

Abstract
The homeodomain and adjacent CVC domain in the visual system homeobox (VSX) proteins are conserved from nematodes to humans. Humans with missense mutations in these regions of VSX2 have microphthalmia, suggesting both regions are critical for function. To assess this, we generated the corresponding mutations in mouse Vsx2. The homeodomain mutant protein lacked DNA binding activity and the knock-in mutant phenocopied the null mutant, ocular retardation J. The CVC mutant protein exhibited weakened DNA binding; and, although the corresponding knock-in allele was recessive, it unexpectedly caused the strongest phenotype, as indicated by severe microphthalmia and hyperpigmentation of the neural retina. This occurred through a cryptic transcriptional feedback loop involving the transcription factors Mitf and Otx1 and the Cdk inhibitor p27Kip1. Our data suggest that the phenotypic severity of the CVC mutant depends on the weakened DNA binding activity elicited by the CVC mutation and a previously unknown protein interaction between Vsx2 and its regulatory target Mitf. Our data also suggest that an essential function of the CVC domain is to assist the homeodomain in high-affinity DNA binding, which is required for eye organogenesis and unhindered execution of the retinal progenitor program in mammals. Finally, the genetic and phenotypic behaviors of the CVC mutation suggest it has the characteristics of a recessive neomorph, a rare type of genetic allele.
Author Summary
Problems with the early development of the mammalian retina can cause congenital eye defects such as microphthalmia, in which the eye is dramatically smaller and functionally compromised. Severe microphthalmia is associated with mutations in the retinal-expressed visual system homeobox 2 (Vsx2) gene, but how Vsx2 controls retinal development, and ultimately eye formation, has remained unclear. We assessed the impact of two missense mutations, discovered in humans, on Vsx2 function and eye development in mice. One mutation altered a highly conserved residue of the homeodomain, and the other altered a highly conserved residue in the CVC domain, a region of unresolved function. Both mutations impacted the DNA binding properties of the protein, although to differing extents. Likewise, both mutations caused microphthalmia and disruptions in retinal development, also to differing extents and by distinct mechanisms. Our data suggest that Vsx2 acts as a gatekeeper of the retinal gene expression program by preventing the activation of interfering or competing gene expression programs. We propose that the evolutionary stable association between the VSX-class homeodomain and CVC domain set the stage for Vsx2 or its archetype to assume a gatekeeper function for retinal development and ultimately eye organogenesis.
Introduction
The homeodomain is a 60 amino acid DNA binding module composed of three alpha helices in a helix-turn-helix configuration. Homeodomain proteins are among the most numerous of transcription factors, second only to C2H2 zinc finger transcription factors in humans [1]. Structural studies of isolated homeodomains and site-directed mutants indicate that the properties needed for DNA binding are encoded within the homeodomain [2], [3], and two recent DNA binding screens of 168 mouse and 84 Drosophila melanogaster homeodomain proteins identified upwards of 16 amino acids occupying specific positions in the homeodomain that confer DNA binding site preferences and may define a general lexicon for predicting or rationally altering binding properties [4]–[6].
Many homeodomains, however, exhibit inherently low sequence specificity or weak binding affinity, characteristics inconsistent with their high degree of functional specificity in vivo. Solutions to this problem include the incorporation of additional DNA binding domains (e.g. Pou, Paired) or protein interaction domains that recruit additional DNA binding proteins (e.g. LIM) [7]–[10]. Other solutions do not incorporate modular domains, but rather utilize non-homeodomain residues or motifs to assist the homeodomain. The DNA binding capacity of several Hox homeodomains is enhanced by a cooperative interaction with PBC homeodomain proteins and is mediated by the hexapeptide/YPWM motif, a stretch of conserved hydrophobic residues near the N-terminus of the Hox homeodomain. This interaction not only increases the complexity of the target sequence since both proteins bind DNA, but it also enhances the DNA binding affinity of the Hox homeodomain [11]. The C-terminal tail in PBC proteins is a helical region adjacent to the C-terminus of the homeodomain that increases the homeodomain's DNA binding affinity, not by acting as a protein interaction motif or by directly contacting DNA, but through an intramolecular interaction that assists in properly positioning the third alpha helix (DNA recognition helix) into the major groove of its DNA binding site [12], [13].
It is unclear whether non-homeodomain motifs are commonly used to enhance homeodomain function. Predicting which non-homeodomain residues or motifs are required for homeodomain function is difficult because these relationships depend on subtle and highly specific differences among homeodomains and to specific structural conformations of DNA sequences that comprise binding sites [14]. However, the hexapeptide and C-terminal tail have two properties in common: they are positioned close to the homeodomain and are evolutionarily conserved in a non-modular fashion, meaning that they are only found in proteins with similar homeodomains.
Another group of homeodomain proteins with a conserved, non-modular motif adjacent to the homeodomain are in the visual system homeobox family (VSX; also referred to as Prd-L:CVC or CVC paired like). These include Vsx1 and Vsx2 (formerly Chx10) in vertebrates and D. melanogaster and ceh-10 in Ceanorhabditis elegans. VSX genes belong to the larger paired-like homeodomain class that include Rx, Arx, and Alx genes [10], but are unique in that they encode a region of approximately 60 amino acids extending from the C-terminus of the homeodomain and named the CVC domain for the genes in which it was initially discovered (Figure 1A) [15]–[17]. Genetic data suggest the CVC domain is essential for VSX function. In C. elegans, two missense mutations in the ceh-10 CVC domain cause embryonic lethality and neuronal differentiation defects similar in severity and timing to those elicited by nonsense mutations [18], [19]. In humans, missense mutations in the VSX1 and VSX2 CVC domains are linked to ocular abnormalities and disease [20]–[25]. While the pathogenicity of the VSX1 CVC variants is unclear [26], [27], evidence for VSX2 is strong. In two consanguineous families, the arginine at position 227, an invariant residue among VSX genes and part of the CVC domain, is substituted with tryptophan (Figure 1A) and this mutation segregates in a homozygous fashion with non-syndromic congenital bilateral microphthalmia (small eye; [20], [24]). A recent case study identified a new missense mutation in the CVC domain (alanine substituted for glycine at position 223), which also segregates in a homozygous fashion with microphthalmia [25]. These mutations are likely to have a profound effect on protein function since microphthalmia occurs in humans with other mutations in VSX2, most notably missense mutations in the homeodomain which substitute glutamine or proline for arginine at position 200 (Figure 1A; [24], [28], [29]. Vsx2-dependent microphthalmia also occurs in homozygous ocular retardation J mice (MGI symbol: orJ), which harbor a nonsense mutation in the homeodomain (Figure 1A). VSX2 protein is not detected from the orJ allele (this study; [30]), and this allele is therefore considered to be a null. Small eye phenotypes are also observed in zebrafish subjected to Vsx2 mRNA knockdown [31]–[33].
Figure 1. DNA binding and transcriptional activities of VSX2 and the VSX2[R200Q] and VSX2[R227W] variants.

(A) ClustalW alignment of the homeodomain and adjacent 60 amino acids in select VSX orthologs and the most similar non-VSX proteins in mice. Only the VSX sequences have a discernable CVC domain. The positions of the orJ, R200Q, and R227W mutations are shown. (B) Left panel: EMSA with in vitro translated VSX2, VSX2[R200Q], and VSX2[R227W] proteins and [32]P-labeled P3 oligo (see Table S1 for sequence). Top right panel: Extended exposure reveals weak binding by VSX2[R227W]. Bottom right panel: Western blot of in vitro translated proteins with VSX2 antibody (Lys, control lysate; -, P3 probe only). (C) Schematic shows five putative Vsx2 binding sites (Hx-6 – Hx-10) in the proximal promoter region (∼0.3 kb) of D-Mitf. Carats and dashed line marks the region of PCR amplification in the ChIP assay shown below schematic (primer set 13; Table S1). Arrowhead points to sequence-verified ChIP product. (D) Luciferase assays in P0 primary retinal cells transfected with the indicated expression vectors (x-axis) and ∼2.2 kb of the D-Mitf promoter region (pGL3P-DMitf). (E) The Hx-9 site was mutated in pGL3P-mDMitf to eliminate DNA binding at that site. Reporter assays were normalized to empty vector controls (white bars). (F) CAT assays in HEK293 cells transfected with the X4G2CAT reporter and VSX2 variants fused to the LexA DNA binding domain. Gal4-Hsf1 was included to stimulate high basal reporter activity [34]. ** P≤0.01; *** P≤0.001.
Addressing whether the CVC domain assists in homeodomain function is complicated by the likelihood that the CVC domain has multiple functions. Its deletion in VSX2 altered DNA binding and transcriptional properties although it is unclear whether these changes were interdependent, and whether they were specific to the CVC domain because other regions were also removed [34]. Its deletion in Vsx1 reduced polyubiquitination suggesting a role in regulating protein stability [35]. Because deleting the entire CVC domain could lead to pleiotropic effects, another approach to identify functional requirements of the CVC domain and its relationship with the homeodomain is to study the effects of the missense mutations on protein function and eye development.
In this study, we generated the homeodomain mutation R200Q and CVC domain mutation R227W in the mouse Vsx2 ortholog and compared their functional properties. A predominant effect of these mutations is to reduce homeodomain-dependent DNA binding but to different degrees. Since Vsx2 regulates eye size and retinal development, we generated knock-in mice and compared their phenotypes to the orJ mouse. Molecular and genetic analyses enabled us to identify the transcriptional circuits driving the phenotypes caused by each mutation. Our data support the model that the proper execution of mammalian eye organogenesis and retinal development is built upon high affinity DNA binding by Vsx2, which is dependent on both the homeodomain and CVC domain. We also provide evidence suggesting that Vsx2 regulates one of its key targets, microphthalmia-associated transcription factor (Mitf) by two mechanisms; direct transcriptional repression and protein∶protein interaction. Both mechanisms may be employed to prevent activation of aberrant gene expression programs that interfere with the execution of the developmental program in retinal progenitor cells (RPCs).
Results
The R200Q and R227W mutations alter the DNA binding affinity of VSX2 protein, but not its ability to repress transcription
We compared the DNA binding properties of in vitro translated VSX2 and the VSX2[R200Q] and VSX2[R227W] variants using an oligonucleotide containing a high affinity Vsx2 binding site by electrophoretic mobility shift assays (EMSA). Consistent with previous studies [28], [32], robust DNA binding was observed for VSX2 whereas binding was not detected with VSX2[R200Q] (Figure 1B, left panel). VSX2[R227W] binding was detectable but weak, and is more visible with a longer exposure time (Figure 1B, top right panel). The reduced DNA binding properties were not due to variations in protein expression (Figure 1B, bottom right panel) or to alterations in the helical organization of the homeodomain as assessed with secondary structure prediction software (http://us.expasy.org; data not shown). These observations indicate that the arginines at positions 200 and 227 are required for high affinity DNA binding.
Of the known candidate targets of Vsx2-mediated transcriptional regulation [32], [36]–[38], the basic helix-loop-helix/leucine zipper (bhlh/zip) gene Mitf is of high importance because its increased expression in the orJ retina contributes to the mutant phenotype [39]–[42]. Regulation of the Mitf locus is complex. Nine promoters have been identified and each produces an RNA transcript with a distinct first exon, several with limited protein-coding information. As a result, multiple Mitf isoforms on the RNA and protein levels are possible, although most if not all isoforms contain the domains and motifs needed for transcription factor activity (see Figure 1 in [39] for a detailed illustration of the mouse Mitf locus and gene products). Depending on the cell type, Mitf isoforms are expressed in different combinations, indicating that promoter utilization is context-dependent [43]. D-Mitf is one of the isoforms upregulated in the orJ retina and has at least 10 putative Vsx2 binding sites within 1 kb upstream of its transcriptional start site (Hx-1 – Hx-10; [39]). Chromatin immunoprecipitation (ChIP) assays revealed VSX2 binding in the vicinity of the Hx-1 – Hx-3 sites, approximately 0.8 kb upstream of the D-Mitf transcriptional start site [39]. We found that VSX2 bound to chromatin in the vicinity of the Hx-6 – Hx-10 sites, less than 0.3 kb upstream D-Mitf transcriptional start site (Figure 1C). Based on these findings, the D-Mitf promoter is likely to be a direct target of Vsx2-mediated transcriptional repression.
To test this further, transcriptional reporter assays were done with a construct containing ∼2.2 kb of the D-Mitf promoter region and SV40 early promoter driving a luciferase reporter (pGL3P-DMitf) and constructs for expressing VSX2, VSX2[R200Q], or VSX2[R227W] proteins in P0 wild-type retinal cells. Inclusion of the SV40 promoter was necessary for reliable basal activity reporter activity. Consistent with our results for the Vsx1 promoter [32], VSX2 repressed reporter activity and repression mediated by VSX2[R200Q] was diminished (Figure 1D). Like Vsx2, VSX2[R227W] also repressed reporter activity and required the presence of the Hx-9 site (Figure 1D, 1E). Similar results were obtained in HEK293 cells (data not shown), indicating that repression of reporter activity was not dependent on additional factors exclusive to retinal cells. The SV40 promoter was not used in these and all other reporter assays in HEK293 cells because it was not required for basal reporter activity. To determine if the mutations interfered with repressor function in addition to DNA binding, the LexA DNA binding domain was fused to the Vsx2 variants and reporter activity was determined in HEK293 cells with X4G2CAT, a choloramphenicol acetyltransferase (CAT) reporter containing multimerized LexA binding sites [34]. In this assay, each of the LexA fusions repressed CAT reporter activity to similar extents (Figure 1F). Thus, the repression mediated by VSX2 and VSX2[R227W] depended on DNA binding and the R200 and R227 residues were not required for repressor activity.
Generation and characterization of Vsx2R200Q and Vsx2R227W knock-in mice
If DNA binding is essential for Vsx2 function, then the VSX2[R200Q] protein should be a functional null and the VSX2[R227W] protein should retain some degree of Vsx2 function. If true, then the simplest predictions for mice with these mutations are that homozygous Vsx2R200Q mutants (R200Q) should phenocopy orJ mutants and homozygous Vsx2R227W mutants (R227W) should exhibit a less severe or hypomorphic phenotype. It is also possible that these alleles could exhibit dominant negative activity if Vsx2 activity requires dimerization, similar to DNA binding mutations in the homeodomain protein Pitx2, which are linked to Axenfeld-Rieger syndrome [44],[45]. We tested these predictions by generating R200Q and R227W knock-in mice by homologous recombination using the ACN targeting vector, which removed all gene targeting elements when the alleles were transmitted through the male germline (Text S1; Figure S1; [46]). The only foreign DNA retained was a 34 base pair sequence containing a remnant lox-p site in intron 3 (Figure S1). Germline transmission was achieved (Figure S1) and the mutants used for this study were established in the129sv genetic background to minimize strain-dependent modifier effects [40], [41], [47], [48].
As in humans, R200Q and R227W mice were microphthalmic, which was apparent by E11.5 and became progressively more severe as development continued (Figure 2A–2L). Relative eye size in orJ and R200Q mutants were similar, but surprisingly, R227W mutants exhibited smaller eyes at E14.5 and beyond (Figure 2E–2L). In contrast to the lack of VSX2 protein in orJ RPCs, both knock-in mutants expressed VSX2 protein in a manner similar to wild-type, suggesting that changes in expression or nuclear localization were not causing the phenotypes (Figure 2M–2P; Figure S2A). Consistent with the EMSA data, ChIP assays with E12.5 retinal lysates showed that VSX2[R200Q] protein was not detectable at the D-Mitf promoter whereas VSX2[R227W] was bound although to a lesser extent than VSX2 (Figure 2Q).
Figure 2. The R200Q and R227W mutations cause non-syndromic congenital microphthalmia.

(A–D) Mice homozygous for the orJ, R200Q, and R227W alleles had smaller eyes than wild-type by E11.5. (E–H) At E14.5, overall embryonic development was unaffected in the mutants, but the failure of the mutant eyes to keep pace with the growth of the wild-type eye was evident. Eye growth in the R227W mutant also failed to keep pace with the orJ and R200Q mutants. (I–L) Dissected E17.5 eyes (right eyes rotated 90°) show similar reductions in eye size in orJ and R200Q homozygotes whereas the reduction in eye size of R227W homozygotes was the most severe. (M–P) VSX2 immunohistochemistry in E12.5 retinas. VSX2 protein was not detected in the orJ retina, confirming it as an expression null. VSX2[R200Q] and VSX2[R227W] were expressed similarly to VSX2[wt], although to a reduced extent in peripheral retina. Dashed lines bound retinas. (Q) ChIP assays with VSX2 antibody reacted with E12.5 native chromatin lysates from wild-type, R200Q, and R227W retinas and amplified using D-Mitf primer set 13 (Table S1). Arrowhead denotes amplification product. Graph shows quantification results of ChIP-qPCR. Scale bars: 0.5 mm (E11.5); 5 mm (E14.5); 1 mm (E17.5).
Histological analysis revealed that mutant eyes had a smaller lens, thickened retinal pigment epithelium (RPE), and a thinner retina compared to wild-type (Figure 3A–3H). Whereas the orJ and R200Q eyes were similar in appearance, R227W eyes were more severely affected as indicated by an even smaller lens, thinner retina, and an infiltration of mesenchymal cells into the vitreal chamber (Figure 3H, asterisks). The smaller retina in the orJ mouse is correlated with a reduction in RPC proliferation [30], [47], [49]–[53] and this was likely the case in the R200Q and R227W retinas as both mutants showed reduced phosphorylated Histone H3 expression (Figure S2B). Consistent with this, VSX2[R200Q] or VSX2[R227W] overexpression in cultured orJ retinal cells was not sufficient to promote proliferation (Figure S2C).
Figure 3. Ocular histology and neurogenesis in Vsx2 mutants.

(A–H) Merged images of cryosections showing DAPI staining (blue) and melanogenic pigmentation (white) for each of the indicated genotypes and ages. Arrowheads in H point to aberrant pigmentation in peripheral retina asterisk denotes ectopic periocular mesenchyme (POM) in vitreal cavity. (I–P) Expression patterns of the neuronal differentiation marker class III β-Tubulin (TUBB3). Neurogenesis lagged behind wild-type and to a similar extent in the orJ and R200Q retinas, but did not initiate in the R227W retina. (Q–T) Merged images of cryosections showing DAPI staining (blue) and melanogenic pigmentation (white) for each of the indicated genotypes at E17.5. The R227W retina was aberrantly pigmented, either partially (T(a)) or completely (T(b)). Arrowheads in T(a) and T(b) point to aberrant pigmentation in peripheral retina, arrows to central retinal regions, and asterisks to ectopic pigmentation in vitreal cavity. (U–W) Pigmented cells expressing VSX2[R227W] were detected in pigmented retinal region. L, lens; RPE, retinal pigment epithelium. Scale bars: 100 µm (A–T), 20 µm (U–W).
The onset of neurogenesis occurs by E11.5 in the central retina and spreads as a wave toward the peripheral retina. This was delayed by 1–2 days in orJ and R200Q retinas and was not observed at all in the embryonic R227W retina (Figure 3I–3P; Figure S3; [47], [52]). The lack of detectable neurogenesis suggested that R227W RPCs underwent a fundamental change in their developmental potential that differed from the other mutants.
Optic cup morphogenesis initiates at approximately E9.5, soon after onset of Vsx2 expression [17], [54]. The Vsx2 expression domain marks the interior layer of the optic cup and gives rise to the retina. Under normal conditions, melanogenic pigmentation does not occur in the retina. In contrast, pigmentation and thinning of the epithelium is pronounced in the peripheral retina of orJ mice, and lineage analysis suggests these pigmented cells arose from retinal-specified progenitor cells [40]. Pigmentation of the central orJ retina is rare in the 129svj genetic background, but is fairly prevalent in mixed 129svj:C57Bl6 mice owing to uncharacterized genetic modifiers [40], [41]. Similar background-dependent effects were observed for the R200Q retina (data not shown). Surprisingly, pigmentation was much more extensive in the R227W retina of mice with the 129sv genetic background (Figure 3Q–3T). This occurred in a progressive manner, which started in the peripheral retina at E14.5 and extended centrally to occupy most or all of the retina by E17.5 (Figure 3H, 3T(a), 3T(b) ). VSX2[R227W] protein was detected in some pigmented cells at E17.5, which indicated that as in the orJ retina, the ectopically pigmented cells arose from RPCs (Figure 3U–3W). In contrast, VSX2[R200Q] protein remained expressed in RPCs at E17.5 (data not shown). These data suggest that the pigmentation of the R227W retina was a direct consequence of the mutant allele.
In agreement with our in vitro data, the R200Q and orJ phenotypes were similar and support the hypothesis that homeodomain-dependent DNA binding is critical for Vsx2 function. That the R227W phenotype was more severe was unexpected since the VSX2[R227W] protein retained properties associated with transcriptional regulation. Our in vivo and in vitro data are consistent in that they indicate the VSX2[R227W] protein was functional, but they differ because the in vivo phenotype indicated that the R227W allele was not hypomorphic. One possibility for this discrepancy is that the VSX2[R227W] protein acquired a novel activity not revealed by the in vitro assays. If this were true, then the R227W allele should be dominant or semi-dominant. This was not the case, however, since eye size, circumference, and retinal histology in R227W/+ mice were indistinguishable between wild-type, orJ/+ or R200Q/+ mice (Figure 4A–4I; Figure S4). Furthermore, hemizygous R227W/orJ mice exhibited an intermediate phenotype compared to the orJ and R227W homozygotes (Figure 4J–4S), which suggested that the failure of the R227W allele to compete with the wild-type allele was not due to reduced expression of the mutant protein. Finally, overexpression of VSX2, VSX2[R200Q], or VSX2[R227W] in newborn wild-type retinal primary cells had minimal effects on proliferation (Figure S2D). These data revealed that even if the VSX2[R227W] protein acquired a novel activity, it is not sufficient to interfere with wild-type Vsx2 function. Thus, even though the R227W phenotype surpasses the null in severity, the allele displayed recessive behavior, consistent with what is observed in humans. Additionally, the recessive nature of the R200Q and R227W alleles suggest that Vsx2 does not require dimerization for function.
Figure 4. The R200Q and R227W alleles and proteins do not exhibit dominant behavior.

(A) Wild-type, orJ/+, R200Q/+, and R227W/+ eyes were indistinguishable at P0. No significant differences in eye circumferences were detected. (B–E) Merged images of cryosections showing DAPI staining (blue) and melanogenic pigmentation (white) for each of the indicated genotypes at P0. (F–I) Expression of the retinal ganglion cell marker POU4F2 in wild-type or Vsx2 heterozygous retinas. (J) Eye circumference of orJ/R227W heterozygotes was intermediate to orJ and R227W homozygotes. (K–S) Expression of VSX2, CCND1, and TUBB3 in orJ, orJ/R227W, and R227W retinas at P0. VSX2 was detected in the orJ/R227W retina only. CCND1 and TUBB3 were detected in orJ or orJ/R227W retinas but not in R227W pigmented retina. (T) Genotype-phenotype correlation of Vsx2 alleles arranged by retinal phenotype. * P≤0.05 Scale bars: 1 mm (A, J); 100 µm (B–I, K–S).
The data presented thus far best fits the genotype-phenotype correlation shown in Figure 4T. In wild-type mice, the RPC program driving retinal development progressed normally and correlated with unhindered DNA binding activity by Vsx2. In orJ and R200Q mice, the RPC program progressed in a suboptimal manner and the RPCs were biased, but not necessarily committed, to expressing a pigmentation program. The orJ and R200Q alleles were recessive and lacked Vsx2-dependent DNA binding activity. In the case of orJ, this is because the protein was not present, and in the case of R200Q because the DNA binding activity was specifically disrupted. In R227W mice, the RPC program initiated, but ultimately failed and was followed by robust expression of a pigmentation program. As with orJ and R200Q, the R227W allele was recessive, but the protein retained DNA binding activity, albeit weakened compared to wild-type.
Multiple regulatory changes at the Mitf locus are associated with pigmentation in the R227W retina
Mitf is initially expressed throughout the optic neuroepithelium at the optic vesicle stage and is downregulated in the presumptive retinal domain soon after Vsx2 is expressed [54]–[56]. Since Mitf is a key regulator of the genetic pathways that drive melanogenic pigmentation, we suspected that Mitf expression levels would correlate with the degree of pigmentation in the mutants. In orJ and R200Q mice, MITF expression at E12.5 was modestly elevated in the central retina and highest in the peripheral retina where pigmentation was most prevalent (Figure 5A–5C). In contrast, MITF was highly expressed throughout the R227W retina (Figure 5D). We also examined the expression of the orthodenticle-related homeodomain proteins OTX1 and OTX2 (OTX) since they are also required for the pigmentation program in the RPE and can directly regulate Mitf expression [57]–[60]. Similar to MITF, OTX expression was modestly increased in the orJ and R200Q central retinas but was highly expressed throughout the R227W retina with the most notable increases in the periphery (Figure 5E–5H; negative control staining shown in Figure S6C). Interestingly, VSX2 was downregulated in the peripheral retina, which correlated with the highest expression levels of MITF and OTX (Figure 5D′, 5H′) and where pigmentation was most apparent at E14.5 (Figure 3H), which suggested a change in fate for these cells. The scattered OTX expression in the wild-type retina is linked to the production of postmitotic Otx2+ cone photoreceptor precursors [61], which were not observed in the mutants, consistent with the delay or absence of neurogenesis.
Figure 5. Phenotypic severity correlates with the expression levels of Mitf and Otx1.

(A–D) MITF expression at E12.5 for the indicated genotypes. The R227W retina expressed MITF at much higher levels compared to the orJ and R200Q mutants. (D′) Merged images of VSX2[R227W] (red) and MITF (green) shows overlap in expression. The lack of VSX2[R227W] expression in the peripheral retina corresponded to the highest levels of MITF. (E–H) OTX expression at E12.5 for the indicated genotypes. OTX expression was highest in the R227W retina. (H′) Merged images of VSX2[R227W] (red) and OTX (green). Like MITF, OTX expression was highest in regions lacking VSX2[R227W]. MITF and OTX were also expressed in RPE (outside lower dashed lines). (I–K) Relative mRNA expression levels of pan-Mitf (I), Otx1 and Otx2 (J), and the D-, H-, A-, J-, and B-Mitf isoforms (K) in E12.5 retinas of the indicated genotypes as determined by qRT-PCR. Samples were normalized to the expression level for each transcript in the orJ retina (white bars). * P≤0.05 Scale bar: 50 µm.
To determine if changes in mRNA levels could account for the differences in MITF and OTX expression, we performed quantitative real-time reverse-transcription PCR (qRT-PCR) on total RNA lysates from E12.5 retinas. Using primers that recognize all Mitf isoforms (pan-Mitf), we found that compared to the orJ retina, wild-type had significantly less expression, R200Q was similar, and R227W had significantly more expression (Figure 5I), all of which is consistent with the immunohistochemical data. We also used gene specific primers for Otx1 and Otx2 to determine their relative expression levels in each genotype (Figure 5J). Compared to orJ, Otx1 expression was lower in wild-type and R200Q and higher in the R227W retina. All of the mutants expressed Otx2 at lower levels than wild-type. These data indicated that the increase in OTX protein expression was specific to Otx1. Furthermore, the low level of Otx1 in the R200Q retina revealed a difference with the orJ retina, which was unexpected in light of the overall similarities in the cellular and tissue phenotypes of the two mutants.
The A, D, H, and J isoforms of Mitf are expressed in the embryonic retinal pigment epithelium (RPE) with A, D, and H being the most abundant [39]. These isoforms are also detected in the retina at much reduced levels with the exception of A, which is expressed at a level comparable to the RPE [39]. We performed qRT-PCR of E12.5 retinal RNA to determine the isoform-profile in the different genotypes (Figure 5K). We found that D and H were higher in the orJ retina than in wild-type, consistent with previous findings [39]. We also observed a significant increase in J, an upward trend in B, and no change in A. In general, the isoform expression profile in the R200Q retina was similar to orJ. In the R227W retina, however, only A and H exhibited higher expression compared to orJ and R200Q. These data suggested the increased Mitf expression in the R227W retina was due to novel changes in the transcriptional regulation of the A and H isoforms.
Periocular mesenchyme (POM) promotes A-Mitf expression, but H-Mitf is the isoform that most likely accounts for the unique increase in the overall Mitf level in the R227W retina
The POM is composed of neural crest and mesoderm-derived progenitors that express Pitx2 and contribute to the formation of ocular structures such as the sclera, choroid, and cornea [62]–[64]. In chick, the POM contains an Activin-like factor that promotes Mitf expression in the nascent RPE domain of the optic vesicle [65]. Interestingly, the most profound changes with respect to tissue morphology, pigmentation, and upregulation of Mitf and Otx1 expression corresponded to regions of the neural retina that were juxtaposed to POM that invaded the vitreal chamber from the retinal peripheral margin after E10.5 (Figure 6A–6F). This was most pronounced in the R227W retina in which the entire vitreal chamber fills with PITX2 positive cells by E17.5 (Figure S5A). This was highly unusual since very few mesenchymal cells normally migrate into the vitreal cavity [63].
Figure 6. Influence of POM on Mitf expression in the R227W retina.

(A–F) PITX2 (red) and MITF (green) expression at E10.5 (A–C) and E12.5 (D–F). Limited PITX2+ cells were detected in the vitreal chamber (between retina and lens) of wild-type eyes. PITX2+ cells were not detected in the vitreal chamber of mutant eyes at E10.5, but were abundant by E12.5 and continuous with POM at the retinal periphery (arrowheads). MITF expression levels were modestly upregulated at E10.5 in the mutant retinas and were clearly elevated by E12.5. (G) Relative expression levels of A-Mitf, H-Mitf, and pan-Mitf in E10.5 whole retina and lens explants cultured for 48 hr. R227W expression levels were normalized to orJ. Invasion of POM into the vitreal chamber did not occur in these cultures (data not shown). (H) Relative expression levels of A-Mitf, H-Mitf, and pan-Mitf in physically manipulated E10.5 R227W whole retina and lens explants cultured for 48 hr after the following manipulations: “−POM” (retina and lens only); “mock” (retina partially separated from lens); “+POM” (retina partially separated from lens with surrounding POM intact); “POM imp” (retina partially separated from lens and POM implanted into vitreal cavity). Expression levels were normalized to the “−POM” condition. * P≤0.05; ** P≤0.01 Scale bar: 50 µm.
To determine whether the POM was responsible for the elevated expression levels of the A- or H-Mitf isoforms in the R227W retina, we cultured retinal explants from E10.5 orJ and R227W embryos with the lens attached but POM and RPE removed for 48 hr and measured transcript levels. Infiltration of POM into the vitreal chamber was not detectable at E10.5 and we found no evidence of POM invasion after 48 hr in culture (data not shown). Under these conditions, A-Mitf was unchanged between the two mutants whereas H-Mitf was significantly higher in the R227W mutant (Figure 6G). To further test the influence of POM, we cultured E10.5 R227W explants containing whole retina and lens in the presence or absence of POM in four different conditions: POM removed (- POM); POM removed with vitreal space physically exposed (mock); retention of POM at the anterior pole (+POM); or POM implanted into the vitreal chamber (POM imp). The POM implant was the only condition in which A-Mitf expression was enhanced further (Figure 6H, left graph). Although the increase in H-Mitf expression was significant in the presence of POM (Figure 6H, middle graph), the magnitude of the change was small (note the difference in scale of the y-axes for the middle graphs in Figure 6G and 6H). The effects of the POM implant on the D- and J- isoforms were not significant (Figure S5B). These data suggested that the elevated level of A-Mitf was under the influence of signals from the POM whereas H-Mitf expression was largely independent of POM-derived signals. Since the high level of pan-Mitf expression also did not depend on the presence of POM (Figure 6H, right graph), H-Mitf appears to be the mRNA isoform primarily responsible for the elevated pan-Mitf level in the R227W retina and its regulation is likely to be cell-autonomous.
Dominant-negative Mitf reveals a positive feedback loop for driving high Mitf expression and pigmentation in the R227W retina
To better understand the role of Mitf in microphthalmia and the retinal defects caused by the Vsx2 mutations, we generated compound mutants homozygous for the Vsx2 alleles and heterozygous for the Mitf mi allele (mi/+). mi is a dominant negative allele and encodes a 3 bp deletion resulting in the loss of an arginine in the basic domain, which disrupts DNA binding in all Mitf isoforms [66], [67]. Early eye development proceeds relatively well in mi/+ mice [68] and orJ; mi/+ mice showed substantial improvements in eye size, tissue histology, and retinal neurogenesis by birth (Figure 7A, 7D, 7G, 7J). As expected, the degree of improvement of eye development in R200Q; mi/+ was similar to orJ; mi/+ (Figure 7B, 7E, 7H, 7K), but interestingly, eye size and retinal development in R227W; mi/+ mice also improved to a level comparable with the other mutants (Figure 7C, 7F, 7I, 7L). Positive effects were observed early in retinal development as evidenced by increased eye size, circumference, and restored neurogenesis (Figure 7M, 7N). These findings were surprising because of the more profound consequences of the R227W mutation on eye development and the elevated levels of Mitf and Otx1. Consistent with the high degree of phenotypic rescue, pan-Mitf and Otx1 expression levels were significantly reduced (Figure 7O). Importantly, pan-Mitf levels decreased disproportionately relative to the allele dosage. This was correlated with large reductions in all isoforms except B-Mitf (Table 1), which is already expressed at low levels [39]. This suggested that the enhanced Mitf expression in the R227W retina is due to positive feedback. This was unique to the R227W retina because Mitf levels were not decreased in the orJ; mi/+ retina (Figure 7P). Our observations thus far reveal the essential and complex role of Mitf misregulation in causing microphthalmia and in interfering with retinal development in the Vsx2 mutant backgrounds.
Figure 7. The dominant negative allele Mitfmi restores retinal development in the Vsx2 mutants.

(A–F) Merged images of cryosections showing DAPI staining (blue) and melanogenic pigmentation (white) in P0 orj, R200Q, and R227W mice that were Mitf wild-type (Mitf+/+; A–C) and mi heterozygous (Mitfmi/+; D–F). Insets show whole eyes. The retina in C was completely transformed into pigmented tissue (bounded by dashed line) and ectopic POM was partially pigmented (asterisk). Eye size and retinal histology were restored to a comparable degree in all Vsx2, mi compound mutants (D–F). Also notable in the R227W, mi compound mutant was the lack of POM in the vitreal chamber (asterisk in F). (G–L) TUBB3 staining at P0. In all cases, lamination patterns were restored in the compound mutants, indicating robust neurogenesis. Retinal tissue in I is bounded by the dashed lines. (M) The reduced eye size in the R227W mutant was partially rescued in the R227W; Mitfmi/+ mutant at E12.5. (N) The expression of TUBB3 was detected in the R227W; Mitfmi/+ retina at E13.5. (O) Mitf and Otx1 transcript levels were much lower in the R227W; Mitfmi/+ retina (black bars) compared to the R227W mutant (white bars). (P) pan-Mitf transcript level in orJ; Mitfmi/+ retina was not lower than that in orJ retina. * P≤0.05; *** P≤0.001 Scale bars: 100 µm (A–F); 1 mm (insets); 50 µm (G–L); 0.5 mm (M); 100 µm (N).
Table 1. Relative retinal mRNA levels in R227W and compound mutants at E12.5.
| R227W | R227W; Mitfmi/+ | R227W, p27+/− | |
| pan- Mitf | 1.00±0.12 | 0.15±0.08*** | 0.52±0.05** ; # |
| D-Mitf | 1.00±0.07 | 0.09±0.04** | 0.27±0.05*** ; # |
| A-Mitf | 1.00±0.12 | 0.14±0.06** | 1.69±0.29## |
| H-Mitf | 1.00±0.11 | 0.07±0.03*** | 0.78±0.23# |
| B-Mitf | 1.00±0.09 | 0.80±0.40 | 0.92±0.20 |
| J-Mitf | 1.00±0.20 | 0.19±0.06** | 1.12±0.29## |
| Otx1 | 1.00±0.11 | 0.37±0.13* | 0.45±0.04** |
P≤0.05;
P≤0.01;
P≤0.001 (compared to R227W).
P≤0.05;
P≤0.01 (compared to R227W; Mitf mi/+).
Genetic reduction of p27Kip1 enhances eye size and restores neurogenesis in the R227W retina
We previously showed that genetic inactivation of the cyclin-dependent kinase inhibitor p27Kip1 (p27; MGI symbol: Cdkn1b) significantly enhances eye size and retinal development in orJ mice [50]. Although we didn't examine the effects of p27 removal on Mitf expression or retinal pigmentation in that study, we have yet to observe pigmentation in regions where retinal histogenesis was restored. This was also true for R200Q; p27 double mutants (data not shown). To determine if p27 also contributed to the alterations in eye development and the pigmentation potential of R227W RPCs, we generated R227W; p27 compound mutants. We analyzed retinas from R227W homozygous, p27 heterozygous (R227W; p27+/−) mice because they expressed p27 at a level similar to that in R227W; mi/+ retinas (Figure 8A). Interestingly, partial reduction in p27 was sufficient to inhibit pigmentation and promote retinal histogenesis (Figure 8B–8D). As in the orJ;p27−/− retina, the peripheral regions of the R227W; p27+/− retina was not rescued to the same extent as the central region. These data revealed that p27 was a key factor in promoting the Vsx2 mutant phenotypes regardless of allele.
Figure 8. p27Kip1 is part of a gene regulatory network promoting pigmentation in the R227W retina.

(A) p27 mRNA was reduced by approximately half in R227W; Mitfmi/+ and R227W; p27+/− compound mutant retinas compared to R227W at E12.5. (B–D) Phenotypes of R227W, p27Kip1+/− compound mutant eyes at P0. (B) Retinal tissue (blue) was restored along with a concomitant loss of pigmentation (white) in the retina. The peripheral retina was not rescued to the same extent as the central region. Eye size was also enhanced in the compound mutant (inset). POU4F2 (C) and the amacrine cell marker SOX2 (D) were expressed in the compound mutant. SOX2 is also expressed in RPCs. (E) Expression of Mitf and Otx1 mRNAs were reduced by approximately half in compound mutant retinas (black bars) compared to R227W (white bars) at E12.5. (F) The expression level of D-Mitf mRNA was reduced in compound mutant retinas, whereas H-Mitf mRNA levels were were not significantly different. (G) p27 overexpression in HEK293 cells increased luciferase activity from pGL3B-DMitf, but to a much lesser extent from pGL3B-HMitf. (H) ChIP assays of E12.5 R227W retinal lysates probed with p27 antibody. ChIP panel on left shows products obtained with primer set 1; panel on right shows products obtained with primer set 13. Sequence-verified products denoted by arrowheads. * P≤0.05; ** P≤0.01; *** P≤0.001 Scale bars: 100 µm (B); 1 mm (inset); 50 µm(C,D).
Vsx2[R227W], p27, H-Mitf, D-Mitf, and Otx1 form a transcriptional positive feedback loop
As with the R227W; mi/+ retina, pan-Mitf and Otx1 mRNA and protein expression levels dropped in the R227W; p27+/− retina (Figure 8E; Figure S6A, S6B; data not shown). There were notable differences, however, between the two rescue models (Table 1). In the R227W; p27+/− retina, the pan-Mitf level was intermediate to the R227W and R227W; mi/+ retinas. Among the Mitf isoforms, only D was reduced in the R227W; p27+/− retina (Figure 8F; Table 1), whereas all Mitf isoforms (except B) dropped well below half in the R227W; mi/+ retina (Table 1). Considering that H-Mitf was the isoform primarily responsible for the elevated increase in the pan-Mitf level in the R227W mutant over orJ (and R200Q), these data show that H-Mitf was not sufficient to drive the R227W phenotype when D-Mitf and p27 levels dropped down. These data are consistent with a genetic pathway in which D-Mitf is downstream of p27, and p27 is downstream of H-Mitf .
Since D-Mitf mRNA expression decreased when one allele of p27 was inactivated in the R227W retina, we asked whether this regulation could be direct. First, we overexpressed p27 with our D-Mitf luciferase reporter (pGL3B-DMitf) in HEK293 cells. In contrast to the repression observed with VSX2 and VSX2[R227W] (Figure 1D), p27 enhanced reporter activity (Figure 8G, left graph). That this reflects a specific interaction is supported by our observation that p27 did not significantly alter reporter activity when Luciferase was expressed under the control of ∼1.2 kb of the H-Mitf promoter region (pGL3B-HMitf; Figure 8G, right graph). Second, we performed ChIP assays with an antibody against p27 and chromatin lysates isolated from E12.5 R227W retinas using 13 primer sets that covered ∼2.2 kb of 5′-intergenic sequence. PCR enrichment was observed with primer set 1 and primer set 13 (Figure 8H). These data suggest p27 acts directly to regulate D-Mitf transcription in R227W RPCs.
Since p27 is an important factor in promoting both the orJ and R227W phenotypes and yet the phenotypes differ in severity, we asked whether p27 is regulated differently in the two mutants. p27 transcript levels were equivalent in wild-type and orJ retinas at P0 [50] and its expression was not significantly different between wild-type, orJ, or R200Q at E12.5 (Figure 9A). In contrast, p27 mRNA and protein expression was higher in the R227W retina (Figure 9A; Figure S6D-S6G), with the highest expression in the peripheral regions, similar to MITF and OTX. These observations correlate p27 expression level with the phenotypic severity of the R227W mutant and indicate a novel mode of p27 regulation.
Figure 9. Molecular interactions between the VSX2 variants and components of the pigmentation circuitry.

(A) p27 mRNA expression in E12.5 retinas of the indicated genotypes as determined by qRT-PCR. Samples were normalized to orJ. Only the R227W retina was significantly different. (B) Luciferase activities from HEK293 cells transfected with the indicated expression vectors (x-axes) and ∼1.1 kb of the p27 promoter region (pGL3B-p27). Graph I: H-MITF repressed reporter activity in a DNA binding-dependent manner. Graph II: VSX2 and VSX2[R227W] enhanced reporter activity. Graph III: H-MITF combined with VSX2[R227W] elicited a specific and synergistic increase in reporter activity that depended on DNA binding as revealed by the abrogated activity of the VSX2[R200Q, R227W] double mutant (RQRW). Graph IV: Expression of the mi version of H-MITF had no effect on reporter activity resulting from VSX2 or its variants. (C) Schematic of p27 5′-intergenic region (∼1.1 kb). Positions of putative Mitf binding sites (M) and homeodomain core sequences (H) are shown. Positions are relative to p27 transcriptional start site. Position of primers that constitute p27 primer set 2 (Table S1) is also shown. Graphs show quantification of ChIP-qPCR assays using MITF or VSX2 antibodies reacted with E12.5 lysates from wild-type, R200Q and R227W retinas. MITF binding was detected in R200Q and R227W lysates. VSX2 binding was detected in R227W lysate. (D) Co-IPs of E12.5 R227W and R200Q retinal protein lysates with a negative control sheep IgG or VSX2 antibodies followed by western blot probed with MITF antibody (top panel). Co-IPs of HEK293 cells transfected with VSX2 or its variants (listed below images) plus H-MITF (middle panel) or its mi variant (bottom panel). IPs were performed with sheep IgG or VSX2 antibodies followed by western blot probed with MITF antibody. input refers to the 20% of whole protein lysate used for co-IP. (E) Luciferase assays in HEK293 cells transfected with the indicated expression vectors (x-axes) and the pGL3B-HMitf. Left graph: effects of VSX2 and its variants on reporter activity were not statistically significant. Right graph: H-MITF repressed reporter activity, whereas OTX1 enhanced reporter activity. Reporter activity in cells co-expressing of OTX1 and H-MITF was significantly higher than the sum of the factors expressed individually (** associated with lines over bars). H-MITF[mi] enhanced reporter activity, but reporter activity in cells co-expressing OTX1 and H- MITF[mi] was not significantly different than the sum of the two factors expressed individually. * P≤0.05; ** P≤0.01; *** P≤0.001.
p27 is a candidate target of transcriptional activation by Mitf in chick RPE [69]. As our data suggested that p27 is downstream of H-Mitf, we tested whether H-Mitf enhances reporter activity using ∼1.1 kb of the p27 promoter region (pGL3B-p27; Figure 9B). This was not the case, however, as H-MITF repressed reporter activity in a DNA binding-dependent manner (Figure 9B, graph I). We also tested VSX2 and its variants and found that both VSX2 and VSX2[R227W] enhanced reporter activity whereas VSX2[R200Q] had no effect (Figure 9B, graph II). Interestingly, when H-MITF was expressed with VSX2[R227W], reporter activity increased further and beyond that observed for VSX2 (Figure 9B, graph III). This appeared to depend on DNA binding by VSX2[R227W] because the enhancement was eliminated in a mutant protein containing both VSX2[R200Q] and VSX2[R227W] (RQRW; Figure 9B, graph III). The enhancement also depended on H-MITF DNA binding by since reporter activity was reduced when VSX2[R227W] was co-expressed with H-MITF[mi] (Figure 9B, graph IV).
The dependence of the enhanced reporter activity on DNA binding suggested that these proteins could bind in the p27 promoter region. To test this, we searched 1.1 kb of the p27 5′-intergenic region directly upstream of the transcriptional start site for Mitf and Vsx2 binding sites. Three Mitf consensus sequences (CANNTG) and five homeodomain core sequences (TAAT) were identified (Figure 9C). However, none of the homeodomain core sequences, when extended, matched the VSX consensus sequence (PyTAATTPuPu; Py, pyrimidine; Pu, Purine). We then performed a ChIP scan using 6 primer pairs that cover this intergenic region and identified one primer pair (primer set 2) that showed enrichment with the VSX2 and MITF antibodies in E12.5 R227W retinal chromatin lysates (data not shown). One of the putative Mitf binding sites was contained within the region amplified by primer set 2 (Figure 9C). Next, we performed ChIP-qPCR to determine the relative occupancies of VSX2 and MITF in this region in E12.5 wild-type, R200Q, and R227W retinal cells. As expected, MITF occupancy was not detected in wild-type but was detected in R200Q and R227W chromatin (Figure 9C, left graph). Importantly, the relative enrichment in MITF occupancy was higher in R227W chromatin and this was coincident with VSX2 occupancy, which was found only in R227W chromatin (Figure 9C, right graph).
The lack of a candidate VSX2 binding site or homeodomain core sequence in the ChIP-enriched region suggested that the interaction of chromatin and the VSX2[R227W] protein could be indirect and mediated by an interaction with MITF. The switch in MITF activity from repressing to enhancing reporter activity in the presence of VSX2[R227W] also suggested that an interaction between the two proteins was influencing p27 transcriptional regulation. Co-immunoprecipitation (co-IP) of E12.5 R227W retinal lysates using the VSX2 antibody for IP and the MITF antibody for western blot revealed that this was indeed the case (Figure 9D, top left panel). Perhaps more surprising, this interaction was not unique to VSX2[R227W] since VSX2[R200Q] also co-immunoprecipitated with MITF in R200Q retinal lysates (Figure 9D, top right panel). Co-IP reactions were negative in the orJ retina, consistent with the lack of Vsx2 protein in this mutant (data not shown). Furthermore, all three VSX2 variants co-immunoprecipitated with H-MITF in transfected HEK293 cells and these interactions were not disrupted by the mi mutation (Figure 9D, bottom panels), which suggests that these interactions were intact in the mi and Vsx2 compound mutants. The sum of these data suggests that the novel regulation of p27 expression in the R227W retina is dependent on the unique convergence of weak DNA binding by VSX2[R227W], the normal DNA binding activity by MITF, and the interaction between the two proteins.
While we now have explanations for the elevated levels of p27 and D-Mitf in the R227W mutant retina, how the H-Mitf level increased was still not clear. Although VSX2 is associated with chromatin in the H-Mitf regulatory region [39], reporter activity in HEK293 cells transfected with pGL3B-HMitf was not different between cells expressing VSX2 or its variants (Figure 9E, left graph). H-MITF was also not sufficient to enhance reporter activity, but rather caused moderate repression (Figure 9E, right graph). In contrast, OTX1 increased reporter activity and this was further enhanced by the presence of H-MITF (Figure 9E, right graph). This interaction was synergistic because the activity of OTX1 and H-MITF expressed together was greater than the sum of the activities of the factors expressed individually, and the P-value (0.004) from a two-way ANOVA supports this conclusion. Unexpectedly, the relative luciferase activity in cells co-expressing OTX1 and the H-MITF[mi] was similar to the co-expression of OTX1 and wild-type H-MITF (Figure 9E, right graph). In this case, however, the effect was not synergistic (P = 0.09, two-way ANOVA), but rather reflected an additive effect since H-MITF[mi] expressed alone also enhanced luciferase activity over the control (Figure 9E, right graph). These observations indicated that the synergistic effect of Otx1 and H-Mitf on the H-Mitf promoter was dependent on DNA binding by H-Mitf. In sum, the uniquely elevated level of H-Mitf in the R227W mutant was not likely due to the direct action of the VSX2[R227W] protein at the H-Mitf promoter, but rather to the collaboration of Otx1 and H-Mitf, and possibly other Mitf isoforms.
Discussion
We show that the homeodomain and CVC mutant proteins VSX2[R200Q] and VSX2[R227W] have diminished DNA binding capacities but retain other functions important for transcriptional activity such as nuclear localization and the ability to regulate transcription when fused to a heterologous DNA binding domain. As in humans, both mutations cause severe non-syndromic bilateral microphthalmia in mice thus confirming their pathogenicity. The recessive nature of these alleles in both species suggests that the alterations in the functional properties of VSX2 caused by each mutation are conserved. Our data argue that the homeodomain is dependent on the CVC domain for high affinity DNA binding highlighting the importance of non-homeodomain residues in mediating canonical homeodomain function. Furthermore, our data suggest that high affinity DNA binding by VSX2 is required to suppress the activation of transcriptional circuits that interfere with the retinal gene expression program in RPCs.
Homeodomain-dependent DNA binding is required for VSX2 function
Two ways in which a mutation can alter the DNA binding properties of a protein are by changing the binding site preference (gain of function) or by reducing overall DNA binding affinity (loss of function). Our data support the contention that the VSX2[R200Q] protein is DNA binding deficient: the VSX2[R200Q] mutant protein failed to bind its preferred sites in vitro and in vivo and the R200Q allele is recessive. Important to note, however, is that the Otx1 transcript level in the R200Q retina was not upregulated, as in the orJ and R227W retinas. On this basis, the R200Q allele behaves as a strong hypomorph since its phenotype at the cell and tissue levels overlaps with the null phenotype, but with at least one difference at the molecular level.
While the low level of Otx1 also suggests that the VSX2[R200Q] protein can still regulate some downstream factors, it is unlikely to accomplish this through direct DNA binding. R200 corresponds to residue 53 of the homeodomain and an arginine at this position (HD-R53) is among the most conserved residues in the homeodomain. HD-R53 forms hydrogen bonds with two phosphates in the DNA backbone and is not directly involved with sequence specificity. Rather it orients the recognition helix into the major groove such that other homeodomain residues can make base-specific contacts, thereby enabling stable and sequence-specific DNA binding [2], [3], [70], [71]. The glutamine substitution likely disrupted the ability of the mutant protein to productively dock in the major groove of the DNA in its binding site, thereby greatly reducing or eliminating its ability to directly regulate downstream target genes. Thus, the lack of Otx1 upregulation in the R200Q retina could be the result of a more complex mechanism, such as by interfering with or altering the transcriptional regulation of an interacting partner such as Mitf.
The CVC domain is required for high-affinity DNA binding by the homeodomain
Genetic data from humans, C. elegans, and now mouse confirm that the CVC domain is essential for the function of Vsx-class proteins. Our EMSA and ChIP data show that a single amino acid substitution in the CVC domain (R227W) weakened the DNA binding capacity of VSX2 to its preferred sites, indicating that optimal homeodomain function depends on the CVC domain. How the CVC domain assists the homeodomain is not clear. One possibility is that it recruits additional proteins required for DNA binding. Alternatively, the CVC domain may directly interact with the homeodomain to overcome a structural constraint on the homeodomain-DNA interaction, similar to that proposed for the C-terminal tail in PBX homeodomain proteins [12]. Structural limitations can be an intrinsic property of the homeodomain or the DNA binding site [14], but since the preferred binding sites of closely related homeodomain proteins such as Rx, Alx, and Arx overlap with the VSX proteins [10], and yet these proteins lack CVC domains, leads us to predict that any structural limitation will be endemic to VSX homeodomains.
The R227W allele is a recessive neomorph that activates a cryptic transcriptional circuit through its interaction with Mitf
Identifying the mechanisms underlying the mutant phenotypes revealed how R227W could surpass orJ and R200Q in severity (Figure 10). During normal eye development, Mitf expression is activated in optic neuroepithelial cells (MitfONC) of the optic vesicle, presumably by signals that promote RPE specification and the pigmentation program [72]. This is followed by activation of Vsx2 in the presumptive retinal domain, which leads to repression of Mitf activity in RPCs (MitfRPC), effectively blocking the execution of the pigmentation program in these cells (Figure 10A). In orJ and R200Q mice, the ability to repress Mitf in RPCs is lost, leading to maintained Mitf activity and an increased probability that a pigmentation program will be activated. The primary differences are that in orJ mice, VSX2 protein is absent (Figure 10B) and in R200Q mice, VSX2[R200Q] protein is present but unable to bind DNA (Figure 10C). Like the orJ and R200Q mutants, Mitf expression also persists in early RPCs of the R227W mutant. Our in vitro reporter data indicates that the VSX2[R227W] protein can still repress D-Mitf, but the in vivo ChIP data suggests that its weak DNA binding may reduce or abrogate its repressive effects on the endogenous D-Mitf promoter. Ultimately, our data suggests the interaction between the VSX2[R227W] and Mitf proteins activates a positive feedback loop that significantly elevates the expression of p27, Otx1 and Mitf, leading to a robust pigmentation program and a blocked retinal program in R227W RPCs (Figure 10D). Our genetic data also suggests that p27 is a novel target specific to the R227W:MITF complex, even though Mitf is proposed to be a direct regulator of p27 expression in chick RPE [69].
Figure 10. Regulation of pigmentation programs in wild-type and mutant RPCs.
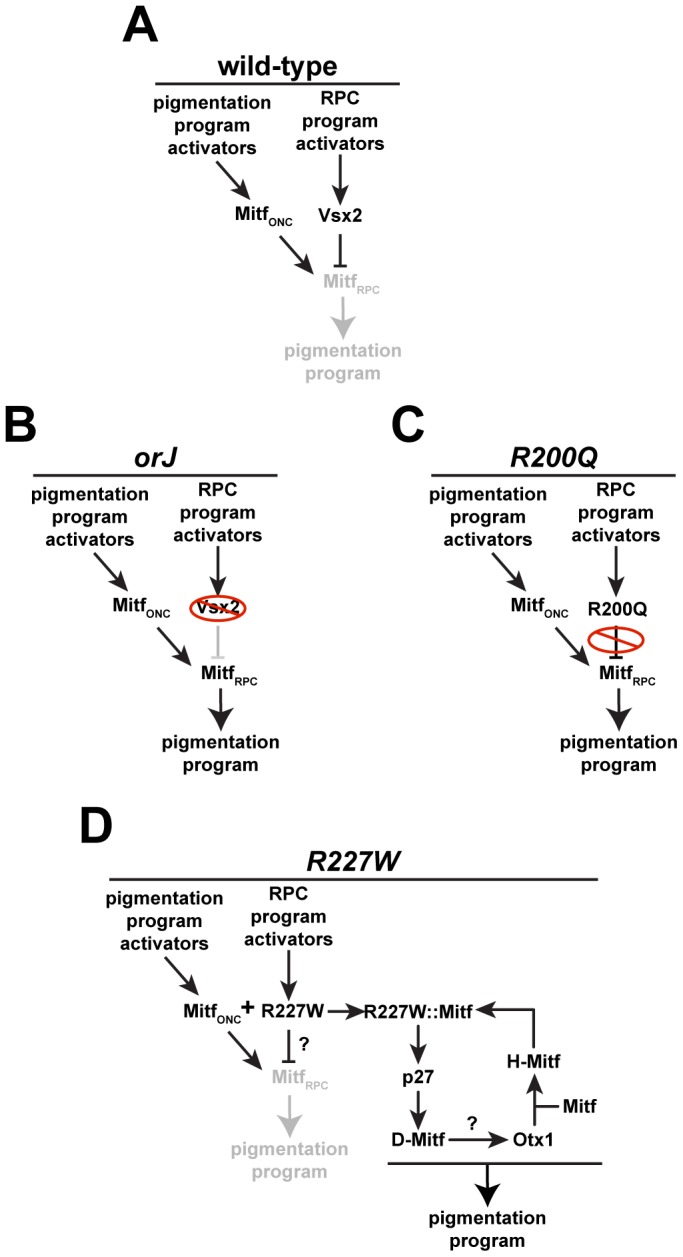
(A) During early eye development, Mitf is expressed in optic neuroepithelial cells (MitfONC) in response to upstream activators. Vsx2 expression is activated in the newly specified retinal domain by upstream activators, which leads to repression of Mitf in RPCs (MitfRPC) and suppression of the pigmentation program. (B) In orJ mice, Mitf persists in RPCs because the VSX2 protein is absent, which increases the probability that pigmentation will occur. (C) In R200Q mice, VSX2[R200Q] protein is present but unable to bind DNA, allowing Mitf to persist in RPCs, increasing the probability of pigmentation. (D) In R227W mice, VSX2[R227W] protein is present and may still suppress the pathway that leads to pigmentation in orJ and R200Q RPCs, but its interaction with Mitf combined with its weak DNA binding activity engages a novel positive feedback loop that activates a robust pigmentation program. Our genetic data place Otx1 downstream of p27 and D-Mitf, but the mechanism causing its elevated expression is not clear.
Mechanistically, manifestation of the R227W phenotype likely occurs in two steps; the weakened DNA binding activity first acts as a partial loss-of-function that is sufficient to allow persistent Mitf expression. This is followed by the activation and maintenance of the feedback loop, which depends on the protein interaction between the VSX2[R227W] and MITF proteins. Since the MITF interaction is shared among the three VSX2 variants, it by definition is not a gain-of-function activity, which suggests there must be another activity unique to the VSX2[R227W] variant. This novel activity could be the result of the unique combination of the weak DNA binding caused by the R227W mutation and the interaction with MITF, ultimately leading to a change in transcriptional regulation of downstream targets, novel or otherwise. The VSX2[R227W] protein may have acquired novel off-target binding properties allowing it regulate a suite of genes not normally regulated by Vsx2. While possible, this is unlikely on its own to explain the phenotype because acquisition of new binding site preferences is typically revealed as a dominant or semi-dominant gain of function phenotype, which was not observed. In addition to its effects on DNA binding, the R227W mutation may have induced a conformational change in the VSX2:MITF complex that alters how the complex interacts with DNA and regulates transcription. It is also possible that another interacting partner is involved and is present only in the R227W mutant. Distinguishing between these possiblities requires further genetic and molecular analyses.
Since the positive feedback loop is not found in the orJ or R200Q mutants, it is by definition a novel mechanism. Combined with the genetic properties then, the R227W allele best fits the classification of a recessive neomorph. Although predicted to exist over 80 years ago by H.J. Muller [73], reports of recessive neomorphic alleles are difficult to find. Recently, strong evidence for another atypical allele, the recessive antimorph, was reported for the CXC domain-containing gene TSO1 in Arabidopsis [74]. It is likely that more recessive neomorphs and antimorphs will be identified as targeted alleles are generated in genetic models that mimic disease-linked missense mutations found in natural populations. Their identification will require direct comparison to a corresponding null allele and may be more easily identified at loci that are highly susceptible to missense mutations and show a wide range of phenotypes.
A novel role for p27 as a regulator of transcription
In identifying the molecular mechanism driving the R227W phenotype, we uncovered evidence that p27 regulates transcription, specifically of the D-Mitf isoform. This was unexpected because D-Mitf levels are not dependent on p27 in the orJ retina (data not shown), indicating that this functional link is context-dependent. Consistent with this, partial genetic reduction in p27 expression in the R227W retina reduced D-Mitf expression, which suggests that the elevated expression of p27 unmasked its transcriptional activity. It is not yet clear if p27 regulates transcription in the wild-type retina or if this is a common feature of p27 function. There are hints, however, that support these possibilities. p27 enhances reporter activity driven by Myelin Basic Protein promoter in an Sp1-dependent manner [75], [76]. p27 also binds to and stabilizes Neurogenin 2, a pro-neurogenic bHLH transcription factor [77]. In the retina, Cyclin D1 was recently shown to bind to chromatin and regulate genes such as Notch1 [78]. As Cyclin D1 and p27 interact genetically and biochemically [79], [80], it is conceivable that misregulation of transcriptional targets could be a causative factor in the alterations in retinal development found in Cyclin D1 and p27 knockout mice [61], [81]–[84].
Vsx2 acts as a gatekeeper allowing the retinal progenitor program to proceed without interference from other gene expression programs
The neuroepithelial-derived components of the eye field develop through a process of progressive specification from the anterior neuroectoderm and Vsx2 expression is arguably the strongest indicator of retinal domain specification [72]. To date, it is the only transcription factor expressed exclusively and comprehensively in the retinal domain and its onset of expression immediately precedes the formation of the optic cup, which coincides with the earliest morphological changes that distinguish RPCs from the remainder of the optic neuroepithelium [17], [54]. Since Vsx2 expression initiates well after eye field specification, it stands to reason that activation of the RPC program is an actively promoted process. Consistent with this, eye field specification occurs and optic vesicles form in the Lhx2 mutant, but Vsx2 is not expressed and the RPC program fails to initiate [54]. Rather, the more likely fate following eye field specification is pigmented epithelium. Mitf is expressed throughout the optic neuroepithelium prior to Vsx2 expression and in mice in which FGF or BMP7 signaling is disrupted, Mitf expression persists in the presumptive retinal domain, which correlates with the failure to express Vsx2 and a high probability of pigmentation where the retina would normally form [72], [85], [86].
Despite these findings, Vsx2 does not act as a master regulator to specify or activate the RPC program because the retinal domain and optic cup still form in all three Vsx2 mutants. Retinal development, although compromised, still occurs in the absence of VSX2 protein (orJ mouse) and this is not due to genetic compensation or functional redundancy by Vsx1 [32]. Importantly, the Vsx2 gene is still expressed in the Vsx2 mutants, indicating that initiation of the RPC program is independent of Vsx2 function, or in the case of the R227W mutant, despite the neomorphic behavior caused by the mutant protein. Thus, a primary role of Vsx2 is not to initiate specification, but rather to allow the RPC program to proceed without interference from competing gene expression programs. A failure to block these programs may not convert RPCs to another well-defined fate per se, but they hinder the RPC program and lead to, in some contexts, to aberrant differentiation as revealed by the pigmentation phenotype.
Antagonizing Mitf activity through transcriptional repression is a key function for Vsx2 in this process. While a likely mechanism for D-Mitf regulation (this study; [39]) other Mitf isoforms are also misexpressed in the Vsx2 mutant retinas and our data suggest that the H and A isoforms are not transcriptionally regulated by Vsx2. Supporting this last point is that A-Mitf is expressed in the wild-type retina during development [39]. Although A-Mitf may be expressed below a threshold needed to disrupt the RPC program, it is noteworthy that in vivo overexpression of Mitf failed to interfere with retinal development as long as Vsx2 was expressed [41]. This suggests that an additional Vsx2-dependent mechanism exists for antagonizing Mitf activity and we propose that it requires VSX2 binding to MITF, preventing MITF from regulating genes important for pigmentation. This mechanism could be important during the initial specification of the retinal domain, when Vsx2 has not had enough time to efficiently downregulate Mitf activity through transcriptional repression (Figure 11A), and during retinal histogenesis for Mitf isoforms that are not transcriptionally repressed by Vsx2 (Figure 11B). In effect, this enables Vsx2 to block Mitf from interfering with the execution of the RPC program by regulating its activity at two levels. Interestingly, the functional significance of the protein interaction between Vsx2 and Mitf may also depend on high affinity DNA binding by Vsx2, which was suggested by the behavior of the VSX2[R200Q] and VSX2[R227W] proteins; both interacted with MITF but were unable to prevent Mitf-dependent alterations in retinal development. In the case of VSX2[R227W], the interaction likely contributed to the more severe Mitf-dependent effects.
Figure 11. Models of Mitf regulation in RPCs during normal retinal development.

(A) In response to RPC program activators, Vsx2 expression is initiated and newly produced protein interacts with preexisting MITF protein, preventing access to targets required for the pigmentation program. (B) Once VSX2 protein expression is established, it regulates Mitf activity by directly repressing Mitf transcription of isoforms such as D-Mitf and binds to MITF proteins produced from promoters that Vsx2 does not efficiently repress such as A-Mitf and possibly H-Mitf.
In sum, this study provides an explanation for the stable association of the homeodomain and CVC domain in the VSX proteins through evolution. Our mutational analysis suggests that an essential function of the CVC domain is to assist the homeodomain in achieving high affinity binding to its preferred sites. Although the specific downstream targets of the VSX proteins may vary in vertebrates and invertebrates, the molecular requirement for the CVC domain in allowing high affinity DNA binding by VSX homeodomains was likely to have been established much earlier in animal evolution than the innovation of the vertebrate eye. In the case of Vsx2, the stability of the homeodomain-CVC domain arrangement may have provided a platform for Vsx2 or its archetype to acquire the ability to efficiently regulate a complex locus such as Mitf, a key step in mammalian eye organogenesis and retinal development. Finally, our study shows that the generation of a small, but targeted allelic series of mutations in mice has the potential to reveal insight into protein function and human disease etiology.
Materials and Methods
Ethics statement
Procedures involving mice were approved by the Institutional Animal Care and Use Committee at the University of Utah and conformed to the standards outlined in the ARVO Statement for the Use of Animals in Ophthalmic and Vision Research.
Mice
Vsx2orJ (129/SvJ genetic background) and Mitfmi (B6C3Fe genetic background) mice were obtained from the Jackson Laboratories (Bar Harbor, ME). p27 knockout mice (129/C57Bl6 hybrid genetic background) were provided by Drs. Matthew Fero and James Roberts (Fred Hutchinson Cancer Center, Seattle, WA). Vsx2R200Q chimeric mice were generated at inGenious Targeting Laboratory (Stony Brook, NY) and Vsx2R227W chimeric mice were generated at the University of Utah Gene Targeting and Transgenic Core Facility (See Text S1 and Figure S1 for details on gene targeting strategy). Germline transmission into the 129/SvJ genetic background was achieved by breeding chimeric mice with mice from the orJ strain. All alleles were identified by PCR genotyping (primers listed in Table S1). For timed matings, females were considered to be at 0.5 days of gestation (E0.5) at noon on the day a vaginal plug was detected.
Proliferation assays
P0 retinal cells from wild-type or orJ mice were dissociated with trypsin and trituration and cultured in DMEM/F12 medium with 1% fetal bovine serum (FBS), growth supplements, and a penicillin/streptomycin mix (Invitrogen) [87]. This age was selected for these and other culture experiments in order to maximize the cell yield on the days the cultures were established. Cells were transfected one day after plating (cell confluence ∼80%) with Lipofectamine and Plus Reagent (Invitrogen). Plasmids (Table S2) were transfected in equal amounts (0.6 µg per well in a 24-well plate, 0.2 µg per well in a 96-well plate). BrdU (10 µg/ml) was added for the last 4.5 hours of the culture period, starting at 44 hr post-transfection. Cultures were fixed with 4% PFA and stained with GFP, VSX2 and BrdU antibodies (Table S3). The percentages of BrdU+ cells in the VSX2+ or GFP+ cell populations were determined. A minimum of three independent trials was performed. Data were graphed as mean±SE. Statistical comparisons were done with the Kruskal-Wallis test. For simplicity, only statistical comparisons of the test conditions (black bars) versus the control (white bar) are shown.
Luciferase assays
Dissociated P0 retinal cells were plated onto poly-D-lysine and 15 µg/ml laminin in the same medium as described above. HEK293 cells were cultured in DMEM supplemented with 10% FBS, 100 U/ml penicillin, and 100 µg/ml streptomycin (Invitrogen). In both cases, cells were transfected one day after plating (cell confluence ∼80%) with Lipofectamine and Plus Reagent. Plasmids are listed in Table S2. To compare reporter activity across conditions, expression vectors were transfected at equimolar concentrations and unless noted, empty expression vectors served as controls. Transfection efficiency was monitored by co-transfection of a Renilla luciferase reporter (Table S2). Cell lysates were prepared 24 h after transfection. Firefly and Renilla luciferase activities were measured with a Dynex Technologies MRX Revelation microplate reader (Dynex Technologies, Denkendorf, Germany) using 100 µl d-luciferin reagent and 100 µl coelenterazine (Biotium, Hayward, CA). Relative luciferase activity was calculated by using the control condition luciferase activity as a normalization factor for each independent trial (performed in duplicate). A minimum of three independent trials was performed for each condition. Data were graphed as mean±SE. Statistical comparisons were done with Fisher's Least Significance Difference (LSD) test and P-value ranges are shown only for test conditions (black bars) versus control (white bar) with the following exceptions: unpaired t-test was used for comparisons of test conditions versus control in Figure 8G and Figure 9E (right graph), and two-way ANOVA was used to test for synergistic interactions between H-Mitf and Otx1, and H-Mitfmi and Otx1 (Figure 9E, right graph).
CAT assays
HEK293 were cultured as described for Luciferase assays. 5 µg of LexA-Vsx2, -R200Q, -R227W or an equimolar amount of LexA were co-transfected with 0.5 µg of Gal4-HSF1 and 0.5 µg of X4G2CAT in 60-mm dishes at ∼50% confluency with Lipofectamine and Plus reagent. CAT assays were performed 24 h post-transfection. The amount of lysate used for CAT analysis was determined by quantifications of western blots of Gal4-HSF1 in ImageJ (http://imagej.nih.gov/ij/). The CAT activity was counted in an LS 6000IC liquid scintillation system (Beckman, Fullerton, CA) 60 min after the addition of [3H]acetyl-CoA (Sigma-Aldrich, St. Louis, MO). Relative CAT activity was calculated by using control condition CAT activity as a normalization factor for each independent trial. Each trial was performed in duplicate and three independent trials were performed. Data were graphed as mean±SE. Statistical comparisons were done with Fisher's Least Significance Difference (LSD) test and P-value ranges are shown only for test conditions (black bars) versus control (white bar).
Explant cultures
Ocular tissues were dissected from E10.5 embryos in HBSS. To implant POM, a local separation of the retina and lens was made with a 32G needle and POM was placed into the separated region. Explants were grown in the culture medium described for dissociated retinal cells. At the end of the culture period (2 days), explants were processed for immuohistology or retinas were dissected free of other tissues and prepared for qRT-PCR.
Immunohistology
Heads, eyes, or retinas were fixed with 4% PFA and cryopreserved [61]. Sections were cut at a thickness of 12 µm. Primary antibodies are listed in Table S3. Primary antibodies were followed with species-specific secondary antibodies conjugated to Alexa Fluor 488 (Invitrogen) or Alexa Fluor 568 (Molecular Probes). Nuclei were stained with 4, 6-diamidino-2-phenylindole (DAPI; Sigma-Aldrich).
Visualization of pigmentation in combination with fluorescence-based images
Images of DAPI stained sections were captured with epi-fluorescence illumination followed by bright-field illumination (BF). BF images were adjusted in Photoshop (Adobe, San Jose, CA) to high contrast and pixel values were converted to their inverse using the invert image function. DAPI images were made transparent with the screen layer setting and the images were merged.
Eye circumference measurements
Eye circumference was assessed in image J and graphed as mean±SE. Statistical comparisons were done with the Kruskal-Wallis test.
Quantitative real-time RT–PCR (qRT–PCR)
Unless noted, retinal tissue was dissected from the surrounding RPE, POM, and lens following treatment with 40 µg/ml dispase (Sigma-Aldrich) in HBSS for 5 min at room temperature. Total RNA was isolated with the RNeasy Mini or Micro Kit according to the manufacturer's instructions (Qiagen, Valencia, CA). cDNAs were prepared with the Superscript RT III Kit (Invitrogen) using 20 ng of total RNA and purified with the PCR purification kit (Qiagen). cDNA corresponding to 2 ng of total template RNA was used for each PCR reaction.
PCR was done on an ABI 7300 Real-Time PCR System with Power SYBR green PCR Master Mix and using the Relative Standard Curve Method for detection and measurement (Applied Biosystems, Invitrogen). Optimization was performed with serial dilutions of cDNA prepared from orJ retina. Gapdh was used as the endogenous control mRNA for all samples. RNA preparations from orJ or R227W retinas were used as calibrator samples. PCR product specificity was confirmed by gel electrophoresis and sequencing. Primer sets are listed in Table S1.
For each trial, measurements were calculated as the expression level (per condition) normalized to the calibrator sample. The mean±SE of the independent trials for each condition was then normalized by the mean of the control group and graphed as “relative to [control]” ([control] = orJ or R227W). Each trial was performed in duplicate and a minimum of three independent trials were performed with the following exceptions: the expression levels of Otx1 and Otx2 in the wild-type retina were measured in two independent samples (Figure 5J). Statistical comparisons were done with the Kruskal-Wallis test and P-value ranges are shown only for test conditions (black bars) versus control (white bar) with the exception of Figure 6H, which shows P-value ranges for all comparisons.
Electrophoretic mobility shift assays (EMSA)
Expression plasmids are listed in Table S2. Proteins were synthesized by in vitro translation (ivt) using the EcoPro T7 System (Novagen, Madison, WI) and relative yields were estimated by western blot analysis probed with anti-VSX2 and detected with SuperSignal West Dura Extended Duration Substrate (Thermo Scientific, Rockford, IL). [32]P end-labeled double stranded P3 oligonucleotide (Table S1) was used to assess DNA binding [32]. The signals for Western blot and EMSA were captured on BioMax Light Film (Kodak, Rochester, NY).
Immunoprecipitations (IP)
Non-denatured native protein lysates from E12.5 retinas with lens or transfected HEK293 cells were prepared RIP buffer (Santa Cruz) on ice for 60 min. ∼40 retinas were pooled for each lysate preparation. Equivalent volumes of cell lysates (input) were incubated with VSX2 antibody or sheep IgG at 4°C for 1 hour or overnight followed by Protein-G agarose (Roche) for 1 hour. Immunoprecipitated proteins were analyzed by Western blot using Mitf antibody. Signals for Western blot were captured by chemiluminescence on a ChemiDoc XRS (Bio-Rad, Hercules, CA).
Chromatin immunoprecipitations (ChIP)
ChIP assays were performed on native retinal lysates as previously described [32]. Retinas with lens were isolated from E12.5 wild-type, R200Q or R227W eyes, and retinas were isolated from P0 wild-type eyes and prepared for ChIP assay. Antibodies are listed in Table S3, primer sets in Table S1. Quantative real time PCR (qPCR) using the Relative Standard Curve Method was performed to assess the relative strengths of protein∶chromatin interactions in each genotype. Results were expressed as the percentage enrichment normalized to the equivalent starting material (input). Statistical comparisons were done with the Kruskal-Wallis test and P-value ranges are shown for test conditions (black bars) versus IgG controls (white bars) in each genotype.
Supporting Information
Generation of R200Q and R227W mutant mice. (A) Sequence tracks of PCR products encompassing the R200Q or R227W mutations. Asterisks denote base substitutions. Template DNA was prepared from mouse-tail DNA (wild-type) and targeted ES clones (mutants). (B) Schematic representation of the targeting strategy. The R200Q and R227W mutations (asterisk) were introduced into exon 4 and the ACN cassette [46] was cloned into intron 3 of Vsx2. These elements were then placed into the pTK1TK2 targeting vector [88]. After homologous recombination, R200Q-ACN and R227W-ACN chimeric mice were generated. The ACN cassette was deleted by Cre recombination in the male germline generating the R200Q and R227W alleles. Chimeric males were crossed with wild-type females to generate germline-transmitted R200Q and R227W heterozygous mice. B: BamHI, H: HindIII, N: NheI. (C) Southern blots of genomic DNA from electroporated ES cell clones containing homologously recombined R200Q-ACN and R227W-ACN insertions. BamHI digestion and 3′ flanking probe (shown in B) were used. The wild-type band is 6.7 kb and the correctly targeted band is 7.6 kb. (D) Genotyping of mutant mice. P1 and P2 primers shown in B (arrows) were used for PCR genotyping. As the mutant allele contains a 34 bp insertion, wild-type and mutant alleles give 258 bp and 292 bp bands, respectively.
(TIF)
Proliferation changes associated with Vsx2[R200Q] and Vsx2[R227W] overexpression. (A) Micrographs showing expression of enhanced Green Fluorescent Protein fused to a nuclear localization signal (nlsGFP), VSX2, VSX2[R200Q], and VSX2[R227W] in transfected P0 orJ retinal cells. (B) Phosphorylated histone H3 (pHH3) expression was reduced in the mutant retinas compared to wild-type. (C) Quantification of BrdU incorporation in P0 orJ retinal cells overexpressing nlsGFP (control), VSX2, VSX2[R200Q], or VSX2[R227W]. (D) Quantification of BrdU incorporation in P0 wild-type retinal cells overexpressing nlsGFP (control), VSX2, VSX2[R200Q], or VSX2[R227W]. Cells were transfected at 24 hr following initial plating and cultured for an additional 48.0 hr. BrdU was added for the last 4.5 hr of the culture period. Only wild-type VSX2 enhanced proliferation over the control. * P≤0.05; ** P≤0.01 Scale bars: 100 µm.
(TIF)
Marker expression in wild-type, orJ, R200Q, and R227W retinas at P0. (A–D) Neurofilament-M (NF-M) expression in inner retinal neurons (differentiated cell layer, DCL) and horizontal cells (horiz), which occupy the neuroblast layer (NBL), was present in all genotypes except R227W. Similar results were obtained for the retinal ganglion cell marker POU4F2 (E–H) and the proliferation/RPC marker PCNA (I–L). Dashed lines bound retinas. Scale bar: 100 µm.
(TIF)
Marker expression in wild-type and R227W/+ retinas at P0. Expression patterns of TUBB3 (A,B), NF-M (C,D), and SOX2 (E,F) in wild-type and R227W/+ retinas were similar. Scale bars: 100 µm.
(TIF)
Invasion of POM into the vitreal chamber in R227W eyes. (A) PITX2+ cells filled the vitreal chamber in the E17.5 R227W eye. (B) The relative expression levels of D-Mitf and J-Mitf were not statistically different between control (−POM) and POM implanted whole retina and lens explant (POM imp) cultures (E10.5+2DIV).
(TIF)
Immunolocalization of MITF and p27 in selected genotypes at E12.5. MITF expression in R227W (A) and R227W; p27+/− (B) eyes. (C) No primary antibody control. p27 expression in wild-type (D), orJ (E), R200Q (F), and R227W (G) eyes. Antigen retrieval was used for p27 staining to reveal staining in neuroblast layers. Bright staining at inner surface of central retina of wild-type is newly generated postmitotic precursors. Bright staining at the periphery of the R200Q retina is also observed occasionally in orJ retina at this age. p27 is also abundantly expressed in lens and surrounding extraocular tissues.
(TIF)
Oligonucleotides.
(DOC)
Plasmids.
(DOC)
Primary antibodies.
(DOC)
Acknowledgments
We wish to thank Rod Bremner, Mario Capecchi, Phil Gage, and Tord Hjalt for reagents; Greg Stoddard for assistance with statistics; and Sabine Fuhrmann and Anthea Letsou for their critical reading of the manuscript. We also thank Amy Kircher, Lai Xue, Anna Clark, Dustin Porter, Alex Swan, and Weiyi Le for technical assistance, and Wei Chen and Sen Wu for their assistance in developing and implementing the gene targeting strategy.
Funding Statement
This work was supported by the U.S. National Institutes of Health [R01-EY013760; R21-EY018392; P30-EY014800; American Recovery and Reinvestment Act; UL1-RR025764 (statistics support); C06-RR11234 (statistics support)] and by Research to Prevent Blindness (unrestricted funding to the Department of Ophthalmology). This work was also supported by a Research to Prevent Blindness Sybil Harrington Research Scholar Award to EML. The funders had no role in study design, data collection and analysis, decision to publish, or preparation of the manuscript.
References
- 1. Vaquerizas JM, Kummerfeld SK, Teichmann SA, Luscombe NM (2009) A census of human transcription factors: function, expression and evolution. Nat Rev Genet 10: 252–263. [DOI] [PubMed] [Google Scholar]
- 2. Fraenkel E, Pabo CO (1998) Comparison of X-ray and NMR structures for the Antennapedia homeodomain-DNA complex. Nat Struct Biol 5: 692–697. [DOI] [PubMed] [Google Scholar]
- 3. Chi YI (2005) Homeodomain revisited: a lesson from disease-causing mutations. Hum Genet 116: 433–444. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4. Affolter M, Slattery M, Mann RS (2008) A lexicon for homeodomain-DNA recognition. Cell 133: 1133–1135. [DOI] [PubMed] [Google Scholar]
- 5. Berger MF, Badis G, Gehrke AR, Talukder S, Philippakis AA, et al. (2008) Variation in homeodomain DNA binding revealed by high-resolution analysis of sequence preferences. Cell 133: 1266–1276. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6. Noyes MB, Christensen RG, Wakabayashi A, Stormo GD, Brodsky MH, et al. (2008) Analysis of homeodomain specificities allows the family-wide prediction of preferred recognition sites. Cell 133: 1277–1289. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7. Holland PW, Booth HA, Bruford EA (2007) Classification and nomenclature of all human homeobox genes. BMC Biol 5: 47. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8. Bach I, Carriere C, Ostendorff HP, Andersen B, Rosenfeld MG (1997) A family of LIM domain-associated cofactors confer transcriptional synergism between LIM and Otx homeodomain proteins. Genes Dev 11: 1370–1380. [DOI] [PubMed] [Google Scholar]
- 9. Phillips K, Luisi B (2000) The virtuoso of versatility: POU proteins that flex to fit. J Mol Biol 302: 1023–1039. [DOI] [PubMed] [Google Scholar]
- 10. Burglin TR (2011) Homeodomain subtypes and functional diversity. Subcell Biochem 52: 95–122. [DOI] [PubMed] [Google Scholar]
- 11. Mann RS, Lelli KM, Joshi R (2009) Hox specificity unique roles for cofactors and collaborators. Curr Top Dev Biol 88: 63–101. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12. Piper DE, Batchelor AH, Chang CP, Cleary ML, Wolberger C (1999) Structure of a HoxB1-Pbx1 heterodimer bound to DNA: role of the hexapeptide and a fourth homeodomain helix in complex formation. Cell 96: 587–597. [DOI] [PubMed] [Google Scholar]
- 13. Farber PJ, Mittermaier A (2011) Concerted dynamics link allosteric sites in the PBX homeodomain. J Mol Biol 405: 819–830. [DOI] [PubMed] [Google Scholar]
- 14. Rohs R, Jin X, West SM, Joshi R, Honig B, et al. (2010) Origins of specificity in protein-DNA recognition. Annu Rev Biochem 79: 233–269. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15. Svendsen PC, McGhee JD (1995) The C. elegans neuronally expressed homeobox gene ceh-10 is closely related to genes expressed in the vertebrate eye. Development 121: 1253–1262. [DOI] [PubMed] [Google Scholar]
- 16. Levine EM, Hitchcock PF, Glasgow E, Schechter N (1994) Restricted expression of a new paired-class homeobox gene in normal and regenerating adult goldfish retina. J Comp Neurol 348: 596–606. [DOI] [PubMed] [Google Scholar]
- 17. Liu IS, Chen JD, Ploder L, Vidgen D, van der Kooy D, et al. (1994) Developmental expression of a novel murine homeobox gene (Chx10): evidence for roles in determination of the neuroretina and inner nuclear layer. Neuron 13: 377–393. [DOI] [PubMed] [Google Scholar]
- 18. Forrester WC, Perens E, Zallen JA, Garriga G (1998) Identification of Caenorhabditis elegans genes required for neuronal differentiation and migration. Genetics 148: 151–165. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19. Altun-Gultekin Z, Andachi Y, Tsalik EL, Pilgrim D, Kohara Y, et al. (2001) A regulatory cascade of three homeobox genes, ceh-10, ttx-3 and ceh-23, controls cell fate specification of a defined interneuron class in C. elegans. Development 128: 1951–1969. [DOI] [PubMed] [Google Scholar]
- 20. Bar-Yosef U, Abuelaish I, Harel T, Hendler N, Ofir R, et al. (2004) CHX10 mutations cause non-syndromic microphthalmia/anophthalmia in Arab and Jewish kindreds. Hum Genet 115: 302–309. [DOI] [PubMed] [Google Scholar]
- 21. Heon E, Greenberg A, Kopp KK, Rootman D, Vincent AL, et al. (2002) VSX1: a gene for posterior polymorphous dystrophy and keratoconus. Hum Mol Genet 11: 1029–1036. [DOI] [PubMed] [Google Scholar]
- 22. Mintz-Hittner HA, Semina EV, Frishman LJ, Prager TC, Murray JC (2004) VSX1 (RINX) mutation with craniofacial anomalies, empty sella, corneal endothelial changes, and abnormal retinal and auditory bipolar cells. Ophthalmology 111: 828–836. [DOI] [PubMed] [Google Scholar]
- 23. Aung T, Lim MC, Wong TT, Thalamuthu A, Yong VH, et al. (2008) Molecular analysis of CHX10 and MFRP in Chinese subjects with primary angle closure glaucoma and short axial length eyes. Mol Vis 14: 1313–1318. [PMC free article] [PubMed] [Google Scholar]
- 24. Iseri SU, Wyatt AW, Nurnberg G, Kluck C, Nurnberg P, et al. (2010) Use of genome-wide SNP homozygosity mapping in small pedigrees to identify new mutations in VSX2 causing recessive microphthalmia and a semidominant inner retinal dystrophy. Hum Genet 128: 51–60. [DOI] [PubMed] [Google Scholar]
- 25. Reis LM, Khan A, Kariminejad A, Ebadi F, Tyler RC, et al. (2011) VSX2 mutations in autosomal recessive microphthalmia. Mol Vis 17: 2527–2532. [PMC free article] [PubMed] [Google Scholar]
- 26. Tanwar M, Kumar M, Nayak B, Pathak D, Sharma N, et al. (2010) VSX1 gene analysis in keratoconus. Mol Vis 16: 2395–2401. [PMC free article] [PubMed] [Google Scholar]
- 27. Watson T, Chow RL (2011) Absence of Vsx1 expression in the normal and damaged mouse cornea. Mol Vis 17: 737–744. [PMC free article] [PubMed] [Google Scholar]
- 28. Ferda Percin E, Ploder LA, Yu JJ, Arici K, Horsford DJ, et al. (2000) Human microphthalmia associated with mutations in the retinal homeobox gene CHX10. Nat Genet 25: 397–401. [DOI] [PubMed] [Google Scholar]
- 29. Faiyaz-Ul-Haque M, Zaidi SH, Al-Mureikhi MS, Peltekova I, Tsui LC, et al. (2007) Mutations in the CHX10 gene in non-syndromic microphthalmia/anophthalmia patients from Qatar. Clin Genet 72: 164–166. [DOI] [PubMed] [Google Scholar]
- 30. Burmeister M, Novak J, Liang MY, Basu S, Ploder L, et al. (1996) Ocular retardation mouse caused by Chx10 homeobox null allele: impaired retinal progenitor proliferation and bipolar cell differentiation. Nat Genet 12: 376–384. [DOI] [PubMed] [Google Scholar]
- 31. Barabino SM, Spada F, Cotelli F, Boncinelli E (1997) Inactivation of the zebrafish homologue of Chx10 by antisense oligonucleotides causes eye malformations similar to the ocular retardation phenotype. Mech Dev 63: 133–143. [DOI] [PubMed] [Google Scholar]
- 32. Clark AM, Yun S, Veien ES, Wu YY, Chow RL, et al. (2008) Negative regulation of Vsx1 by its paralog Chx10/Vsx2 is conserved in the vertebrate retina. Brain Res 1192: 99–113. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33. Vitorino M, Jusuf PR, Maurus D, Kimura Y, Higashijima S, et al. (2009) Vsx2 in the zebrafish retina: restricted lineages through derepression. Neural Dev 4: 14. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34. Dorval KM, Bobechko BP, Ahmad KF, Bremner R (2005) Transcriptional activity of the paired-like homeodomain proteins CHX10 and VSX1. J Biol Chem 280: 10100–10108. [DOI] [PubMed] [Google Scholar]
- 35. Kurtzman AL, Gregori L, Haas AL, Schechter N (2000) Ubiquitination and degradation of the zebrafish paired-like homeobox protein VSX-1. J Neurochem 75: 48–55. [DOI] [PubMed] [Google Scholar]
- 36. Dorval KM, Bobechko BP, Fujieda H, Chen S, Zack DJ, et al. (2006) CHX10 targets a subset of photoreceptor genes. J Biol Chem 281: 744–751. [DOI] [PubMed] [Google Scholar]
- 37. Reichman S, Kalathur RK, Lambard S, Ait-Ali N, Yang Y, et al. (2010) The homeobox gene CHX10/VSX2 regulates RdCVF promoter activity in the inner retina. Hum Mol Genet 19: 250–261. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38. Rowan S, Cepko CL (2005) A POU factor binding site upstream of the Chx10 homeobox gene is required for Chx10 expression in subsets of retinal progenitor cells and bipolar cells. Dev Biol 281: 240–255. [DOI] [PubMed] [Google Scholar]
- 39. Bharti K, Liu W, Csermely T, Bertuzzi S, Arnheiter H (2008) Alternative promoter use in eye development: the complex role and regulation of the transcription factor MITF. Development 135: 1169–1178. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40. Rowan S, Chen CM, Young TL, Fisher DE, Cepko CL (2004) Transdifferentiation of the retina into pigmented cells in ocular retardation mice defines a new function of the homeodomain gene Chx10. Development 131: 5139–5152. [DOI] [PubMed] [Google Scholar]
- 41. Horsford DJ, Nguyen MT, Sellar GC, Kothary R, Arnheiter H, et al. (2005) Chx10 repression of Mitf is required for the maintenance of mammalian neuroretinal identity. Development 132: 177–187. [DOI] [PubMed] [Google Scholar]
- 42. Konyukhov BV, Sazhina MV (1966) Interaction of the genes of ocular retardation and microphthalmia in mice. Folia Biol (Praha) 12: 116–123. [PubMed] [Google Scholar]
- 43. Steingrimsson E, Copeland NG, Jenkins NA (2004) Melanocytes and the Microphthalmia Transcription Factor Network. Annu Rev Genet 38: 365–411. [DOI] [PubMed] [Google Scholar]
- 44. Saadi I, Semina EV, Amendt BA, Harris DJ, Murphy KP, et al. (2001) Identification of a dominant negative homeodomain mutation in Rieger syndrome. J Biol Chem 276: 23034–23041. [DOI] [PubMed] [Google Scholar]
- 45. Chaney BA, Clark-Baldwin K, Dave V, Ma J, Rance M (2005) Solution structure of the K50 class homeodomain PITX2 bound to DNA and implications for mutations that cause Rieger syndrome. Biochemistry 44: 7497–7511. [DOI] [PubMed] [Google Scholar]
- 46. Bunting M, Bernstein KE, Greer JM, Capecchi MR, Thomas KR (1999) Targeting genes for self-excision in the germ line. Genes Dev 13: 1524–1528. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 47. Bone-Larson C, Basu S, Radel JD, Liang M, Perozek T, et al. (2000) Partial rescue of the ocular retardation phenotype by genetic modifiers. J Neurobiol 42: 232–247. [DOI] [PubMed] [Google Scholar]
- 48. Wong G, Conger SB, Burmeister M (2006) Mapping of genetic modifiers affecting the eye phenotype of ocular retardation (Chx10or-J) mice. Mamm Genome 17: 518–525. [DOI] [PubMed] [Google Scholar]
- 49. Dhomen NS, Balaggan KS, Pearson RA, Bainbridge JW, Levine EM, et al. (2006) Absence of chx10 causes neural progenitors to persist in the adult retina. Invest Ophthalmol Vis Sci 47: 386–396. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 50. Green ES, Stubbs JL, Levine EM (2003) Genetic rescue of cell number in a mouse model of microphthalmia: interactions between Chx10 and G1-phase cell cycle regulators. Development 130: 539–552. [DOI] [PubMed] [Google Scholar]
- 51. Livne-Bar I, Pacal M, Cheung MC, Hankin M, Trogadis J, et al. (2006) Chx10 is required to block photoreceptor differentiation but is dispensable for progenitor proliferation in the postnatal retina. Proc Natl Acad Sci U S A 103: 4988–4993. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 52. Sigulinsky CL, Green ES, Clark AM, Levine EM (2008) Vsx2/Chx10 ensures the correct timing and magnitude of Hedgehog signaling in the mouse retina. Dev Biol 317: 560–575. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 53. Konyukhov BV, Sazhina MV (1971) Genetic control over the duration of G 1 phase. Experientia 27: 970–971. [DOI] [PubMed] [Google Scholar]
- 54. Yun S, Saijoh Y, Hirokawa KE, Kopinke D, Murtaugh LC, et al. (2009) Lhx2 links the intrinsic and extrinsic factors that control optic cup formation. Development 136: 3895–3906. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 55. Nguyen M, Arnheiter H (2000) Signaling and transcriptional regulation in early mammalian eye development: a link between FGF and MITF. Development 127: 3581–3591. [DOI] [PubMed] [Google Scholar]
- 56. Bora N, Conway SJ, Liang H, Smith SB (1998) Transient overexpression of the Microphthalmia gene in the eyes of Microphthalmia vitiligo mutant mice. Dev Dyn 213: 283–292. [DOI] [PubMed] [Google Scholar]
- 57. Martinez-Morales JR, Rodrigo I, Bovolenta P (2004) Eye development: a view from the retina pigmented epithelium. Bioessays 26: 766–777. [DOI] [PubMed] [Google Scholar]
- 58. Westenskow PD, McKean JB, Kubo F, Nakagawa S, Fuhrmann S (2010) Ectopic Mitf in the embryonic chick retina by co-transfection of beta-catenin and Otx2. Invest Ophthalmol Vis Sci 51: 5328–5335. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 59. Martinez-Morales JR, Signore M, Acampora D, Simeone A, Bovolenta P (2001) Otx genes are required for tissue specification in the developing eye. Development 128: 2019–2030. [DOI] [PubMed] [Google Scholar]
- 60. Martinez-Morales JR, Dolez V, Rodrigo I, Zaccarini R, Leconte L, et al. (2003) OTX2 activates the molecular network underlying retina pigment epithelium differentiation. The Journal of biological chemistry 278: 21721–21731. [DOI] [PubMed] [Google Scholar]
- 61. Das G, Choi Y, Sicinski P, Levine EM (2009) Cyclin D1 fine-tunes the neurogenic output of embryonic retinal progenitor cells. Neural Dev 4: 15. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 62. Evans AL, Gage PJ (2005) Expression of the homeobox gene Pitx2 in neural crest is required for optic stalk and ocular anterior segment development. Hum Mol Genet 14: 3347–3359. [DOI] [PubMed] [Google Scholar]
- 63. Gage PJ, Rhoades W, Prucka SK, Hjalt T (2005) Fate maps of neural crest and mesoderm in the mammalian eye. Invest Ophthalmol Vis Sci 46: 4200–4208. [DOI] [PubMed] [Google Scholar]
- 64. Hjalt TA, Semina EV, Amendt BA, Murray JC (2000) The Pitx2 protein in mouse development. Dev Dyn 218: 195–200. [DOI] [PubMed] [Google Scholar]
- 65. Fuhrmann S, Levine EM, Reh TA (2000) Extraocular mesenchyme patterns the optic vesicle during early eye development in the embryonic chick. Development 127: 4599–4609. [DOI] [PubMed] [Google Scholar]
- 66. Hodgkinson CA, Moore KJ, Nakayama A, Steingrimsson E, Copeland NG, et al. (1993) Mutations at the mouse microphthalmia locus are associated with defects in a gene encoding a novel basic-helix-loop-helix-zipper protein. Cell 74: 395–404. [DOI] [PubMed] [Google Scholar]
- 67. Hemesath TJ, Steingrimsson E, McGill G, Hansen MJ, Vaught J, et al. (1994) microphthalmia, a critical factor in melanocyte development, defines a discrete transcription factor family. Genes Dev 8: 2770–2780. [DOI] [PubMed] [Google Scholar]
- 68. Scholtz CL, Chan KK (1987) Complicated colobomatous microphthalmia in the microphthalmic (mi/mi) mouse. Development 99: 501–508. [DOI] [PubMed] [Google Scholar]
- 69. Tsukiji N, Nishihara D, Yajima I, Takeda K, Shibahara S, et al. (2009) Mitf functions as an in ovo regulator for cell differentiation and proliferation during development of the chick RPE. Dev Biol 326: 335–346. [DOI] [PubMed] [Google Scholar]
- 70. Laughon A (1991) DNA binding specificity of homeodomains. Biochemistry 30: 11357–11367. [DOI] [PubMed] [Google Scholar]
- 71. Hanes SD, Brent R (1991) A genetic model for interaction of the homeodomain recognition helix with DNA. Science 251: 426–430. [DOI] [PubMed] [Google Scholar]
- 72. Fuhrmann S (2010) Eye morphogenesis and patterning of the optic vesicle. Current topics in developmental biology 93: 61–84. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 73. Muller HJ (1932) Further Studies on the Nature and Causes of Gene Mutations. Proc Int Congr Genet 6: 213–252. [Google Scholar]
- 74. Sijacic P, Wang W, Liu Z (2011) Recessive antimorphic alleles overcome functionally redundant loci to reveal TSO1 function in Arabidopsis flowers and meristems. PLoS Genet 7: e1002352 doi:10.1371/journal.pgen.1002352. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 75. Miskimins R, Srinivasan R, Marin-Husstege M, Miskimins WK, Casaccia-Bonnefil P (2002) p27(Kip1) enhances myelin basic protein gene promoter activity. J Neurosci Res 67: 100–105. [DOI] [PubMed] [Google Scholar]
- 76. Wei Q, Miskimins WK, Miskimins R (2003) The Sp1 family of transcription factors is involved in p27(Kip1)-mediated activation of myelin basic protein gene expression. Mol Cell Biol 23: 4035–4045. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 77. Nguyen L, Besson A, Heng JI, Schuurmans C, Teboul L, et al. (2006) p27kip1 independently promotes neuronal differentiation and migration in the cerebral cortex. Genes Dev 20: 1511–1524. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 78. Bienvenu F, Jirawatnotai S, Elias JE, Meyer CA, Mizeracka K, et al. (2010) Transcriptional role of cyclin D1 in development revealed by a genetic-proteomic screen. Nature 463: 374–378. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 79. Tong W, Pollard JW (2001) Genetic evidence for the interactions of cyclin D1 and p27(Kip1) in mice. Mol Cell Biol 21: 1319–1328. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 80. Geng Y, Yu Q, Sicinska E, Das M, Bronson RT, et al. (2001) Deletion of the p27Kip1 gene restores normal development in cyclin D1-deficient mice. Proc Natl Acad Sci U S A 98: 194–199. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 81. Dyer MA, Cepko CL (2001) p27Kip1 and p57Kip2 regulate proliferation in distinct retinal progenitor cell populations. J Neurosci 21: 4259–4271. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 82. Levine EM, Close J, Fero M, Ostrovsky A, Reh TA (2000) p27(Kip1) regulates cell cycle withdrawal of late multipotent progenitor cells in the mammalian retina. Dev Biol 219: 299–314. [DOI] [PubMed] [Google Scholar]
- 83. Cunningham JJ, Levine EM, Zindy F, Goloubeva O, Roussel MF, et al. (2002) The cyclin-dependent kinase inhibitors p19(Ink4d) and p27(Kip1) are coexpressed in select retinal cells and act cooperatively to control cell cycle exit. Mol Cell Neurosci 19: 359–374. [DOI] [PubMed] [Google Scholar]
- 84. Das G, Clark AM, Levine EM (2012) Cyclin D1 inactivation extends proliferation and alters histogenesis in the postnatal mouse retina. Developmental dynamics : an official publication of the American Association of Anatomists 241: 941–952. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 85. Cai Z, Feng GS, Zhang X (2010) Temporal requirement of the protein tyrosine phosphatase Shp2 in establishing the neuronal fate in early retinal development. J Neurosci 30: 4110–4119. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 86. Morcillo J, Martinez-Morales JR, Trousse F, Fermin Y, Sowden JC, et al. (2006) Proper patterning of the optic fissure requires the sequential activity of BMP7 and SHH. Development 133: 3179–3190. [DOI] [PubMed] [Google Scholar]
- 87. Levine EM, Roelink H, Turner J, Reh TA (1997) Sonic hedgehog promotes rod photoreceptor differentiation in mammalian retinal cells in vitro. J Neurosci 17: 6277–6288. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 88. Capecchi MR (1980) High efficiency transformation by direct microinjection of DNA into cultured mammalian cells. Cell 22: 479–488. [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Supplementary Materials
Generation of R200Q and R227W mutant mice. (A) Sequence tracks of PCR products encompassing the R200Q or R227W mutations. Asterisks denote base substitutions. Template DNA was prepared from mouse-tail DNA (wild-type) and targeted ES clones (mutants). (B) Schematic representation of the targeting strategy. The R200Q and R227W mutations (asterisk) were introduced into exon 4 and the ACN cassette [46] was cloned into intron 3 of Vsx2. These elements were then placed into the pTK1TK2 targeting vector [88]. After homologous recombination, R200Q-ACN and R227W-ACN chimeric mice were generated. The ACN cassette was deleted by Cre recombination in the male germline generating the R200Q and R227W alleles. Chimeric males were crossed with wild-type females to generate germline-transmitted R200Q and R227W heterozygous mice. B: BamHI, H: HindIII, N: NheI. (C) Southern blots of genomic DNA from electroporated ES cell clones containing homologously recombined R200Q-ACN and R227W-ACN insertions. BamHI digestion and 3′ flanking probe (shown in B) were used. The wild-type band is 6.7 kb and the correctly targeted band is 7.6 kb. (D) Genotyping of mutant mice. P1 and P2 primers shown in B (arrows) were used for PCR genotyping. As the mutant allele contains a 34 bp insertion, wild-type and mutant alleles give 258 bp and 292 bp bands, respectively.
(TIF)
Proliferation changes associated with Vsx2[R200Q] and Vsx2[R227W] overexpression. (A) Micrographs showing expression of enhanced Green Fluorescent Protein fused to a nuclear localization signal (nlsGFP), VSX2, VSX2[R200Q], and VSX2[R227W] in transfected P0 orJ retinal cells. (B) Phosphorylated histone H3 (pHH3) expression was reduced in the mutant retinas compared to wild-type. (C) Quantification of BrdU incorporation in P0 orJ retinal cells overexpressing nlsGFP (control), VSX2, VSX2[R200Q], or VSX2[R227W]. (D) Quantification of BrdU incorporation in P0 wild-type retinal cells overexpressing nlsGFP (control), VSX2, VSX2[R200Q], or VSX2[R227W]. Cells were transfected at 24 hr following initial plating and cultured for an additional 48.0 hr. BrdU was added for the last 4.5 hr of the culture period. Only wild-type VSX2 enhanced proliferation over the control. * P≤0.05; ** P≤0.01 Scale bars: 100 µm.
(TIF)
Marker expression in wild-type, orJ, R200Q, and R227W retinas at P0. (A–D) Neurofilament-M (NF-M) expression in inner retinal neurons (differentiated cell layer, DCL) and horizontal cells (horiz), which occupy the neuroblast layer (NBL), was present in all genotypes except R227W. Similar results were obtained for the retinal ganglion cell marker POU4F2 (E–H) and the proliferation/RPC marker PCNA (I–L). Dashed lines bound retinas. Scale bar: 100 µm.
(TIF)
Marker expression in wild-type and R227W/+ retinas at P0. Expression patterns of TUBB3 (A,B), NF-M (C,D), and SOX2 (E,F) in wild-type and R227W/+ retinas were similar. Scale bars: 100 µm.
(TIF)
Invasion of POM into the vitreal chamber in R227W eyes. (A) PITX2+ cells filled the vitreal chamber in the E17.5 R227W eye. (B) The relative expression levels of D-Mitf and J-Mitf were not statistically different between control (−POM) and POM implanted whole retina and lens explant (POM imp) cultures (E10.5+2DIV).
(TIF)
Immunolocalization of MITF and p27 in selected genotypes at E12.5. MITF expression in R227W (A) and R227W; p27+/− (B) eyes. (C) No primary antibody control. p27 expression in wild-type (D), orJ (E), R200Q (F), and R227W (G) eyes. Antigen retrieval was used for p27 staining to reveal staining in neuroblast layers. Bright staining at inner surface of central retina of wild-type is newly generated postmitotic precursors. Bright staining at the periphery of the R200Q retina is also observed occasionally in orJ retina at this age. p27 is also abundantly expressed in lens and surrounding extraocular tissues.
(TIF)
Oligonucleotides.
(DOC)
Plasmids.
(DOC)
Primary antibodies.
(DOC)