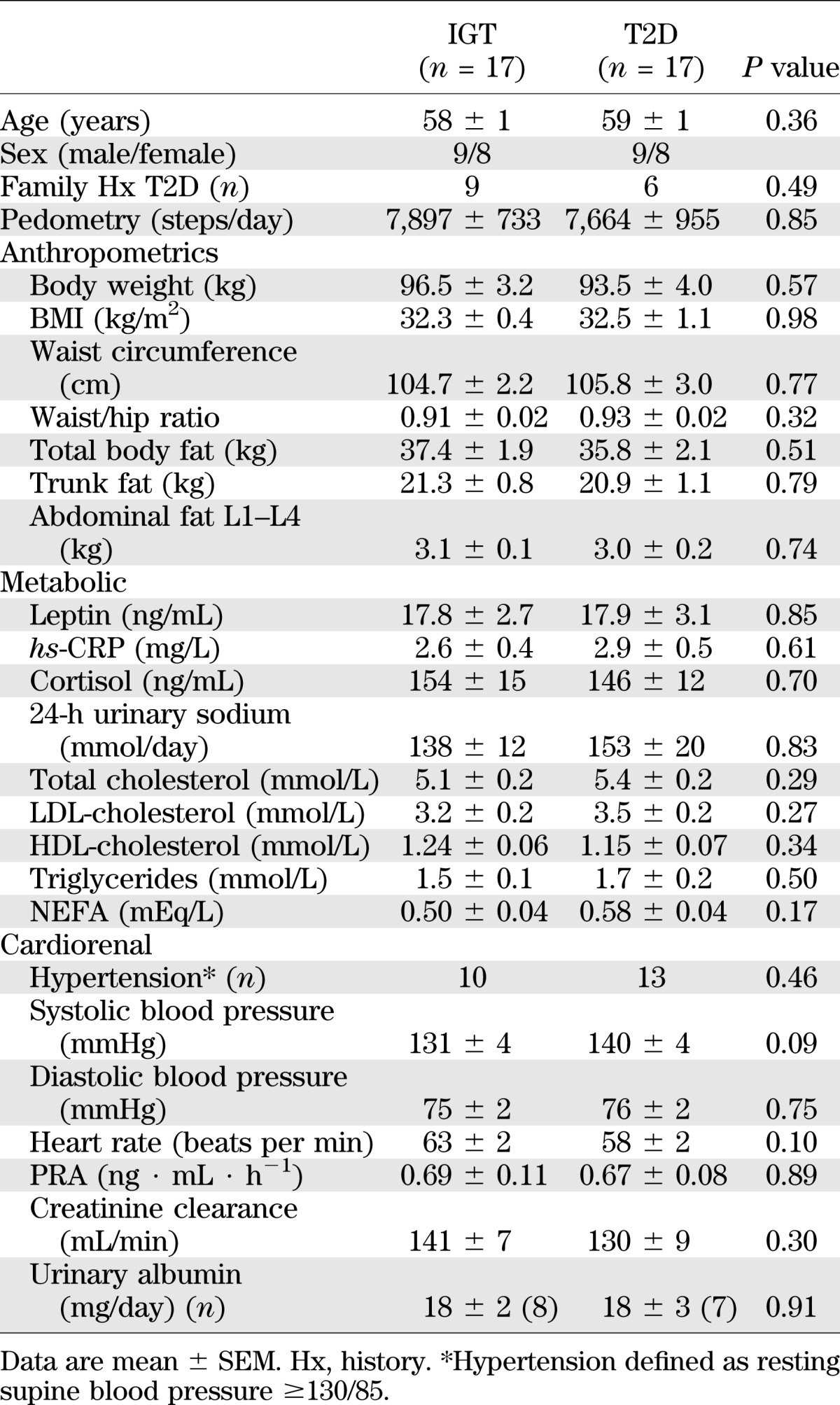
FIG. 1.

Scatter plots of glucose tolerance and sympathetic nervous function in subjects with IGT (n = 17) and treatment-naive T2D (n = 17). A: Two-hour plasma glucose concentration following 75-g oral glucose load. B: MSNA burst incidence. C: Arterial norepinephrine (NE) concentration. D: Whole-body NE spillover rate. E: NE plasma clearance. F: Neuronal uptake of endogenous NE estimated as the ratio of plasma tritiated DHPG to NE concentration. Lines represent group mean values. hb, heartbeat. *P ≤ 0.05 and ***P < 0.001 vs. IGT group.
TABLE 2.
Glucose tolerance, insulin sensitivity, and pancreatic β-cell function

Within the T2D group, nine subjects were classified as insulin hyposecretors and eight as hypersecretors (insulin AUC0–120 averaged 6,386 ± 288 and 12,310 ± 1,405 mU/L ∙ min−1, respectively). Hypo- and hypersecretors had similar age, body weight, BMI, waist circumference, blood pressure, and family history of diabetes (n = 3 positive per group). Hypersecretors were more insulin-sensitive as per clamp M value (10.4 ± 1.2 versus 6.9 ± 0.8 mg ∙ kg FFM ∙ min−1; P = 0.03)
Resting SNS activity.
Successful microneurographic recordings were obtained in 31 subjects (15 T2D and 16 IGT). Analysis revealed higher MSNA burst incidence in the T2D compared with the IGT group (67 ± 4 versus 55 ± 3 bursts/100 heartbeats; P = 0.05; Fig. 1B). Burst frequency averaged 37 ± 3 versus 32 ± 2 bursts/min (P = 0.18) and median burst amplitude 50 ± 3 versus 52 ± 2% (P = 0.57), respectively. Within the T2D group, insulin hypersecretors had markedly augmented sympathetic drive as quantified by burst incidence, median burst amplitude, and total MSNA versus the hyposecretors (all P < 0.05; Fig. 2).
FIG. 2.

Measures of MSNA in treatment-naive T2D subjects, subclassified as insulin hypersecretors (Hyper; n = 6) or hyposecretors (Hypo; n = 9). A: Insulin AUC during OGTT. B: MSNA burst frequency (P = 0.06). C: MSNA burst incidence. D: Median burst amplitude. E: Total MSNA burst frequency. F: Total MSNA burst incidence. hb, heartbeat. *P < 0.05, **P < 0.01, and ***P < 0.001 vs. Hyper group.
Norepinephrine kinetics data are presented in Fig. 1 and show elevated arterial noradrenaline concentrations (by 58%; P = 0.02) and significantly reduced plasma norepinephrine clearance in the T2D group (by 17%; P = 0.03). Calculated whole-body norepinephrine spillover rates averaged 484 ± 63 and 398 ± 51 ng/min in the T2D and IGT groups, respectively (P = 0.27). Norepinephrine spillover rate was 28% higher in T2D insulin hypersecretors compared with hyposecretors, but this was not statistically significant. The rate of uptake of tritiated noradrenaline into sympathetic nerves was significantly reduced in the T2D group, as indicated by a lower [3H]DHPG to [3H]norepinephrine ratio (P = 0.04; Fig. 1F). Spontaneous cardiac baroreflex sensitivity was similar in the two groups, averaging 15.0 ± 1.8 and 15.8 ± 2.1 ms/mmHg, respectively.
Correlates of resting sympathetic activity.
Univariate correlates of sympathetic neural variables are presented in Table 3. Whole-body norepinephrine spillover was most strongly associated with clamp-derived M/I values (inverse relationship; Fig. 3A) and plasma renin activity. MSNA was positively associated with total body fat mass, plasma leptin, NEFA, and hs-CRP concentrations (all P ≤ 0.05). Plasma norepinephrine clearance was inversely related to 2-h plasma glucose during OGTT (Fig. 3B) and to fasting NEFA (both P <0.05). Endogenous DHPG to norepinephrine ratio was positively associated with measures of insulin sensitivity (M and M/I values) and inversely with measures of insulin resistance (fasting insulin, homeostasis model assessment of insulin resistance [HOMA-IR]). Tritiated DHPG to norepinephrine ratio correlated inversely with waist circumference and measures of insulin resistance and positively with HDL-cholesterol and clinic heart rate. Both endogenous and tritiated ratios were inversely related to total dietary fat intake (grams per day) (both P < 0.05).
TABLE 3.
Univariate correlates of sympathetic nervous system activity in the study cohort

FIG. 3.

Correlates of SNS activity and function. A: Insulin sensitivity assessed with glucose clamp (M/I value, mg · kg FFM · min−1 · mU/L × 100) versus whole-body norepinephrine (NE) spillover. B: Two-hour plasma glucose concentration versus NE plasma clearance rate. C and D: Indices of neuronal NE reuptake. C: Fasting plasma insulin versus endogenous plasma DHPG to NE ratio. D: Fasting plasma insulin versus plasma tritiated DHPG to tritiated NE ratio.
The inverse relationship between whole-body norepinephrine spillover and M/I values was significant in both T2D (r = −0.56; P = 0.046) and IGT groups (r = −0.54; P = 0.02). In T2D subjects, MSNA burst incidence was most strongly related to insulin AUC0–120 (r = 0.70; P = 0.004) and the insulin AUC0–30 to glucose AUC0–30 ratio (r = 0.56; P = 0.03). In the IGT group, trunk fat mass (r = 0.50; P = 0.047) and hs-CRP (r = 0.69; P = 0.003) were the only significant correlates of MSNA.
In stepwise regression analyses of pooled data (Table 4), MSNA was independently predicted by hs-CRP and NEFA, which together accounted for 33% of the variance. Whole-body norepinephrine spillover was independently predicted by clamp-derived M/I value (inverse), which accounted for 22% of the variance.
TABLE 4.
Stepwise regression analyses of sympathetic nervous system parameters

Norepinephrine kinetics during OGTT.
Norepinephrine kinetics data during the course of the OGTT are illustrated in Fig. 4. In subjects with IGT, arterial plasma norepinephrine concentration and calculated norepinephrine spillover rates increased significantly at all time points after glucose ingestion. In contrast, the sympathetic response in T2D subjects was blunted, with no statistically significant increment versus baseline. Percent change in norepinephrine spillover averaged 14 ± 10 versus 41 ± 9% at 60 min, 14 ± 6 versus 42 ± 11% at 90 min, and 15 ± 6 versus 39 ± 9% at 120 min in T2D and IGT groups, respectively (all P < 0.05). Plasma norepinephrine clearance was lower at all time points in the T2D group (group effect P = 0.005). Differences in nutritional sympathetic responsiveness could not be explained by differences in baroreflex sensitivity or the postprandial vasodilatory response to insulin, as these variables were comparable in IGT and T2D groups during the OGTT (Fig. 4E and F). Arterial systolic and diastolic blood pressures and the ECG-derived heart rate response to glucose ingestion were also similar in the two groups. In the pooled data, the maximal increase in norepinephrine spillover correlated with resting baroreflex sensitivity (r = 0.43; P = 0.016).
FIG. 4.

Neuroadrenergic and cardiovascular responses during a 75-g OGTT in subject groups with IGT (n = 17) and T2D (n = 17). A: Arterial norepinephrine concentration: time effect, P < 0.001; group effect, P = 0.036; group × time interaction, P = 0.019. B: Calculated whole-body norepinephrine (NE) spillover rate: time effect, P < 0.001. C: NE plasma clearance: group effect, P = 0.005. D: Neuronal reuptake of endogenous NE estimated as the ratio of plasma tritiated DHPG to NE concentration: time effect, P = 0.002; group effect, P = 0.055. E: Spontaneous cardiac baroreflex sensitivity: time effect, P < 0.001. F: Calf blood flow: time effect, P < 0.001. **P < 0.01 and ***P < 0.001 vs. time 0.
Within the T2D group, insulin hypersecretors vasodilated more readily in response to oral glucose ingestion, as evidenced by a significant group by time interaction (P = 0.04) for calf blood flow. However, arterial norepinephrine levels, norepinephrine spillover rate, plasma norepinephrine clearance, and the baroreflex response during OGTT did not differ significantly between groups.
DISCUSSION
In this study, we sought to compare neuroadrenergic function in a cohort of obese MetS subjects with either IGT or treatment-naive T2D in order to better understand the pathophysiological changes that accompany the transition to diabetes. Our findings demonstrate disturbed sympathetic neurobiology in T2D, characterized by augmented resting sympathetic nervous activity (MSNA, arterial noradrenaline levels) and blunted sympathetic responsiveness to oral glucose ingestion relative to subjects with IGT. Furthermore, for the first time, we have demonstrated reductions in both the plasma clearance of norepinephrine and its neuronal reuptake in T2D. Regression analyses revealed that insulin resistance (M/I value) was the key independent driver of whole-body norepinephrine spillover, whereas elevated fasting insulin predicted decreased neuronal norepinephrine reuptake. Our data were obtained in subjects who were untreated and matched for key factors that may affect sympathetic activity (age, sex, BMI, family history of diabetes, dietary sodium intake, and exercise level). These findings have broad clinical implications for cardiovascular risk, target organ damage, and postprandial thermogenesis.
Our results concerning resting sympathetic drive concur with those published previously by Huggett et al. (25,34) showing higher multiunit and single-unit MSNA in T2D compared with overweight matched control subjects. Within our T2D group, hyperinsulinemic subjects had markedly augmented central sympathetic outflow, evidenced by a 42% greater MSNA burst incidence, 26% higher median burst amplitude, and 28% higher norepinephrine spillover rate compared with hyposecretors in the later stage of disease. Previously published studies in middle-aged lean and healthy obese populations have reported MSNA burst incidence in the range 37–43 bursts per 100 heartbeats (7,34), highlighting that both the IGT and T2D groups had substantially higher MSNA compared with metabolically healthy counterparts. Clamp-derived measures of insulin resistance did not correlate with MSNA, whereas M/I was a strong and independent inverse predictor of whole-body norepinephrine spillover overall and in both subgroups. The lack of a relationship between MSNA and glucose utilization in the current study is somewhat surprising because skeletal muscle is the key site of glucose uptake during euglycemic hyperinsulinemia, and experimental maneuvers that increase SNS activity (reflex sympathetic activation or norepinephrine infusion) are known to antagonize insulin-stimulated muscle glucose disposal (18,35). Within the T2D group, MSNA was, however, strongly associated with insulin hypersecretion. It is possible that other pathophysiological factors such as activation of the renin-angiotensin system, oxidative stress, and prevailing inflammatory status may have modulated the interrelationship of MSNA and insulin resistance (36).
As previously demonstrated by our group and others (21–23), insulin resistance in obese subjects is associated with blunted sympathetic neural responsiveness to physiological hyperinsulinemia and may involve both central and peripheral mechanisms. The sympathetic response to insulin is mediated via hypothalamic melanocortin, phosphatidylinositol 3-kinase, and mitogen-activated protein kinase pathways (37,38). Animal studies and limited data in humans demonstrate reduced brain insulin uptake across the blood–brain barrier (39–41) and impaired brain insulin signaling (42,43) in the insulin-resistant state and in T2D (43). One viewpoint has been that preserved sensitivity to the sympathoexcitatory effects of insulin causes the sustained sympathetic activation found in chronically hyperinsulinemic obese subjects and precludes the demonstration of further enhancement in sympathetic activity during acute increases in plasma insulin (i.e., the notion of a ceiling effect) (21). With diminished pancreatic reserve, and lower insulin levels, there is marked attenuation in resting MSNA and nutritional sympathetic responsiveness as reported in insulin-dependent patients with diabetes without autonomic neuropathy (44,45). This is consistent with our subgroup analyses of T2D subjects. Endothelial dysfunction is a feature of insulin-resistant states and T2D and may contribute to blunted baroreflex-mediated activation of the SNS postprandially (46). In our data set, basal cardiac baroreflex sensitivity was positively associated with the maximal increment in norepinephrine spillover postglucose, albeit we did not demonstrate differences in the calf blood flow or baroreflex response to glucose between T2D and IGT groups. Hypothalamic insulin signaling is important in initiating a catabolic response including reduction in food intake and increase in energy expenditure, with approximately one-third of the thermic effect of food being accounted for by the meal-induced increment in SNS activity (facultative thermogenesis). A blunted sympathetic response would therefore favor maintenance of the obese state (47) and may also contribute to the postprandial hypotension commonly documented in patients with diabetes mellitus (48).
Approximately 10% of norepinephrine released by sympathetic nerves escapes the synaptic cleft and spills over into plasma (30). The other 90% undergoes neuronal reuptake via NET (uptake-1) and is repackaged in storage vesicles or transformed by monoamine oxidase to its deaminated metabolite, DHPG. In the current study, we report alterations in norepinephrine disposition in T2D at two levels: decreased neuronal reuptake (by 46%) and decreased plasma clearance rates (by 17%). Neuronal reuptake of norepinephrine, as estimated by the [3H]DHPG to [3H]NA ratio, was independently predicted by two metabolic factors: fasting insulin (inverse) and HDL-cholesterol levels. There is a growing body of evidence that insulin modulates NET and hence synaptic norepinephrine levels. In rodents, acute and chronic insulin treatment results in significantly decreased brain NET expression (49,50). Cardiac scintigraphic studies using 123I-metaiodobenzylguanidine, a tracer taken up by sympathetic nerves via NET, show reduced cardiac metaiodobenzylguanidine uptake in experimental models of diabetes, which correlated with decreased NET density (51). Moreover, lower activity of monoamine oxidase A has also been demonstrated in vascular tissues of T2D patients compared with nondiabetic tissues (52).
Plasma norepinephrine clearance occurs via both neuronal and extraneuronal uptake (uptake-2), with the liver, kidneys, lungs, and muscle being quantitatively the most important sites. Factors such as organ perfusion, determined by cardiac output and regional blood flow, affinity for extraneuronal organic cation transporters such as monoamine transporter, as well as NET function, may importantly influence plasma clearance of norepinephrine (30). In our study, 2-h plasma glucose level was an inverse, independent predictor of total body plasma clearance, suggesting that glycemic control and/or insulin resistance impact significantly on the removal of norepinephrine from the circulation. Greater availability of norepinephrine within the synaptic cleft is clinically relevant to obesity-related target organ damage including cardiac function and structure, endothelial dysfunction, hypertension, and renal impairment (53) and also favors the development/maintenance of an insulin-resistant state (11,18,35).
The limitations of our study merit emphasis. First, we only investigated subjects with IGT and T2D and had no normal glucose tolerant or lean control subjects. Second, we did not measure HbA1c, and classification of subjects was based on a single OGTT. Third, our sample size was relatively small, particularly for subgroup analyses within the T2D group. Finally, arterial blood sampling does not permit differentiation of norepinephrine spillover in the systemic and pulmonary circulations or at the level of individual organs (30). In conclusion, our findings show that obese patients who are in the early phase of T2D have elevated central sympathetic outflow, altered norepinephrine disposition, and blunted sympathetic responsiveness to carbohydrate load, relative to obese subjects with IGT. Insulin resistance and fasting insulin were key modulators of neuroadrenergic function. Further research is warranted to investigate the differences between T2D subjects who are insulin hyper- and hyposecretors and whether enhancement of insulin sensitivity through weight loss or drug treatment improves sympathetic function within such cohorts.
ACKNOWLEDGMENTS
M.D.E. and M.P.S. have been investigators in studies supported by Medtronic and have received speaker’s fees from pharmaceutical companies. G.W.L. has acted as a consultant for Medtronic and has received honoraria from Medtronic, Pfizer, and Wyeth Pharmaceuticals for presentations. M.D.E. serves on scientific advisory boards of Abbott (formerly Solvay) Pharmaceuticals and Medtronic (formerly Ardian). M.P.S. serves on scientific advisory boards for Abbott (formerly Solvay) Pharmaceuticals, Novartis Pharmaceuticals, and Medtronic and has received honoraria from Abbott, Servier, Novartis, Boehringer Ingelheim, and Medtronic for presentations. The authors’ laboratory currently receives research funding from Medtronic (formerly Ardian), Abbott (formerly Solvay) Pharmaceuticals, Allergan, and Servier. This study was funded by a Heart Foundation Grant-in-Aid (G11M5892 to N.E.S.). E.A.L., M.D.E., J.B.D., M.P.S., and G.W.L. are supported by National Health and Medical Research Council Fellowships. These organizations played no role in the design, analysis, or interpretation of data described in this study, nor in the preparation, review, or approval of the manuscript. No other potential conflicts of interest relevant to this article were reported.
N.E.S. designed the study, performed experiments, analyzed the data, and wrote the manuscript. M.T.G. and C.I.S. performed experiments and analyzed the data. N.E. and R.C. performed laboratory analyses. E.A.L. performed experiments and contributed to writing of the manuscript. P.J.N. and G.W.L. assisted with data collection and study design and contributed to the writing of the manuscript. M.D.E. and J.B.D. contributed to the writing of the manuscript. A.J.T. and M.P.S. assisted with data collection and contributed to the writing of the manuscript. N.E.S. is the guarantor of this work and, as such, had full access to all the data in the study and takes responsibility for the integrity of the data and the accuracy of the data analysis.
The authors thank the study participants for cooperation and effort and research nurses Donna Vizi and Jenny Starr (Alfred Baker Medical Unit, Baker IDI Heart and Diabetes Institute) for excellent assistance. The authors also thank the Victorian Government’s Operational Infrastructure Support Program.
Footnotes
Clinical trial reg. no. NCT00408850, clinicaltrials.gov.
REFERENCES
- 1.Kelly T, Yang W, Chen CS, Reynolds K, He J. Global burden of obesity in 2005 and projections to 2030. Int J Obes (Lond) 2008;32:1431–1437 [DOI] [PubMed] [Google Scholar]
- 2.Shaw JE, Sicree RA, Zimmet PZ. Global estimates of the prevalence of diabetes for 2010 and 2030. Diabetes Res Clin Pract 2010;87:4–14 [DOI] [PubMed] [Google Scholar]
- 3.Dunstan D, Zimmet P, Welborn T, et al. Diabesity and Associated Disorders in Australia 2000. The Australian Diabetes, Obesity and Lifestyle Study (AusDiab). Melbourne, Australia, Baker International Diabetes Institute, 2001, p. 5–6 [Google Scholar]
- 4.Magliano DJ, Shaw JE, Shortreed SM, et al. Lifetime risk and projected population prevalence of diabetes. Diabetologia 2008;51:2179–2186 [DOI] [PubMed] [Google Scholar]
- 5.Meigs JB. The metabolic syndrome. BMJ 2003;327:61–62 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Alberti KGMM, Eckel RH, Grundy SM, et al. International Diabetes Federation Task Force on Epidemiology and Prevention. Hational Heart, Lung, and Blood Institute. American Heart Association. World Heart Federation. International Atherosclerosis Society. International Association for the Study of Obesity Harmonizing the metabolic syndrome: a joint interim statement of the International Diabetes Federation Task Force on Epidemiology and Prevention; National Heart, Lung, and Blood Institute; American Heart Association; World Heart Federation; International Atherosclerosis Society; and International Association for the Study of Obesity. Circulation 2009;120:1640–1645 [DOI] [PubMed] [Google Scholar]
- 7.Grassi G, Dell’Oro R, Quarti-Trevano F, et al. Neuroadrenergic and reflex abnormalities in patients with metabolic syndrome. Diabetologia 2005;48:1359–1365 [DOI] [PubMed] [Google Scholar]
- 8.Beske SD, Alvarez GE, Ballard TP, Davy KP. Reduced cardiovagal baroreflex gain in visceral obesity: implications for the metabolic syndrome. Am J Physiol Heart Circ Physiol 2002;282:H630–H635 [DOI] [PubMed] [Google Scholar]
- 9.Straznicky NE, Lambert EA, Lambert GW, Masuo K, Esler MD, Nestel PJ. Effects of dietary weight loss on sympathetic activity and cardiac risk factors associated with the metabolic syndrome. J Clin Endocrinol Metab 2005;90:5998–6005 [DOI] [PubMed] [Google Scholar]
- 10.Esler M, Rumantir M, Wiesner G, Kaye D, Hastings J, Lambert G. Sympathetic nervous system and insulin resistance: from obesity to diabetes. Am J Hypertens 2001;14:304S–309S [DOI] [PubMed] [Google Scholar]
- 11.Masuo K, Mikami H, Ogihara T, Tuck ML. Sympathetic nerve hyperactivity precedes hyperinsulinemia and blood pressure elevation in a young, nonobese Japanese population. Am J Hypertens 1997;10:77–83 [DOI] [PubMed] [Google Scholar]
- 12.Flaa A, Aksnes TA, Kjeldsen SE, Eide I, Rostrup M. Increased sympathetic reactivity may predict insulin resistance: an 18-year follow-up study. Metabolism 2008;57:1422–1427 [DOI] [PubMed] [Google Scholar]
- 13.Carnethon MR, Golden SH, Folsom AR, Haskell W, Liao D. Prospective investigation of autonomic nervous system function and the development of type 2 diabetes: the Atherosclerosis Risk In Communities study, 1987-1998. Circulation 2003;107:2190–2195 [DOI] [PubMed] [Google Scholar]
- 14.Carnethon MR, Jacobs DR, Jr, Sidney S, Liu K, CARDIA study Influence of autonomic nervous system dysfunction on the development of type 2 diabetes: the CARDIA study. Diabetes Care 2003;26:3035–3041 [DOI] [PubMed] [Google Scholar]
- 15.Fiorentini A, Perciaccante A, Paris A, Serra P, Tubani L. Circadian rhythm of autonomic activity in non diabetic offsprings of type 2 diabetic patients. Cardiovasc Diabetol 2005;4:15. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Frontoni S, Bracaglia D, Baroni A, et al. Early autonomic dysfunction in glucose-tolerant but insulin-resistant offspring of type 2 diabetic patients. Hypertension 2003;41:1223–1227 [DOI] [PubMed] [Google Scholar]
- 17.Julius S, Gudbrandsson T, Jamerson K, Tariq Shahab S, Andersson O. The hemodynamic link between insulin resistance and hypertension. J Hypertens 1991;9:983–986 [DOI] [PubMed] [Google Scholar]
- 18.Lembo G, Capaldo B, Rendina V, et al. Acute noradrenergic activation induces insulin resistance in human skeletal muscle. Am J Physiol 1994;266:E242–E247 [DOI] [PubMed] [Google Scholar]
- 19.Randle PJ, Hales CN. garland PB, Newsholme EA. The glucose fatty-acid cycle. Its role in insulin sensitivity and the metabolic disturbances of diabetes mellitus. Lancet 1963;i:785–789 [DOI] [PubMed] [Google Scholar]
- 20.Anderson EA, Balon TW, Hoffman RP, Sinkey CA, Mark AL. Insulin increases sympathetic activity but not blood pressure in borderline hypertensive humans. Hypertension 1992;19:621–627 [DOI] [PubMed] [Google Scholar]
- 21.Vollenweider P, Randin D, Tappy L, Jéquier E, Nicod P, Scherrer U. Impaired insulin-induced sympathetic neural activation and vasodilation in skeletal muscle in obese humans. J Clin Invest 1994;93:2365–2371 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Straznicky NE, Lambert GW, Masuo K, et al. Blunted sympathetic neural response to oral glucose in obese subjects with the insulin-resistant metabolic syndrome. Am J Clin Nutr 2009;89:27–36 [DOI] [PubMed] [Google Scholar]
- 23.Straznicky NE, Lambert GW, Lambert EA. Neuroadrenergic dysfunction in obesity: an overview of the effects of weight loss. Curr Opin Lipidol 2010;21:21–30 [DOI] [PubMed] [Google Scholar]
- 24.Golay A, Schutz Y, Felber JP, de Fronzo RA, Jéquier E. Lack of thermogenic response to glucose/insulin infusion in diabetic obese subjects. Int J Obes 1986;10:107–116 [PubMed] [Google Scholar]
- 25.Huggett RJ, Scott EM, Gilbey SG, Stoker JB, Mackintosh AF, Mary DASG. Impact of type 2 diabetes mellitus on sympathetic neural mechanisms in hypertension. Circulation 2003;108:3097–3101 [DOI] [PubMed] [Google Scholar]
- 26.Tack CJJ, Smits P, Willemsen JJ, Lenders JWM, Thien T, Lutterman JA. Effects of insulin on vascular tone and sympathetic nervous system in NIDDM. Diabetes 1996;45:15–22 [DOI] [PubMed] [Google Scholar]
- 27.World Health Organization Definition and Diagnosis of Diabetes Mellitus and Intermediate Hyperglycaemia: Report of WHO/IFD Consultation. Geneva, Switzerland, World Health Organization, 2006, p. 1–46 [Google Scholar]
- 28.Paradisi G, Smith L, Burtner C, et al. Dual energy X-ray absorptiometry assessment of fat mass distribution and its association with the insulin resistance syndrome. Diabetes Care 1999;22:1310–1317 [DOI] [PubMed] [Google Scholar]
- 29.DeFronzo RA, Tobin JD, Andres R. Glucose clamp technique: a method for quantifying insulin secretion and resistance. Am J Physiol 1979;237:E214–E223 [DOI] [PubMed] [Google Scholar]
- 30.Eisenhofer G. Sympathetic nerve function—assessment by radioisotope dilution analysis. Clin Auton Res 2005;15:264–283 [DOI] [PubMed] [Google Scholar]
- 31.Eisenhofer G, Goldstein DS, Kopin IJ. Plasma dihydroxyphenylglycol for estimation of noradrenaline neuronal re-uptake in the sympathetic nervous system in vivo. Clin Sci (Lond) 1989;76:171–182 [DOI] [PubMed] [Google Scholar]
- 32.Herzberg-Schafer SA, Staiger H, Heni M, et al. Evaluation of fasting state-/oral glucose tolerance test-derived measures of insulin release for the detection of genetically impaired β-cell function. PLoS ONE 2010;5:e14194. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.Parati G, Saul JP, Castiglioni P. Assessing arterial baroreflex control of heart rate: new perspectives. J Hypertens 2004;22:1259–1263 [DOI] [PubMed] [Google Scholar]
- 34.Huggett RJ, Scott EM, Gilbey SG, Bannister J, Mackintosh AF, Mary DASG. Disparity of autonomic control in type 2 diabetes mellitus. Diabetologia 2005;48:172–179 [DOI] [PubMed] [Google Scholar]
- 35.Jamerson KA, Smith SD, Amerena JV, Grant E, Julius S. Vasoconstriction with norepinephrine causes less forearm insulin resistance than a reflex sympathetic vasoconstriction. Hypertension 1994;23:1006–1011 [DOI] [PubMed] [Google Scholar]
- 36.Manrique C, Lastra G, Gardner M, Sowers JR. The renin angiotensin aldosterone system in hypertension: roles of insulin resistance and oxidative stress. Med Clin North Am 2009;93:569–582 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37.Ward KR, Bardgett JF, Wolfgang L, Stocker SD. Sympathetic response to insulin is mediated by melanocortin 3/4 receptors in the hypothalamic paraventricular nucleus. Hypertension 2011;57:435–441 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38.Rahmouni K, Morgan DA, Morgan GM, et al. Hypothalamic PI3K and MAPK differentially mediate regional sympathetic activation to insulin. J Clin Invest 2004;114:652–658 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39.Kaiyala KJ, Prigeon RL, Kahn SE, Woods SC, Schwartz MW. Obesity induced by a high-fat diet is associated with reduced brain insulin transport in dogs. Diabetes 2000;49:1525–1533 [DOI] [PubMed] [Google Scholar]
- 40.Stein LJ, Dorsa DM, Baskin DG, Figlewicz DP, Porte D, Jr, Woods SC. Reduced effect of experimental peripheral hyperinsulinemia to elevate cerebrospinal fluid insulin concentrations of obese Zucker rats. Endocrinology 1987;121:1611–1615 [DOI] [PubMed] [Google Scholar]
- 41.Kern W, Benedict C, Schultes B, et al. Low cerebrospinal fluid insulin levels in obese humans. Diabetologia 2006;49:2790–2792 [DOI] [PubMed] [Google Scholar]
- 42.Carvalheira JBC, Ribeiro EB, Araújo EP, et al. Selective impairment of insulin signalling in the hypothalamus of obese Zucker rats. Diabetologia 2003;46:1629–1640 [DOI] [PubMed] [Google Scholar]
- 43.Liu Y, Liu F, Grundke-Iqbal I, Iqbal K, Gong CX. Deficient brain insulin signalling pathway in Alzheimer’s disease and diabetes. J Pathol 2011;225:54–62 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44.Fagius J, Berne C. The increase in sympathetic nerve activity after glucose ingestion is reduced in type I diabetes. Clin Sci (Lond) 2000;98:627–632 [PubMed] [Google Scholar]
- 45.Hoffman RP, Sinkey CA, Kienzle MG, Anderson EA. Muscle sympathetic nerve activity is reduced in IDDM before overt autonomic neuropathy. Diabetes 1993;42:375–380 [DOI] [PubMed] [Google Scholar]
- 46.Young CN, Deo SH, Chaudhary K, Thyfault JP, Fadel PJ. Insulin enhances the gain of arterial baroreflex control of muscle sympathetic nerve activity in humans. J Physiol 2010;588:3593–3603 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 47.Straznicky NE, Eikelis N, Nestel PJ, et al. Baseline sympathetic nervous system activity predicts dietary weight loss in obese metabolic syndrome subjects. J Clin Endocrinol Metab 2012;97:605–613 [DOI] [PubMed] [Google Scholar]
- 48.Tanakaya M, Takahashi N, Takeuchi K, et al. Postprandial hypotension due to a lack of sympathetic compensation in patients with diabetes mellitus. Acta Med Okayama 2007;61:191–197 [DOI] [PubMed] [Google Scholar]
- 49.Figlewicz DP, Szot P, Israel PA, Payne C, Dorsa DM. Insulin reduces norepinephrine transporter mRNA in vivo in rat locus coeruleus. Brain Res 1993;602:161–164 [DOI] [PubMed] [Google Scholar]
- 50.Robertson SD, Matthies HJG, Owens WA, et al. Insulin reveals Akt signaling as a novel regulator of norepinephrine transporter trafficking and norepinephrine homeostasis. J Neurosci 2010;30:11305–11316 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 51.Kiyono Y, Iida Y, Kawashima H, et al. Norepinephrine transporter density as a causative factor in alterations in MIBG myocardial uptake in NIDDM model rats. Eur J Nucl Med Mol Imaging 2002;29:999–1005 [DOI] [PubMed] [Google Scholar]
- 52.Nunes SF, Figueiredo IV, Pereira JS, et al. Monoamine oxidase and semicarbazide-sensitive amine oxidase kinetic analysis in mesenteric arteries of patients with type 2 diabetes. Physiol Res 2011;60:309–315 [DOI] [PubMed] [Google Scholar]
- 53.Lambert E, Sari CI, Dawood T, et al. Sympathetic nervous system activity is associated with obesity-induced subclinical organ damage in young adults. Hypertension 2010;56:351–358 [DOI] [PubMed] [Google Scholar]