

Abstract
Objective
Integrins mediate platelet adhesion and transmit “outside in” signals, leading to platelet spreading. Phosphoinositide 3-Kinases (PI3Ks) play a critical role in outside-in signaling and platelet spreading; however, the mechanisms of PI3K activation and function in outside-in signaling are unclear. We sought to determine the role of the Akt family of serine/threonine kinases and activation mechanisms of the PI3K/Akt pathway in outside-in signaling.
Methods and Results
Akt inhibitors and Akt3 knockout inhibited platelet spreading on fibrinogen, indicating that Akt is important in integrin outside-in signaling. Akt inhibitors and Akt3 knockout also diminished integrin-dependent phosphorylation of glycogen synthase kinase-3β (GSK-3β). Inhibition of GSK-3β reversed the inhibitory effects of Akt3 knockout and inhibitors of Akt or PI3K on platelet spreading, indicating that GSK-3β is a downstream target of Akt in outside-in signaling. Integrin-dependent activation of the PI3K-Akt pathway requires Src Family Kinase (SFK). Akt phosphorylation is also significantly inhibited in ADP receptor P2Y12 knockout platelets and further inhibited in P2Y12 knockout platelets treated with a P2Y1 antagonist. Consistently, P2Y12 knockout and P2Y1 inhibition together reduced platelet spreading.
Conclusion
These results demonstrate that integrin outside-in signaling and platelet spreading requires SFK-dependent and ADP receptor-amplified activation of the PI3K-Akt-GSK-3β pathway.
Keywords: platelet, integrin, Akt, outside-in signaling, ADP
Integrins are a family of cell adhesion receptors that regulate numerous cellular processes such as adhesion, migration, and proliferation. Integrin αIIbβ3 is the major integrin subtype expressed in blood platelets. Integrin signaling is bidirectional in that intracellular signaling mechanisms induce changes in the extracellular ligand binding domain of integrins to an activated state, and ligand binding to the activated integrins conversely transmits “outside-in” signals,1-3 which are critically important in stable platelet adhesion, spreading, and clot retraction.4 Cell spreading is an early consequence of integrin outside-in signaling and represents the outward movement of the cell membrane, characterized by formation of lamellipodia and filipodia. Understanding the mechanisms and signaling proteins involved in “outside-in” signal transduction may facilitate the development of pharmacological inhibitors for regulation of these processes. We have recently shown that the most proximal event following integrin ligation is the binding of the G protein subunit Gα13 to the cytoplasmic domain of β3 integrin.5 This interaction then leads to activation of SFKs, particularly c-Src which is associated with β3.6, 7 A downstream pathway of the Gα13-c-Src-mediated signal is the Src-dependent phosphorylation of p190 RhoGAP, which leads to inactivation of RhoA and subsequent inhibition of the RhoA contractile pathway, thus promoting cell spreading.8, 9 c-Src is also important for the phosphorylation of the β3 cytoplasmic domain and for the activation of the ITAM and Syk signaling pathway, which are also important in facilitating cell spreading.6, 10
Numerous groups have demonstrated an important role for PI3K in mediating or facilitating integrin outside-in signaling.11-15 Accordingly, PI3K inhibitors diminish cell spreading in platelets and other cell types;11, 16 however, the mechanism by which integrin outside-in signaling activates PI3K remains unclear. Also, the downstream mechanism by which PI3Ks exert their effects is unknown. A well known downstream effector of PI3K is the serine/threonine kinase Akt.17 Akt isoforms are expressed in platelets and play important roles in promoting granule secretion and the second wave of platelet aggregation.18-22 It has been shown that Akt is phosphorylated and activated downstream from integrin ligation.14 However, the functional importance of Akt in mediating integrin outside-in signaling remains unknown.
In the present study, we address the following questions with regard to PI3K-Akt signaling during integrin outside-in signaling: (1) what is the upstream mechanism that activates the PI3K-Akt signaling pathway; (2) whether and how Akt isoforms play a role as downstream mediators of PI3K signaling, and (3) if Akt is important in integrin signaling, what is the downstream mechanism mediating the PI3K-Akt signaling pathway. Here, we show that Akt isoforms are important in mediating PI3K signaling induced by integrin outside-in signaling. We further show that a downstream mechanism important in mediating the PI3K-Akt signaling is the Akt-mediated phosphorylation and inhibition of GSK-3β. In addition, we have discovered that the integrin-mediated full activation of the PI3K/Akt/GSK-3β signaling pathway requires the ADP receptor P2Y12 and involves P2Y1. These findings provide evidence for an important role of a SFK-dependent and ADP-amplified PI3K/Akt/GSK-3β signaling pathway in integrin outside-in signaling leading to platelet spreading.
Methods
Materials
Src family kinase inhibitor PP2, Akt inhibitor AktX, PI3K inhibitors LY294002 and wortmannin were purchased from Calbiochem. P2Y12 antagonist 2-MeSAMP (2-methylthioadenosine 5’-monophosphate triethylammonium salt), P2Y1 antagonist A3P5P (adenosine-3’-phosphate-5’phosphate), BSA (bovine serum albumin), and GSK-3β inhibitor SB216763 were purchased from Sigma. Fibrinogen was purchased from Enzyme Research Laboratories.
Animals
The generation of Akt1, Akt2, or Akt3 knockout23-25 and P2Y12 knockout mice26 were previously described. Mice used in this study were 8-15 week-old on C57BL/6 background. Animal usage and protocol was approved by the institutional animal care committee of the University of Illinois at Chicago.
Preparation of platelets
For studies using human platelets, fresh blood was drawn by venipuncture from healthy volunteers and anticoagulated with 1-seventh volume of acid-citrate dextrose (ACD) as previously described.27 Institutional Review Board approval was obtained from the University of Illinois at Chicago, and informed consent was provided according to the Declaration of Helsinki. For the preparation of mouse platelets, fresh blood was drawn from mouse inferior vena cava and anti-coagulated with ACD as previously described.27 Blood from 5 to 6 mice of either genotype was pooled and platelets were isolated by differential centrifugation of whole blood with 0.1 μg/mL prostaglandin E1 and 1 U/mL apyrase (Sigma-Aldrich).27 Platelets were washed twice with modified Tyrode’s buffer containing 5 mM EDTA and allowed to rest for at least 1 hour at room temperature before use.28
Immunoblot detection
Washed human or mouse platelets (3×108/mL) were allowed to spread on fibrinogen coated polystyrene dishes (100 μg/mL in 0.1M NaHCO3, pH 8.3 overnight at room temperature) for various lengths of time. Non-adherent platelets were aspirated and the reaction was stopped by addition of sample buffer containing 2% SDS, 0.1 M Tris, 2% glycerol, 2 mM PMSF, 2 mM Na3VO4, 2 mM NaF, and Complete Protease Inhibitor Cocktail (Roche Molecular Biochemicals). Proteins were separated by sodium dodecyl sulfate polyacrylamide gel electrophoresis (SDS-PAGE) on a 4-15% polyacrylamide gel and then transferred to polyvinylidene difluoride membranes. The membranes were immunoblotted with an antibody recognizing GSK-3β phosphorylated at Ser9, an antibody recognizing Akt (all isoforms) phosphorylated at Ser473, or antibodies against GSK-3β (Cell Signaling Technology), or Akt (Santa Cruz Biotechnology, Inc) (non-phosphorylated and phosphorylated).
Platelet spreading on immobilized fibrinogen
Microscope cover glasses (Fisher Scientific) were coated with 100 μg/mL fibrinogen (Enzyme Research Laboratories) in 0.1 M NaHCO3 (pH 8.3) and blocked with 5% bovine serum albumin (BSA) in phosphate-buffered saline (PBS). Washed mouse platelets (2×107/mL) were allowed to adhere and spread on fibrinogen-coated wells (300 μL/well) at 37°C for 2 hours. Washed human platelets (1×107/mL) were allowed to adhere and spread on fibrinogen-coated wells (300 μL/well) at 37°C for 1 hour. Slides were aspirated to remove non-adherent platelets and fixed with 4% paraformaldehyde, permeabilized, and stained with Alexa Fluor FITC conjugated phalloidin (Invitrogen) as previously described.16 Adherent platelets were observed with a Leica DM IRB fluorescence microscope (Leica Microsystems) using 100X/1.30 NA oil objective (1.5X magnification factor for mouse platelets, 1.0X for human platelets). Images were acquired using a Cool SNAP HQ CCD camera (Photometrics) and processed with RS Image version 1.4 software (Photometrics) or Micro-Manager version 1.4.
ATP secretion
Black, 96-well plates were coated with 100 μg/mL fibrinogen or BSA in 0.1 M NaHCO3 (pH 8.3). Plates were blocked with BSA and washed human platelets (100μL of 3×108/mL) were added to fibrinogen-coated wells or BSA (control) coated wells at 37°C for various time points. ENLITEN luciferase reagent (Promega) was added to wells and luminescence was immediately measured using Wallac Victor2 1420 multilabel counter (Perkin Elmer).
RESULTS
The role of Akt isoforms as downstream effectors of PI3K in mediating platelet spreading
We sought to determine whether the PI3K effector, Akt, plays an important role in integrin outside-in signaling. To inhibit the functions of all Akt isoforms, a pan Akt inhibitor, AktX, was used. AktX is a phenoxazine that selectively inhibits the phosphorylation of Akt, thus suppressing its kinase reactivity.29 This Akt inhibitor is not a phosphoinositide analog, and thus does not directly affect functions of other phosphoinositide-regulated proteins.29 This excludes the possible nonspecific inhibition of other PI3K effectors, which is a potential complication with previously used Akt inhibitors such as SH-6, a competitive inhibitor of phosphoinositide binding to Akt. Human platelets were treated with increasing doses of AktX, and allowed to spread on immobilized fibrinogen. AktX dose-dependently inhibited human platelet spreading (Fig. 1, A and B). At 10 μM, AktX completely abolished platelet spreading on fibrinogen. A similar dose-dependent defect in spreading was observed using mouse platelets treated with increasing concentrations of AktX (Fig. 1, C and D), suggesting that platelets from humans and mice similarly require Akt to promote integrin outside-in signaling and platelet spreading. To confirm the effectiveness of AktX on Akt activation and function in our experimental system, wild type mouse platelets were treated with increasing doses of AktX and allowed to spread on fibrinogen. After 30 minutes of spreading, platelets were harvested for western blot analysis of Akt phosphorylation at Ser473. Akt is phosphorylated following platelet spreading on a fibrinogen-coated surface, indicating that Akt becomes activated during integrin outside-in signaling. AktX dose-dependently inhibited phosphorylation of Akt at Ser473 (Fig. 1E). Altogether, these data suggest Akt is activated and plays an important role in mediating human and mouse platelet spreading.
Figure 1.
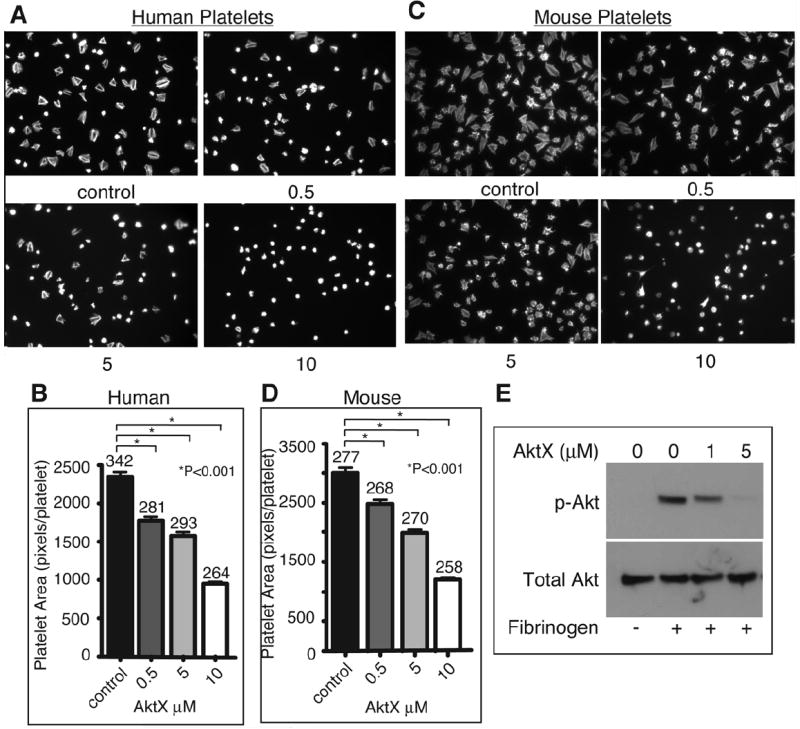
Effect of an Akt inhibitor on platelet spreading. (A) Human platelets spread on fibrinogen for 1 hour, were then fixed, permeabilized, stained, and observed with a fluorescence microscope. Representative pictures. (B) Surface area of single platelets from (A) was measured (mean surface area ± SE). Data from 3 experiments. Difference between groups was analyzed using Student’s t test. Numbers of platelets analyzed are indicated above the bars. (C) Mouse platelets spread on fibrinogen for 2 hours, and were analyzed as described in (A). (D) Quantitation of data from (C) for platelet surface area as in (B). (E) Human platelets were incubated with increasing concentrations of AktX and spread on fibrinogen for 30 minutes or kept in suspension. Adherent platelets or control platelets were solubilized, and analyzed by western blot.
Platelets express three different Akt isoforms: Akt1, Akt2 and Akt3. Our recent results suggest that Akt3 is a major Akt isoform in platelets (~70% of total Akt in mouse platelets).21 We therefore evaluated whether Akt3 plays a role in the integrin outside-in signaling pathway. Fig. 2A shows that Akt is robustly phosphorylated in wild type platelets spread on a fibrinogen-coated surface as detected by an anti-phospho-Akt S473 antibody recognizing all three Akt isoforms. Integrin-dependent phosphorylation of Akt was significantly inhibited in Akt3-/- platelets, indicating that Akt3 is a major Akt isoform phosphorylated downstream of integrin signaling in mouse platelets (Fig. 2A). Phosphorylation of Akt at T308 was also significantly reduced in Akt3-/- mouse platelets adherent on fibrinogen (Fig. 2A). Thus, we further investigated the role of Akt3 in platelet spreading using Akt3-/- mice. As shown in Fig. 2, B and C, Akt3-/- partially, but significantly reduced platelet spreading on fibrinogen, suggesting that Akt3 is important in promoting platelet spreading. However, the incomplete inhibition by Akt3-/- is in contrast to the complete inhibition of platelet spreading by a higher dose of AktX, which inhibits all Akt isoforms. We also assessed the spreading of Akt1-/- or Akt2-/- mouse platelets. Akt1-/- mouse platelets showed a partial reduction in spreading compared to WT, while no difference was observed using Akt2-/- mouse platelets (Supplemental Fig. I A and B). Combined with the data obtained with pan Akt inhibitors, our results suggest that Akt3 is an important isoform mediating integrin outside-in signaling, but other isoforms of Akt, particularly Akt1, may also be involved in mediating integrin outside-in signaling.
Figure 2.
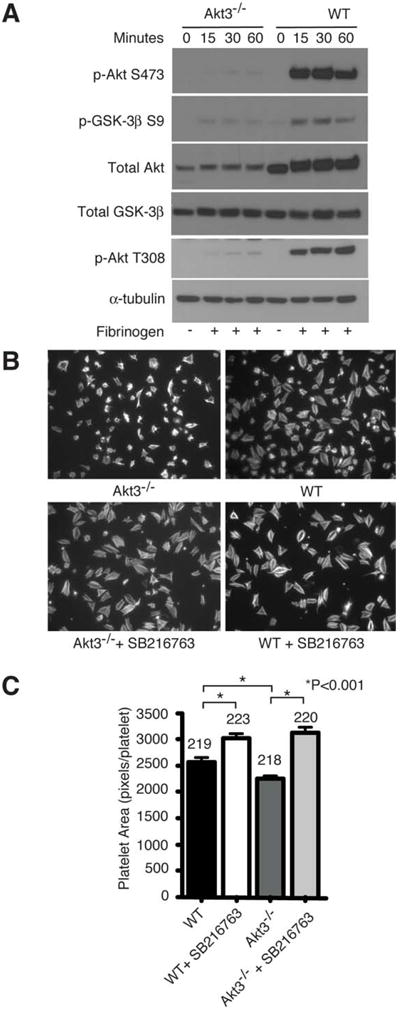
Effects of Akt3 knockout on integrin outside-in signaling and the role of GSK-3β. (A) WT or Akt3-/- mouse platelets were plated on fibrinogen for indicated lengths of time or kept in suspension. Adherent or control platelets were solubilized. and analyzed by western blot. (B) Platelets from Akt3-/- or WT mice were preincubated with GSK-3β inhibitor SB216763 or control DMSO, plated on fibrinogen for 2 hours, fixed, permeabilized, stained, and observed with a fluorescence microscope. (C) The surface area of single platelets in (B) was measured (mean±SE). Difference between groups was analyzed using Student’s t test. Shown in figure are representative pictures from one of three experiments. Numbers of platelets analyzed are indicated above the bars.
Akt-dependent phosphorylation of GSK-3β is a downstream mechanism of integrin outside-in signaling
Akt is known to phosphorylate GSK-3β, which negatively regulates GSK-3β function.30 GSK-3β was shown to negatively regulate thrombin receptor-mediated platelet activation and in vivo thrombosis.31 To determine whether Akt3 mediates integrin outside-in signaling and platelet spreading by phosphorylating and inactivating GSK-3β, wild type and Akt3-/- platelets were allowed to spread on fibrinogen and phosphorylation of Ser9 of GSK-3β was assessed by immunoblot with an antibody specifically recognizing phosphorylation at Ser9 site. Compared to wild type platelets spread on fibrinogen, Akt3-/- platelets showed a significant reduction in GSK-3β phosphorylation, indicating that Akt3 is important for mediating GSK-3β phosphorylation during integrin outside-in signaling (Fig. 2A). Akt1-/- mouse platelets also showed a trend of reduced GSK-3β phosphorylation, but was not statistically significant (Supplemental Fig. I C and D). In order to further investigate whether negative regulation of GSK-3β is important in promoting integrin-dependent platelet spreading, we assessed the effect of GSK-3β selective inhibitor SB216763 on platelet spreading on fibrinogen-coated surfaces. Treatment of platelets with SB216763 significantly promoted platelet spreading compared to DMSO control, indicating that GSK-3β negatively regulates platelet spreading and thus, inhibition of GSK-3β promotes platelet spreading (Fig. 2, B and C). Because Akt3-/- has reduced GSK-3β phosphorylation, which presumably results in increased GSK-3β activity, we sought to determine whether the spreading defect of Akt3-/- platelets could be corrected by treatment with GSK-3β inhibitor SB216763. Akt3-/- platelets were treated with the GSK-3β inhibitor and allowed to spread on fibrinogen. Indeed, the inhibitory effect of Akt3 knockout on platelet spreading was reversed by GSK-3β inhibitor SB216763 (Fig. 2, B and C). Interestingly, spreading of platelets treated with the pan Akt inhibitor AktX or with the PI3K inhibitor LY294002 was partially rescued by treatment with SB216763 (Fig. 3, A and B). Thus, negative regulation of GSK-3β is a likely mechanism downstream of PI3K and Akt3 that promotes integrin-dependent platelet spreading, although we do not exclude possible additional mechanisms, particularly for other Akt isoforms.
Figure 3.
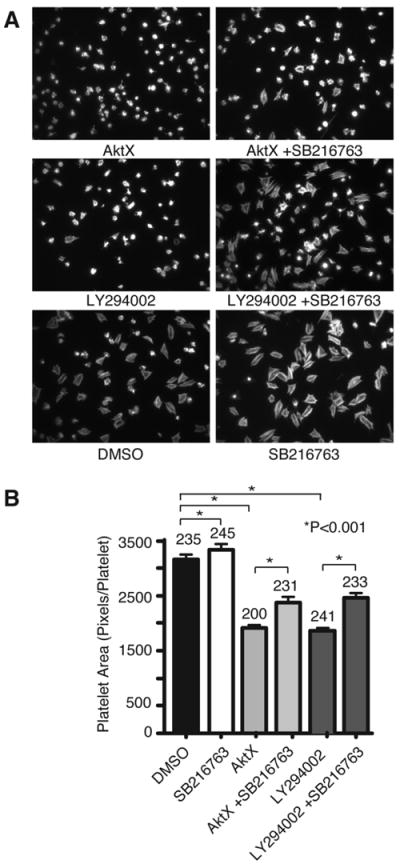
Reversal of the inhibitory effect of PI3K/Akt inhibitors on platelet spreading by GSK-3β inhibitor. (A) WT mouse platelets were preincubated with AktX (5 μM), LY294002 (20 μM), or DMSO control in the presence and absence of GSK-3β inhibitor SB216763 (10 μM) and plated on immobilized fibrinogen for 2 hours. Adherent platelets were fixed, permeabilized, stained, and observed with a fluorescence microscope. (B) The surface area of single platelets was quantitated (mean ± SE). Statistical significance was analyzed using Student’s t test. Shown in figure are representative pictures from one of three experiments. Numbers of platelets analyzed are indicated above the bars.
Src and ADP receptor-dependent activation of the PI3K-Akt signaling pathway during integrin signaling
Thus far, our experiments reveal an important role for the PI3K-Akt-GSK-3β signaling pathway in promoting platelet spreading. To understand the molecular mechanisms that are responsible for the activation of this pathway during integrin outside-in signaling, we examined whether phosphorylation of Akt can be affected by inhibitors of intracellular signaling molecules involved in integrin outside-in signaling. As expected, Akt is phosphorylated following spreading of human platelets on integrin ligand fibrinogen. PI3K inhibitor wortmannin or LY294002 totally abolished Akt phosphorylation, indicating that PI3K is indeed responsible for Akt phosphorylation and thus, Akt phosphorylation is an indicator of PI3K activation under our experimental conditions (Fig. 4, A and B). PP2, an inhibitor of Src family kinases (SFKs), also abolished integrin-dependent Akt phosphorylation, indicating that SFK is required for integrin-induced activation of the PI3K/Akt signaling pathway (Fig. 4C). In order to determine whether activation of the ITAM and Syk signaling pathway, which are important in amplifying integrin signaling, are required for integrin-induced PI3K-Akt activation, we tested the effect of the Syk inhibitor piceatannol on phosphorylation of Akt. Akt phosphorylation in human platelets spread on fibrinogen was not affected by piceatannol (10 μM) (Fig. 4D), indicating the ITAM/Syk pathway is not required for Akt activation. Thus, the PI3K/Akt pathway is downstream from SFKs, but not ITAM/Syk, in the integrin outside-in signaling pathway.
Figure 4.
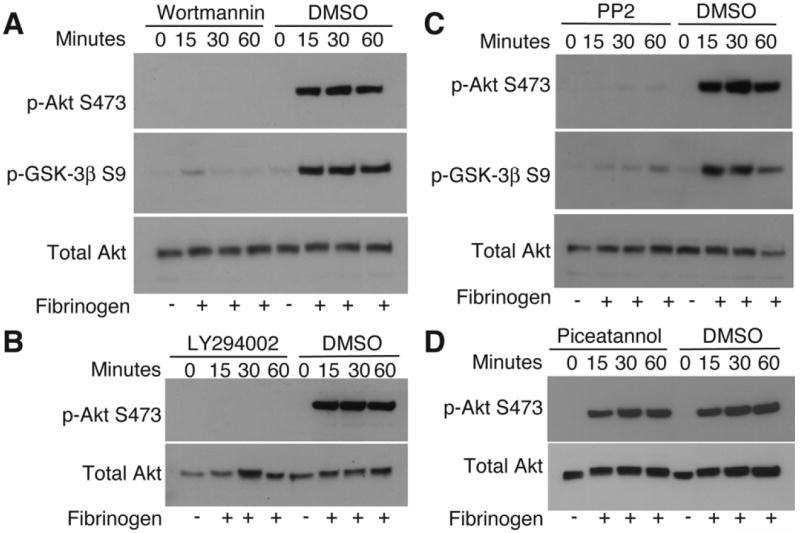
Upstream molecules important in Akt activation. Human platelets were treated with (A) PI3K inhibitor wortmannin (100 nM) (B) PI3K inhibitor LY294002 (20 μM), (C) SFK inhibitor PP2 (10 μM), (D) Syk inhibitor piceatannol (10 μM), or DMSO control, and plated on immobilized fibrinogen or kept in suspension., solubilized and western blot analysis was performed.
Interestingly, integrin-mediated Akt phosphorylation was dramatically reduced in human platelets by an antagonist of ADP receptor P2Y12, 2-MeSAMP, and was further reduced when 2-MeSAMP was used together with A3P5P, an antagonist of the other platelet ADP receptor, P2Y1 (Fig. 5A). To exclude the possible nonspecific effects of ADP receptor antagonists, we also examined integrin-dependent Akt phosphorylation in mouse platelets deficient in the P2Y12 receptor (P2Y12 knockout). Indeed, Akt phosphorylation in P2Y12-/- platelets was also dramatically reduced (Fig. 5B). P2Y12-/- mouse platelets treated with A3P5P further inhibited phosphorylation of Akt, as well as GSK-3β (Fig. 5C). Thus, we conclude that ADP receptor signaling, particularly P2Y12 signaling, is important in integrin-induced and Src-dependent activation of the PI3K/Akt signaling pathway. It is important to note that there is still residual phosphorylation of Akt and GSK-3β in the presence of P2Y1/P2Y12 blockade, indicating a small pool of Akt and GSK-3β may be activated by ADP-independent mechanisms.
Figure 5.
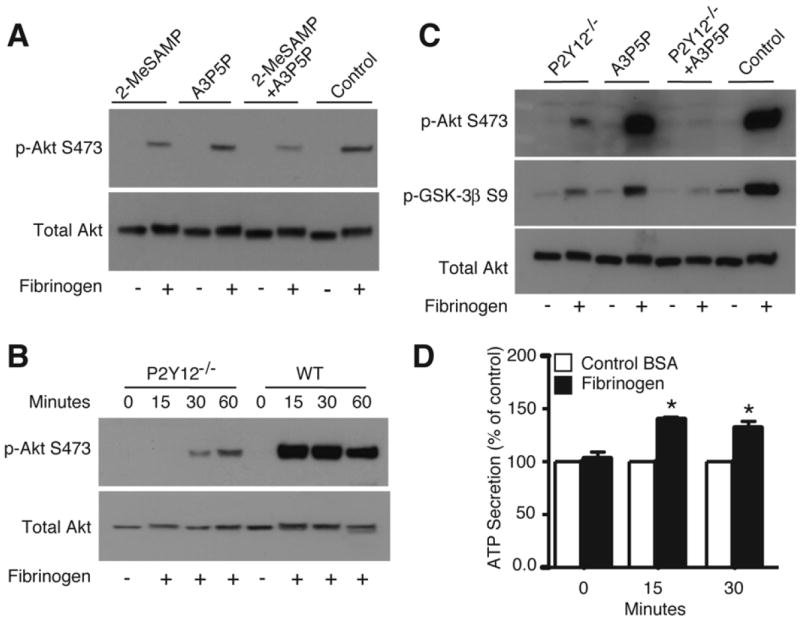
Effect of P2Y12 knockout and ADP receptor antagonists on integrin-dependent Akt activation. (A) Human platelets were pre-incubated with 2-MeSAMP (50 μM), A3P5P (0.5 mM), both, or control, and plated on immobilized fibrinogen for 30 minutes or kept in suspension, solubilized, and analyzed by western blot. (B) WT or P2Y12-/- mouse platelets spread on immobilized fibrinogen and were analyzed by western blot as in (A). (C) WT or P2Y12-/- mouse platelets were treated with A3P5P (0.5 mM) or control, and spread on fibrinogen for 30 min. or kept in suspension. Western blot was performed as in (A). (D) Human platelets were added to BSA or fibrinogen coated wells for indicated times. Luciferase was added and ATP secretion was detected using a luminometer. The relative quantity of ATP secretion is expressed as the percentage increase of BSA control (mean ± SE, n=3). The difference between control and fibrinogen at 15 and 30 minutes time point are significant (P<0.03), as determined by student’s t-test.
To directly address whether integrin ligation with fibrinogen induces dense granule secretion, we measured ATP release of platelets spread on fibrinogen using a luciferase assay. There was a significant increase in ATP release that was detected in platelets spread on fibrinogen, compared to platelets added to BSA control wells (Fig. 5D). Integrin-dependent platelet adhesion does not require prior stimulation by exogenous agonists; therefore, our data indicates that ADP receptor signaling that stimulates activation of the PI3K/Akt pathway is likely to be induced by endogenously secreted ADP during platelet spreading on integrin ligands.
To determine whether the effects of ADP receptor signaling on PI3K/Akt pathway is functionally important in integrin-dependent platelet spreading, we compared the spreading of P2Y12-/- platelets and P2Y12 antagonist 2-MeSAMP treated platelets with wild type platelets. P2Y12-/- platelets and P2Y12 antagonist-treated platelets showed a significant, but partial inhibition of platelet spreading compared to control platelets, indicating that P2Y12 signaling is important in promoting integrin outside-in signaling leading to platelet spreading (Fig. 6, A-D). We also tested the effect of the P2Y1 antagonist A3P5P on spreading of control or P2Y12-/- mouse platelets. Consistent with the relatively minor defects caused by A3P5P in the activation of the PI3K-Akt pathway, only a minor spreading defect was observed in wild type or human platelets treated with A3P5P. However, A3P5P significantly further inhibited platelet spreading in P2Y12-/- mouse platelets (Fig. 6, A and B). Human platelets treated with ADP receptor P2Y12 antagonist 2-MeSAMP (and to a much less degree A3P5P) similarly inhibited platelet spreading on fibrinogen (Fig. 6, C and D). These results suggest an important role for P2Y12 and P2Y1 receptors in promoting integrin outside-in signaling leading to platelet spreading.
Figure 6.

Effect of P2Y12 knockout and ADP receptor antagonists on platelet spreading. (A) WT or P2Y12-/- mouse platelets were treated with A3P5P (0.5 mM) or control and spread on fibrinogen for 2 hours, fixed, permeabilized, stained, and observed as previously described. Representative pictures. (B) The surface areas of individual platelets were measured (mean±SE). Difference between groups was analyzed using Student’s t test. Numbers of platelets analyzed are indicated above the bars. Data are from 3 experiments. (C) Human platelets were treated with 2-MeSAMP (50 μM), A3P5P (0.5 mM), both, or control, and spread on fibrinogen for 1 hour as described in (A). (D) Quantitation of (C) performed as in (B). (E) A schematic depicting the regulatory mechanisms and roles of the PI3K/Akt pathway in integrin-mediated platelet spreading.
Discussion
In this study, we show that (1) Akt, as a downstream effector of PI3K, plays an important role in mediating integrin outside-in signaling leading to platelet spreading; (2) Akt promotes platelet spreading by phosphorylating and inhibiting GSK-3β; and (3) activation of PI3K-Akt-GSK-3β pathway is downstream from SFK and requires platelet secretion of ADP and ADP receptor signaling. Taken together, our results reveal a novel outside-in signaling pathway involving sequential activation of Src, ADP secretion, P2Y12/P2Y1-dependent activation of PI3K and Akt, and Akt-mediated inhibition of GSK-3β, leading to stimulation of platelet spreading (Fig. 6E). Our study also demonstrates a novel mutually amplifying relationship between the G-protein-coupled P2Y12/P2Y1 receptor signaling pathways and integrin outside-in signaling pathways.
It has been shown by several groups that PI3Ks, including both type I and type II, play an important role in integrin signaling.12-15, 32 However, the downstream effector that is important in mediating PI3K signals promoting platelet spreading is unclear. Among potential PI3K effectors, all three Akt isoforms have been shown to be expressed in platelets and to be important in mediating platelet granule secretion, and secretion-dependent amplification of platelet aggregation.19-22 However, it is unclear whether Akt isoforms are also important in integrin outside-in signaling. In this study, we have shown that treatment of human or mouse platelets with a pan Akt inhibitor, AktX, that is a non-phosphoinositide competitor, potently inhibits platelet spreading on fibrinogen (Fig. 1). Furthermore, the spreading of Akt3-/- platelets was also partially inhibited (Fig. 2, B and C). Therefore, we conclude that Akt is an important downstream effector that mediates PI3K-dependent integrin outside-in signaling. Our data further indicate that the Akt3 isoform is important in outside-in signaling, but also suggest the involvement of other Akt isoforms.
Our data suggest that phosphorylation and inhibition of GSK-3β, a known Akt substrate, is an important downstream mechanism responsible for the role of Akt3 in promoting platelet spreading. This is supported by the data that Akt3-/- platelets spread on fibrinogen showed a significant reduction in GSK-3β phosphorylation (Fig. 2A) and that GSK-3β inhibition by SB216763 was sufficient to completely rescue the defect in Akt3-/- platelet spreading. These data indicate that the role of Akt3 in platelet spreading is mainly mediated by GSK-3β (Fig. 2, B and C). Consistently, inhibition of GSK-3β also partially rescued spreading defects of platelets treated with pan-Akt inhibitor AktX or PI3K inhibitor LY294002 (Fig. 3, A and B). However, partial rescue by SB216763 suggest the possible presence of other integrin-dependent Akt-mediated signaling pathways that are independent of GSK-3β. Further study is needed to resolve how GSK-3β negatively regulates platelet spreading.
It is known that ADP secretion can promote platelet spreading on immobilized fibrinogen and scavengers of ADP such as apyrase can inhibit platelet spreading.33, 34 It has also been reported that platelets from a patient deficient in ADP receptor P2Y12 have defective spreading on fibrinogen.35 Correspondingly, our data demonstrating defective spreading in P2Y12-/- mouse platelets and P2Y12 or P2Y1 inhibitor treated platelets support the implication of ADP in amplifying integrin outside-in signaling and spreading (Fig. 6). However, the mechanism by which ADP promotes platelet spreading has been unclear. It is both possible that the roles of ADP can be due to increasing either integrin activation (inside-out signaling) or outside-in signaling. While we do not exclude the well-established role for ADP in inside-out signaling and integrin activation, our data clearly show that one mechanism by which ADP promotes platelet spreading is its amplification of the PI3K-Akt-GSK-3β signaling pathway during outside-in signaling (Fig. 5). It is important to note that because integrin-dependent platelet adhesion does not require prior stimulation by agonists and we have used carefully prepared resting platelets without exogenous agonists, ADP receptor signaling that stimulates activation of the PI3K/Akt pathway is likely to be induced by endogenously secreted ADP during platelet spreading on integrin ligands. Indeed, we, for the first time, detected granule secretion from platelets adherent on a fibrinogen coated surface (Fig. 5D). Thus, we conclude that granule secretion induced by integrin outside-in signaling plays an important role in stimulating activation of the PI3K/Akt pathway and thus platelet spreading.
Interestingly, our data also show that, while ADP receptors play a major role in PI3K/Akt activation, activation of a small pool of Akt is independent of ADP receptors, but clearly requires the function of SFKs (Fig. 4C). The spreading defect in platelets treated with a high dose of AktX (Fig. 1) also appears to be greater than that in P2Y12-/- platelets treated with P2Y1 antagonists (Fig. 6), which is consistent with an ADP-independent activation of Akt in promoting spreading. In other platelet signaling pathways, such as thrombin receptor and GPIb-IX pathways, it has previously been shown that SFKs are important in stimulating the activation of PI3K/Akt pathway.36-38 Furthermore, in the platelet activation pathways induced by thrombin and thromboxane A2, PI3K/Akt activation involves a P2Y12-independent mechanism that accounts for a small fraction of Akt activation and a major amplification role of P2Y12/Gi signaling.27, 28 Thus, a small, initial SFK-dependent activation of PI3K/Akt and an ADP-dependent amplification of PI3K/Akt activation may be a general mechanism in which the PI3K/Akt pathway becomes fully functional during platelet activation induced by not only soluble agonist receptors, but also integrins and other adhesion receptors. It is interesting to note that a major role of Akt isoforms in regulating platelet function downstream from soluble agonists is to promote platelet dense granule secretion.16, 19-22 Thus, we hypothesized that one of the roles of SFK-dependent initial activation of Akt isoforms in regulating platelet spreading may be to induce platelet granule secretion. Secreted ADP then greatly augments the PI3K-Akt-GSK-3β pathway, resulting in stronger amplification of integrin outside-in signaling and enhancement of platelet spreading. Nonetheless, the precise mechanism by which the PI3K-Akt-GSK-3β pathway promotes outward movement of the platelet membrane and spreading requires further examination.
Supplementary Material
Spreading of Akt isoform knockout mouse platelets. (A) WT, Akt1-/-, Akt2-/-, or Akt3-/- mouse platelets (2×107/mL) were plated on immobilized fibrinogen for 2 hours, fixed, permeabilized, stained with phalloidin, and observed with a fluorescence microscope. Representative pictures are shown. (B) The surface area of single platelets was quantitated (average surface area ± SE). Statistical significance of the difference between groups was analyzed using Student’s t test. Numbers of platelets analyzed for each group (from three experiments) are indicated above the bars (*p<0.05). (C) WT or Akt1-/- mouse platelets were plated on fibrinogen for indicated time or kept in suspension as a control. Adherent platelets or control platelets were analyzed by Western blot for phosphorylation of GSK-3β at S9, and total GSK-3β levels. (D) Western blots from 3 experiments were scanned and quantified using NIH Image J for uncalibrated optical density. The relative quantity of phospho-GSK-3β of Akt1-/- vs. WT platelets spread on fibrinogen is expressed as the % of WT (mean ± SE). No statistical significance was obtained.
Acknowledgments
Sources of Funding
This work is supported by grants from NHLBI/NIH, HL068819, HL062350, and HL080264 to XD; and from CA090764, AG016927, and AG025953 to NH.
Footnotes
Disclosures
None.
References
- 1.Ginsberg MH, Partridge A, Shattil SJ. Integrin regulation. Curr Opin Cell Biol. 2005;17:509–516. doi: 10.1016/j.ceb.2005.08.010. [DOI] [PubMed] [Google Scholar]
- 2.Ma YQ, Qin J, Plow EF. Platelet integrin alpha(IIb)beta(3): activation mechanisms. J Thromb Haemost. 2007;5:1345–1352. doi: 10.1111/j.1538-7836.2007.02537.x. [DOI] [PubMed] [Google Scholar]
- 3.Li Z, Delaney MK, O’Brien KA, Du X. Signaling during platelet adhesion and activation. Arterioscler Thromb Vasc Biol. 2010;30:2341–2349. doi: 10.1161/ATVBAHA.110.207522. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Shattil SJ, Newman PJ. Integrins: dynamic scaffolds for adhesion and signaling in platelets. Blood. 2004;104:1606–1615. doi: 10.1182/blood-2004-04-1257. [DOI] [PubMed] [Google Scholar]
- 5.Gong H, Shen B, Flevaris P, Chow C, Lam SC, Voyno-Yasenetskaya TA, Kozasa T, Du X. G protein subunit Galpha13 binds to integrin alphaIIbbeta3 and mediates integrin “outside-in” signaling. Science. 2010;327:340–343. doi: 10.1126/science.1174779. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Obergfell A, Eto K, Mocsai A, Buensuceso C, Moores SL, Brugge JS, Lowell CA, Shattil SJ. Coordinate interactions of Csk, Src, and Syk kinases with [alpha]IIb[beta]3 initiate integrin signaling to the cytoskeleton. J Cell Biol. 2002;157:265–275. doi: 10.1083/jcb.200112113. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Arias-Salgado EG, Lizano S, Sarkar S, Brugge JS, Ginsberg MH, Shattil SJ. Src kinase activation by direct interaction with the integrin beta cytoplasmic domain. Proc Natl Acad Sci U S A. 2003;100:13298–13302. doi: 10.1073/pnas.2336149100. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Flevaris P, Stojanovic A, Gong H, Chishti A, Welch E, Du X. A molecular switch that controls cell spreading and retraction. J Cell Biol. 2007;179:553–565. doi: 10.1083/jcb.200703185. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Arthur WT, Burridge K. RhoA inactivation by p190RhoGAP regulates cell spreading and migration by promoting membrane protrusion and polarity. Mol Biol Cell. 2001;12:2711–2720. doi: 10.1091/mbc.12.9.2711. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Boylan B, Gao C, Rathore V, Gill JC, Newman DK, Newman PJ. Identification of FcgammaRIIa as the ITAM-bearing receptor mediating alphaIIbbeta3 outside-in integrin signaling in human platelets. Blood. 2008;112:2780–2786. doi: 10.1182/blood-2008-02-142125. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Heraud JM, Racaud-Sultan C, Gironcel D, Albiges-Rizo C, Giacomini T, Roques S, Martel V, Breton-Douillon M, Perret B, Chap H. Lipid products of phosphoinositide 3-kinase and phosphatidylinositol 4’,5’-bisphosphate are both required for ADP-dependent platelet spreading. J Biol Chem. 1998;273:17817–17823. doi: 10.1074/jbc.273.28.17817. [DOI] [PubMed] [Google Scholar]
- 12.Canobbio I, Stefanini L, Cipolla L, Ciraolo E, Gruppi C, Balduini C, Hirsch E, Torti M. Genetic evidence for a predominant role of PI3Kbeta catalytic activity in ITAM- and integrin-mediated signaling in platelets. Blood. 2009;114:2193–2196. doi: 10.1182/blood-2009-03-208074. [DOI] [PubMed] [Google Scholar]
- 13.Jackson SP, Yap CL, Anderson KE. Phosphoinositide 3-kinases and the regulation of platelet function. Biochem Soc Trans. 2004;32:387–392. doi: 10.1042/bst0320387. [DOI] [PubMed] [Google Scholar]
- 14.Banfic H, Tang X, Batty IH, Downes CP, Chen C, Rittenhouse SE. A novel integrin-activated pathway forms PKB/Akt-stimulatory phosphatidylinositol 3,4-bisphosphate via phosphatidylinositol 3-phosphate in platelets. J Biol Chem. 1998;273:13–16. doi: 10.1074/jbc.273.1.13. [DOI] [PubMed] [Google Scholar]
- 15.Zhang J, Banfic H, Straforini F, Tosi L, Volinia S, Rittenhouse SE. A type II phosphoinositide 3-kinase is stimulated via activated integrin in platelets. A source of phosphatidylinositol 3-phosphate. J Biol Chem. 1998;273:14081–14084. doi: 10.1074/jbc.273.23.14081. [DOI] [PubMed] [Google Scholar]
- 16.Yin H, Stojanovic A, Hay N, Du X. The role of Akt in the signaling pathway of the glycoprotein Ib-IX induced platelet activation. Blood. 2008;111:658–665. doi: 10.1182/blood-2007-04-085514. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Bhaskar PT, Hay N. The two TORCs and Akt. Dev Cell. 2007;12:487–502. doi: 10.1016/j.devcel.2007.03.020. [DOI] [PubMed] [Google Scholar]
- 18.Woulfe DS. Akt signaling in platelets and thrombosis. Expert Rev Hematol. 2010;3:81–91. doi: 10.1586/ehm.09.75. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Chen J, De S, Damron DS, Chen WS, Hay N, Byzova TV. Impaired platelet responses to thrombin and collagen in AKT-1-deficient mice. Blood. 2004;104:1703–1710. doi: 10.1182/blood-2003-10-3428. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Woulfe D, Jiang H, Morgans A, Monks R, Birnbaum M, Brass LF. Defects in secretion, aggregation, and thrombus formation in platelets from mice lacking Akt2. J Clin Invest. 2004;113:441–450. doi: 10.1172/JCI20267. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.O’Brien KA, Stojanovic-Terpo A, Hay N, Du X. An important role for Akt3 in platelet activation and thrombosis. Blood. 2011;118:4215–4223. doi: 10.1182/blood-2010-12-323204. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Stojanovic A, Marjanovic JA, Brovkovych VM, Peng X, Hay N, Skidgel RA, Du X. A Phosphoinositide 3-Kinase-AKT-Nitric Oxide-cGMP Signaling Pathway in Stimulating Platelet Secretion and Aggregation. J Biol Chem. 2006;281:16333–16339. doi: 10.1074/jbc.M512378200. [DOI] [PubMed] [Google Scholar]
- 23.Chen WS, Xu PZ, Gottlob K, Chen ML, Sokol K, Shiyanova T, Roninson I, Weng W, Suzuki R, Tobe K, Kadowaki T, Hay N. Growth retardation and increased apoptosis in mice with homozygous disruption of the Akt1 gene. Genes Dev. 2001;15:2203–2208. doi: 10.1101/gad.913901. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Garofalo RS, Orena SJ, Rafidi K, Torchia AJ, Stock JL, Hildebrandt AL, Coskran T, Black SC, Brees DJ, Wicks JR, McNeish JD, Coleman KG. Severe diabetes, age-dependent loss of adipose tissue, and mild growth deficiency in mice lacking Akt2/PKB beta. J Clin Invest. 2003;112:197–208. doi: 10.1172/JCI16885. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Easton RM, Cho H, Roovers K, Shineman DW, Mizrahi M, Forman MS, Lee VM, Szabolcs M, de Jong R, Oltersdorf T, Ludwig T, Efstratiadis A, Birnbaum MJ. Role for Akt3/protein kinase Bgamma in attainment of normal brain size. Mol Cell Biol. 2005;25:1869–1878. doi: 10.1128/MCB.25.5.1869-1878.2005. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Foster CJ, Prosser DM, Agans JM, Zhai Y, Smith MD, Lachowicz JE, Zhang FL, Gustafson E, Monsma FJ, Jr, Wiekowski MT, Abbondanzo SJ, Cook DN, Bayne ML, Lira SA, Chintala MS. Molecular identification and characterization of the platelet ADP receptor targeted by thienopyridine antithrombotic drugs. J Clin Invest. 2001;107:1591–1598. doi: 10.1172/JCI12242. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Li Z, Zhang G, Le Breton GC, Gao X, Malik AB, Du X. Two waves of platelet secretion induced by thromboxane A2 receptor and a critical role for phosphoinositide 3-kinases. J Biol Chem. 2003;278:30725–30731. doi: 10.1074/jbc.M301838200. [DOI] [PubMed] [Google Scholar]
- 28.Liu J, Pestina TI, Berndt MC, Jackson CW, Gartner TK. Botrocetin/VWF-induced signaling through GPIb-IX-V produces TxA2 in an alphaIIbbeta3- and aggregation-independent manner. Blood. 2005;106:2750–2756. doi: 10.1182/blood-2005-04-1667. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Thimmaiah KN, Easton JB, Germain GS, Morton CL, Kamath S, Buolamwini JK, Houghton PJ. Identification of N10-substituted phenoxazines as potent and specific inhibitors of Akt signaling. J Biol Chem. 2005;280:31924–31935. doi: 10.1074/jbc.M507057200. [DOI] [PubMed] [Google Scholar]
- 30.Cross DA, Alessi DR, Cohen P, Andjelkovich M, Hemmings BA. Inhibition of glycogen synthase kinase-3 by insulin mediated by protein kinase B. Nature. 1995;378:785–789. doi: 10.1038/378785a0. [DOI] [PubMed] [Google Scholar]
- 31.Li D, August S, Woulfe DS. GSK3beta is a negative regulator of platelet function and thrombosis. Blood. 2008;111:3522–3530. doi: 10.1182/blood-2007-09-111518. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Kovacsovics TJ, Bachelot C, Toker A, Vlahos CJ, Duckworth B, Cantley LC, Hartwig JH. Phosphoinositide 3-kinase inhibition spares actin assembly in activating platelets but reverses platelet aggregation. J Biol Chem. 1995;270:11358–11366. doi: 10.1074/jbc.270.19.11358. [DOI] [PubMed] [Google Scholar]
- 33.Haimovich B, Lipfert L, Brugge JS, Shattil SJ. Tyrosine phosphorylation and cytoskeletal reorganization in platelets are triggered by interaction of integrin receptors with their immobilized ligands. J Biol Chem. 1993;268:15868–15877. [PubMed] [Google Scholar]
- 34.Jirouskova M, Jaiswal JK, Coller BS. Ligand density dramatically affects integrin alpha IIb beta 3-mediated platelet signaling and spreading. Blood. 2007;109(12):5260–5269. doi: 10.1182/blood-2006-10-054015. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35.Shiraga M, Miyata S, Kato H, Kashiwagi H, Honda S, Kurata Y, Tomiyama Y, Kanakura Y. Impaired platelet function in a patient with P2Y12 deficiency caused by a mutation in the translation initiation codon. J Thromb Haemost. 2005;3:2315–2323. doi: 10.1111/j.1538-7836.2005.01554.x. [DOI] [PubMed] [Google Scholar]
- 36.Yin H, Liu J, Li Z, Berndt MC, Lowell CA, Du X. Src family tyrosine kinase Lyn mediates VWF/GPIb-IX-induced platelet activation via the cGMP signaling pathway. Blood. 2008;112:1139–1146. doi: 10.1182/blood-2008-02-140970. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37.Li Z, Zhang G, Liu J, Stojanovic A, Ruan C, Lowell CA, Du X. An important role of the SRC family kinase Lyn in stimulating platelet granule secretion. J Biol Chem. 2010;285:12559–12570. doi: 10.1074/jbc.M109.098756. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38.Cho MJ, Pestina TI, Steward SA, Lowell CA, Jackson CW, Gartner TK. Role of the Src family kinase Lyn in TxA2 production, adenosine diphosphate secretion, Akt phosphorylation, and irreversible aggregation in platelets stimulated with gamma-thrombin. Blood. 2002;99:2442–2447. doi: 10.1182/blood.v99.7.2442. [DOI] [PubMed] [Google Scholar]
- 39.Xiang B, Zhang G, Liu J, Morris AJ, Smyth SS, Gartner TK, Li Z. A G(i) - independent mechanism mediating Akt phosphorylation in platelets. J Thromb Haemost. 2010;8:2032–2041. doi: 10.1111/j.1538-7836.2010.03969.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Supplementary Materials
Spreading of Akt isoform knockout mouse platelets. (A) WT, Akt1-/-, Akt2-/-, or Akt3-/- mouse platelets (2×107/mL) were plated on immobilized fibrinogen for 2 hours, fixed, permeabilized, stained with phalloidin, and observed with a fluorescence microscope. Representative pictures are shown. (B) The surface area of single platelets was quantitated (average surface area ± SE). Statistical significance of the difference between groups was analyzed using Student’s t test. Numbers of platelets analyzed for each group (from three experiments) are indicated above the bars (*p<0.05). (C) WT or Akt1-/- mouse platelets were plated on fibrinogen for indicated time or kept in suspension as a control. Adherent platelets or control platelets were analyzed by Western blot for phosphorylation of GSK-3β at S9, and total GSK-3β levels. (D) Western blots from 3 experiments were scanned and quantified using NIH Image J for uncalibrated optical density. The relative quantity of phospho-GSK-3β of Akt1-/- vs. WT platelets spread on fibrinogen is expressed as the % of WT (mean ± SE). No statistical significance was obtained.