

Abstract
Specialized chromatin containing CENP-A nucleosomes instead of H3 nucleosomes is found at all centromeres. However, the mechanisms that specify the locations at which CENP-A chromatin is assembled remain elusive in organisms with regional, epigenetically regulated centromeres. It is known that normal centromeric DNA is transcribed in several systems including the fission yeast, Schizosaccharomyces pombe. Here, we show that factors which preserve stable histone H3 chromatin during transcription also play a role in preventing promiscuous CENP-ACnp1 deposition in fission yeast. Mutations in the histone chaperone FACT impair the maintenance of H3 chromatin on transcribed regions and promote widespread CENP-ACnp1 incorporation at non-centromeric sites. FACT has little or no effect on CENP-ACnp1 assembly at endogenous centromeres where CENP-ACnp1 is normally assembled. In contrast, Clr6 complex II (Clr6-CII; equivalent to Rpd3S) histone deacetylase function has a more subtle impact on the stability of transcribed H3 chromatin and acts to prevent the ectopic accumulation of CENP-ACnp1 at specific loci, including subtelomeric regions, where CENP-ACnp1 is preferentially assembled. Moreover, defective Clr6-CII function allows the de novo assembly of CENP-ACnp1 chromatin on centromeric DNA, bypassing the normal requirement for heterochromatin. Thus, our analyses show that alterations in the process of chromatin assembly during transcription can destabilize H3 nucleosomes and thereby allow CENP-ACnp1 to assemble in its place. We propose that normal centromeres provide a specific chromatin context that limits reassembly of H3 chromatin during transcription and thereby promotes the establishment of CENP-ACnp1 chromatin and associated kinetochores. These findings have important implications for genetic and epigenetic processes involved in centromere specification.
Author Summary
Centromeres are the chromosomal locations at which kinetochores, the machinery that directs chromosome segregation, are assembled. In most eukaryotes, centromere location is epigenetically determined, meaning that the underlying DNA sequence does not dictate where they are formed. The genome is packaged in particles called nucleosomes, composed of histone proteins. Centromere DNA is wrapped around unusual nucleosomes that differ from those elsewhere in the genome because the histone H3-like CENP-A replaces the normal histone H3 component. We used fission yeast to investigate where CENP-ACnp1 nucleosomes are formed in cells containing excess CENP-ACnp1 and how the formation of these non-centromeric CENP-A nucleosomes is controlled. H3 nucleosomes are disassembled and reassembled during transcription by RNA polymerase II (RNAPII). We show that the normal process of reassembling robust H3 chromatin on RNAPII genes is required to prevent CENP-ACnp1 assembly in its place. Centromeres allow a low level of RNAPII transcription, and our analyses suggest that DNA sequences and chromatin contexts at centromeres may limit the activities required to stabilize and reassemble H3 chromatin during transcription in order to promote the establishment of CENP-ACnp1 chromatin and associated kinetochores.
Introduction
Centromere formation is influenced by both genetic and epigenetic processes (reviewed in [1], [2], [3], [4], [5], [6]). The fundamental feature that defines active centromeres resides at the chromatin level; the presence of specialized chromatin in which histone H3 is replaced by the conserved H3 variant CENP-A (CenH3). CENP-A is highly enriched at active centromeres and is indispensible for centromere function. CENP-A chromatin provides a platform for recruiting kinetochore proteins which in turn direct CENP-ACnp1 targeting and retention; thus CENP-A chromatin serves as an epigenetic mark allowing propagation of centromeres at specific loci [1], [5], [7].
Fission yeast (Schizosaccharomyces pombe) centromeres provide an excellent model for dissecting the mechanism of CENP-A chromatin assembly [8]. CENP-ACnp1 chromatin assembles on 7-10 kb central domain regions which, as at mammalian centromeres, are surrounded by heterochromatin formed by methylation of histone H3 lysine 9 [5]. We previously showed that flanking heterochromatin is required to establish CENP-ACnp1 on the central domain of naïve plasmid-based minichromosome DNA [9], [10]. However, once established, CENP-ACnp1 chromatin is propagated in the absence of the adjacent heterochromatin. The epigenetic nature of CENP-ACnp1 assembly in fission yeast is underscored by the findings that neocentromeres can form at subtelomeric regions and centromeres can be inactivated on dicentric chromosomes [11], [12]. The assembly of CENP-ACnp1 chromatin at novel secondary sites could be beneficial for the rescue of acentric chromosomes but detrimental if activated on normal chromosomes. Dicentric chromosomes are highly unstable and thus mechanisms must operate to suppress assembly of CENP-ACnp1 chromatin at such secondary sites (reviewed in [6]). Related to this, CENP-A is overexpressed and more CENP-A is incorporated at centromeres in some tumor cells [13], . CENP-A overexpression can trigger neocentromere formation resulting in dicentric chromosomes and consequential genome instability that drives tumor progression [15], [16], [17]. Thus mechanisms that prevent promiscuous deposition of CENP-A into non-centromeric chromatin are important for preventing genome instability and provide insight into the processes that normally allow CENP-A deposition.
Accumulating evidence in several organisms indicates that transcription occurs at centromeres and neocentromeres [18], [19], [20], [21], [22], [23], however it is unclear how transcription might influence CENP-A deposition mechanistically. Recently, we demonstrated that non-coding RNAs are transcribed by RNA polymerase II from the central CENP-ACnp1 chromatin domains of fission yeast centromeres [24]. It is well known that advancing RNAPII promotes the disassembly of nucleosomes in its path and this mediates the eviction of H3.1 and its replacement with H3.3 in metazoa (reviewed in [25], [26]). Similarly, transcription-coupled nucleosome disassembly may allow the exchange of H3 for CENP-A within centromeres. At promoters, chromatin remodeling and histone acetylation destabilize or remove nucleosomes to allow transcription factors and RNAPII access to the DNA template. During transcriptional elongation, nucleosomes in front of RNAPII are transiently disassembled and histone chaperones, such as FACT (Facilitates chromatin transcription) and Spt6, act to recycle the dissociated histones so that the same histones are reassembled in nucleosomes behind elongating RNAPII [27], [28], [29]. Moreover, acetylation of nucleosomes ahead of RNAPII appears to facilitate the passage of RNAPII through chromatin, but this acetylation must be removed to restore chromatin to its original stable state within coding regions. In Saccharomyces cerevisiae, the Rpd3S histone deacetylase (HDAC) complex deacetylates histones in coding regions [30], [31]. The integrity of transcribed chromatin has been shown to be weakened in cells with defective FACT, Spt6 or Rpd3S and as a consequence cryptic transcription initiates from within coding regions to produce spurious intragenic transcripts [28], [31], [32]. S. pombe Clr6 Complex II (Clr6-CII) performs the same function as S. cerevisiae Rpd3S and is composed of related subunits (Clr6, Pst2, Cph1, Cph2, Alp13, Prw1; Clr6 = Rpd3) [33].
Here we investigate the contribution of factors that govern nucleosome dynamics during RNAPII transcription to CENP-ACnp1 deposition. We find that defects in FACT and Clr6-CII, which normally restore H3 chromatin integrity during RNAPII transcription, cause increased CENP-ACnp1 incorporation at non-centromeric loci when CENP-ACnp1 levels are elevated. FACT mutants show a strong phenotype, severely disrupt H3 chromatin integrity on RNAPII genes and promote widespread mis-incorporation of CENP-ACnp1. In contrast, cells with defective Clr6-CII display a more subtle disturbance of H3 chromatin integrity and allow increased CENP-ACnp1 incorporation at more specific locations, including subtelomeric regions. Remarkably, Clr6-CII mutants allow the de novo assembly of CENP-ACnp1 and kinetochore proteins on plasmids carrying centromeric central domain DNA alone and thus circumvent the requirement for flanking heterochromatin in the establishment of CENP-ACnp1 chromatin. We propose that CENP-ACnp1 chromatin can assemble de novo on particular genomic regions when the integrity of H3 chromatin fails to be restored during transcription-induced chromatin reconfiguration. By extrapolation, at endogenous centromeres, specific chromatin contexts such as flanking heterochromatin (provided by repetitive elements) or pre-established centromeric chromatin may alter nucleosome dynamics during RNAPII transcription to facilitate the replacement of H3 by CENP-ACnp1.
Results
FACT mutants are hypersensitive to CENP-ACnp1 overexpression
FACT is composed of two evolutionarily conserved subunits, Spt16 and Pob3 (SSRP1 in human) [34], [35]. In budding yeast both FACT subunits are essential, but fission yeast requires only Spt16 for survival [36]. Thus, Spt16 performs the major functions of FACT in fission yeast. To fully assess whether FACT modulates CENP-ACnp1 deposition in fission yeast, we generated temperature sensitive (ts) alleles of spt16 +. Cells with a defect in mechanisms that prevent inappropriate CENP-ACnp1 deposition in non-centromeric chromatin are expected to be sensitive to CENP-ACnp1 overexpression. We noticed that elevated CENP-ACnp1 levels (OE-CENP-ACnp1) exacerbate the ts phenotypes in spt16-18 cells (Figure 1A; for generation of spt16-ts alleles see Figure S1A). Since different spt16 alleles and pob3Δ also exhibit sensitivity, this phenotype is not allele or subunit specific (Figure 1B and 1C). The effect was greater when CENP-ACnp1 was expressed from the stronger promoter nmt41 (nmt promoters are described in [37]). The nmt41 promoter produces 4-fold more GFP-CENP-ACnp1 than the weaker nmt81 version (Figure 1D). The level of OE-CENP-ACnp1 expressed from nmt41-GFP-cnp1 + and nmt81-GFP-cnp1 + is comparable in wild-type (wt) and spt16-18 cells (Figure 1D). This interaction is specific to CENP-ACnp1 since spt16-18 cells are not sensitive to elevated expression of histone H3 (Figure 1A; see [38]).
Figure 1. Overexpression of CENP-ACnp1 causes toxicity in FACT mutants.
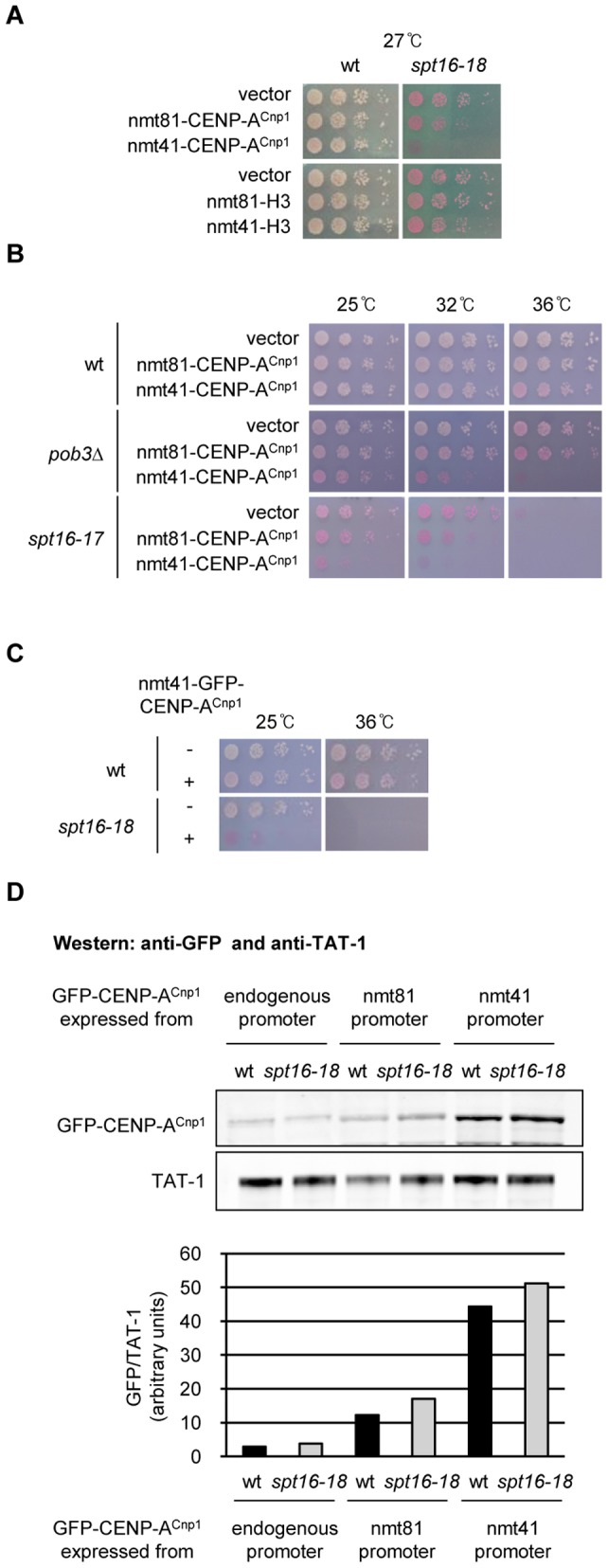
(A) Viability of wild-type (wt) and spt16-18 cells expressing additional CENP-ACnp1 or H3 at low (nmt81-CENP-ACnp1, nmt81-H3) or medium (nmt41-CENP-ACnp1, nmt41-H3) levels compared to empty vector. Cells were grown at 27°C which is semi-permissive for spt16-18. Phloxine B plates stain dead cells red. (B) Viability of wt, pob3Δ and spt16-17 cells expressing additional CENP-ACnp1 at low (nmt81-CENP-ACnp1) or medium (nmt41-CENP-ACnp1) levels compared to empty vector at indicated temperatures. (C) Viability of wt and spt16-18 cells expressing GFP-CENP-ACnp1 from integrated pREP41-GFP-cnp1 + (nmt41-GFP-CENP-Acnp1) compared to no GFP-CENP-ACnp1 control. (D) Western analysis of GFP-CENP-ACnp1 levels in wt and spt16-18 cells expressing GFP-CENP-ACnp1 under endogenous, nmt81 or nmt41 promoter (upper panel). The intensities of GFP-CENP-ACnp1 and TAT-1 (alpha-tubulin) signals were measured using LICOR Odyssey Infrared Imaging System software (Li-COR Bioscience) and the relative intensities of GFP-CENP-ACnp1/TAT-1 were quantified (bottom panel). GFP-CENP-ACnp1 was expressed for 24 h at 25°C before harvest.
Marker genes placed in the central domain of fission yeast centromeres are normally silenced and mutations affecting CENP-ACnp1 deposition exhibit decreased silencing [39]. Silencing of ura4 + in the central domain (cnt1:ura4 +) is largely unaffected in pob3Δ cells and in spt16-ts alleles at the permissive temperature (25°C) (Figure S1B and S1C). Factors involved in CENP-ACnp1 deposition at centromeres often show genetic interactions when combined with defects in CENP-ACnp1 itself [39]. However, pob3Δ does not display reduced growth when combined with cnp1-87 which has a relatively weak mutation in CENP-ACnp1 (Figure S1D). One explanation for the sensitivity of FACT mutants to elevated CENP-ACnp1 levels but the lack of an interaction with CENP-ACnp1 mutants is that FACT is required to prevent the promiscuous incorporation of CENP-ACnp1 in place of H3 at non-centromeric locations and is not directly involved in maintaining CENP-ACnp1 chromatin at centromeres. The mis-incorporation of CENP-ACnp1 at other locations in FACT mutants may cause cell lethality by interfering with normal chromatin-based processes such as transcription or possibly due to the induction of ectopic kinetochores.
Spt16 is required to maintain the integrity of H3 chromatin on RNAPII genes
It is well known that FACT is required for the RNAPII transcription-coupled reassembly of chromatin on transcription units in S. cerevisiae [40], [41]. However, although the reassembly of chromatin on transcribed templates with recycled histones is defective in spt16 mutants, the assembly of chromatin from the free histone pool remains active so that new histones can be incorporated. Indeed, the elevated loss of nucleosomes from transcription units in spt16 mutants enhances their replenishment with new histones from the free pool [40]. Thus FACT normally prevents the incorporation of free histones by recycling pre-existing histones in template-associated nucleosomes during transcription. Our previous analyses indicate that CENP-ACnp1 and H3 can compete for incorporation into chromatin at centromeres [38]. If FACT mutants cause elevated turnover of H3 nucleosomes on RNAPII-transcribed templates in S. pombe then this might provide the opportunity for CENP-ACnp1 to replace H3 in non-centromeric transcribed chromatin.
We first tested if, as in S. cerevisiae, FACT is also required for the reassembly of chromatin on genes transcribed by RNAPII in S. pombe. The detection of shorter sense and antisense transcripts initiated from within open reading frames (ORFs) is a hallmark of defective RNAPII transcription-coupled chromatin reassembly [28], [31], [32]. Clr6-CII is the S. pombe equivalent of Rpd3S in S. cerevisiae and microarray expression profiling previously indicated the presence of antisense transcripts from a set of genes in S. pombe cells with defective Clr6-CII [33]. We reasoned that the same genes may display a similar defect in FACT mutants and selected three genes (SPBC197.11, pot1+ and msh1+), for northern analyses in wild-type (wt), spt16-18 and pst2Δ (Pst2; Sin3-related Clr6-CII subunit) cells (Figure 2A). Probes complementary to 3′ region of these genes detect short, abnormal transcripts in cells with defective Spt16 or Pst2 (Figure 2B and 2C) and the transcript sizes suggest that they originate from within the ORFs. Clr6-CII preferentially targets transcribed regions to suppress aberrant transcription initiation, whereas Clr6 complex I (Clr6-CI) acts at promoters [33]. Consistent with this, spurious intragenic transcripts are detected in cells lacking other Clr6-CII subunits (cph1Δ, alp13Δ) but not in mutants that specifically affect Clr6-CI (pst1-1; Figure 2C).
Figure 2. Spt16 is required to suppress cryptic transcription initiation and nucleosome loss at RNAPII genes.

(A) Schematic of genes and positions of RNA probes (arrow) used in Northern analysis. (B) Northern analyses of transcripts from SPBC19C7.11, pot1 + and msh1 + genes. RNA was extracted from cells grown at 25°C (wt, spt16-18), 32°C for 6 h (wt, pst2Δ) or 36°C for 1 h (wt, spt16-18) after shift from 25°C. Arrow indicates full-length transcripts. (C) Northern analysis of transcripts from SPBC19C7.11. Cells were grown at 36°C for 2 h after shift from 25°C (wt, spt16-ts) or at 32°C for 6 h after shift from 25°C (wt, pst2Δ, cph1Δ, alp13Δ, pst1-1), as indicated. (D) ChIP analysis of H3 levels at act1 + and pot1 + in wt and spt16-18 cells grown at 36°C for 1 h after shift from 25°C (top). ChIP analysis of H2B levels at act1 + and pot1 + in wt and spt16-18 cells expressing H2B-FLAG or untagged H2B (bottom). Enrichment is reported as % IP. Error bar indicates S.D. from 3 biological replicates. (E) Genome browser view showing ChIP-chip occupancy profiles for H3 in wt (blue) and spt16-18 cells (red). The relative ratio (spt16-18/wt) is indicated in black. Data on the Y-axis are presented in log2 scale and the X-axis shows genome positions in base pairs. Open reading frames (ORFs) are displayed as boxes and colored according to transcription levels (highly transcribed genes in red, medium transcribed genes in green and low transcribed genes in blue). (F) Average gene analysis for the ratio of H3 occupancy in spt16-18 mutants versus wt. Genes are aligned at transcription start site and divided into four groups dependent of their transcription levels. Data on the Y-axis are presented in log2 scale and the X-axis shows position relative to start (bp). Values for gene expression were calculated using Podbat based on the RNA data from a previous study [61], [62]. The gene expression value ranged between 5 and 15, and genes were assigned into categories based on this value. Five categories were made: very high (>14) (n = 37), high (12–14) (n = 591), medium (10–12) (n = 1726), low (8–10) (n = 1904) and very low (<8) (n = 815). n = number of genes in each group. Error bars represent 99% confidence intervals.
To directly determine if chromatin integrity is compromised in S. pombe cells with defective FACT we measured the levels of H3 and H2B on the pot1 + gene where aberrant intragenic transcripts were readily detected in spt16-18 cells. We also analyzed the act1 + gene, which is highly transcribed and thus associated nucleosomes are likely to be more dynamic. ChIP analyses indicated that the levels of histones are dramatically reduced on act1 + in spt16-18 cells compared to wild-type cells (Figure 2D). A modest reduction of histone levels is also observed on pot1 + in spt16-18 cells. This suggests that Spt16 is required to maintain H3 nucleosomes on act1 + and pot1 +. To further assess loss of chromatin integrity from RNAPII genes in spt16-18 cells, we performed genome-wide analyses of H3 association in wt and spt16-18 cells. We observed a widespread decrease in the relative levels of H3 on RNAPII genes in spt16-18 cells (Figure 2E). The reduction of H3 levels on RNAPII genes in spt16-18 cells is correlated with their level of transcription (Figure 2F). Thus, as in S. cerevisiae, FACT is required to maintain canonical H3 nucleosomes on genes during transcription by RNAPII.
CENP-ACnp1 accumulates at non-centromeric locations in cells with defective FACT
Normally, cells express low levels of CENP-A and this together with robust and specific mechanisms involving centromere associated assembly factors maintains CENP-A exclusively at centromeres. In order to determine if defective FACT function allows the incorporation of CENP-ACnp1 at non-centromeric locations, we compared the levels of CENP-ACnp1 on act1 + and pot1 + genes in wild-type and spt16-18 cells expressing additional CENP-ACnp1. CENP-ACnp1 incorporation into the chromatin covering pot1 +, but not act1, was substantially increased in spt16-18 relative to wild-type cells expressing excess CENP-ACnp1 (nmt41-cnp1 +; Figure 3A). The spt16-18 mutation alone or OE-CENP-ACnp1 in wild-type cells does not significantly affect the levels of CENP-ACnp1 on pot1 +. The increased accumulation of CENP-ACnp1 on pot1 + compared to act1 + in spt16-18 cells is intriguing since H3 occupancy is more severely reduced on act1 + (Figure 2D). The act1 + gene is very highly expressed and even in wild-type cells it retains a relatively low level of H3. We surmise that, like H3, CENP-ACnp1 cannot stably assemble on highly transcribed genes such as act1 + in spt16-18 cells due to its continual removal. But on genes such as pot1 + that are transcribed at intermediate levels, H3 or CENP-ACnp1 can be stably incorporated from the free histone pool.
Figure 3. Spt16 prevents promiscuous incorporation of CENP-ACnp1.

(A) ChIP analysis of CENP-ACnp1 levels at endogenous centromeres (cc1/3), act1 + and pot1 + in wt and spt16-18 cells in the absence or presence of OE-CENP-ACnp1 (nmt41-cnp1 +; pREP41-cnp1 + integrated at ars1 locus). Cells were grown at 36°C for 1 h after shift from 25°C. (B) ChIP analysis of CENP-CCnp3 in the same samples. (C) ChIP-chip: relative CENP-ACnp1 levels in spt16-18 cells compared to wt. ORFs are displayed as grey boxes. Regions of at least 1 kb in length and with >2-fold increase in CENP-ACnp1 signal above genome-wide average are colored red. Data on the Y-axis are presented in linear scale. Blue: running average signal/100 probes. Grey: signal for individual probes. (D) ChIP analyses of CENP-ACnp1 and CENP-CCnp3 levels at prm1 + and tip41 +. Error bars indicate S.D. from 3 biological replicates. (E) Average gene analysis for the ratio of CENP-ACnp1 occupancy in spt16-18 versus wt cells (with OE-CENP-ACnp1). Genes within 100 kb of centromeres were selected and divided into four groups dependent of their relative transcription levels and aligned at their transcriptional start sites. Data on the Y-axis are presented in log2 scale and the X-axis shows position relative to start (bp). Values for gene expression were calculated using Podbat based on the RNA data from a previous study [61], [62]. The gene expression value ranged between 5 and 15, and genes were assigned into categories based on this value. Five categories were made: very high (>14) (n = 37), high (12–14) (n = 591), medium (10–12) (n = 1726), low (8–10)(n = 1904) and very low (<8) (n = 815). n = number of genes in each group. Error bars represent 99% confidence intervals. (F) Genome browser view showing relative enrichment of CENP-ACnp1 at cen1-proximal regions (spt16-18/wt; with OE-CENP-ACnp1). Data on the Y-axis are presented in linear scale. Boxes are ORFs; red: genes with very high expression levels. (G) ChIP analysis of CENP-ACnp1 levels at prm1 +, pot1 +, endogenous centromeres (cc1/3) and a neocentromere region (tel1R) in wt, clr4Δ and cd60 (neocentromere strain; cen1 DNA deleted) cells containing either spt16 + or spt16-18 allele and all with OE-CENP-ACnp1 (nmt41-cnp1 +). Note: centromere primers (cc1/3) detect both cen1 and cen3 and thus CENP-ACnp1 enrichment at cc1/3 in cd60 represents CENP-ACnp1 levels at cen3 only. In all ChIP analyses, enrichment is reported as % IP.
It is well established that CENP-A chromatin serves as a platform for assembly of kinetochore proteins at centromeres [42]. The expression of additional CENP-ACnp1 in FACT defective spt16-18 cells results in a modest but reproducible reduction in the association of the CENP-CCnp3 kinetochore protein with centromeres (Figure 3B). One explanation is that the pool of endogenous CENP-CCnp3 is limited and the promiscuous incorporation of CENP-ACnp1 at many non-centromeric locations leads to the redistribution of this CENP-CCnp3. However, CENP-CCnp3 levels were not observed to increase on act1 + or pot1 + in spt16-18 expressing additional CENP-ACnp1 (nmt41-cnp1 +; Figure 3B). Other regions in the genome may accumulate higher levels of CENP-ACnp1 allowing them to attract CENP-CCnp3 away from centromeres. To explore this possibility, we compared the genome-wide distribution of overexpressed CENP-ACnp1 in wild-type and spt16-18 cells. The absolute levels of CENP-ACnp1 association were quantified across the genome using ChIP-chip. In spt16-18 cells expressing excess CENP-ACnp1 (nmt41-cnp1 +), a 1.7 fold global increase in chromosomal levels of CENP-ACnp1 occurs. The most notable accumulation relative to wild-type cells is observed on the euchromatic regions adjacent to centromeric heterochromatin (Figure 3C and Figure S2; also see Table S1 for CENP-ACnp1 and H3 enrichment at selected genes). To further evaluate this, we conducted a genome-wide data-driven search for regions of CENP-ACnp1-chromatin association. Regions of at least 1 kb in length and with more than 2-fold increase in CENP-ACnp1 signal above genome-wide average were selected. 168 regions were identified, and 96 of these were within 100 kb of centromeres (Red boxes; Figure 3C), i.e. 57% of regions found in 4.8% of the genome. This was found to be highly significant (p<10-84, hypergeometric distribution). ChIP-qPCR analyses confirm that high levels of CENP-ACnp1 accumulate on prm1 + (high expression class) and tip41 + (very low expression class) which are proximal to cen1 and cen3, respectively (Figure 3D). Increased association of CENP-CCnp3 is detected on tip41 + in spt16-18 OE-CENP-ACnp1 cells, indicating that other kinetochore proteins can be recruited to some non-centromeric sites (Figure 3D). Only genes with internal cryptic transcriptional start sites will generate short aberrant intragenic transcripts. Indeed, short cryptic transcripts are not readily detected from prm1 + and tip41 + in spt16-18 cells and pst2Δ cells (Figure S3A). Also, short cryptic transcripts were not evident from act1 + (Figure S3B). Thus, it is unlikely that cryptic transcription itself is the primary cause of H3 loss or CENP-ACnp1 incorporation in FACT mutants. ChIP-chip analyses show that the levels of H3 and CENP-ACnp1 are lowest on transcribed genes with high levels of expression and this decreases further in spt16-18 cells compared to wild-type cells (Figure S4). Even within centromere proximal regions where overexpressed CENP-ACnp1 is preferentially assembled in spt16-18 cells, very highly transcribed genes do not allow CENP-ACnp1 accumulation (Figure 3E and 3F). This supports the conclusion that genes with low to intermediate levels of RNAPII transcription permit more stable incorporation of CENP-ACnp1 in the absence of FACT function.
Centromere activity is required for the incorporation of excess CENP-ACnp1 in centromere proximal regions
The preferential accumulation of CENP-ACnp1 close to centromeres in FACT defective spt16-18 cells overexpressing CENP-ACnp1 may be dependent on centromeric heterochromatin or on the presence of an active centromere. Centromeric heterochromatin is completely dependent on clr4 +, encoding the only H3K9 methyltransferase in S. pombe, which is non-essential. We have previously shown that heterochromatin is required for the de novo assembly of CENP-ACnp1 on nearby centromeric DNA. However, increased CENP-ACnp1 association was still detected on prm1 + in spt16-18 cells overexpressing CENP-ACnp1 that lack Clr4 (clr4Δ; Figure 3G). Therefore to determine if the accumulation of CENP-ACnp1 is directed by the presence of an active centromere, we used cd60 cells in which the normal cen1 was deleted and a neocentromere formed in the tel1R subtelomeric region [12]. In these cells, enrichment of CENP-ACnp1 on prm1 + (∼20 kb away from the cen1 deletion) is significantly reduced in spt16-18 cells overexpressing CENP-ACnp1 but the association of CENP-ACnp1 with the non-centromeric pot1 + gene is unaffected (Figure 3G). The 40 kb deletion in cd60 cells, completely removes both the central CENP-ACnp1/kinetochore and heterochromatin domains from cen1. This indicates that it is the pre-assembled centromere-kinetochore, rather than heterochromatin, that promotes the assembly of CENP-ACnp1 over adjacent euchromatin in FACT defective spt16-18 cells. Pre-assembled centromeres must attract excess CENP-ACnp1 and deposition factors which aid the assembly of CENP-ACnp1 chromatin over nearby euchromatin when it becomes permissive for CENP-ACnp1 incorporation in cells with compromised FACT function.
Cells with defective Spt6 are sensitive to CENP-ACnp1 overexpression
If RNAPII transcription-coupled nucleosome reassembly acts to exclude CENP-ACnp1 from incorporation at non-centromeric regions, then other mutants affecting this process should also allow mis-incorporation of CENP-ACnp1. In S. cerevisiae, the Spt6 chaperone also acts to reassemble nucleosomes within ORFs following transcription [28]. Our analyses demonstrate that S. pombe cells bearing the spt6-1 mutation [43] are also sensitive to elevated CENP-ACnp1 levels (Figure S5A). In addition, spt6-1 also allows this CENP-ACnp1 to accumulate at non-centromeric regions (Figure S5B). Thus both the FACT and Spt6 chaperones, which are required to maintain chromatin integrity on genes following transcription by RNAPII, are implicated in preventing the misincorporation of CENP-ACnp1 when it is expressed at elevated levels.
FACT is required for the preferential incorporation of CENP-ACnp1 at specific locations
Neocentromeres can form at subtelomeric regions in S. pombe following removal of the normal centromere [12]. The expression of additional CENP-ACnp1 in wild-type cells also allows its accumulation over these telomere adjacent regions (Castillo et al in preparation; see Figure S6A). This suggests that subtelomeric regions possess particular features that make them favorable substrates for CENP-ACnp1 assembly. Interestingly, our ChIP-chip analyses reveal that CENP-ACnp1 does not accumulate over subtelomeric regions in FACT defective spt16-18 cells expressing additional CENP-ACnp1, even though they are normally preferred sites for the accumulation of additional CENP-ACnp1 in wild-type cells (Figure 3C - subtelomeric CENP-ACnp1 accumulation does not increase in spt16-18 relative to wild-type cells). This suggests that FACT is not normally active, or is unable to prevent CENP-ACnp1 deposition, at wild-type subtelomeric regions.
To explore the relationship between FACT and CENP-ACnp1 permissive regions further we examined the effect of defective FACT function on the association of CENP-ACnp1 with centromeric DNA in both its normal context and at an ectopic location. CENP-ACnp1 normally assembles on the central domain regions of centromeres due to kinetochore-mediated CENP-ACnp1 recruitment and/or maintenance mechanisms. Centromeric heterochromatin also aids the assembly of CENP-ACnp1 chromatin at centromeres [9], [10]. However, in addition to these extrinsic influences, central domain DNA itself may possess intrinsic sequence-driven features that promote CENP-ACnp1 assembly. To examine this, we constructed a strain in which the entire 8.6 kb from the central domain of cen2 was inserted at the euchromatic ura4 + locus (ura4 + -int-cc2). This separates central domain DNA on which CENP-ACnp1 chromatin normally assembles from flanking heterochromatin (Figure S6B). In cells expressing additional CENP-ACnp1 we detected substantially greater levels of CENP-ACnp1 over ura4 + -int-cc2 than on the non-centromeric act1 + or pot1 + loci (Figure S6C). This indicates that even when central domain DNA is placed outside the context of a normal centromere it has an innate ability to attract CENP-ACnp1. We conclude that cc2 central domain DNA must possess particular features that promote CENP-ACnp1 incorporation. However, in FACT defective spt16-18 cells the central domain loses its ability to preferentially attract CENP-ACnp1 so that it is drawn away and accumulates on pot1 + and ura4 + -int-cc2 at similar levels (Figure S6D). This implies that when FACT function is compromised some genes present the same features as those presented by the central domain regions so that they become equivalently competent in attracting excess CENP-ACnp1. The central domain and subtelomeric regions must share specific features that attract CENP-ACnp1 and both lose this exclusivity when FACT is defective.
To further examine the role of FACT in restricting the sequences on which CENP-ACnp1 is normally incorporated, we used strains with small (1.7 kb; cnt1:ura4 +) or large (4.7 kb; cnt1:bigura4 +) gene insertions of non-centromeric DNA in the central domain of cen1 (Figure 4A) [38]. In wild-type cells expressing normal levels of CENP-ACnp1, CENP-ACnp1 was highly enriched on the small ura4 + gene (cnt1:ura4 +; Figure 4B), whilst four-fold less CENP-ACnp1 was detected over ura4 + in cnt1:bigura4 +. Thus, the large non-centromeric DNA insertion is a relatively poor substrate for CENP-ACnp1 deposition even though it is placed in an environment conducive for CENP-ACnp1 assembly. However, in FACT defective cells increased CENP-ACnp1 assembles on cnt1:bigura4 +; similar to the levels detected on the smaller cnt1:ura4 + insertion or the endogenous cen2 central domain (cc2). The relative enrichment of CENP-CCnp3 on ura4 + and cc2 is essentially identical to that of CENP-ACnp1 in all cases (Figure 4C). RT-PCR analysis shows that transcription of ura4 + from cnt1:ura4 + or cnt1:bigura4 + is not significantly affected in spt16-18 cells (Figure S7). This suggests that altered nucleosome dynamics rather than transcriptional activity causes increased CENP-ACnp1 incorporation on bigura4 + in FACT mutants. These analyses demonstrate that FACT usually acts to prevent the incorporation of CENP-ACnp1 on non-centromeric DNA such as RNAPII genes and in its absence the sequence specificity for the assembly of CENP-ACnp1 on central domain DNA and subtelomeric regions is abolished. The observation that FACT does not prevent CENP-ACnp1 incorporation at endogenous cen2 central domain suggests that FACT activity is normally inhibited or limited at native centromeres to favor CENP-ACnp1 deposition within centromeric DNA. Similarly FACT activity must be excluded from, or counteracted in, subtelomeric regions to allow CENP-ACnp1 incorporation.
Figure 4. Spt16 prevents efficient assembly of CENP-ACnp1 chromatin on large non-centromeric DNA inserted within the central domain.

(A) Schematic of cnt1:ura4 + and cnt1:bigura4 +. (B) ChIP analysis of CENP-ACnp1 at ura4 + and endogenous centromere (cc2) in the indicated strains. ura4 +/cc2: relative enrichment of CENP-ACnp1 at ura4 + compared to cc2. Cells were grown at 36°C for 1 h after shift from 25°C. (C) ChIP analysis of CENP-CCnp3 in the same samples. Data for two biological replicates (#1 and #2) are presented. Enrichment is reported as % IP.
Stable maintenance of CENP-ACnp1 chromatin at centromeres requires functional FACT
Our above analyses indicate that FACT activity blocks the deposition of CENP-ACnp1 at non-centromeric locations. However, CENP-ACnp1 remains at centromeres in FACT defective cells and is not dispersed across the genome unless additional CENP-ACnp1 is expressed (Figure 3A and 3D). Thus, a strong propagation mechanism must remain operational at centromeres to maintain CENP-ACnp1 and prevent the redistribution of this limited CENP-ACnp1 pool when FACT function is compromised. The Mis6 and Mis18 kinetochore proteins have been shown to be required to replenish CENP-ACnp1 at centromeres [44], [45]. Mutations in Mis6 (mis6-302) or Mis18 (mis18-262) exhibited synthetic lethality when combined with a deletion of the gene encoding the small subunit FACT (pob3Δ) at semi-permissive temperatures (Figure 5A). In contrast, defective Mis12 (mis12-537), an essential kinetochore protein not involved in CENP-ACnp1 maintenance [45], does not genetically interact with pob3Δ. In addition, mutations in CENP-ACnp1 (cnp1-87) or its chaperone Scm3 (scm3-15) do not exhibit synergistic phenotypes in combination with pob3Δ since they must impair the deposition of CENP-ACnp1 regardless of the genomic location [38], [46], [47]. We also found that deletion of the gene encoding Sim3 (sim3Δ), the NASP (N1/N2)-related CENP-ACnp1 chaperone [48], relieves the lethal effect of CENP-ACnp1 overexpression in FACT defective pob3Δ cells (Figure S8). This implies that Sim3 participates in the promiscuous deposition of CENP-ACnp1 in FACT defective cells. Since Pob3 is also involved in heterochromatin integrity, the synergistic phenotype of pob3Δ mis6-302 and pob3Δ mis18-262 double mutants could be due to defective heterochromatin rather than altered CENP-ACnp1 deposition [36]. However, this possibility is excluded because mis6-302 does not display synergistic phenotypes when combined with clr4Δ which abolishes heterochromatin (Figure 5B).
Figure 5. Pob3 genetically interacts with Mis6 and Mis18 and is required to maintain CENP-ACnp1 at endogenous centromeres in mis6-302 cells.
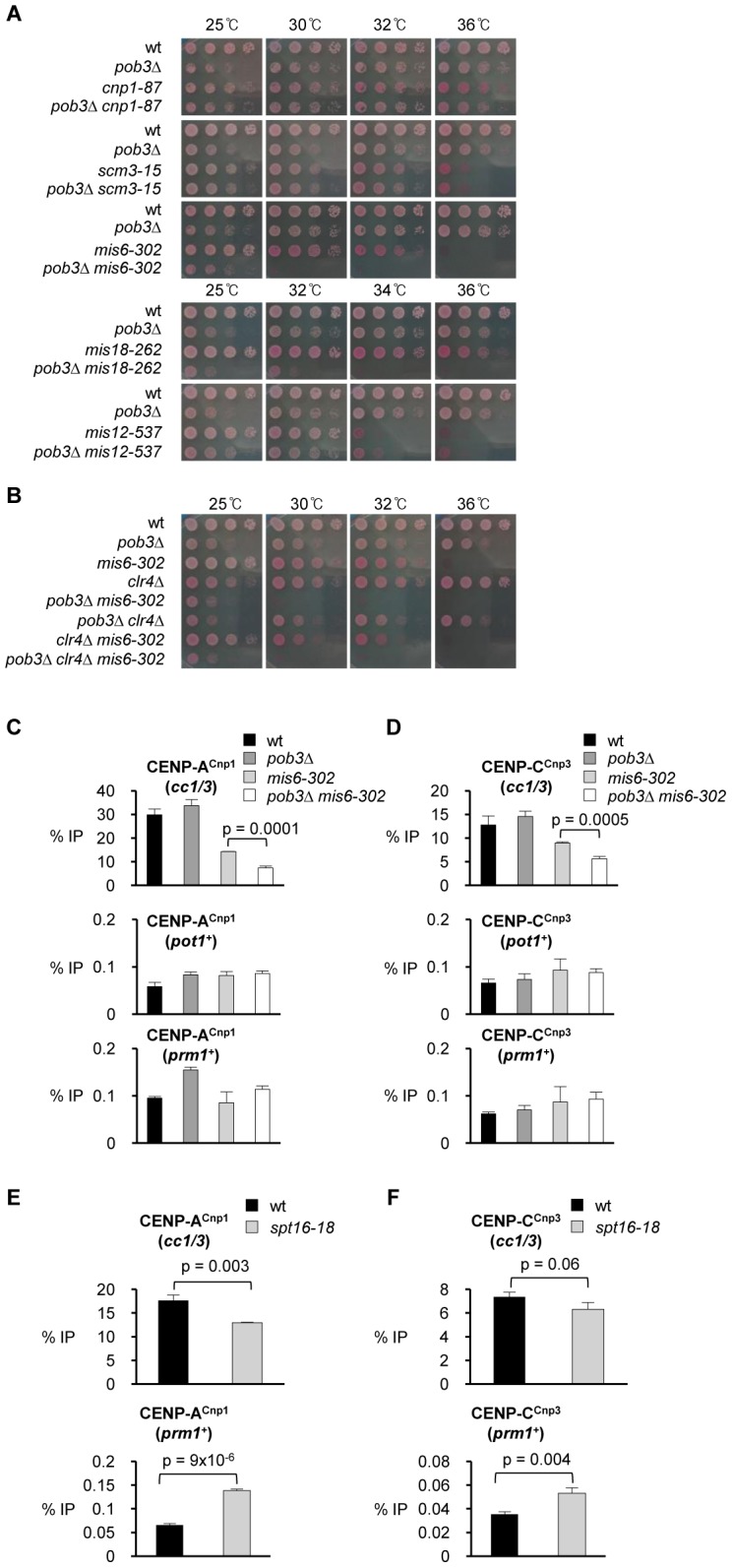
(A) Viability of cells bearing pob3Δ combined with mutants affecting centromere function (cnp1-87, scm3-15, mis6-302 and mis12-537) relative to wt cells and single mutants. Cells were spotted on plates containing Phloxine B at indicated temperatures. (B) Viability of wt and mutant cells bearing a combination of mutations in Pob3, Mis6 and Clr4 (pob3Δ, mis6-302, clr4Δ, pob3Δ mis6-302, pob3Δ clr4Δ, clr4Δ mis6-302 and pob3Δ clr4Δ mis6-302). (C) ChIP analysis of CENP-ACnp1 at endogenous centromeres (cc1/3), pot1 + and prm1 + in the indicated strains. Cells were grown at 25°C, shifted to 30°C for 17 h after shift from 25°C. (D) ChIP analysis of CENP-CCnp3 in the same samples. (E) ChIP analysis of CENP-ACnp1 levels at endogenous centromeres (cc1/3) and prm1 + in wt and spt16-18 cells grown at 27°C (semi-permissive for spt16-18) for 24 h. (F) ChIP analysis of CENP-CCnp3 in the same samples. Enrichment is reported as % IP. Error bars indicate S.D. from 3 biological replicates.
Consistent with the observed genetic interaction between Mis6 and Pob3 mutants, the levels of CENP-ACnp1 and CENP-CCnp3 at centromeres were found to be significantly reduced in pob3Δ mis6-302 cells compared to the pob3Δ or mis6-302 single mutants (Figure 5C and 5D). Thus, when the mechanism for CENP-ACnp1 maintenance at centromeres is compromised, CENP-ACnp1 is released and the lack of FACT function allows its redistribution to non-centromeric sites. However, in pob3Δ mis6-302 cells, the association of CENP-ACnp1 with non-centromeric genes such as pot1+ and prm1 + is not elevated, probably because the redistribution and dilution of the limited endogenous pool of CENP-ACnp1 over many non-centromeric sites is below the level of detectability. A low level of additional CENP-ACnp1 (nmt81-CENP-ACnp1) partially rescues the lethality of pob3Δ mis6-302 cell at 32°C (Figure S9; Note: additional CENP-ACnp1 reduces pob3Δ viability). This reinforces the conclusion that the observed synthetic lethality of the pob3Δ mis6-302 double mutant cells results from reduced levels of CENP-ACnp1 at centromeres.
Accumulation of CENP-ACnp1 at non-centromeric regions was not detectable in FACT defective spt16-18 cells expressing normal CENP-ACnp1 levels after short-term inactivation (1 h at 37°C; Figure 3D). The redistribution of CENP-ACnp1 in spt16-18 cells may require a longer time period or cell cycle progression. To test this possibility, we examined the CENP-ACnp1 redistribution in spt16-18 cells expressing normal CENP-ACnp1 levels after prolonged incubation at a semi-permissive temperature (24 h at 27°C). Under these conditions we detect reduced levels of CENP-ACnp1 and CENP-CCnp3 at centromeres and an increased association of both proteins with the cen1 proximal gene prm1 + (Figure 5E and 5F). Thus endogenous CENP-ACnp1 can be redistributed from centromeres in cells not overexpressing CENP-ACnp1 when FACT function is impaired for a sustained period. We conclude that FACT contributes to the stable maintenance of CENP-ACnp1 chromatin at centromeres by preventing the aberrant deposition of CENP-ACnp1 at non-centromeric locations.
Loss of Clr6 complex II (Rpd3S) allows limited redistribution of CENP-ACnp1
Our analyses indicate that both FACT and Clr6-CII are required to maintain chromatin integrity on transcribed genes, as indicated by the appearance of short aberrant intragenic transcripts, when their function is compromised (Figure 2B and 2C). To determine if Clr6-CII is also required to maintain H3 chromatin and prevent CENP-ACnp1 mis-incorporation ChIP analysis was performed on cells lacking Clr6-CII subunits (pst2Δ and cph1Δ). Unlike FACT defective spt16-18 cells, no reduction in H3 occupancy was observed on highly transcribed genes in pst2Δ cells compared to wild-type cells (Figure 6A and Figure S10A). Loss of Clr6-CII function may only affect H3 chromatin accessibility and not nucleosome occupancy. In S. cerevisiae lack of Rpd3S HDAC has also been found to affect chromatin integrity but not histone occupancy [31], [49]. Thus Clr6-CII and Rpd3S have a more subtle impact on the integrity of chromatin associated with RNAPII transcribed genes in both yeasts. Consistent with this, S. pombe pst2Δ cells are not hypersensitive to the expression of excess CENP-ACnp1 (Figure 6B). Thus defective Clr6-CII may not induce the widespread mis-incorporation of CENP-ACnp1 with associated loss of viability. Indeed in pst2Δ cells expressing additional CENP-ACnp1, high levels of CENP-ACnp1 and CENP-CCnp3 were detected on tip41 +, but not on the pot1 + or prm1 + genes (Figure 6C). Furthermore, analysis of the genome-wide distribution of CENP-ACnp1 in pst2Δ versus wild-type cells overexpressing CENP-ACnp1 revealed that the mis-incorporation of CENP-ACnp1 is far less prevalent in pst2Δ than spt16-18 cells (Figure S10B). However, as with FACT defective spt16-18 cells, the most notable accumulation of CENP-ACnp1 in pst2Δ Clr6-CII deficient cells was observed over the euchromatic regions proximal to centromeres. Interestingly, a low but consistent increase in CENP-ACnp1 levels also occurred over subtelomeric regions of chromosomes 1 and 2 where FACT was found not to affect CENP-ACnp1 incorporation (Figure S10B and Figure 3C). It seems likely that Clr6-CII activity is generally not required to suppress replacement of H3 nucleosome with CENP-ACnp1 nucleosome. However, in particular regions such as subtelomeric domains, where the activity of FACT in promoting H3 assembly appears limited, or in regions proximal to centromeres which are prone to CENP-ACnp1 assembly; the activity of the Clr6-CII HDAC may normally be required to suppress histone exchange by transcription-coupled deacetylation of resident H3 nucleosomes.
Figure 6. Loss of Clr6-CII function promotes assembly of CENP-ACnp1 chromatin at specific loci.

(A) ChIP analysis of H3 levels at act1 + in the indicated strains grown at 32°C. Note: spt16-17 (but not spt16-18) cells are viable at 32°C and thus are grown in parallel with other mutants as a positive control in this experiment. (B) Viability of wt and pst2Δ cells expressing additional CENP-ACnp1 at low (nmt81-CENP-ACnp1) or medium (nmt41-CENP-ACnp1) levels compared to empty vector at 32°C and 36°C. (C) ChIP analyses of CENP-ACnp1 and CENP-CCnp3 levels in wt and pst2Δ cells at pot1 +, prm1 +, tip41 + and endogenous centromeres (cc1/3). Cells were grown at 32°C. (D) Schematic of pcc2 plasmid. pcc2 plasmid contains 8.6 kb cen2 central domain (cc2), ura4 + and sup3-5. (E) ChIP analysis of CENP-ACnp1 levels at cc2 in pcc2 plasmid and at endogenous centromere (cc1/3) in wt and pst2Δ cells carrying pcc2. The relative enrichment of CENP-ACnp1 at cc2 compared to endogenous centromere (cc1/3) is presented (cc2 relative to cc1/3). Enrichment of CENP-ACnp1 at ura4 + in pcc2 is also measured. (F) ChIP analysis of CENP-CCnp3 levels in the same samples. (G) ChIP analysis of Sim4 levels in the same samples. ChIP was performed after 30 and 50 cell doublings at 32°C from the introduction of pcc2. Enrichment is reported as % IP. Error bars indicate S.D. from at least 3 biological replicates.
Clr6 Complex II (Rpd3S) prevents the assembly of CENP-ACnp1 chromatin on centromeric central domain DNA
The endogenous levels of CENP-ACnp1 in wild type cells are insufficient to permit its accumulation at non-centromeric regions. The expression of additional CENP-ACnp1 allows its incorporation at more non-centromeric locations in cells with defective FACT or Clr6-CII. However, wild-type CENP-ACnp1 levels are sufficient to allow CENP-ACnp1 chromatin and functional kinetochores to be assembled de novo over extra centromeric central domain DNA introduced on plasmid-based minichromosomes. In these de novo establishment assays heterochromatin is normally required to promote the assembly of CENP-ACnp1 chromatin on adjacent central domain DNA [10]. We tested if mutations that allow CENP-ACnp1 incorporation at non-centromeric locations when CENP-ACnp1 is overexpressed alter the requirements for de novo CENP-ACnp1 assembly on centromeric DNA in the absence of CENP-ACnp1 overexpression. Plasmids bearing only central domain DNA with no heterochromatin forming repeat sequences are usually unable to assemble CENP-ACnp1 upon introduction into wild-type cells (pcc2; Figure 6D) [10]. Surprisingly, CENP-ACnp1 and the kinetochore proteins CENP-CCnp3 and CENP-KSim4 were readily detected over the central domain of pcc2 following its transformation into pst2Δ cells (Figure 6E, 6F, 6G). This effect is specific for centromeric central domain DNA since CENP-ACnp1 and CENP-CCnp3 were not significantly enriched on the plasmid borne ura4 + gene (Figure 6E and 6F). It is also specific to Clr6-CII mutants since CENP-ACnp1 and CENP-CCnp3 assemble on the central domain of pcc2 in cells lacking subunits of Clr6-CII/Rpd3S (pst2Δ and cph1Δ), but not Clr6-CI (pst1-1; Figure S10C). Defects in FACT/Spt16 or Spt6 function did not permit the assembly of CENP-ACnp1 on pcc2 (Figure S10D and S10E). Importantly, ChIP analysis shows that pst2Δ does not induce H3K9 methylation on the pcc2 plasmid, indicating that the de novo CENP-ACnp1 chromatin assembly pcc2 in pst2Δ cells is not induced by aberrant heterochromatin formation (Figure S11).
The distinct impact of mutations in Clr6-CII compared to FACT or Spt6 in the pcc2 de novo assembly assay is indicative of competition between the distinct genomic loci affected by these mutations for the limited pool of endogenous CENP-ACnp1. Defective FACT function results in widespread loss of H3 chromatin and this renders a large fraction of the genome receptive for CENP-ACnp1 incorporation. Under these circumstances the normal limited pool of free CENP-ACnp1 is broadly distributed and CENP-ACnp1 cannot be specifically recruited and assembled de novo on the newly introduced pcc2 (Figure 7A and 7B). In contrast, cells with defective Clr6-CII function have a more subtle alteration in H3 chromatin integrity and this allows CENP-ACnp1 to be incorporated into particular regions that possess intrinsic properties which favor CENP-ACnp1 deposition, such as pcc2 and subtelomeric regions (Figure 7C). We conclude that the defect in restoring H3 chromatin integrity after RNAPII transcription that results from loss of Clr6-CII function removes an impediment to the efficient CENP-ACnp1 assembly onto its preferred substrate, central domain DNA, and this bypasses the need for flanking heterochromatin. It follows that flanking centromeric heterochromatin may impose particular constraints on chromatin disassembly/reassembly events associated with central domain transcription in order to reduce H3 nucleosome stability and promote CENP-ACnp1 assembly in its place.
Figure 7. Summary on the role of factors that promote the integrity of H3 chromatin during transcription in preventing promiscuous CENP-ACnp1 deposition.

(A) In wild-type cells, a limited amount of free CENP-ACnp1 is available to accumulate outside endogenous centromeres at which kinetochore proteins act to attract CENP-ACnp1. This limited pool of free CENP-ACnp1 can be preferentially deposited to specific sites such as centromeric central domain (CC) and subtelomeric regions (ST). Intensity of red color on the chromosome and the plasmid represents relative “receptiveness” of the locus for CENP-ACnp1 incorporation. Bold arrow indicates regions where CENP-ACnp1 incorporation normally occurs without overexpression (i.e. centromeres). Dashed arrows indicate regions where de novo assembly of CENP-ACnp1 is expected under conditions where CENP-ACnp1 deposition is stimulated (e.g. when CENP-ACnp1 is overexpressed or flanking heterochromatin is provided). (B) In cells with defective FACT, non-centromeric regions become permissive to CENP-ACnp1 (indicated by red color all over the chromosome and the plasmid). Endogenous centromeres drive CENP-ACnp1 assembly at proximal euchromatic regions when they become permissive to CENP-ACnp1 assembly (indicated by dark red color at centromere-proximal regions). CENP-ACnp1 incorporation when overexpressed is not significantly elevated at CC and ST in FACT mutants compared to wild-type cells, suggesting that FACT action may be already limited at these sites. Without CENP-ACnp1 overexpression, the limited pool of free CENP-ACnp1 is distributed to non-centromeric regions and cannot accumulate at normally preferred sites such as CC and ST. (C) In cells with defective Clr6-CII, only specific regions such as centromere-proximal euchromatic regions, CC and ST become permissive to CENP-ACnp1. Clr6-CII has a weaker impact on H3 chromatin compared to FACT and thus loss of Clr6-CII function promotes CENP-ACnp1 incorporation only at regions where CENP-ACnp1 assembly is predisposed (centromere-proximal regions) or FACT action is limited (CC and ST). In Clr6-CII mutants, limited pool of free CENP-ACnp1 is not distributed and can accumulate at preferred sites, allowing de novo assembly of CENP-ACnp1 chromatin on a plasmid bearing CC in the absence of flanking heterochromatin.
Discussion
In most eukaryotic organisms, centromere identity is determined epigenetically. However, certain DNA elements such as human α-satellite repeats are the sites where endogenous centromeres are normally located [1], [5]. Moreover, the introduction of α-satellite repeat DNA allows the de novo assembly of functional centromeres [50]. Thus these repeat elements represent preferred substrates, suggesting that their underlying DNA sequence plays some role in specifying the location of centromeres [6]. Previously, we demonstrated that central domain DNA at normal centromeres in fission yeast is transcribed by RNAPII [24]. Here, we show that factors such as FACT and Clr6-CII, which actively promote the integrity of H3 chromatin during RNAPII transcription, suppress the incorporation of excess CENP-ACnp1. Importantly, we find that FACT blocks the incorporation of CENP-ACnp1 over large portions of the genome. However this restraint does not operate in genomic regions where CENP-ACnp1 assembly is normally favored i.e centromeres and subtelomeric regions. These regions accumulate higher levels of CENP-ACnp1 in Clr6-CII mutants, suggesting a role for transcription-coupled processes in CENP-ACnp1 assembly at these sites. We propose a model in which certain DNA sequences and chromatin contexts have the ability to restrict FACT activity during transcription and consequently represent hotspots for CENP-ACnp1 assembly (summarized in Figure 7).
Competition between CENP-ACnp1 and H3 for incorporation into chromatin
CENP-ACnp1 competes with histone H3 for incorporation into centromeric chromatin [38]. At centromeres, mechanisms which ensure the maintenance of CENP-ACnp1 act to prevent incorporation of H3. Similarly, we find that at non-centromeric regions, mechanisms that maintain H3 nucleosomes during the intrinsically disruptive process of transcription act to prevent incorporation of CENP-ACnp1. FACT is involved in several chromatin-based processes in addition to transcription, and thus it is possible that promiscuous CENP-ACnp1 incorporation in FACT mutants is caused by defects in other processes rather than transcription-coupled nucleosome reassembly [51]. Although this possibility cannot be ruled out completely, we have observed similar CENP-ACnp1 phenotypes in cells with defective Spt6 (Figure S5A and S5B). This supports a direct connection between defective H3 chromatin assembly during RNAPII transcription and promiscuous CENP-ACnp1 incorporation. We propose that CENP-ACnp1 is opportunistic in nature and its assembly into chromatin is strongly affected by its availability relative to histone H3 and processes that promote transcription-coupled recycling of H3 nucleosomes. When excess free CENP-ACnp1 is available and transcription-coupled H3 nucleosome assembly is defective, CENP-ACnp1 gains access to regions from which it is normally excluded. In this context it should be noted that the N-terminal tail of CENP-ACnp1 is distinct from that of H3; it lacks the key lysine residues, K4, K9, K14, K18, K23, K27, and K36. Thus, CENP-ACnp1 is undoubtedly managed differently than H3 with respect to transcription.
What gives CENP-ACnp1 a selective advantage for incorporation into non-centromeric regions in FACT defective (spt16-18) cells? Our observation that H3 occupancy decreases in spt16-18 cells in proportion to transcription rates suggests that H3 incorporation from the free histone pool is not sufficiently effective to maintain a steady-state level of H3 on transcribed chromatin templates in spt16-18 cells (Figure 2F). Thus, the maintenance of H3 chromatin within transcribed regions must be largely dependent on replication-coupled assembly in S phase and histone chaperone-mediated reassembly during transcription. It is known that CENP-A incorporation at centromeres is uncoupled from replication in human cells and in fission yeast CENP-A incorporation can occur in S phase or in G2 phase independently of replication [48], [52]. It therefore seems likely that transcription-coupled loss of H3 nucleosomes in spt16-18 cells may favor the incorporation of CENP-A. Given the differences between CENP-A and H3, CENP-A nucleosomes are likely to react differently to transcription and persist in situations that cause H3 nucleosomes to disassemble during transcription. The HDAC Clr6-CII preferentially targets RNAPII-transcribed regions [33]. In cells that are defective in Clr6-CII function (pst2Δ, cph2Δ), the persistence of transcription-associated histone acetylation destabilizes H3 nucleosomes (as indicated by the exposure of cryptic transcription initiation sites) and thereby enhances their replacement with CENP-ACnp1 nucleosomes. The distinct N-terminal tail of CENP-ACnp1 presumably provides CENP-ACnp1 nucleosomes with greater stability than H3 nucleosomes during transcription in Clr6-CII mutant cells. It is also possible that once assembled, CENP-ACnp1 nucleosomes repress transcription and that this reduces nucleosome turnover and consequently stabilizes CENP-ACnp1 chromatin. Further analyses are required to reveal exactly how CENP-A replaces H3 and is stabilized on transcription units.
Genetic and epigenetic processes influencing CENP-ACnp1 chromatin assembly
The data that we present suggest that defective FACT function diminishes the distinction between centromeric and non-centromeric regions, allowing widespread incorporation of CENP-ACnp1 into transcribed DNA when CENP-ACnp1 is overexpressed. The central domains from centromeres (ectopically placed cc2 DNA; Figure S6C) and potential sites of neocentromeres in subtelomeric regions (Figure S6A) clearly have an innate ability to incorporate CENP-ACnp1 in wild-type cells that express additional CENP-ACnp1. However, in FACT mutants, many other genomic locations become permissive for CENP-ACnp1 incorporation, therefore the pool of additional free CENP-ACnp1 is distributed over many chromosomal regions so that preferential incorporation at normal secondary sites such as ectopic cc2 or subtelomeric regions is reduced (Figure 3C and Figure S6D). We conclude that central domain and subtelomeric regions, which naturally favor CENP-ACnp1 deposition, possess features that reduce or evade FACT action so that H3 is more readily replaced by CENP-ACnp1. Consistent with a role for cis-acting elements, FACT does not suppress CENP-ACnp1 assembly on the central domain DNA at endogenous centromeres while it does prevent efficient CENP-ACnp1 assembly on a large non-centromeric reporter gene insertion at the endogenous centromere (Figure 4A, 4B, 4C).
Genetic interactions exhibited between Pob3 and Mis6 or Mis18, along with ChIP analyses, demonstrate that FACT operates to retain CENP-ACnp1 at centromeres when the CENP-ACnp1 maintenance mechanism is weakened because it prevents CENP-ACnp1 incorporation elsewhere (Figure 5A, 5B, 5C, 5D). However, even in the presence of an intact CENP-ACnp1 maintenance mechanism, prolonged attenuation of FACT/Spt16 function causes redistribution of CENP-ACnp1 and CENP-CCnp3 (Figure 5E and 5F). Thus, the CENP-ACnp1 maintenance mechanism operated by the kinetochore is not sufficient to allow CENP-ACnp1 maintenance at centromeres when FACT function is impaired for long periods; under these conditions, the sequence-driven preference for CENP-ACnp1 assembly at centromeres is compromised.
Perspective
Human FACT has been shown to interact with CENP-A nucleosomes [42]. In chicken DT40 cells FACT has been shown to be required for the deposition of newly synthesized CENP-A, but not for the maintenance of pre-existing CENP-A, at centromeres [53]. Although we cannot exclude a direct role for FACT in CENP-A chromatin assembly at centromeres, our analyses in S. pombe suggests that defective FACT function indirectly affects CENP-ACnp1 deposition at centromeres by allowing CENP-ACnp1 mis-incorporation at non-centromeric locations. This raises the possibility that the depletion of FACT in vertebrate cells may result in newly synthesized CENP-A being dispersed throughout the genome so that its incorporation at centromeres is reduced. In this regard, we predict that factors such as FACT, which are involved in transcription-coupled chromatin reassembly, will have a conserved role in preventing the mis-incorporation of CENP-A at non-centromeric locations in higher eukaryotes. It is possible that at centromeres CENP-A sequesters/inhibits FACT to reduce its activity in recycling H3 nucleosomes during RNAPII transcription through centromeric chromatin. Spt16 is known to directly bind the N-terminal tails and globular core domain of H3 [54]; it will be interesting to determine if CENP-ACnp1 competes with H3 for binding to Spt16.
In C. elegans, CENP-A and centromere activity is distributed along chromosomes. Recent analyses show that transcription in the germline acts to exclude CENP-A incorporation in progeny [55]. In contrast, RNAPII/transcription has been found to be essential for the efficient binding of CENP-C and normal mitotic kinetochore function in human cells [56]. Our analyses in S. pombe suggest a model that reconciles these apparently disparate findings; RNAPII transcription may normally prevent CENP-A deposition at genes through the action of H3 nucleosome reassembly machineries such as FACT, however, when FACT function is defective, RNAPII transcription may promote CENP-A deposition. Thus, RNAPII transcription may act either positively or negatively on CENP-A deposition depending on the functionality of FACT. In monocentric organisms, it is possible that the function of FACT or other H3 nucleosome reassembly pathways is limited at centromeres so that RNAPII transcription at centromeres promotes CENP-A deposition.
Neocentromeres are rare in most systems, but they can form at novel locations in both natural and experimental situations [4], [12], [57] Given the link between transcription-coupled nucleosome dynamics and CENP-ACnp1 assembly highlighted here, it is possible that neocentromeres tend to arise at locations where H3 nucleosomes are less robustly maintained, so that CENP-A and other histones are more frequently incorporated from the free pool, rather than being recycled. Telomeric chromatin may affect the dynamics of H3 nucleosomes on sub-telomeric transcription units so that they are particularly prone to replacement with CENP-A. Likewise, at centromeres heterochromatin may impose constraints on H3 nucleosomes stability during transcription to promote its replacement with CENP-A. The elevated levels of CENP-A, caused by loss of regulation in cancer cells, may increase the frequency at which CENP-A chromatin is established, inducing additional neocentromere formation with resulting in genome instability [14], [17].
Materials and Methods
Cell growth and manipulation
Standard genetic and molecular techniques were followed. Fission yeast methods were as described [58]. For the strains used in the experiments, see Table S2. The cnt1:bigura4 + strain contains the ura4 + embedded within additional DNA consisting of ade6 + sequences inserted within the central domain of cen1 [38]. Note: The sequence of the ade6 + and ura4 + genes is 61% A/T which is close to the average A/T content of 64% for the S. pombe genome.
ChIP
ChIP was performed as described using anti-H3 antibody (ab1791, Abcam), anti-FLAG M2 affinity gel (F2426, Sigma), anti-CENP-ACnp1 antibody, anti-CENP-CCnp3 antibody and anti-H3K9me2 antibody and subsequently analyzed by quantitative PCR (qPCR) [59]. For primers used in qPCR, see Table S3.
Growth of cells overexpressing CENP-ACnp1 for ChIP analyses
Cells expressing additional CENP-ACnp1 from integrated pREP41-cnp1+ (nmt41-CENP-ACnp1) or cells with integrated empty vector were initially grown in rich medium which contains thiamine to repress the expression of additional CENP-ACnp1. The cells were then streaked on minimal (PMG) plates which lack thiamine to allow expression of nmt41-CENP-ACnp1. Subsequently, cells were grown in PMG liquid medium (without thiamine) at 25°C and shifted to 36°C for 1 h to inactivate Spt16 function before ChIP analyses.
Plasmid-based assay for assembly of CENP-ACnp1 chromatin
A plasmid (pcc2) carrying central domain sequence (cc2) but not outer repeat sequence (otr) is introduced into wild-type, pst2Δ, cph1Δ, pst1-1, spt16-6 or spt6-1 cells by electroporation. Transformants were selected on PMG-ura plates supplemented with low adenine (1/50th) at 32°C which allow to distinguish cells with episomal plasmids from those containing integrated plasmids by the colony color (cells with integrated plasmids form white colonies whereas those with episomal plasmids form light pink colonies). The resulting transformants were grown in PMG-ura liquid medium and analyzed by ChIP-qPCR. To confirm that cells maintain episomal plasmids and do not accumulate integrated plasmids, a plasmid stability test was performed at the time of fixation. Cells (200∼2000) were plated onto PMG-ura supplemented with low adenine (1/50th) and allowed to form colonies. Samples exhibiting less than 2% of integrations (i.e. white colonies) were used for ChIP. To extend the number of cell doublings (to 50 doublings) in wild-type or pst2Δ cells carrying pcc2 without increasing the proportion of cells with integrated plasmid, the cells grown in PMG-ura liquid medium (30 doublings after transformation) were plated onto PMG-ura supplemented with low adenine (1/50th). Colonies with light pink color which maintain episomal plasmids without integration were selected and pooled together (∼200 colonies in total) in PMG-ura liquid medium. Cells were grown for additional 8 h (50 doublings after transformation) and subject to ChIP analyses. To confirm that cells maintain episomal plasmids and do not accumulate integrated plasmids, a plasmid stability test was performed at the time of fixation as described above.
Generation of temperature sensitive alleles of spt16 +
To screen spt16-ts alleles, DNA fragment containing either 5′ or 3′ half of spt16 + ORF was mutagenized in vitro using Gene Morph II random mutagenesis kit (Stratagene). Each end of the mutagenized fragments was fused with a kanMX6 marker gene or the upstream (for 5′ half; spt16-1 to spt16-12)/downstream sequences (for 3′ half; spt16-13 to spt16-25) of spt16 + ORF by fusion PCR. The resulting fusion PCR products were further amplified using nested primers and introduced into wild-type cells by electroporation. Transformants were selected on plates containing G418 and temperature sensitive (ts) mutants were identified among G418-resistant colonies by lethality at 36°C after replica-plating to plates containing Phloxine B. To confirm that the temperature sensitivity is caused by mutations in spt16 +, a plasmid rescue experiment was performed. Mutants whose ts phenotypes are rescued by plasmid expressing wild-type spt16 + were selected for further analyses and the causative mutations were identified by sequencing. For detailed information on the spt16-ts alleles, see Table S4.
Northern analysis
Northern analysis was performed as described previously using in vitro transcribed RNA probes [24]. For details on the primers used to create the probes, see Table S3.
Western analysis
Western analysis was performed as described previously using anti-GFP antibody (gift from Kevin Hardwick) and anti-TAT-1 antibody (alpha-tubulin - gift from Keith Gull) [47]. The intensities of GFP and TAT-1 signals were quantified using LICOR Odyssey Infrared Imaging System software (Li-COR Bioscience).
ChIP–chip
DNA was immunoprecipitated as described earlier using 10 µl of anti-CENP-ACnp1 and 1.5 µg of anti-H3 (ab1791, abcam) antibody per 100 µl chromatin extract [60]. For microarrays with spiked-in controls Affymetrix GeneChip Eukaryotic Poly-A RNA Control Kit was used. RNA from the kit was transcribed into cDNA using SuperScript II Reverse Transcriptase (invitrogen) and oligo(dT) primers (invitrogen). The cDNA was diluted 10,000 times and added to the immunoprecipitated samples before round A and B amplification. Fragmentation, labeling and hybridization to the Affymetrix GeneChip S. pombe Tiling 1.0FR was performed by Affymetrix core facility at Novum (BEA) according to Affymetrix standard protocols. Raw data from Affymetrix (.CEL format) were normalized with Affymetrix Tiling Analysis Software (TAS) v1.1 and analyzed and visualized using Podbat [61].
RT–PCR analysis
RT–PCR using total RNAs prepared with RNeasy mini kit (Qiagen) was performed as described [24].
Supporting Information
FACT is not required for central core silencing and does not show genetic interaction with CENP-ACnp1. (A) Schematic of targeted random mutagenesis in 5′ or 3′ regions within spt16 +. See additional details in Materials and Methods. (B) Viability of wt, cnp1-87 and spt16-ts cells with cnt1:ura4 + on N/S (non-selective), -Ura (uracil lacking) and FOA (counterselective drug for ura4 + expression) plates at 25°C. (C) Viability of wt, pob3Δ and cnp1-87 cells with cnt1:ura4 + on N/S (non-selective), -Ura and FOA plates at 32°C. (D) Viability of wt, pob3Δ, cnp1-87 and pob3Δ cnp1-87 cells at indicated temperatures.
(TIF)
CENP-ACnp1 accumulates preferentially at centromere proximal regions in spt16-18 cells with overexpression of CENP-ACnp1. ChIP-chip analyses of relative levels of CENP-ACnp1 in spt16-18 cells compared to wt in the presence of OE-CENP-ACnp1 (nmt41-cnp1 +) at centromere proximal regions. ORFs are displayed as grey boxes. Regions of at least 1 kb in length and with >2-fold increase in CENP-ACnp1 signal above genome-wide average are depicted with red boxes. Data on the Y-axis are presented in linear scale. Blue: running average signal/100 probes. Grey: signal for individual probes.
(TIF)
Detection of cryptic shorter transcripts from prm1 +, tip41 + and act1 +. (A) Northern analyses of transcripts from prm1 + and tip41 + gene. (B) Northern analyses of transcripts from act1 + gene. RNA was extracted from cells grown at 25°C (wt, spt16-18), 32°C for 6 h (wt, pst2Δ) or 36°C for 1 h (wt, spt16-18) after shift from 25°C. Arrow indicates full-length transcripts.
(TIF)
H3 and CENP-ACnp1 are preferentially incorporated in genes expressed at low to intermediate levels in spt16-18 cells. Moving average plots (window size = 100, step size = 1) of H3 (upper panel) and CENP-ACnp1 (lower panel) plotted as a function of RNA expression in WT (arbitrary units a.u.). H3/CENP-ACnp1 association in spt16-18 cells (red) and WT (black) at 36°C.
(TIF)
Effects of CENP-ACnp1 overexpression in cells with defective Spt6. (A) Viability of wt, spt6-1 and spt16-20 cells expressing additional CENP-ACnp1 at low (nmt81-CENP-ACnp1) and medium (nmt41-CENP-ACnp1) levels compared to empty vector at 32°C. Note: spt16-20 cells have a semi-permissive temperature similar to that of spt6-1 and thus are used as a positive control in this experiment. (B) ChIP analysis of CENP-ACnp1 levels at prm1 +, tip41 + and endogenous centromeres (cc1/3) in wt and spt6-1 cells in the absence or presence of OE-CENP-ACnp1 (nmt41-cnp1 +). Cells were grown at 36°C for 1 h after shift from 25°C.
(TIF)
CENP-ACnp1 preferentially accumulates at subtelomeric regions and ectopically placed central domain DNA when overexpressed. (A) ChIP-chip analyses of relative levels of CENP-ACnp1 in wt cells with OE-CENP-ACnp1 (nmt41-cnp1 +) compared to wt cells without OE-CENP-ACnp1. Cells were grown at 36°C for 1 h after shift from 25°C. Data on the Y-axis are presented in log2 scale. (B) Schematic of ectopic cc2 inserted at ura4 + locus (ura4 + -int-cc2). (C) ChIP analysis of CENP-ACnp1 levels at act1 +, pot1 + and ura4 + -int-cc2 in wt cells in the absence or presence of OE-CENP-ACnp1 (nmt41-cnp1 +) grown at indicated temperatures. (D) ChIP analysis of CENP-ACnp1 levels at act1 +, pot1 + and ura4 + -int-cc2 in wt and spt16-18 cells in the absence or presence of OE-CENP-ACnp1 (nmt41-cnp1 +). Cells were grown at 36°C for 1 h after shift from 25°C.
(TIF)
Expression of ura4 + from cnt1:ura4 + or cnt1:bigura4 + is not significantly affected in spt16-18 cells. qRT-PCR analyses to measure the levels of ura4 + transcripts from cnt1:ura4 + or cnt1:bigura4 + in wild-type and spt16-18 cells. Cells were grown at 36°C for 1 h after shift from 25°C. The relative expression levels were calculated as the value of ura4 + expression relative to act1 +. These values (ura4 +/act1 +) were further normalized to those of respective wild-type (relative to wt). Error bars indicate S.D. from 2 biological replicates.
(TIF)
Loss of Sim3 relieves the toxic effects of CENP-ACnp1 overexpression in pob3Δ cells. Viability of wt, pob3Δ, sim3Δ and pob3Δ sim3Δ cells expressing additional CENP-ACnp1 at low (nmt81-CENP-ACnp1) or medium (nmt41-CENP-ACnp1) levels compared to empty vector. Cells were grown at 25°C, 32°C or 36°C. Phloxine B plates stain dead cells red.
(TIF)
Low level overexpression of CENP-ACnp1 partially rescues the lethality of pob3Δ mis6-302 cells. Viability of wt, pob3Δ, mis6-302, pob3Δ mis6-302 and cnp1-1 strains expressing additional CENP-ACnp1 at medium (nmt41-CENP-ACnp1) or low (nmt81-CENP-ACnp1) levels compared to empty vector at 25°C or 32°C.
(TIF)
Defective function of Clr6-CII allows assembly of CENP-ACnp1 chromatin at specific locations. (A) Average gene analysis for the ratio of H3 occupancy in pst2Δ mutants versus wt. Genes are aligned at transcription start site and divided into four groups dependent of their transcription levels. n = number of genes in each group. Error bars represent 99% confidence intervals. (B) ChIP-chip analyses of relative CENP-ACnp1 levels in pst2Δ cells compared to wt in the presence of OE-CENP-ACnp1 (nmt41-cnp1 +). ORFs are displayed as grey boxes. Regions of at least 1 kb in length and with >2-fold increase in CENP-ACnp1 signal above genome-wide average are colored red. Data on the Y-axis are presented in linear scale. Blue: running average signal/100 probes. Grey: signal for individual probes. (C) ChIP analyses of CENP-ACnp1 and CENP-CCnp3 levels at pot1 + and cc2 in pcc2 plasmid compared to endogenous centromere (cc1/3) in wt, pst2Δ, cph1Δ and pst1-1 cells carrying pcc2. Cells were collected after 30 cell doublings at 32°C from the introduction of pcc2. Error bar indicates standard deviation from 3-4 independent biological experiments. (D) ChIP analyses of CENP-ACnp1 and CENP-CCnp3 levels at pot1 + and cc2 in pcc2 plasmid compared to endogenous centromere (cc1/3) in wt and spt16-6 cells carrying pcc2. Error bar indicates standard deviation from 3 independent biological experiments. (Note: we find that most of spt16-ts alleles including spt16-18 do not allow efficient propagation of pcc2 plasmid and thus a specific allele (spt16-6) which allows propagation of pcc2 is used in this particular assay.) (E) ChIP analyses of CENP-ACnp1 and CENP-CCnp3 levels at pot1 + and cc2 in pcc2 plasmid compared to endogenous centromere (cc1/3) in wt and spt6-1 cells carrying pcc2. Error bar indicates standard deviation from 3 independent biological experiments.
(TIF)
Loss of Clr6-CII function does not induce H3K9 methylation on pcc2 plasmid. (A) Schematic of pcc2 plasmid. Regions amplified by primer pairs used in ChIP-qPCR (cc2, ura4 + and vector - a region on the plasmid backbone) are indicated as short black bars. (B) ChIP analyses of H3K9 methylation (H3K9me2) levels at cc2, ura4 + and vector in pcc2 in wt and pst2Δ cells carrying pcc2. (C) ChIP analyses of H3K9me2 levels at chromosomal act1 + and dg in the same samples. dg represents a part of heterochromatic centromere outer repeats and thus serves as a positive control for H3K9me2 ChIP. ChIP was performed after 30 and 50 cell doublings at 32°C from the introduction of pcc2. Enrichment is reported as % IP. Error bars indicate S.D. from 4 biological replicates.
(TIF)
Relative enrichment of CENP-ACnp1 and H3 in spt16-18 versus wild-type cells (at 36°C) at selected genes from ChIP-chip data and their relative RNA expression levels (at 30°C; transcription levels were categorized as in Figure 2F).
(DOC)
List of strains.
(DOC)
List of primers.
(DOC)
List of spt16-ts alleles.
(DOC)
Acknowledgments
We are grateful to the following colleagues for strains: S. Grewal, K. Ishii, F. Winston, and M. Yanagida. A special thanks to F. Winston for providing the spt6-1 strain prior to publication.
Funding Statement
This research was supported by the Wellcome Trust (065061/Z; Principal Research Fellowship to RCA), EMBO (Long Term Fellowship to ESC), the Swedish Research Council (Vetenskapsrådet) to KE, and Cancer Society (Cancerfonden) to KE. The Wellcome Trust Centre for Cell Biology is supported by core funding from the Wellcome Trust (092076). The funders had no role in study design, data collection and analysis, decision to publish, or preparation of the manuscript.
References
- 1. Black BE, Cleveland DW (2011) Epigenetic centromere propagation and the nature of CENP-a nucleosomes. Cell 144: 471–479. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2. Stimpson KM, Sullivan BA (2010) Epigenomics of centromere assembly and function. Current opinion in cell biology 22: 772–780. [DOI] [PubMed] [Google Scholar]
- 3. Malik HS, Henikoff S (2009) Major evolutionary transitions in centromere complexity. Cell 138: 1067–1082. [DOI] [PubMed] [Google Scholar]
- 4. Marshall OJ, Chueh AC, Wong LH, Choo KH (2008) Neocentromeres: new insights into centromere structure, disease development, and karyotype evolution. American journal of human genetics 82: 261–282. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5. Allshire RC, Karpen GH (2008) Epigenetic regulation of centromeric chromatin: old dogs, new tricks? Nature reviews Genetics 9: 923–937. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6. Buscaino A, Allshire R, Pidoux A (2010) Building centromeres: home sweet home or a nomadic existence? Current opinion in genetics & development 20: 118–126. [DOI] [PubMed] [Google Scholar]
- 7. Mendiburo MJ, Padeken J, Fulop S, Schepers A, Heun P (2011) Drosophila CENH3 is sufficient for centromere formation. Science 334: 686–690. [DOI] [PubMed] [Google Scholar]
- 8. Ishii K (2009) Conservation and divergence of centromere specification in yeast. Current opinion in microbiology 12: 616–622. [DOI] [PubMed] [Google Scholar]
- 9. Kagansky A, Folco HD, Almeida R, Pidoux AL, Boukaba A, et al. (2009) Synthetic heterochromatin bypasses RNAi and centromeric repeats to establish functional centromeres. Science 324: 1716–1719. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10. Folco HD, Pidoux AL, Urano T, Allshire RC (2008) Heterochromatin and RNAi are required to establish CENP-A chromatin at centromeres. Science 319: 94–97. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11. Sato H, Masuda F, Takayama Y, Takahashi K, Saitoh S (2012) Epigenetic inactivation and subsequent heterochromatinization of a centromere stabilize dicentric chromosomes. Current biology : CB 22: 658–667. [DOI] [PubMed] [Google Scholar]
- 12. Ishii K, Ogiyama Y, Chikashige Y, Soejima S, Masuda F, et al. (2008) Heterochromatin integrity affects chromosome reorganization after centromere dysfunction. Science 321: 1088–1091. [DOI] [PubMed] [Google Scholar]
- 13. Sullivan LL, Boivin CD, Mravinac B, Song IY, Sullivan BA (2011) Genomic size of CENP-A domain is proportional to total alpha satellite array size at human centromeres and expands in cancer cells. Chromosome research : an international journal on the molecular, supramolecular and evolutionary aspects of chromosome biology 19: 457–470. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14. Tomonaga T, Matsushita K, Yamaguchi S, Oohashi T, Shimada H, et al. (2003) Overexpression and mistargeting of centromere protein-A in human primary colorectal cancer. Cancer research 63: 3511–3516. [PubMed] [Google Scholar]
- 15. Olszak AM, van Essen D, Pereira AJ, Diehl S, Manke T, et al. (2011) Heterochromatin boundaries are hotspots for de novo kinetochore formation. Nature cell biology 13: 799–808. [DOI] [PubMed] [Google Scholar]
- 16. Amato A, Schillaci T, Lentini L, Di Leonardo A (2009) CENPA overexpression promotes genome instability in pRb-depleted human cells. Mol Cancer 8: 119. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17. Heun P, Erhardt S, Blower MD, Weiss S, Skora AD, et al. (2006) Mislocalization of the Drosophila centromere-specific histone CID promotes formation of functional ectopic kinetochores. Developmental cell 10: 303–315. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18. Chan FL, Marshall OJ, Saffery R, Kim BW, Earle E, et al. (2012) Active transcription and essential role of RNA polymerase II at the centromere during mitosis. Proceedings of the National Academy of Sciences of the United States of America 109: 1979–1984. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19. Bergmann JH, Rodriguez MG, Martins NM, Kimura H, Kelly DA, et al. (2011) Epigenetic engineering shows H3K4me2 is required for HJURP targeting and CENP-A assembly on a synthetic human kinetochore. The EMBO journal 30: 328–340. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20. Chueh AC, Northrop EL, Brettingham-Moore KH, Choo KH, Wong LH (2009) LINE retrotransposon RNA is an essential structural and functional epigenetic component of a core neocentromeric chromatin. PLoS Genet 5: e1000354 doi:10.1371/journal.pcbi.1000354. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21. Carone DM, Longo MS, Ferreri GC, Hall L, Harris M, et al. (2009) A new class of retroviral and satellite encoded small RNAs emanates from mammalian centromeres. Chromosoma 118: 113–125. [DOI] [PubMed] [Google Scholar]
- 22. Wong LH, Brettingham-Moore KH, Chan L, Quach JM, Anderson MA, et al. (2007) Centromere RNA is a key component for the assembly of nucleoproteins at the nucleolus and centromere. Genome research 17: 1146–1160. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23. Topp CN, Zhong CX, Dawe RK (2004) Centromere-encoded RNAs are integral components of the maize kinetochore. Proceedings of the National Academy of Sciences of the United States of America 101: 15986–15991. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24. Choi ES, Stralfors A, Castillo AG, Durand-Dubief M, Ekwall K, et al. (2011) Identification of noncoding transcripts from within CENP-A chromatin at fission yeast centromeres. The Journal of biological chemistry 286: 23600–23607. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25. Henikoff S (2008) Nucleosome destabilization in the epigenetic regulation of gene expression. Nature reviews Genetics 9: 15–26. [DOI] [PubMed] [Google Scholar]
- 26. Li B, Carey M, Workman JL (2007) The role of chromatin during transcription. Cell 128: 707–719. [DOI] [PubMed] [Google Scholar]
- 27. Saunders A, Werner J, Andrulis ED, Nakayama T, Hirose S, et al. (2003) Tracking FACT and the RNA polymerase II elongation complex through chromatin in vivo. Science 301: 1094–1096. [DOI] [PubMed] [Google Scholar]
- 28. Kaplan CD, Laprade L, Winston F (2003) Transcription elongation factors repress transcription initiation from cryptic sites. Science 301: 1096–1099. [DOI] [PubMed] [Google Scholar]
- 29. Belotserkovskaya R, Oh S, Bondarenko VA, Orphanides G, Studitsky VM, et al. (2003) FACT facilitates transcription-dependent nucleosome alteration. Science 301: 1090–1093. [DOI] [PubMed] [Google Scholar]
- 30. Keogh MC, Kurdistani SK, Morris SA, Ahn SH, Podolny V, et al. (2005) Cotranscriptional set2 methylation of histone H3 lysine 36 recruits a repressive Rpd3 complex. Cell 123: 593–605. [DOI] [PubMed] [Google Scholar]
- 31. Carrozza MJ, Li B, Florens L, Suganuma T, Swanson SK, et al. (2005) Histone H3 methylation by Set2 directs deacetylation of coding regions by Rpd3S to suppress spurious intragenic transcription. Cell 123: 581–592. [DOI] [PubMed] [Google Scholar]
- 32. Mason PB, Struhl K (2003) The FACT complex travels with elongating RNA polymerase II and is important for the fidelity of transcriptional initiation in vivo. Molecular and cellular biology 23: 8323–8333. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33. Nicolas E, Yamada T, Cam HP, Fitzgerald PC, Kobayashi R, et al. (2007) Distinct roles of HDAC complexes in promoter silencing, antisense suppression and DNA damage protection. Nature structural & molecular biology 14: 372–380. [DOI] [PubMed] [Google Scholar]
- 34. Wittmeyer J, Joss L, Formosa T (1999) Spt16 and Pob3 of Saccharomyces cerevisiae form an essential, abundant heterodimer that is nuclear, chromatin-associated, and copurifies with DNA polymerase alpha. Biochemistry 38: 8961–8971. [DOI] [PubMed] [Google Scholar]
- 35. Orphanides G, Wu WH, Lane WS, Hampsey M, Reinberg D (1999) The chromatin-specific transcription elongation factor FACT comprises human SPT16 and SSRP1 proteins. Nature 400: 284–288. [DOI] [PubMed] [Google Scholar]
- 36. Lejeune E, Bortfeld M, White SA, Pidoux AL, Ekwall K, et al. (2007) The chromatin-remodeling factor FACT contributes to centromeric heterochromatin independently of RNAi. Current biology : CB 17: 1219–1224. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37. Forsburg SL (1993) Comparison of Schizosaccharomyces pombe expression systems. Nucleic acids research 21: 2955–2956. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38. Castillo AG, Mellone BG, Partridge JF, Richardson W, Hamilton GL, et al. (2007) Plasticity of fission yeast CENP-A chromatin driven by relative levels of histone H3 and H4. PLoS Genet 3: e121 doi:10.1371/journal.pgen.0030121. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39. Pidoux AL, Richardson W, Allshire RC (2003) Sim4: a novel fission yeast kinetochore protein required for centromeric silencing and chromosome segregation. The Journal of cell biology 161: 295–307. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40. Jamai A, Puglisi A, Strubin M (2009) Histone chaperone spt16 promotes redeposition of the original h3-h4 histones evicted by elongating RNA polymerase. Molecular cell 35: 377–383. [DOI] [PubMed] [Google Scholar]
- 41. Schwabish MA, Struhl K (2004) Evidence for eviction and rapid deposition of histones upon transcriptional elongation by RNA polymerase II. Molecular and cellular biology 24: 10111–10117. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42. Foltz DR, Jansen LE, Black BE, Bailey AO, Yates JR 3rd, et al. (2006) The human CENP-A centromeric nucleosome-associated complex. Nature cell biology 8: 458–469. [DOI] [PubMed] [Google Scholar]
- 43. Kiely CM, Marguerat S, Garcia JF, Madhani HD, Bahler J, et al. (2011) Spt6 is required for heterochromatic silencing in the fission yeast Schizosaccharomyces pombe. Molecular and cellular biology [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44. Hayashi T, Fujita Y, Iwasaki O, Adachi Y, Takahashi K, et al. (2004) Mis16 and Mis18 are required for CENP-A loading and histone deacetylation at centromeres. Cell 118: 715–729. [DOI] [PubMed] [Google Scholar]
- 45. Takahashi K, Chen ES, Yanagida M (2000) Requirement of Mis6 centromere connector for localizing a CENP-A-like protein in fission yeast. Science 288: 2215–2219. [DOI] [PubMed] [Google Scholar]
- 46. Williams JS, Hayashi T, Yanagida M, Russell P (2009) Fission yeast Scm3 mediates stable assembly of Cnp1/CENP-A into centromeric chromatin. Molecular cell 33: 287–298. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 47. Pidoux AL, Choi ES, Abbott JK, Liu X, Kagansky A, et al. (2009) Fission yeast Scm3: A CENP-A receptor required for integrity of subkinetochore chromatin. Molecular cell 33: 299–311. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 48. Dunleavy EM, Pidoux AL, Monet M, Bonilla C, Richardson W, et al. (2007) A NASP (N1/N2)-related protein, Sim3, binds CENP-A and is required for its deposition at fission yeast centromeres. Molecular cell 28: 1029–1044. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 49. Govind CK, Qiu H, Ginsburg DS, Ruan C, Hofmeyer K, et al. (2010) Phosphorylated Pol II CTD recruits multiple HDACs, including Rpd3C(S), for methylation-dependent deacetylation of ORF nucleosomes. Molecular cell 39: 234–246. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 50. Okada T, Ohzeki J, Nakano M, Yoda K, Brinkley WR, et al. (2007) CENP-B controls centromere formation depending on the chromatin context. Cell 131: 1287–1300. [DOI] [PubMed] [Google Scholar]
- 51. Winkler DD, Luger K (2011) The Histone Chaperone FACT: Structural Insights and Mechanisms for Nucleosome Reorganization. The Journal of biological chemistry 286: 18369–18374. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 52. Jansen LE, Black BE, Foltz DR, Cleveland DW (2007) Propagation of centromeric chromatin requires exit from mitosis. The Journal of cell biology 176: 795–805. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 53. Okada M, Okawa K, Isobe T, Fukagawa T (2009) CENP-H-containing complex facilitates centromere deposition of CENP-A in cooperation with FACT and CHD1. Molecular biology of the cell 20: 3986–3995. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 54. Stuwe T, Hothorn M, Lejeune E, Rybin V, Bortfeld M, et al. (2008) The FACT Spt16 “peptidase” domain is a histone H3-H4 binding module. Proceedings of the National Academy of Sciences of the United States of America 105: 8884–8889. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 55. Gassmann R, Rechtsteiner A, Yuen KW, Muroyama A, Egelhofer T, et al. (2012) An inverse relationship to germline transcription defines centromeric chromatin in C. elegans. Nature 484: 534–537. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 56. Chan FL, Marshall OJ, Saffery R, Kim BW, Earle E, et al. (2012) Active transcription and essential role of RNA polymerase II at the centromere during mitosis. Proceedings of the National Academy of Sciences of the United States of America 109: 1979–1984. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 57. Ketel C, Wang HS, McClellan M, Bouchonville K, Selmecki A, et al. (2009) Neocentromeres form efficiently at multiple possible loci in Candida albicans. PLoS Genet 5: e1000400 doi:10.1371/journal.pgen.1000400. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 58. Moreno S, Klar A, Nurse P (1991) Molecular genetic analysis of fission yeast Schizosaccharomyces pombe. Methods in enzymology 194: 795–823. [DOI] [PubMed] [Google Scholar]
- 59. Nelson JD, Denisenko O, Bomsztyk K (2006) Protocol for the fast chromatin immunoprecipitation (ChIP) method. Nature protocols 1: 179–185. [DOI] [PubMed] [Google Scholar]
- 60. Durand-Dubief M, Ekwall K (2009) Chromatin immunoprecipitation using microarrays. Methods in molecular biology 529: 279–295. [DOI] [PubMed] [Google Scholar]
- 61. Sadeghi L, Bonilla C, Stralfors A, Ekwall K, Svensson JP (2011) Podbat: a novel genomic tool reveals Swr1-independent H2A.Z incorporation at gene coding sequences through epigenetic meta-analysis. PLoS Comput Biol 7: e1002163 doi:10.1371/journal.pcbi.1002163. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 62. Durand-Dubief M, Persson J, Norman U, Hartsuiker E, Ekwall K (2010) Topoisomerase I regulates open chromatin and controls gene expression in vivo. The EMBO journal 29: 2126–2134. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Supplementary Materials
FACT is not required for central core silencing and does not show genetic interaction with CENP-ACnp1. (A) Schematic of targeted random mutagenesis in 5′ or 3′ regions within spt16 +. See additional details in Materials and Methods. (B) Viability of wt, cnp1-87 and spt16-ts cells with cnt1:ura4 + on N/S (non-selective), -Ura (uracil lacking) and FOA (counterselective drug for ura4 + expression) plates at 25°C. (C) Viability of wt, pob3Δ and cnp1-87 cells with cnt1:ura4 + on N/S (non-selective), -Ura and FOA plates at 32°C. (D) Viability of wt, pob3Δ, cnp1-87 and pob3Δ cnp1-87 cells at indicated temperatures.
(TIF)
CENP-ACnp1 accumulates preferentially at centromere proximal regions in spt16-18 cells with overexpression of CENP-ACnp1. ChIP-chip analyses of relative levels of CENP-ACnp1 in spt16-18 cells compared to wt in the presence of OE-CENP-ACnp1 (nmt41-cnp1 +) at centromere proximal regions. ORFs are displayed as grey boxes. Regions of at least 1 kb in length and with >2-fold increase in CENP-ACnp1 signal above genome-wide average are depicted with red boxes. Data on the Y-axis are presented in linear scale. Blue: running average signal/100 probes. Grey: signal for individual probes.
(TIF)
Detection of cryptic shorter transcripts from prm1 +, tip41 + and act1 +. (A) Northern analyses of transcripts from prm1 + and tip41 + gene. (B) Northern analyses of transcripts from act1 + gene. RNA was extracted from cells grown at 25°C (wt, spt16-18), 32°C for 6 h (wt, pst2Δ) or 36°C for 1 h (wt, spt16-18) after shift from 25°C. Arrow indicates full-length transcripts.
(TIF)
H3 and CENP-ACnp1 are preferentially incorporated in genes expressed at low to intermediate levels in spt16-18 cells. Moving average plots (window size = 100, step size = 1) of H3 (upper panel) and CENP-ACnp1 (lower panel) plotted as a function of RNA expression in WT (arbitrary units a.u.). H3/CENP-ACnp1 association in spt16-18 cells (red) and WT (black) at 36°C.
(TIF)
Effects of CENP-ACnp1 overexpression in cells with defective Spt6. (A) Viability of wt, spt6-1 and spt16-20 cells expressing additional CENP-ACnp1 at low (nmt81-CENP-ACnp1) and medium (nmt41-CENP-ACnp1) levels compared to empty vector at 32°C. Note: spt16-20 cells have a semi-permissive temperature similar to that of spt6-1 and thus are used as a positive control in this experiment. (B) ChIP analysis of CENP-ACnp1 levels at prm1 +, tip41 + and endogenous centromeres (cc1/3) in wt and spt6-1 cells in the absence or presence of OE-CENP-ACnp1 (nmt41-cnp1 +). Cells were grown at 36°C for 1 h after shift from 25°C.
(TIF)
CENP-ACnp1 preferentially accumulates at subtelomeric regions and ectopically placed central domain DNA when overexpressed. (A) ChIP-chip analyses of relative levels of CENP-ACnp1 in wt cells with OE-CENP-ACnp1 (nmt41-cnp1 +) compared to wt cells without OE-CENP-ACnp1. Cells were grown at 36°C for 1 h after shift from 25°C. Data on the Y-axis are presented in log2 scale. (B) Schematic of ectopic cc2 inserted at ura4 + locus (ura4 + -int-cc2). (C) ChIP analysis of CENP-ACnp1 levels at act1 +, pot1 + and ura4 + -int-cc2 in wt cells in the absence or presence of OE-CENP-ACnp1 (nmt41-cnp1 +) grown at indicated temperatures. (D) ChIP analysis of CENP-ACnp1 levels at act1 +, pot1 + and ura4 + -int-cc2 in wt and spt16-18 cells in the absence or presence of OE-CENP-ACnp1 (nmt41-cnp1 +). Cells were grown at 36°C for 1 h after shift from 25°C.
(TIF)
Expression of ura4 + from cnt1:ura4 + or cnt1:bigura4 + is not significantly affected in spt16-18 cells. qRT-PCR analyses to measure the levels of ura4 + transcripts from cnt1:ura4 + or cnt1:bigura4 + in wild-type and spt16-18 cells. Cells were grown at 36°C for 1 h after shift from 25°C. The relative expression levels were calculated as the value of ura4 + expression relative to act1 +. These values (ura4 +/act1 +) were further normalized to those of respective wild-type (relative to wt). Error bars indicate S.D. from 2 biological replicates.
(TIF)
Loss of Sim3 relieves the toxic effects of CENP-ACnp1 overexpression in pob3Δ cells. Viability of wt, pob3Δ, sim3Δ and pob3Δ sim3Δ cells expressing additional CENP-ACnp1 at low (nmt81-CENP-ACnp1) or medium (nmt41-CENP-ACnp1) levels compared to empty vector. Cells were grown at 25°C, 32°C or 36°C. Phloxine B plates stain dead cells red.
(TIF)
Low level overexpression of CENP-ACnp1 partially rescues the lethality of pob3Δ mis6-302 cells. Viability of wt, pob3Δ, mis6-302, pob3Δ mis6-302 and cnp1-1 strains expressing additional CENP-ACnp1 at medium (nmt41-CENP-ACnp1) or low (nmt81-CENP-ACnp1) levels compared to empty vector at 25°C or 32°C.
(TIF)
Defective function of Clr6-CII allows assembly of CENP-ACnp1 chromatin at specific locations. (A) Average gene analysis for the ratio of H3 occupancy in pst2Δ mutants versus wt. Genes are aligned at transcription start site and divided into four groups dependent of their transcription levels. n = number of genes in each group. Error bars represent 99% confidence intervals. (B) ChIP-chip analyses of relative CENP-ACnp1 levels in pst2Δ cells compared to wt in the presence of OE-CENP-ACnp1 (nmt41-cnp1 +). ORFs are displayed as grey boxes. Regions of at least 1 kb in length and with >2-fold increase in CENP-ACnp1 signal above genome-wide average are colored red. Data on the Y-axis are presented in linear scale. Blue: running average signal/100 probes. Grey: signal for individual probes. (C) ChIP analyses of CENP-ACnp1 and CENP-CCnp3 levels at pot1 + and cc2 in pcc2 plasmid compared to endogenous centromere (cc1/3) in wt, pst2Δ, cph1Δ and pst1-1 cells carrying pcc2. Cells were collected after 30 cell doublings at 32°C from the introduction of pcc2. Error bar indicates standard deviation from 3-4 independent biological experiments. (D) ChIP analyses of CENP-ACnp1 and CENP-CCnp3 levels at pot1 + and cc2 in pcc2 plasmid compared to endogenous centromere (cc1/3) in wt and spt16-6 cells carrying pcc2. Error bar indicates standard deviation from 3 independent biological experiments. (Note: we find that most of spt16-ts alleles including spt16-18 do not allow efficient propagation of pcc2 plasmid and thus a specific allele (spt16-6) which allows propagation of pcc2 is used in this particular assay.) (E) ChIP analyses of CENP-ACnp1 and CENP-CCnp3 levels at pot1 + and cc2 in pcc2 plasmid compared to endogenous centromere (cc1/3) in wt and spt6-1 cells carrying pcc2. Error bar indicates standard deviation from 3 independent biological experiments.
(TIF)
Loss of Clr6-CII function does not induce H3K9 methylation on pcc2 plasmid. (A) Schematic of pcc2 plasmid. Regions amplified by primer pairs used in ChIP-qPCR (cc2, ura4 + and vector - a region on the plasmid backbone) are indicated as short black bars. (B) ChIP analyses of H3K9 methylation (H3K9me2) levels at cc2, ura4 + and vector in pcc2 in wt and pst2Δ cells carrying pcc2. (C) ChIP analyses of H3K9me2 levels at chromosomal act1 + and dg in the same samples. dg represents a part of heterochromatic centromere outer repeats and thus serves as a positive control for H3K9me2 ChIP. ChIP was performed after 30 and 50 cell doublings at 32°C from the introduction of pcc2. Enrichment is reported as % IP. Error bars indicate S.D. from 4 biological replicates.
(TIF)
Relative enrichment of CENP-ACnp1 and H3 in spt16-18 versus wild-type cells (at 36°C) at selected genes from ChIP-chip data and their relative RNA expression levels (at 30°C; transcription levels were categorized as in Figure 2F).
(DOC)
List of strains.
(DOC)
List of primers.
(DOC)
List of spt16-ts alleles.
(DOC)