

Abstract
Cardiac α-tropomyosin (Tm) single site mutations, D175N and E180G, cause familial hypertrophic cardiomyopathy (FHC). Previous studies have shown that these mutations increase both Ca2+-sensitivity and residual contractile activity at low Ca2+, which causes incomplete relaxation during diastole resulting in hypertrophy and sarcomeric disarray. However, the molecular basis for the cause and the difference in severity of the manifested phenotypes of disease is not known. In this work we have: 1) used ATPase studies on reconstituted thin filaments in solution to show that these FHC mutants result in an increase in Ca2+-sensitivity and increased residual ATPase; 2) shown that both FHC mutants increase the rate of cleavage at R133, about 45 residues N-terminal to the mutations, when free and bound to actin; 3) shown that for Tm-E180G, the increase in the rate of cleavage is greater than for D175N; 4) shown that for E180G, cleavage also occurs at a new site 53 residues C-terminal to E180G, in parallel with cleavage at R133. The long-range decreases in dynamic stability due to these two single site mutations, suggests increases in flexibility that may decrease the ability of Tm to inhibit activity at low Ca2+ for D175N and to a greater degree for E180G, which may contribute to differences in the severity of FHC.
Familial hypertrophic cardiomyopathy (FHC) is a genetically transmitted disease that causes progressive remodeling of the myocardium and may result in sudden death. FHC is caused by mutations in sarcomeric proteins including the thin filament regulatory proteins troponin (Tn) and tropomyosin (Tm) (1-3). The single site mutations, D175N and E180G, in cardiac α-tropomyosin have been the subject of several early studies (4, 5), including force measurements on fibers (6, 7), in vitro motility assays (8), ATPase measurements on Tm mutants exchanged into myofibrils (9), physiological measurements on mouse model hearts (10) and myosin subfragment 1 (S1) binding kinetics using reconstituted thin filaments (11). These studies suggest that incomplete relaxation of the heart at the low Ca2+ concentrations present during diastole coupled with the increased Ca2+-sensitivity can cause some of the phenotypes observed in the disease. In most cases the Ca+2 -sensitivity increase was greater for E180G than D175N. However, the molecular basis for the in vivo abnormal functional properties of these Tm mutants is not known.
Our early study on the properties of the D175N and E180G Tm mutants showed that: 1) the binding of these mutants to actin in the absence and presence of Tn is weakened (12), (note label mixup, (13)); 2) the thermal stability of E180G is decreased; 3) the region of the molecule near position 190 is destabilized. The greatest effects occur for the E180G FHC mutant. More recent biochemical studies have verified the weakened binding to actin (14). These mutations are located outside of the hydrophobic region of the coiled-coil but may be involved in ionic interactions across the chains (15). Thus, the single site mutations D175N and E180G dramatically affect the global properties of Tm, but the underlying mechanisms are not clear. It is also unknown how these effects cause the FHC phenotype.
Tm is a coiled-coil molecule stabilized primarily by hydrophobic side-chain interactions between the two parallel α-helical polypeptide chains. In the actinTmTn thin filament, Tm acts in two ways: 1) by sterically inhibiting the interaction of myosin heads with actin at low Ca2+ and 2) by inhibiting the transition of myosin heads on actin to the force-generating state. This is the basis of the 3-state model consisting of Blocked (− Ca2+), Closed (Ca+2-indiuced) and Open (myosin-induced) (16) and is correlated with 3 positions of Tm on actin (17). The 3-state model has provided a useful framework for understanding normal muscle regulation. Recently, we proposed the inclusion of a 4th state, an active state present in the absence of Ca2+ produced by strong-binding myosin heads (18) in order to help understand how mutations, involving most of the sarcomeric muscle proteins, can cause cardiac diseases such as familial hypertrophic cardiomyopathy (FHC) and dilated cardiomyopathy (DCM). The FHC effects are mimicked by NEM-S1 (19), a strong binding analogue of S1 which does not dissociate from actin in the presence of ATP, indicating that the increase in Ca2+-sensitivity involves myosin.
In this work, we showed that substitution of Gly and Gln neutral amino acids for negatively charged Glu and Asp, respectively, on the outside of the Tm coiled coil affects the protein local stability about 50 residues away, a phenomenon of long range information transfer. The decreased stability of the FHC mutants suggests a greater flexibility of Tm on the thin filament which may be important in their malfunction. The results are discussed within the framework of the 4-state model recently published whereby the contribution of an active state in the absence of Ca2+ can explain several effects produced by strong binding myosin heads (18).
Experimental Proceedures
Protein preparations
The recombinant FHC Tms, WT, E180G and D175N, were prepared from rat cardiac αTm by recombinant DNA methods as described earlier (14, 20). In addition to an Ala-Ser N-terminal extension to facilitate binding to actin, a K279N mutation was included to change the rat sequence to match the rabbit/human sequences. Transfected cells were lysed using the freeze-thaw method. Buffer (50 mM Tris pH 8.0, 1 mM EDTA, 100 mM NaCl and 1 mM phenylmethylsulfonyl fluoride) was added to the frozen pellet, suspended and spun to remove cell debris and the supernatant was boiled for a minute to precipitate unwanted proteins, then spun and the supernatant was acidified to pH 4.6 to isoelectrically precipitate Tm. The resulting precipitate was dissolved in 10 mM HEPES pH 7.5, 100 mM NaCl and purified with a HiTrap Q column (AKTA) using a linear salt gradient from 100 mM to 1 M NaCl. Purity was confirmed by mass spectrometry and SDS-PAGE gel analysis. Protein concentrations were determined spectrophotometrically using E277nm (1 mg/mL) = 0.24. Actin was prepared from rabbit skeletal muscle acetone powder (21, Lehrer, 1972 #1070). Myosin subfragment 1 (S1) was prepared from rabbit skeletal muscle using standard procedures (22). Rabbit skeletal Tn was purified from ether-dried muscle powder by standard methods (23).
Methods
Trypsin digestion was performed on Tm at 0.5 mg/mL in 10 mM HEPES buffer, pH 7.5, 0.1 M NaCl, 5 mM MgCl2 and 10 mM β-mercaptoethanol, treated with 0.001 mg/mL porcine trypsin (G-Biosciences #786-245). For digestion of actin-Tm, 0.003 mg/mL trypsin was mixed with 1.5 mg/mL F-actin and 0.3 mg/mL Tm (excess actin) in the same buffer solution. The reactions were quenched with 20× excess soybean trypsin inhibitor (STI) by weight with respect to trypsin. All digestions were done at 26°C. 12.5% polyacrylamide handcast gels were used to visualize Tm digestion and 12% Mini-Protean TGX Precast Gels (Bio-Rad #456-1043) were used for actin-Tm digestion. Coomassie stained gels were scanned and analyzed with ImageJ software. The band density of Tm at 0 time was used to normalize all band densities.
Circular dichroism spectropolarimeter Aviv 60DS CD was used for monitoring thermal unfolding. 1 mg/mL Tm in buffer (10 mM HEPES pH 7.5 and 100 mM NaCl), in a 1 mm cuvet, was incubated with 1 mM dithiothreitol (DTT) and heated to 40 °C for 15 min to reduce possible S-S bonds. The CD signal at 222 nm was recorded during temperature scans (at 1 deg/min) between 25 °C and 70 °C.
Mass spectrometry was performed on protein samples 1:1 with 20 mg/mL sinapinic acid in 50% CH3CN and 0.1% TFA and analyzed with a Perceptive Voyager MALDI-TOF.
N-terminal amino acid sequence analyses were performed by Dr. Michael Berne (Tufts University). Tm digests were analyzed by SDS-PAGE then transferred onto Immobilon-PSQ PVDF 0.2 μm Membrane (Millipore ISEQ26260).
Acto-S1 ATPase activity was measured spectrophotometrically using the EnzCheck Phosphate Assay Kit (Molecular Probes). The continuous linear production of Pi vs. time was monitored on a Cary 50 UV-Vis for each addition of sample to low salt buffer (20 mM HEPES pH 7.5, 15 mM NaCl, 3 mM MgCl2, 1 mM DTT) containing 0.1 mM CaCl2 from stock solutions in the following order: 1mM ATP. 0.2 – 0.5 μM S1, 4 μM actin, 1-2 μM Tm, 1-2 μM Tn and 1 mM ethylene glycol tetraacetic acid (EGTA) + 1 mM nitrilotriacetic acid (NTA). Monitoring the linear A360nm vs. time gave relative ATPase values for S1 background, actin activation, Tm inhibition, +Ca2+ Tn activation, −Ca2+ Tn inhibition. For the ATPase vs. [Ca2+] measurements the samples contained 0.5 μM S1, 4 μM actin, 1 μM Tm and 1 μM rabbit skeletal Tn. The proteins were mixed into low salt buffer as above with addition of 0.5 mM EGTA and 0.5 mM nitrilotriacetic acid . 1 mM ATP and S1 background activity was obtained and subtracted from all calculations. The reaction solution was titrated with 20 mM CaCl2. [Ca2+]free was calculated using the program Maxchelator (http://maxchelator.stanford.edu). All ATPase experiments were done at room temperature (25°C).
Results
S1-Actin ATPase
ATPase measurements with reconstituted thin filaments were performed using the continuous colorimetric measurement of phosphate liberation during successive mixing in of myosin subfragment 1 (S1), actin, Tm, Tn +Ca2+ and Tn −Ca2+ (Fig. 1a, see Methods). Tm and Tn bound fast to actin and actinTm, respectively, as evidenced by the quick linear dependence of phosphate liberation after each addition. In the absence of Tn and Ca2+, WT and D175N inhibited the actin-S1 activity by about 80±3%, whereas E180G only inhibited by 64±5%. Similarly, in the presence of TmTn+Ca2+, the S1-actin ATPase activity was inhibited by 45±1% and 47±8% for WT and D175N, respectively, whereas E180G inhibited somewhat less, (37±7%). Finally, the S1- actinTmTn ATPase in the absence of Ca2+ for both WT and D175N was inhibited by 85±3%, whereas the inhibition for E180G was less (74±6%). Doubling the concentration of Tm or/and Tn did not change the results within experimental error. These data show that there is less inhibition of ATPase for E180G under all conditions.
Fig. 1.
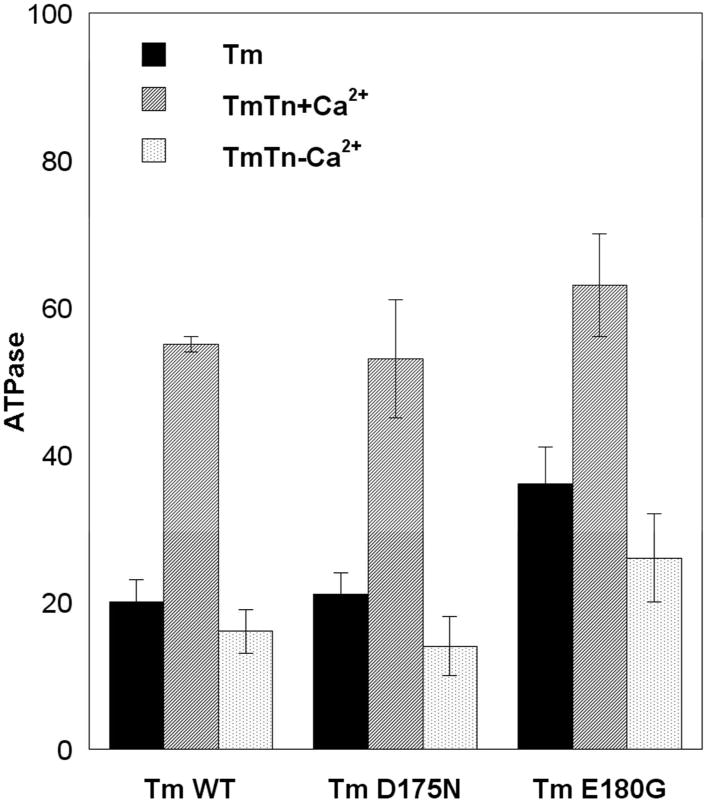

Effects of Tm-FHC mutants on actin-S1 ATPase activity. Data obtained by continuously monitoring Pi liberation (see Methods). A. Effects of Tm and Tn +/− Ca2+. ATPase was monitored during successive additions of S1, actin, Tm, Tn, EGTA to a solution containing, 0.1mM CaCl2, 15 mM NaCl, 3 mM MgCl2, 1 mM DTT and 1 mM ATP in buffer (10 mM HEPES, pH 7.5). Data were normalized to actin-S1 activity after subtracting the background S1 ATPase. B. ATPase vs. [Ca2+]. CaCl2 was titrated into the reconstituted actinTmTn thin filament in buffer containing 1mM
EGTA and 1 mM NTA. Data were normalized to the high [Ca2+] values fitted to the Hill equation. Background S1 alone ATPase was subtracted.
Titrations with Ca2+ in an EGTA/Nitrilotriacetic acid buffered system showed increased Ca2+-sensitivity of S1-actinTmTn ATPase for D175N and E180G indicated by the shift of the midpoint of the ATPase curves to lower [Ca2+]. We also obtained greater residual activity at very low [Ca2+] for E180G (Fig. 1b). E180G showed the largest effect of enhanced residual ATPase in agreement with Fig.1a, and greatest Ca2+-sensitivity. The Hill coefficients were 2.11 +/− 0.08, 1.62 +/− 0.04, 1.89 +/− 0.08 for WT, D175N and E180G, respectively. Considering the different systems and measurements, these data obtained with reconstituted thin filaments in solution reasonably agree qualitatively with measurements of ATPase in fibers and with physiological experiments of pCa vs. force (6, 7).
Thermal unfolding
To compare the global stability of the samples we studied the thermal unfolding transitions with circular dichroism (Fig. 2). The WT and D175N showed a major transition at 47 °C and a shoulder at 55°. The E180G was markedly less stable with the early transition shifted 5 °C to lower temperature without an effect on the high temperature transition. These data confirm our earlier results on different constructs of the same mutants (12), corrected for the sample mislabeling (see erratum (13)) except for a small shift of the transitions to greater temperature probably due to different solution conditions. More recent studies using differential scanning calorimetry obtained similar results (14). Thus, the single site mutant E180G destabilizes a very large part of Tm, whereas the D175N has little effect on the thermal stability.
Fig. 2.

CD thermal unfolding profiles at 222 nm (Θ vs T) and derivatives (dΘ/dT) of theTm-FHC mutants and WT control. Samples contained 1 mg/mL protein in 100 mM NaCl, 10 mM HEPES buffer pH 7.5, 1mM DTT.
Trypsin digestion
Using the limited trypsin digestion approach on Tm (24, 25) we studied the early cleavage progress on WT, D175N and E180G. This treatment produced a dimer of 17-18 kDa chains and a dimer of 15 kDa from the intact Tm dimer of 33 kDa seen on gels (Fig. 3, Table I). Each purified peptide was shown to be largely α-helical at room temperature (25). In a separate CD experiment during similar treatment of rabbit cardiac Tm with trypsin we noted that only about 13% of the Tm helix was lost at 30 minutes where most of the Tm was cleaved. For all Tms, the 17 – 18 kDa C-terminal peptide seen in gels (34 kDa – 36 kDa dimer) was further cleaved, faster than the 15 kDa peptide (30 kDa dimer), in agreement with its lower stability (25). Cleavage of the dimer (17-18 kDa peptide) produces a 13 – 14 kDa peptide as seen by the lag in its appearance and its increase as the 18 kDa decreases (Fig. 3). A slightly lower MW species in the main Tm band (33 kDa) was observed to appear with time. This is due to simultaneous truncation of 7 amino acids at the N-terminus as shown earlier (24).
Fig. 3.

Kinetics of trypsin digestion of FHC Tms and WT control. Top, 12.5% polyacrylamide SDS-gels quenched at indicated times; Bottom, densitometric analyses showing kinetics of change of gel bands. The densities were normalized to the density of Tm at 0 time.
Table 1.
Identification of tryptic peptides with N-terminal sequencing and MALDI TOF molecular weight determination. MW of Tms (including AS extension): WT = 32839; D175N = 32838; E180G = 32762. [ ] denotes tentative sequence.
| Mutant | Gel band | MW (obs) (kDa) | MW (calc) (kDa) | N-terminal sequence (obs) | Peptide Sequence |
|---|---|---|---|---|---|
| WT & D175N | 18kD | 17.66 | 17.568 | (R) AQKDE | 134 - 284 |
| 15kD | 15.37 | 15.289 | ASMDA | 1 - 133 | |
| 13kD | 13.67 | (R) KLVII | 168 - 284 | ||
| (K) HIAED | 153 - 284 | ||||
| (K) MQMLK | 8 - 133 | ||||
| E180G | 27kD | 26.83 | [26.90] | ---- | [1 - 233] |
| 17kD | 17.47 | 17.496 | (R) AQKDE | 134 - 284 | |
| 15kD | 15.21 | 15.289 | ASMDA | 1 - 133 | |
| 13- | 13.56 | (R) KLVII | 168 - 284 | ||
| 14kD | 14.46 | (K) MQMLK | 8 - 133 | ||
| (R) AQKDE | [134 - 233] |
N-terminal sequencing verified that the initial cleavage site was at Arg 133 (Table I). The rate of cleavage of Tm was greater for the FHC mutants as judged by the decrease in the half time of Tm loss from 11 min for the WT to 6 and 3 min for the D175N and E180G, respectively (Fig. 3). Thus, the two FHC mutations increased the rate of cleavage of a residue located about 50 residues N-terminal from the mutation site.
For E180G, initial cleavage also took place at another site with a similar rate as cleavage at R133 as evidenced by the early appearance of a band at about 27 kDa in parallel with the appearance of the 18 kDa band (Fig. 3). The site of cleavage was tentatively identified by comparing the molecular weight determined by MALDI-TOF with the calculated molecular weight of fragments cleaved at Arg or Lys in the C-terminal region of Tm. Thus, the single site mutation from Asp to Gly at 180 produced another region of dynamic instability at K233, a site located 53 residues C-terminal to the mutation site.
When bound to actin the rate of Tm cleavage was slowed 16× for WT, 20× for D175N and about 30× for E180G, correcting for the difference in trypsin concentration. A similar slowing by actin was observed in our work with native Tm (24). Despite the overall slowing of cleavage, the increased rate for the FHC mutants still persisted (t ½ = 60 min, 40 min and 32 min for WT, D175N and E180G, respectively). Evidence that Tm was cleaved when bound to actin and not cleaved when free in solution was obtained by the lack of the presence of a faster moving Tm band on gels due to truncation of 7 amino acid residues from the N-terminus (9 including the added Ala-Ser) as occurs in parallel with cleavage for free Tm and for these samples in the absence of actin (24). This protection is probably due to Tm end-to-end interactions or shielding by actin. As noted earlier (24), the actin was not cleaved by trypsin over the 1 ½ hour time period. As for Tm alone, a 15 kDa gel band was present at early times, but a 17 - 18 kDa band and a 27 kDa was not observed in the gels even at short times. This appears to be due to loss of binding to actin of the cleaved peptides (26) and the more rapid cleavage of the 18 kDa and 27 kDa peptides than the 15 kDa peptide when free in solution especially at 3× the trypsin concentration.
N-terminal sequencing of the blotted peptides from gels and MALDI TOF MS allowed identification of most of the peptides (Table I). For WT and D175N, the N-terminal half (15 kDa) and the C-terminal half (17 kDa for E180G and 18 kDa for WT and D175N) were as identified earlier (24). The 18 kDa was degraded further to produce species between 13 kDa and 15 kDa. N-terminal sequencing of the broad gel band in that region showed the presence of peptides 168-284, 153-284 and a C-terminal peptide truncated at Lys 7, 8-133. For E180G, the same 2 initially cleaved halves at 15 kDa and 17 kDa were identified. However, for E180G, a new gel band at 27 kDa (54 kDa dimer) was tentatively identified at Lys 233. At longer reaction times peptides due to further cleavage of the 27 kDa of E180G were observed below 15 kDa. Some of these peptides from E180G corresponded to the small peptides observed for the other 2 Tms. An additional peptide of about 13 kDa seems to be from A134 to K 233. It is tentatively identified as the product of the 17 kDa peptide cleaved at K233.
The pathway of cleavage for WT, D175N and E180G is shown in Fig. 5. Peptides smaller than 15 kDa are shown as single chains due to their instability. From the above results it is seen that there is a “hot spot” which allows preferential cleavage at R133 for all 3 Tms and an additional “hot spot” at K233 for E180G.
Fig. 5.
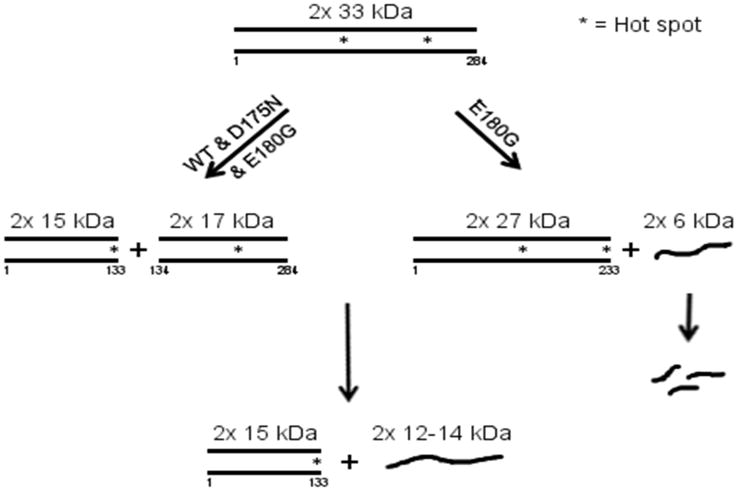
Schematic of the course of cleavage by trypsin. Note the common pathway for all via initial cleavage at R133 and an additional pathway for E180G with cleavage at K233. Helical peptides are indicated as dimers.
* indicates a “hot spot” where cleavage occurs preferentially.
Discussion
Our principal finding is that the FHC-Tm mutations D175N and E180G, located about 2/3 from the N-terminus of the molecule, increase the initial rate of cleavage by trypsin at R133, located near the middle of Tm. The increase in rate was 2× for D175N and 4× for E180G. Our previous work showed that native ααTm is initially cleaved by trypsin at R133, due to the presence of a charged Asp in the hydrophobic ridge at nearby D137, first observed by Pato and Smillie (25). Substitution of canonical Leu for Asp 137 markedly inhibited the cleavage (24). To cleave an α-helix by trypsin a transient unfolding of about 8 continuous amino acid residues of the polypeptide chain (∼2 turns of α-helix) is required (27), which is a very small fraction of the Tm molecule. Because of the strong interchain interactions in the Tm coiled-coil, both chains may unfold simultaneously. The limited tryptic digestion is therefore a test for local dynamic instability that is not observed by equilibrium measurements. The region that transiently unfolds to allow trypsin cleavage represents a “hot spot” in the sequence (Fig. 5) which would provide flexibility around that region. A recent study also used the method of limited trypsin cleavage at R133 to study the effect of another amino acid substitution (28).
Over the years there have been several studies that have obtained evidence for flexibility of Tm. Early studies showed that a small helix pretransition in the 35°C to 40°C range is associated with unfolding of about 10% - 20% of the helix in the middle of the molecule (29-32) suggesting non-uniform stability contributing to flexibiity. This is seen in this work as a shoulder on the derivative curves (Fig. 2). Fluorescence probe studies with the label at Cys 190 have indicated that the probe responds to the unfolding in the pretransition temperature region (12, 13, 33). The main transition at 47°C is due to unfolding of the C-terminal region and the transition at about 54°C is due to unfolding in the N-terminal region (25, 26, 32). The relative areas under the derivative curves in Fig. 2 can be associated with the relative size of the unfolding domains, and indicates that as much as 2× as many residues are unfolded in the C-terminal domain as in the more stable N-domain. It thus appears, for Tm, in order of stability, the unfolding domains approximately involve: 20% of the residues in the middle of the molecule, 55% of the C-terminal domain and 25% of the N-terminal domain. The unfolding transitions are cooperative, which suggests that the corresponding regions behave as independent structural domains. These domains are at odds with the seven quasi-equivalent regions identified in the amino acid-sequence of Tm, which appear to act as sites of interaction with the actin monomers (34). These regions have somewhat differing actin binding properties (35), but no apparent correspondence between the thermal stability and the strength of the interaction with actin can be inferred. Finally the region containing the D175N and E180G mutations is of special interest due to its presumed involvement in interaction with troponin e.g., preliminary data showed that a crosslink could be formed between Tm at position 176 and TnT between residues 171-174 (36). Thus, the mutations could affect the properties of the thin filament in several different ways.
Further decreased stability of the C-terminal domain, observed for E180G, and the dynamic unfolding revealed by limited trypsin digestion for both FHC mutants, indicate flexibility which could be manifested at physiological temperature when subjected to a perturbation such as may occur during the movement of Tm during regulation.
Recent theoretical and experimental studies involving persistence length, a measure of flexibility, obtained different values ranging from 20 nm (37), 50 nm (38) to about 400 nm (39). These studies assume a relatively uniform semi-flexible molecule. Another measure of flexibility is the cooperative unit size, n, which involves the ability of strong binding myosin heads to move Tm, opening n actin subunits. We obtained values about 7 to 14 actin subunits uncovered by one rigor myosin head depending upon the absence or presence of Tn or the source of the Tm (12, 40, 41). Other regions which may introduce flexibility have been suggested from X-ray structural studies. Regions containing several Ala residues in hydrophobic positions have been associated with bends in the coiled-coil (42). The presence of Glu residues in the hydrophobic ridge appear to be near regions of local coiled-coil chain separation allowing penetration by water molecules (43). Surprisingly, trypsin does not cleave in these regions.
It is difficult to understand how the E180G single site mutation can cause the large domain instability and how both E180G and D175N single site mutations cause the increase of long-range dynamic instability measured by rate of trypsin cleavage, particularly in the rod-shaped Tm molecule, where interactions between non-adjacent regions are not possible. Both mutants involve the loss of a negative charge in the native Tm, located at e (E180) and g (D175) positions in the 7 residue repeat of the helix, abcdefg. These sites are involved in e-g ionic interactions across and within the chains (15) and the loss of the favorable charge interactions could be the cause of the instablity. E180G exhibits more drastic disease phenotypes and this may be related to its greater flexibility caused by the instability. It appears that the local instability or unfolding generated in the mutation region is transmitted to the R133 region and in the case of the E180G to both the R133 and D233 regions. Recently, long-range conformational changes in 2 coiled-coils have been proposed involving a shift in register of heptad repeats (44-46). Local unfolding could be transmitted via thermal fluctuations analogous to the movement of imperfections in a crystal lattice (47) to distant unstable regions (hot spots).
It should be noted that these FHC mutations are located in a region of Tm that interacts with Tn. Fluorescent probes at position 190 are sensitive to Tn binding (48) and more recently a 9A crosslink between a Cys substituted at Tm176 was found on TnT in the region of residues 171-174 (36). It is therefore possible that the interaction with Tn is perturbed due to the mutations, which could cause the increased Ca2+-sensitiity.
Recently a 4th state, a substate of the 3-state regulation model has been proposed to aid in understanding FHC and other phenomena that involve activation by myosin (18). The involvement of the 4th state can explain the increased Ca2+-sensitivity and increased basal activity at low Ca2+. In the 4th state, Tm is in an Open or active state where it does not block myosin-nucleotide binding or the strong-binding myosin head force-generating transition, but this Open state does not have Ca2+ bound to TnC. In this proposed 4th state TnI is bound to TnC after dissociation from actinTm caused by strong-binding myosin heads (49, 50). It has been shown that TnI binding to TnC increases the Ca2+ affinity to TnC-TnI (51-53). Thus, the presence of a 4th state explains both the residual force in the absence of Ca2+ and the increased Ca2+-sensitivity. We have shown that the strong binding of 2-3 myosin heads per Tm was sufficient to release TnI-TnC (absence of Ca2+) and TnI (49) from actinTm and TnI in actinTmTn (absence of Ca2+) (50). We postulated that a TnI binding site involving both actin and Tm was lost when Tm was moved from the Blocked to the Open state by the binding of only a few heads per Tm. The increase in flexibility of the FHC Tm mutants suggested in this work, could increase the contribution of the activation pathway involving the 4th state because the flexible FHC Tm mutants do not interact with the thin filament as strongly as WT resulting in less blocking of myosin head binding, allowing more heads to enter the force-generating state via the 4th state. Weaker binding of these mutants to actin has been observed (12, 14). We also obtained here a decreased inhibition of ATPase in the absence of Tn (actinTm) for the E180G mutant, indicating weaker interaction. Evidence for a slightly lower occupation of the Blocked state for E180G was obtained from S1 binding kinetics (11) and for a greater interaction of myosin to actin (54). Many of the sarcomeric proteins have mutations that produce different severity of FHC and DHC and show variable amounts of Ca2+-sensitivity and basal activity. Thus, any change in: myosin crossbridge interaction with actin, Tm interaction with actin as seen here, affinity of TnI and Ca2+ to TnC; TnI to actinTm, due to mutations, could change the contribution of the 4th state resulting in increases (in FHC) or decreases (in DHC) of Ca2+-sensitivity.
In addition to FHC, other phenomena, such as the effect of sarcomere length on Ca2+affinity (55), stretch activation of cardiac (56) and of insect flight muscle (57) also appear to be related to thin filament activation by myosin heads.
In conclusion, the work presented here, for Tm FHC mutations D180G and E175N, provides a possible link between the severity of FHC and the degree of molecular flexibility manifested as a result of the mutation. Clearly further studies will be needed to help explain the molecular details of these long-range effects and how they are related to the severity of these myopathies.
Fig. 4.
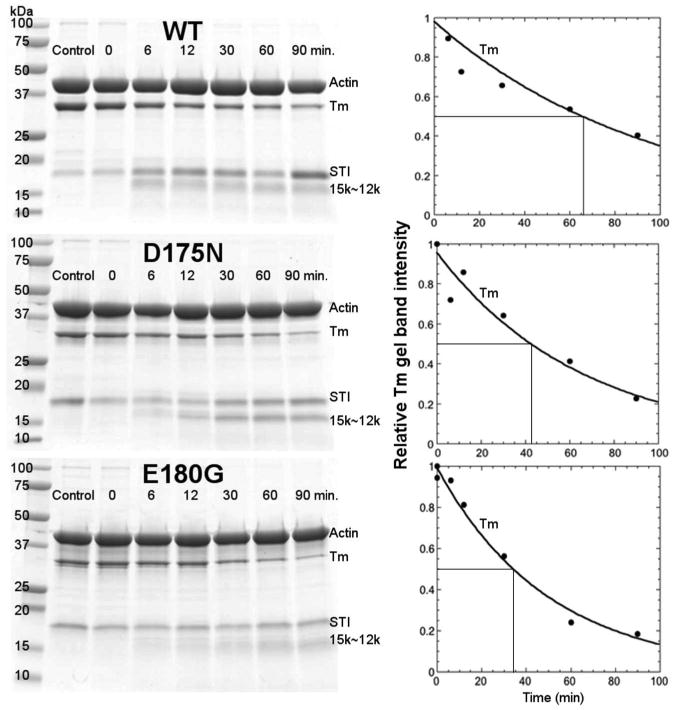
Kinetics of trypsin digestion of Tm-FHC mutants and WT control bound to actin. Left, 12.5% polyacriylamide SDS-gels quenched at indicated times: Right, densitometric analysis of kinetics of cleavage of Tm normalized to 0 time.
Acknowledgments
We are grateful to Dr. Zenon Grabarek for helpful suggestions during the course of the experiments and for critical reading of drafts of the manuscript. We also thank Drs. Sarah Learman, F.Timur Senguen, and Ms. Betty Gowell for experimental help and to Dr. Robin Maytum, University of Bedfordshire, UK for constructs and transformed cells.
Supported by NIH HL 91162
Abbreviations
- Tm
tropomyosin
- Tn
troponin
- TnI, TnC, TnT
Tn subunits
- ActinTmTn
reconstituted thin filament
- S1
myosin subfragment 1
- FHC
familial hypertrophic cardiomyopathy)
- DHC
dilated hypertrophic cardiomypathy
References
- 1.Watkins H, Seidman JG, Seidman CE. Familial hypertrophic cardiomyopathy: a genetic model of cardiac hypertrophy. Hum Mol Genet. 1995;4 Spec No:1721–1727. doi: 10.1093/hmg/4.suppl_1.1721. [DOI] [PubMed] [Google Scholar]
- 2.Tardiff JC. Sarcomeric proteins and familial hypertrophic cardiomyopathy: linking mutations in structural proteins to complex cardiovascular phenotypes. Heart Fail Rev. 2005;10:237–248. doi: 10.1007/s10741-005-5253-5. [DOI] [PubMed] [Google Scholar]
- 3.Redwood CS, Moolman-Smook JC, Watkins H. Properties of mutant contractile proteins that cause hypertrophic cardiomyopathy. Cardiovasc Res. 1999;44:20–36. doi: 10.1016/s0008-6363(99)00213-8. [DOI] [PubMed] [Google Scholar]
- 4.Thierfelder L, Watkins H, MacRae C, Lamas R, McKenna W, Vosberg HP, Seidman JG, Seidman CE. α-Tropomyosin and cardiac troponin T mutations cause familial hypertrophic cardiomyopathy. Cell. 1994;77:701–712. doi: 10.1016/0092-8674(94)90054-x. [DOI] [PubMed] [Google Scholar]
- 5.Nakajima-Taniguchi C, Matsui H, Nagata S, Kishimoto T, Yamauchi-Takihara K. Novel missense mutation in alpha-tropomyosin gene found in Japanese patients with hypertrophic cardiomyopathy. J Mol Cell Cardiol. 1995;27:2053–2058. doi: 10.1016/0022-2828(95)90026-8. [DOI] [PubMed] [Google Scholar]
- 6.Bottinelli R, Coviello DA, Redwood CS, Pellegrino MA, Maron BJ, Spirito P, Watkins H, Reggiani C. A mutant tropomyosin that causes hypertrophic cardiomyopathy is expressed in vivo and associated with an increased calcium sensitivity. Circ Res. 1998;82:106–115. doi: 10.1161/01.res.82.1.106. [DOI] [PubMed] [Google Scholar]
- 7.Bai F, Weis A, Takeda AK, Chase PB, Kawai M. Enhanced active cross-bridges during diastole: molecular pathogenesis of tropomyosin's HCM mutations. Biophys J. 2011;100:1014–1023. doi: 10.1016/j.bpj.2011.01.001. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Bing W, Knott A, Redwood C, Esposito G, Purcell I, Watkins H, Marston S. Effect of hypertrophic cardiomyopathy mutations in human cardiac muscle alpha -tropomyosin (Asp175Asn and Glu180Gly) on the regulatory properties of human cardiac troponin determined by in vitro motility assay. J Mol Cell Cardiol. 2000;32:1489–1498. doi: 10.1006/jmcc.2000.1182. [DOI] [PubMed] [Google Scholar]
- 9.Chang AN, Harada K, Ackerman MJ, Potter JD. Functional consequences of hypertrophic and dilated cardiomyopathy-causing mutations in alpha-tropomyosin. J Biol Chem. 2005;280:34343–34349. doi: 10.1074/jbc.M505014200. [DOI] [PubMed] [Google Scholar]
- 10.Prabhakar R, Petrashevskaya N, Schwartz A, Aronow B, Boivin GP, Molkentin JD, Wieczorek DF. A mouse model of familial hypertrophic cardiomyopathy caused by a alpha-tropomyosin mutation. Mol Cell Biochem. 2003;251:33–42. [PubMed] [Google Scholar]
- 11.Boussouf SE, Maytum R, Jaquet K, Geeves MA. Role of tropomyosin isoforms in the calcium sensitivity of striated muscle thin filaments. J Muscle Res Cell Motil. 2007;28:49–58. doi: 10.1007/s10974-007-9103-z. [DOI] [PubMed] [Google Scholar]
- 12.Golitsina N, An Y, Greenfield NJ, Thierfelder L, Iizuka K, Seidman JG, Seidman CE, Lehrer SS, Hitchcock-DeGregori SE. Effects of two familial hypertrophic cardiomyopathy-causing mutations on alpha-tropomyosin structure and function. Biochemistry. 1997;36:4637–4642. doi: 10.1021/bi962970y. [DOI] [PubMed] [Google Scholar]
- 13.Golitsina N, An Y, Greenfield NJ, Thierfelder L, Iizuka K, Seidman JG, Seidman CE, Lehrer SS, Hitchcock-DeGregori SE. Effects of two familial hypertrophic cardiomyopathy-causing mutations on alpha-tropomyosin structure and function. Biochemistry. 1999;38:3850. doi: 10.1021/bi9950701. [DOI] [PubMed] [Google Scholar]
- 14.Kremneva E, Boussouf S, Nikolaeva O, Maytum R, Geeves MA, Levitsky DI. Effects of two familial hypertrophic cardiomyopathy mutations in alpha-tropomyosin, Asp175Asn and Glu180Gly, on the thermal unfolding of actin-bound tropomyosin. Biophys J. 2004;87:3922–3933. doi: 10.1529/biophysj.104.048793. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.McLachlan AD, Stewart M. Tm Coiled-coil Interactions: Evidence for an Unstaggered Structure. J Mol Biol. 1975;98:293–304. doi: 10.1016/s0022-2836(75)80119-7. [DOI] [PubMed] [Google Scholar]
- 16.McKillop DFA, Geeves MA. Regulation of the interaction between actin and myosin subfragment 1: evidence for three states of the thin filament. Biohys J. 1993;65:693–701. doi: 10.1016/S0006-3495(93)81110-X. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Vibert P, Craig R, Lehman W. Steric-model for activation of muscle thin filaments. J Mol Biol. 1997;266:8–14. doi: 10.1006/jmbi.1996.0800. [DOI] [PubMed] [Google Scholar]
- 18.Lehrer SS. The 3-state model of muscle regulation revisited: is a fourth state involved? J Muscle Res Cell Motil. 2011;32:203–208. doi: 10.1007/s10974-011-9263-8. [DOI] [PubMed] [Google Scholar]
- 19.Moss RL, Razumova M, Fitzsimons DP. Myosin crossbridge activation of cardiac thin filaments: implications for myocardial function in health and disease. Circ Res. 2004;94:1290–1300. doi: 10.1161/01.RES.0000127125.61647.4F. [DOI] [PubMed] [Google Scholar]
- 20.Sumida JP, Wu E, Lehrer SS. Conserved Asp-137 imparts flexibility to tropomyosin and affects function. J Biol Chem. 2008;283:6728–6734. doi: 10.1074/jbc.M707485200. [DOI] [PubMed] [Google Scholar]
- 21.Spudich J, Watt S. The regulation of rabbit skeletal muscle contraction. I. Biochemical studies of the interaction of the tropomyosin-troponin complex with actin and the proteolytic fragments of myosin. J Biol Chem. 1971;246:4866–4871. [PubMed] [Google Scholar]
- 22.Margossian S, Lowey S. Preparation of myosin and its subfragments. Methods Enzymol. 1982;85:55–71. doi: 10.1016/0076-6879(82)85009-x. [DOI] [PubMed] [Google Scholar]
- 23.Van Eerd J, Kawasaki Y. Effect of calcium on the interaction between subunits of troponin and tropomyosin. Biochemistry. 1973;12:4972–4980. doi: 10.1021/bi00748a024. [DOI] [PubMed] [Google Scholar]
- 24.Sumida JP, Hayes D, Langsetmo K, Lehrer SS. Tropomyosin: charge effects in the hydrophobic ridge. Biophysical Journal. 2006;(90):923a. [Google Scholar]
- 25.Pato MD, Mak AS, Smillie LB. Fragments of rabbit striated muscle alpha-tropomyosin. I. Preparation and characterization. J Biol Chem. 1981;256:593–601. [PubMed] [Google Scholar]
- 26.Woods EF. Stability of segments of rabbit a-tropomyosin. Aust J Biol Sci. 1977;30:527–542. doi: 10.1071/bi9770527. [DOI] [PubMed] [Google Scholar]
- 27.Park C, Marqusee S. Probing the high energy states by proteolysis. J Mol Biol. 2004;343:1467–1476. doi: 10.1016/j.jmb.2004.08.085. [DOI] [PubMed] [Google Scholar]
- 28.Nevzorov IA, Nikolaeva OP, Kainov YA, Redwood CS, Levitsky DI. Conserved noncanonical residue Gly-126 confers instability to the middle part of the tropomyosin molecule. J Biol Chem. 2011;286:15766–15772. doi: 10.1074/jbc.M110.209353. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Lehrer SS. Effects of an interchain disulfide bond on tropomyosin structure: intrinsic fluorescence and circular dichroism studies. J Mol Biol. 1978;118:209–226. doi: 10.1016/0022-2836(78)90413-8. [DOI] [PubMed] [Google Scholar]
- 30.Ueno H. Local structural changes in tropomyosin detected by a trypsin-probe method. Biochemistry. 1984;23:4791–4798. doi: 10.1021/bi00315a040. [DOI] [PubMed] [Google Scholar]
- 31.Betteridge DR, Lehrer SS. Two conformational states of didansylcystine-labeled rabbit cardiac tropomyosin. J Mol Biol. 1983;167:481–496. doi: 10.1016/s0022-2836(83)80346-5. [DOI] [PubMed] [Google Scholar]
- 32.Ishii Y, Hitchcock-DeGregori S, Mabuchi K, Lehrer SS. Unfolding domains of recombinant fusion alpha alpha-tropomyosin. Protein Sci. 1992;1:1319–1325. doi: 10.1002/pro.5560011011. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.Ishii Y. The local and global unfolding of coiled-coil tropomyosin. Eur J Biochem. 1994;221:705–712. doi: 10.1111/j.1432-1033.1994.tb18783.x. [DOI] [PubMed] [Google Scholar]
- 34.Phillips GN, Jr, Fillers JP, Cohen C. Tropomyosin crystal structure and muscle regulation. J Mol Biol. 1986;192:111–131. doi: 10.1016/0022-2836(86)90468-7. [DOI] [PubMed] [Google Scholar]
- 35.Hitchcock-DeGregori S, Varnell T. Tropomyosin has discrete actin-binding sites with sevenfold and fourteen-fold periodicity. J Mol Biol. 1990;214:885–890. doi: 10.1016/0022-2836(90)90343-K. [DOI] [PubMed] [Google Scholar]
- 36.Mudalige AW, Lehrer SS. What Region of Tropomyosin Interacts with the N-terminal Half of Troponin T? Biophysical Journal. 2010;98:351a. [Google Scholar]
- 37.Geeves M, Griffiths H, Mijailovich S, Smith D. Cooperative [Ca(2)+]-dependent regulation of the rate of myosin binding to actin: solution data and the tropomyosin chain model. Biophys J. 2011;100:2679–2687. doi: 10.1016/j.bpj.2011.04.020. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38.Loong CK, Zhou HX, Chase PB. Persistence Length of Human Cardiac alpha-Tropomyosin Measured by Single Molecule Direct Probe Microscopy. PLoS One. 2012;7:e39676. doi: 10.1371/journal.pone.0039676. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39.Li XE, Holmes KC, Lehman W, Jung H, Fischer S. The shape and flexibility of tropomyosin coiled coils: implications for actin filament assembly and regulation. J Mol Biol. 2009;395:327–339. doi: 10.1016/j.jmb.2009.10.060. [DOI] [PubMed] [Google Scholar]
- 40.Geeves MA, Lehrer SS. Dynamics of the muscle thin filament regulatory switch: the size of the cooperative unit. Biophys J. 1994;67:273–282. doi: 10.1016/S0006-3495(94)80478-3. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41.Maytum R, Konrad M, Lehrer SS, Geeves MA. Regulatory properties of tropomyosin effects of length, isoform, and N-terminal sequence. Biochemistry. 2001;40:7334–7341. doi: 10.1021/bi010072i. [DOI] [PubMed] [Google Scholar]
- 42.Li XE, Lehman W, Fischer S, Holmes KC. Curvature variation along the tropomyosin molecule. J Struct Biol. 2009;170:307–312. doi: 10.1016/j.jsb.2009.12.017. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43.Minakata S, Maeda K, Oda N, Wakabayashi K, Nitanai Y, Maeda Y. Two-crystal structures of tropomyosin C-terminal fragment 176-273: exposure of the hydrophobic core to the solvent destabilizes the tropomyosin molecule. Biophys J. 2008;95:710–719. doi: 10.1529/biophysj.107.126144. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44.Gibbons IR, Garbarino JE, Tan CE, Reck-Peterson SL, Vale RD, Carter AP. The affinity of the dynein microtubule-binding domain is modulated by the conformation of its coiled-coil stalk. J Biol Chem. 2005;280:23960–23965. doi: 10.1074/jbc.M501636200. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 45.Kon T, Imamula K, Roberts AJ, Ohkura R, Knight PJ, Gibbons IR, Burgess SA, Sutoh K. Helix sliding in the stalk coiled coil of dynein couples ATPase and microtubule binding. Nat Struct Mol Biol. 2009;16:325–333. doi: 10.1038/nsmb.1555. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 46.Croasdale R, Ivins FJ, Muskett F, Daviter T, Scott DJ, Hardy T, Smerdon SJ, Fry AM, Pfuhl M. An undecided coiled coil: the leucine zipper of Nek2 kinase exhibits atypical conformational exchange dynamics. J Biol Chem. 2011;286:27537–27547. doi: 10.1074/jbc.M110.196972. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 47.Kittel C. Solid State Physics. Wiley; New York: 2004. [Google Scholar]
- 48.Ishii Y, Lehrer SS. Effects of Troponin in the Conformation of Pyrene-Tropomyosin. Biophys J. 1986;49:258a. [Google Scholar]
- 49.Zhou X, Morris EP, Lehrer SS. Troponin I and troponin I-C binding to actin-tropomyosin and dissociation by myosin S1. Bophys J. 1995;68:A167. doi: 10.1021/bi992327m. [DOI] [PubMed] [Google Scholar]
- 50.Mudalige WA, Tao TC, Lehrer SS. Ca2+-dependent photocrosslinking of tropomyosin residue 146 to residues 157-163 in the C-terminal domain of troponin I in reconstituted skeletal muscle thin filaments. J Mol Biol. 2009;389:575–583. doi: 10.1016/j.jmb.2009.04.027. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 51.Potter JD, Gergely J. Troponin, tropomyosin and actin interactions in the regulation of muscle contraction. Biochemistry. 1974;13:2697–2703. doi: 10.1021/bi00710a007. [DOI] [PubMed] [Google Scholar]
- 52.Wang CK, Cheung HC. Energetics of the binding of calcium and troponin I to troponin C from rabbit skeletal muscle. Biophys J. 1985;48:727–739. doi: 10.1016/S0006-3495(85)83831-5. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 53.Liao R, Wang CK, Cheung HC. Coupling of calcium to the interaction of troponin I with troponin C from cardiac muscle. Biochemistry. 1994;33:12729–12734. doi: 10.1021/bi00208a026. [DOI] [PubMed] [Google Scholar]
- 54.Rysev NA, Karpicheva OE, Redwood CS, Borovikov YS. The effect of the Asp175Asn and Glu180Gly TPM1 mutations on actin-myosin interaction during the ATPase cycle. Biochim Biophys Acta. 1824:366–373. doi: 10.1016/j.bbapap.2011.11.004. [DOI] [PubMed] [Google Scholar]
- 55.Hoffman PA, Fuchs F. Effect of length and cross-bridge attachment on Ca2+ binding to cardiac troponin C. Am J Physiol. 1987;253:C90–C96. doi: 10.1152/ajpcell.1987.253.1.C90. [DOI] [PubMed] [Google Scholar]
- 56.Stelzer JE, Larsson L, Fitzsimons DP, Moss RL. Activation dependence of stretch activation in mouse skinned myocardium: implications for ventricular function. J Gen Physiol. 2006;127:95–107. doi: 10.1085/jgp.200509432. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 57.Bullard B, Pastore A. Regulating the contraction of insect flight muscle. J Muscle Res Cell Motil. 2011;32:303–313. doi: 10.1007/s10974-011-9278-1. [DOI] [PubMed] [Google Scholar]