

Abstract
In comparison to imidazole (IMZ) and 1,2,4-triazole (1,2,4-TRZ) the isosteric 1,2,3-triazole (1,2,3-TRZ) is unrepresented among CYP inhibitors. This is surprising because 1,2,3-TRZs are easily obtained via ‘click’ chemistry. To understand this underrepresentation of 1,2,3-TRZs among CYP inhibitors, thermodynamic and DFT computational studies were performed with unsusbstituted IMZ, 1,2,4-TRZ, and 1,2,3-TRZ. The results indicate that the lower affinity of 1,2,3-TRZ for the heme iron includes a large unfavorable entropy term likely originating in solvent – 1,2,3-TRZ interactions; the difference is not solely due to differences in the enthalpy of heme – ligand interactions. In addition, the 1,2,3-TRZ fragment was incorporated into a well-established CYP3A4 substrate and mechanism based inactivator, 17-α-ethynylestradiol (17EE), via click chemistry. This derivative, 17-click, yielded optical spectra consistent with low spin ferric heme iron (type II) in contrast to 17EE, which yields a high spin complex (type I). Furthermore, the rate of CYP3A4-mediated metabolism of 17-click was comparable to 17EE, and with different regioselectivity. Surprisingly, CW EPR and HYSCORE EPR spectroscopy indicate that the 17-click does not displace water from the 6th axial ligand position of CYP3A4 as expected for a type II ligand. We propose a binding model where 17-click pendant 1,2,3-TRZ hydrogen bonds with the 6th axial water ligand. The results demonstrate the potential for 1,2,3-TRZ to form metabolically labile water-bridged low spin heme complexes, consistent with recent evidence that nitrogenous type II ligands of CYPs can be efficiently metabolized. The specific case of [CYP3A4•17-click] highlights the risk of interpreting CYP-ligand complex structure on the basis of optical spectra.
Cytochrome P450 (CYP)-heme interactions with nitrogenous drugs contribute to the pharmacology of a wide range of therapeutic agents. Collectively, CYPs represent both therapeutic targets and critical determinants of drug metabolism and drug-drug interactions. For example, drugs targeted to aromatase (CYP19) for use in breast cancer(1), to lanosterol demethylase (CYP51A1) for antifungal therapy(2), or to other CYPs expressed in infectious pathogens(3), exploit heme-imidazole or heme-triazole interactions. Similar heme-nitrogen interactions are often considered to be a component of inhibition of the drug-metabolizing CYPs, and such inhibitors can alter the clearance of other drugs(4). However, recent studies(5–7) demonstrate that the heme-nitrogen interactions are not exclusively ‘inhibitory,’ and our fundamental understanding of the interaction between nitrogenous drugs and the heme group of CYPs is incomplete.
Surprisingly, all marketed azole drugs designed to exploit heme-nitrogen interactions with CYPs are either imidazole (IMZ), 1,2,4-triazole (1,2,4-TRZ), or thiazole-based despite the extraordinary synthetic advantages of 1,2,3-triazoles (1,2,3-TRZs)(8, 9). Although several studies have suggested(10, 11) that, despite their lower basicity, the 1,2,3-TRZs can ligate to the heme iron of CYPs, they are conspicuously absent among marketed CYP inhibitors. In the absence of significant confounding problems, 1,2,3-TRZs, in principle, could be a powerful element of fragment-based anti-CYP drug discovery. In order to determine whether 1,2,3-TRZs have unique properties among isosteric nitrogen heterocycles, we have compared the spectroscopic and thermodynamic behavior of 1,2,3-TRZ binding to CYPs with that of IMZ and 1,2,4-TRZ. We also provide a spectral and functional characterization of a well-established CYP3A4 inhibitor after installation of a 1,2,3-TRZ on its molecular framework. The results indicate that heme interactions with 1,2,3-TRZs include unusual structural features that could explain the limitations of this fragment in CYP inhibitor discovery, but which could be exploited to ‘steer’ regiospecific metabolism of other drugs. They further emphasize that ligands that yield apparent type II optical spectra are not necessarily directly ligated to the heme iron nor are they necessarily inhibitory, as recently revealed by Jones and co-workers(5, 6).
Materials and Methods
Chemicals
1H-1,2,3-triazole, imidazole, 1H-1,2,4-triazole, and 17α-ethynylestradiol used during binding and turnover analysis were obtained from Sigma Chemical Co. (St. Louis, MO, USA) and used without further purification. All reagents, purification solvents, and chemical precursors used during synthesis of 17-click and 2-OH/4-OH 17-click were also obtained from Sigma. Deuterated solvents for NMR analysis were purchased from Cambridge Isotopes (Andover, MA, USA). Solvents for LC-MS were of Optima grade from Fisher. Water not employed as LC-MS solvent was of Milli-Q quality from a Barnstead nanopure UV dispenser. Supersomes co-expressing cytochrome b5 and P450 reductase, were purchased from BD Biosciences (San Jose, CA).
1H NMR
Characterization of synthesized 17-click and 2-OH/4-OH 17-click by NMR was performed on a Varian (Palo Alto, CA) Inova 500 (500 MHz) spectrometer equipped with a 5mm HCN z-axis PFG triple resonance probe.
Protein expression and purification
CYP3A4 was expressed and purified as previously described(12). CYP2C9 (hepta mutant) was constructed as previously described(13) with the exeption of a hexa His rather than a tetra His C-terminal sequence to facilitate purification. The enzyme was expressed in DL39 E. coli and purified as described for CYP3A4.
Synthesis of 17α-(2H-2,3,4-triazolyl)-estradiol (17-click)
17α-(2H-2,3,4-triazolyl)-estradiol (1) was obtained using a one step click reaction between 17-α-ethynylestradiol and azidotrimethylsilane using a published procedure for preparation of N-unsubstituted 1,2,3-triazoles(14). Purification was achieved using silica gel (0.035–0.07 mm, 6 nm pore dia.) chromatography with 8% CH3OH in CH2Cl2 isochratic mobile phase to afford pure (1) in 60% yield. (1): NMR (δ ppm, DMSO, d6): 0.93 (3H, s, 18-CH3), 6.40 (1H, d, J = 2.4 Hz, 4-H), 6.46 (1H, dd, J = 8.4, 2.4 Hz, 2-H), 6.95 (1H, d, J= 8.4 Hz, 1H), 8.95 (1H, s, triazole –CH), 14.69 (1H, broad s, triazole –NH). MS: [M+H] (theoretical) = 340.203, [M+H] (observed) = 340.202.
Synthesis of 2-OH and 4-OH 17-click
19-Nor-17α-pregna-1,3,5(10)-trien-20-yne-2,3,17β-triol (2), and 19-nor-17α-pregna-1,3,5(10)-trien-20-yne-3,4,17β-triol (3), were synthesized using the protocol of Xie et al.(15) with minor modifications. Notably, during electrophilic aromatic substitution (formylation reaction, step 1), the Grignard reagent, EtMgBr, was used as a 1 M solution in THF delivered to the reaction after 1:6 dilution in anhydrous diethylether. For successful acylation reaction, triethylphosphate was used as suggested. It was observed during the Dakin oxidation (step 2), that the molar ratio of NaOH and H2O2 is critical to limiting the formation of unwanted byproducts. Therefore, standardized 1M NaOH (0.75 equiv.) and 3% H2O2 (1.1 equiv.; ε240nm = 43.6 M−1 cm−1(16)) solutions were prepared and added to an argon purged THF solution of the 2-formyl and 4-formyl intermediates for a final aqueous concentration of 8.6%/vol. The catechol products were separated on silica gel (0.035–0.07 mm, 6nm pore dia.) with 2% CH3OH in CH2Cl2 mobile phase under isocratic conditions: (2) was isolated in 48% overall yield and (3) isolated in 8% overall yield, both were off-white solids; NMR chemical shifts and MS data for all intermediates conformed to that published previously(15). To generate the 2-OH (4) and 4-OH (5) 17- click, click reactions were conducted in microscale using a slightly modified procedure to that used to obtain (1): to a small conical pressure vial were added 400 μL DMF:H2O (4:1) solution containing 11 mg (35 μmol) of intermediate (2) or (3), followed by addition of 7 mg of sodium ascorbate (35 μmol), and 1 mg of CuSO4·5H2O (4 μmol Cu(II)). The solution was purged with argon while stirring at room temperature before initiation of the reaction with 7.5 μL (57 μmol) of azidotrimethylsilane. The reaction was heated (100°C) overnight with vigorous stirring. The mixture was diluted with water prior to extraction with ethylacetate. The organic layer was washed with several equivalents of water prior to treatment with brine and anhydrous MgSO4. The organic layer was evaporated to dryness and the residue purified over silica gel (70–230 mesh, 100 Å pore) using a 5′ disposable glass pipette with 5–8 % CH3OH in CH2Cl2 mobile phase. Both (4) and (5) were isolated as slightly yellow solids in ~30% yield. (4): NMR (δ ppm, CD3OD): 1.05 (3H, s, 18-CH3), 6.43 (1H, s, 4-H), 6.61 (1H, s, 1-H), 7.65 (1H, broad s, triazole –CH). MS: [M+H] (theoretical) = 356.197, [M+H] (observed) = 356.196. (5) NMR (δ ppm, CD3OD): 1.05 (3H, s, 18-CH3), 6.53 (1H, d, J = 8.8Hz, 2-H), 6.55 (1H, d, J = 8.8Hz 1-H), 7.64 (1H, broad s, triazole –CH). MS: [M+H] (theoretical) = 356.197, [M+H] (observed) = 356.197.
Kinetic assay
Kinetic parameters kcat and KM for total 17-click turnover were estimated by LC-MS/MS using the methods of substrate depletion(17, 18). Briefly, typical incubation mixtures were prepared by combining 198 pmol of rCYP3A4 in Supersomes co-expressing P450 reductase and b5 (198 μL; 50 nM working P450 concentration based on 0.5 nmol P450/0.5 mL as determined by the manufacturer), 79.2 μL of appropriate 50X 17-click stock solution (0.005–10 mM) in ethanol (2 % v/v final EtOH), and 3286.8 μL KPi (0.1 M, pH =7.4; 6 mM MgCl2) buffer. Triplicate 90 μL aliquots were dispensed in 96 well plate format prior to pre-incubation of the plates at 37 °C for 5 min. Wells representing the t = 0 time point received 10 μL of KPi buffer while the reactions were initiated by addition of 10 μL of 10 mM NADPH solution (1 mM final). Reactions were quenched every 30 seconds up to 3.5 minutes, and then every 1–2 minutes up to 16 min with 150 μL of acetonitrile containing 1 μM carbamazapine (CBZ) as internal standard (IS). The quenched reactions were centrifuged at 3480 x g at 4°C for 10 min. to remove precipitated protein. LC-MS/MS analysis of the samples was conducted on an API 4000 triple quadrupole mass spectrometer in positive electrospray mode (MDS SCIEX, Concord, ON), coupled to LC-10ADvp pumps and a SCL-10ADvp controller (Shimadzu, Columbia, MD) with a CTC PAL (Leap Technologies, Carrboro, NC) autosampler. For LC, an Aquasil (Thermo Scientific) C18, 30 × 2.1 mm I.D. (3 μm pore size) was used with water (solvent A) and acetonitrile (solvent B) mobile phases containing 0.1 % formic acid. Data acquisition was conducted in multiple reaction monitoring (MRM) scan mode monitoring the transitions m/z 340.2/159.1 (Q1/Q3) for the 17-click parent, and m/z 237.2/193.1 (Q1/Q3) for the CBZ internal standard. The desolvation temperature was 650 °C, the spray voltage was 2.5 kV, declustering potential 55 V, a collision energy of 38 V, entrance potential was 10 V, and the exit potential was 13 V. Data analysis was conducted with analyte:IS peak area ratios determined using Analyst software (AB SCIEX, Framingham, MA) as described previously(17).
Incubation mixtures for measurement of 17EE depletion were prepared in an identical fashion to that described for 17-click. 17EE reactions were quenched every 30 seconds (up to four minutes) with 200 μL of ethyl acetate containing 0.1 μM β-estradiol IS. The quenched plates were placed in a dry ice/acetone bath to separate the aqueous layer and the organic layer was removed and evaporated to dryness in a new plate under a gentle stream of nitrogen at 45°C. To facilitate detection of 17EE by ESI-MS/MS, the reaction residue was resuspended in 100 μL 50 mM sodium carbonate buffer (pH = 11.0) and 100 μL of 1 mg/mL dansyl chloride in acetone solution. The derivatization reactions were heated (50 °C) for up to 40 minutes. The derivatized reaction mixtures were assayed directly by MRM after automatic tune optimization with infused standards in the positive ion mode: m/z 530.4/171.1 (Q1/Q3) to detect parent 17EE, and m/z 506.4/171.1 (Q1/Q3) for β-estradiol IS.
Measurement of 17-click IC50
Median inhibitory concentration (IC50) values of 17-click were determined against testosterone 6β-hydroxylation by CYP3A4 in Supersomes co-expressing P450 reductase and b5. Incubations in 100mM KPi (pH = 7.4) contained varying concentrations of 17-click (0–60 μM), 10 nM enzyme, and 50 μM testosterone, and were preincubated for 5 min. prior to initiation of the reaction with NADPH (10 mM; 1 mM final). Triplicate reactions were quenched after 20 min using an equal volume (100 μL) of 1 μM tolbutamide (IS) in acetonitrile. Quantitation of percent remaining activity was conducted using the MRM scan mode of a 4000Q-Trap (MDS SCIEX, Concord, ON) connected to a Shimadzu HPLC system. 6β-hydroxy testosterone was monitored using m/z 305.0 and m/z 269.0 (Q1/Q3) with a declustering potential of 55 and collision energy of 25. Tolbutamide was monitored using m/z 271.2 and m/z 91.1 (Q1/Q3), using a declustering potential of 66 and collision energy of 39. The following instrument settings were applied: dwell time, 500 ms; curtain gas, 10; spray voltage, 4.5 kV; source, 400 °C; ion spray gas 1 and 2, 40.
HPLC-qTOF MS analysis of 17-click metabolites
Liquid chromatography was performed using a Waters (Milford, MA) ACQUITY UPLC coupled to a SYNAPT High Resolution Quadrupole TOF mass spectrometer via an ESI interface. Chromatographic separation was achieved on a Hypersil GOLD (ThermoFisher) UPLC column (150 × 2.1 mm, 1.9 μm particle). A gradient program consisting of Solvent A (H2O containing 0.1% formic acid (v/v) and Solvent B (ACN containing 0.1% (v/v) formic acid) was used. Solvent A was held at 90% for 3 min, followed by a linear gradient to 48% B over 12 min. Solvent B was then increased using a linear gradient to 95% B at 17 min and held for 5 min. Pre-equilibration of the column at 90% A was conducted for 5 min prior to each injection. The parent ions were detected in full scan mode (50–800 m/z) with a cone voltage of 17 eV, a collision energy of 6 eV, and transfer cell energy set 4 eV. For fragmentation analysis, data were aquired in MS expression (MSE) mode, with the cone voltage set to 17 eV, the collision cell set to 4 eV, and trap cell set to 6 eV. Fragmentation in the MSE mode was induced using a collision energy ramp of 15–45 eV. In both modes, the capillary voltage was set to 3.5 kV with a source temperature of 120 °C and desolvation temperature of 350°C. Calibration of the instrument was performed using sodium formate over a range of 50–800 prior to use. Leucine enkephalin was used as a lockspray reference with a sampling interval of 30 s and scan rate of 1.0 s. Spectra were averaged for the FWHM of the metabolite peaks using Masslynx software and subjected to mass defect filtering with Metabolynx software using a parent mass tolerance of 50 mDa. All elemental compositions were determined using Masslynx. The Masslynx software MassFragment was used to obtain structural proposals for the fragments.
Ion trap LC-MSn analysis of 17-click metabolites
Further characterization of the primary 17-click metabolite (M4) was conducted using a Finnigan LTQ ion trap instrument (Thermo Electron Corp., San Jose, CA, USA) connected to an Agilent 1100 series HPLC system equipped with autoinjector. Ionization of the metabolites was achieved by ESI in the positive ion mode. The capillary temperature was 300 °C; sheath gas flow, 40; auxiliary gas flow, 10; source voltage, 3 kV. MS4 was conducted using two distinct normalized collision energy sequences: (1) m/z 356 @ 8 %, m/z 338 @ 15 %, and m/z 213 @ 20 %; (2) m/z 356 @ 8 %, m/z 338 @ 15 %, and m/z 320 @ 20%; activation time of 30 ms was used throughout both sequences. LC conditions were as described above for qTOF analysis.
Computational methods
Density Functional Theory (DFT) calculations were performed on a simplified heme macrocycle consisting of an unsubstituted porphine, ferric iron, thiolate (-SH) proximal ligand complex with a variable neutral sixth distal ligand (e.g. water, IMZ, 1,2,3-TRZ, or 1,2,4-TRZ). Structures were initially built in Avagadro. DFT calculations were performed with GAMESS (version number 1 October, 2010 (R1)). Calculations used unrestricted DFT (M06) methods(19). We chose the charge of the molecule to be zero; Fe(III) with balancing negative charges on the macrocycle (−2) and thiolate (−1); and the Fe(III) spin to be S = ½ corresponding to a single unpaired electronic configuration. GAMESS implemented DFT used a Euler-Maclaurin quadrature with 96 radial points, with theta and phi set to 12 and 24 for the number of angle grids in the Gauss-Legendre quadrature. The molecular coordinates used were systematically generated delocalized internal coordinates generated from Cartesian coordinates in the GAMESS input file. Basis sets for geometry optimization and energy calculations were s6-31G(d)(20), implemented as spherical harmonics unless indicated otherwise. Convergence criteria were loosened in all heme calculations. All calculation parameters used GAMESS default settings unless otherwise mentioned. GAMESS output was visualized using MacMolPlt(21). The relative binding energy of the fragment azoles was evaluated in silico by assessing the equilibrium between resting state heme complex (Fe(III)-H2O) and free azole fragment on the reactant side of the equilibrium and azole bound heme (Fe(III)-azole) and free H2O on the product side of the equilibrium. The differences in the sum of energies on the product and reactant side were used to calculate ΔEbinding for each azole fragment, which corresponds to the difference in energy between the water-ligated and azole-ligated model heme. Bond orders were calculated using Mullikan population analysis of the canonical Kohn-Sham orbitals.
UV/vis absorbance analysis of ligand binding
Absorbance measurements were conducted on an Olis Modernized Aminco DW-2 (Olis, Inc., Bogart, GA) dual beam spectrophotometer equipped with a Julabo F30-C compact refrigerated circulator (Julabo USA, inc., Allentown, PA). Each binding experiment required 500 μL initial sample volume using a 0.1 × 1 cm path length quartz cuvette, and the typical sample consisted of 1–2 μM purified CYP in 100 mM KPi buffer + 20% glycerol. Spectra were recorded in the absolute mode (270–650 nm) in between 1 μL aliquots of the appropriate ligand stock solution added to the sample cuvette. For titrations of lipophilic species 17EE and 17-click, ethanol stock solutions were used with a final concentration of organic kept below 2%/vol. During temperature dependent measurements (van ’t Hoff analysis), the protein samples were allowed to equilibrate at the desired temperature for a minimum of ten minutes prior to initiation of the experiment, and the sample was allowed to equilibrate for 5 minutes between ligand aliquots prior to data acquisition. The temperature was precisely monitored inside the cuvette by thermocouple and fluctuations larger than ± 0.4 °C during the course of the experiment resulted in a discarding of the data. Corrections were applied to the data to account for baseline drift and sample dilution over the course of the binding experiments. Difference spectra were generated by subtracting the ligand free spectrum from all ligand-containing spectra containing, and binding isotherms were constructed using the spectral peak minus spectral trough intensity values plotted as a function of ligand concentration. Affinity, KD, and Bmax parameters were estimated from non-linear regression analysis in IGOR pro 6.1 (Wavemetrics, Lake Oswego, OR, USA) using equation (1). Equations (2), (3), and (4) were used when curvature was observed in the Eadie-Hofstee transform of the data.
| (1) |
| (2) |
| (3) |
| (4) |
To accurately compare UV/vis binding spectral characteristics for azole fragments and for 17-click, the spectra for a given titration were normalized to 1 μM CYP concentration using the Soret band absorbance measured in the absence of ligand (CYP3A4 ε417nm = 115 mM−1·cm−1(22); and estimated for CYP2C9 ε417nm ~ 133 mM−1·cm−1). For CYP3A4 binding spectra, comparison of trough intensity at 390 nm in calculated difference spectra was used as an estimate of type IIa character(23, 24). In cases where complete saturation had not been achieved at the highest experimental ligand concentration, principal component analysis (PCA) was conducted as described previously(25–27) using software written in the Python programming language (version 2.6) and adapted to IGOR Pro 6.1 (Wavemetrics, Lake Oswego, OR, USA) to obtain the ‘saturated’ type II binding spectrum and facilitate more accurate spectral comparisons.
van ’t Hoff data analysis
The natural logarithm of the equilibrium association constant (lnKa = lnKD−1) obtained by non-linear regression analysis of the UV/vis generated binding isotherms, was plotted as a function of inverse experimental temperature (Kelvin). At each temperature Ka was measured in duplicate and the associated standard error from the fit was assigned to each point in the van ’t Hoff plot to be weighted during regression analysis using the linear form of the van ’t Hoff equation: lnKa = − ΔH°/R(1/T) + ΔS°/R. ΔH° and ΔS° were calculated from the best fit slope and intercept, respectively, using a value R = 1.9859 cal/mol/K.
Corrections to [IMZ] during van ’t Hoff analysis
Due to the mixture of charge states anticipated at pH = 7.4 for basic IMZ, a correction procedure was applied to the absolute IMZ concentration when determining Ka and is based on several assumptions: 1) Only the neutral fraction of IMZ fragment is involved in CYP binding; 2) The temperature dependence of pKa for the IMZ ionization reaction is accurately described by published values for the thermodynamic parameters ΔH° and ΔC°p measured under dilute aqueous conditions(28); and 3) The IMZ·H+ activity coefficient (γIMZ·H+) can be calculated at constant ionic strength determined by the buffer (I ≈ 240 mM [KPi]) without accounting for short-range non-electrostatic interactions using the extended Debye-Hückel expression, equation (5):
| (5) |
Here, Am represents the Debye-Hückel constant (temperature dependent values between 0.494–0.513 (kg·mol−1)1/2 taken from(29)), zi, the charge of the ith ion, I, the ionic strength of the solution, and B, the so-called ‘ion size’ parameter (set equal to 1.6 kg1/2mol−1/2 as described in ref.(30)). First, published values for pKa298K = 7.09 ± 0.1, ΔH° = 36.59 ± 0.06 kJ/mol, and ΔC°p = −16 ± 5 J/mol/K, reported in the study by Fukawa and Takahashi(28) for the IMZ proton dissociation reaction (Ka) occurring in the presence of 100 mM potassium chloride, were used to calculate pKa within the range of experimental temperatures used in this study via the methods of Clarke and Glew (equation 6)(31).
| (6) |
From the expression: (10pH−pKa · γIMZ•H+) = [IMZ]/[IMZ·H+], the molar [IMZ] fraction was obtained using calculated values for pKa and γIMZ·H+, along with the initial measured buffer pH. The [IMZ] concentrations were then corrected accordingly prior to non-linear regression analysis of the binding isotherms to estimate IMZ affinity (KD). It was observed that addition of up to 6 mM IMZ to the buffered solutions only minimally effected the final solution pH (+ 0.03 units). It is noted that ionic strength corrections to ΔH°, and ΔC°p parameters describing deprotonation reactions for charge symmetric systems, such as the case of the deprotonation of amines, are zero at this level of theory(30).
Pulsed/CW EPR
EPR measurements were conducted at X-band on an ELEXSYS E680 EPR spectrometer (Bruker-Biospin, Billerica, MA) equipped with a Flexline ER 4118 CF cryostat and Flexline ER 4118X-MD4 ENDOR resonator. A two-dimensional pulsed EPR technique, HYSCORE, was used to probe the interaction of nearby nuclei. The pulse sequence is π/2–τ–π/2-t1-π-t2-π/2–τ-echo where the τ, t1 and t2 indicate delays between pulses whose nominal turning angles are π/2 or π. A 16-step phase cycle was used to reject unwanted responses and correct the baseline as explained in(32). In HYSCORE, ENDOR frequencies (the hyperfine-shifted NMR frequencies) from the nearby nucleus are correlated with each other and dispersed in two dimensions, allowing resolution of peaks that overlap in simple 1D spectra. HYSCORE spectra were collected at the gz (low field) peak of the EPR spectrum. HYSCORE spectra from protons are analyzed using the contour lineshape analysis(33, 34) followed by complete spectral simulation with orientation selection.
Conventional continuous-wave (CW) EPR spectra were measured on a Bruker ELEXSYS E540 X-band spectrometer with ER 4102 ST resonator and either a liquid nitrogen quartz insertion dewar or a Bruker ER 4112 HV helium flow cryostat. EPR samples were prepared by mixing 50 μl of protein solution containing 20% glycerol with 225 μM of 17-click and 50mM 1H-1,2,3-triazole respectively. These samples were placed in 3mm OD (outer diameter) EPR tubes, frozen and stored in liquid nitrogen. The spectral g-values were obtained by fitting the CW-EPR spectra using Easyspin software(35).
Results
Models for type II CYP binding: 1,2,3-TRZ-heme energetics calculated by DFT
Gas phase DFT calculations (s6-31G(d) basis set; U-M06 functional(19, 20, 36)) were conducted to examine the binding of a model Fe(III) heme with 1,2,3-TRZ, 1,2,4-TRZ, and IMZ fragments to assess the potential for 1,2,3-TRZbased ligands to form type II complexes with CYPs (Table 1). The M06 functional was chosen, in part, because it has been parameterized using transition metals in the training set and correctly predicts the Fe-azole vs. Fe-H2O bond energies whereas the popular B3LYP hybrid functional fails (data not shown)(19). This non-isodesmic comparison is necessary because the resting state of the enzyme is sixcoordinate with water as the distal axial ligand, although the isodesmic comparisons within the different N-Fe cases should show better correlation with experiment. All computational comparisons are isogyric as the water-ligated and azole-ligated heme structures are both low spin with S = 1/2. The energies for the gas phase reaction between H2O-ligated low spin model heme-thiolate and the free azole fragments (Materials and Methods) are presented in Table 1 with IMZ (−5.3 kcal/mol) > 1,2,4-TRZ (−3.2 kcal/mol) > 1,2,3-TRZ (−1.7 kcal/mol). This result suggests that 1,2,3-TRZ-heme interactions are slightly weaker energetically than the other azole complexes but still favorable relative to the H2O-ligated state. Our results correlate with the solution pKa of the fragments: 6.99 (IMZ) > 2.45 (1,2,4-TRZ) > 1.15 (1,2,3-TRZ)(37), suggesting basicity is a major determinant for heme coordination. Thus, 1,2,3-TRZ was anticipated to induce similar type II optical signatures in CYP, albeit with lower affinity (KD) relative to IMZ and 1,2,4-TRZ. Interestingly, interactions with the heme of both N1 and N2 of 1,2,3-TRZ are comparable.
Table 1.
Results of gas phase DFT calculations.#
| Fragment-Heme Complex | ΔErxn (kcal/mol) | Bond Order | Bond Length Å |
|---|---|---|---|
| IMZ | −5.3 | 0.311 | 2.081 |
| 1,2,4-TRZ | −3.2 | 0.285 | 2.088 |
| 1,2,3-TRZ (N1) | −1.7 | 0.258 | 2.101 |
| 1,2,3-TRZ (N2) | −2.8 | 0.248 | 2.093 |
s6-31G(d) basis set; U-M06 functional.
Apparent type II binding of 1,2,3-TRZ fragment to multiple CYPs
Despite the DFT results, which indicate that 1,2,3-TRZs could interact with heme, an extensive analysis of the literature revealed no CYP-targeted azole drugs that possess the fragment. Therefore we explored experimentally the possibility that 1,2,3-TRZ can form type II complexes, and how these complexes compare spectrally with established drug-like azole fragments, 1,2,4-TRZ, and IMZ. Two mammalian CYP isoforms, CYP3A4 and a CYP2C9 variant (N-terminal truncation and hepta mutant(13), CYP2C9dh; hereafter referred to as CYP2C9), were expressed and purified(13, 38). Figures 1 & 2 display the calculated difference spectra and resultant binding isotherms obtained from UV/vis absorbance titration of CYP3A4 and CYP2C9, respectively, with 1,2,3-TRZ, 1,2,4-TRZ, and IMZ at 20°C in 100 mM KPi (pH = 7.4, + 20% glycerol). The pertinent spectral features of each titration are summarized in Table 2. All three fragments yield type II spectral complexes, which suggest that they displace the axial water molecule from resting state heme and coordinate to ferric iron(39). The magnitude of the Soret band red shift varies in CYP3A4: IMZ (8 nm) > 1,2,4-TRZ (6 nm) = 1,2,3-TRZ (6 nm), with similar shifts observed in the alpha/beta region. In CYP2C9 the Soret shifts vary: IMZ (9 nm) > 1,2,4-TRZ (5 nm) > 1,2,3-TRZ (2 nm), also with similar shifts in the alpha/beta spectral region. The affinity was markedly lower for 1,2,3-TRZ fragment binding both CYP3A4 (KD = 14.3 ± 0.5 mM; ~38 fold lower vs. IMZ and up to ~367 fold lower vs. 1,2,4-TRZ; Table 2) and CYP2C9 (KD = 10.0 ± 0.4 mM; ~up to 50 fold lower vs. 1,2,4-TRZ, and ~101 fold lower vs. IMZ; Table 2). Curiously, these results corroborate a previous report(23) of complexity inherent in the binding of 1,2,4-TRZ to CYP3A4 and CYP2C9, which manifests as pronounced curvature in Eadie-Hofstee plots (Figures 1 & 2, inset). In both cases, the data fit best to a two site binding model (Materials and Methods, equation 4): the first molecule of 1,2,4-TRZ binds CYP3A4 with KD1 = 0.039 ± 0.007 mM and is responsible for 13% of the total absorbance change observed at saturation, followed by binding of a second low affinity equivalent with KD2 = 4.80 ± 0.21 mM; in CYP2C9, both 1,2,4-TRZ binding sites are responsible for nearly equal shift toward the low spin azole-ligated spectrum (51% vs. 49% total spectral shift at saturation for high affinity and low affinity site, respectively), and the first molecule of 1,2,4-TRZ binds with KD1 = 0.198 ± 0.040 mM followed by a second low affinity equivalent with KD2 = 2.85 ± 0.82 mM. In contrast to a previous report(23), multiple binding of IMZ was not observed in our titrations with CYP2C9. Possibly, the difference lies in the use of the wild type enzyme vs. the variant used in our studies.
Figure 1.

Optical titrations and binding isotherms for nitrogen heterocycles binding to CYP3A4. 1,2,3-TRZ binding CYP3A4 shows type II behavior. UV/Vis equilibrium absorbance titration (20°C) of purified CYP3A4 (1–2 μM; 0.1 M KPi, pH=7.4, + 20% glycerol) with 1,2,3-TRZ (top), 1,2,4-TRZ (middle), and IMZ (bottom). Shown are calculated difference spectra (left), with the α and β region of the recorded absolute spectra as inset, and the resultant binding isotherms (right), generated by plotting Δ(peaktrough) intensities as a function of ligand concentration, with best fits used to estimate KD (Table 2). Interestingly, 1,2,4-TRZ displays complex equilibrium binding behavior (see text) that is most evident as curvature in the Eadie-Hofstee transform (inset), shown here fitted to a two site binding model (Material and Methods, equation 4).
Figure 2.

Optical titrations and binding isotherms for nitrogen heterocycles binding to CYP2C9. 1,2,3- TRZs binding to CYP2C9 yield type II behavior. UV/Vis equilibrium absorbance titration (20°C) of purified CYP2C9h (1–2μM; 0.1 M KPi, pH = 7.4, + 20% glycerol) with 1,2,3-TRZ (top), 1,2,4-TRZ (middle), and IMZ (bottom). Shown are calculated difference spectra (left), with the α and β region of the recorded absolute spectra as inset, and the resultant binding isotherms (right), generated by plotting Δ(peaktrough) intensities as a function of ligand concentration, with best fits used to estimate KD (Table 2). As with CYP3A4, 1,2,4-TRZ displays complex equilibrium binding to CYP2C9, which is most evident as curvature in the Eadie-Hofstee transforms (inset). The data fit best to a two site model (Material and Methods, equation 4) where binding at each site induces similar spin-shifts.
Table 2.
Tabulated UV/Vis absorbance data for azole fragment binding CYP3A4 and CYP2C9.
| Soret (nm) | α (nm) | β (nm) | α:β | Difference: Peak (nm) | Difference: Trough (nm) | Absmax (1μM)# | Abs390 (1μM)% | KD (mM) | |
|---|---|---|---|---|---|---|---|---|---|
| CYP3A4* | 416 | 565 | 534 | 0.94 | --- | --- | --- | --- | --- |
| 1,2,3-TRZ | 422 | 565 | 538 | 0.77 | 430 | 394 | 0.058 | −0.023 | 14.3 +/− 0.5 |
| 1,2,4-TRZ | 422 | 569 | 537 | 0.74 | 432 | 395 | 0.0065 0.050 |
−0.025 | KD1=0.0390±0.0071 KD2 = 4.80 ± 0.22 |
| IMZ | 424 | 570 | 538 | 0.73 | 432 | 407 | 0.065 | −0.027 | 0.381 +/− 0.011 |
| CYP2C9* | 418 | 569 | 537 | 1.1 | --- | --- | --- | --- | --- |
| 1,2,3-TRZ | 420 | 570 | 540 | 0.84 | 433 | 411 | 0.041 | --- | 10.0 +/− 0.4 |
| 1,2,4-TRZ | 423 | 572 | 542 | 0.78 | 437 | 411 | 0.023 0.044 |
--- | KD1=0.198±0.040 KD2 = 2.85 ± 0.82 |
| IMZ | 427 | 574 | 544 | 0.77 | 439 | 413 | 0.066 | --- | 0.0989 +/− 0.0018 |
20°C
Maximum peak trough intensity normalized to 1 μM
Used as an estimate of efficiency of type II ligand to decrease inherent high spin enzyme fraction (type IIa)(24).
Regardless of these system-dependent differences, the binding studies and DFT calculations demonstrate that type II binding is favorable in the case of 1,2,3-TRZ. The DFT calculations correctly predict the rank order affinity of the three ligands for CYP2C9, but they fail to correctly predict the rank order affinity for CYP3A4 binding (1,2,4-TRZ > IMZ > 1,2,3-TRZ). This is not an altogether surprising result, as the DFT calculations do not include zero point energy, solvent interactions, and possible contribution of the protein itself to binding free energies.
Comparative van ’t Hoff analysis of 1,2,3-TRZ and IMZ binding CYP3A4
The thermodynamic basis for the inferior CYP3A4 binding energetics of 1,2,3-TRZ relative to IMZ was investigated by van ’t Hoff analysis to estimate ΔH°binding and ΔS°binding. 1,2,4-TRZ was excluded from the analysis due to the additional binding complexity observed during the initial binding screen. The temperature dependence of the equilibrium association constant (Ka) for azole binding to CYP3A4 was measured via UV/vis absorbance titration within a temperature range of 5–30 °C in 100 mM KPi (pH = 7.4, +20% glycerol) and the results are shown in Figure 3. In order to account for the relatively high basicity of IMZ and accurately define its thermodynamic quantities for CYP binding, an additional correction procedure (Materials and Methods) was used to calculate the effective IMZ concentration present during binding analysis, with the assumption that only neutral IMZ species binds CYP. Similar corrections were unnecessary for 1,2,3-TRZ due to the fact that it is expected to exist in essentially 100% neutral form at pH = 7.4 (pKa = 1.15(37)). From the fits to the linear form of the van ‘t Hoff equation (Materials and Methods), estimates of ΔH°binding and ΔS°binding for both species were calculated from the slope and y-intercept, respectively. In 100 mM KPi, binding of both fragments is enthalpically favorable, as would be expected for azole fragments that directly coordinate heme iron and presumably make little or no contact with active site residues. Values of ΔH°binding were similar for IMZ (−8.1 ± 0.3 kcal/mol) and 1,2,3-TRZ (−8.8 ± 0.1 kcal/mol), clearly demonstrating that the main determinant for the low ΔG°binding for 1,2,3-TRZ relative to IMZ is entropy, which is much more unfavorable for 1,2,3-TRZ (1,2,3-TRZ: ΔS°binding = −22 ± 0.4 cal/mol/K; IMZ: ΔS°binding = −10 ± 1 cal/mol/K). It is speculated here that this discrepancy for ΔS°binding between IMZ and 1,2,3-TRZ may be due, in part, to the unusual nature of 1,2,3-TRZ tautomeric equilibria. Specifically, the well-documented dependencies of these equilibria on solvent polarity, solute concentration, and temperature(40) (see Discussion) suggest that protein binding and heme ligation may further influence the distribution of 1,2,3-TRZ tautomers, which have unique electronic properties such as differing dipole moments(41) and NMR chemical shifts(40).
Figure 3.

van ’t Hoff plots demonstrating the temperature dependence (5–30°C) of the equilibrium affinity constant (Ka) for 1,2,3-TRZ (top), and IMZ (Middle) binding CYP3A4 (1μM) in 100 mM KPi (pH = 7.4, +20% glycerol) as measured by UV/vis absorbance titration. Fit results provide estimates for ΔH°binding and ΔS°binding and are summarized in the bar graph (bottom) with values of ΔG° at T = 298 K. Ka values plotted for IMZ represent those obtained after correction of the IMZ titrant concentration to reflect only neutral species (See Materials and Methods).
Influence of 1H-1,2,3-triazole fragment on CYP-inhibitor/substrate interactions: the case of 17α-ethynylestradiol
Equilibrium Binding
The results above demonstrate the potential for the isolated 1,2,3-TRZ fragment to form a type II complex with multiple CYPs, albeit with lower affinity than other nitrogenous isosteres. Thus, it remains unclear whether the interactions between this fragment and the heme could be strong enough to influence binding of a well-characterized CYP3A4 substrate. To investigate this possibility, 17α-ethynylestradiol (17EE), a type I substrate/mechanism based inhibitor was chosen for derivatization due to its well-characterized metabolic profile, which suggests the molecule prefers to dock with its steroid A-ring nearest heme (see Discussion), and thus provides an opportunity to test the energetics of 1,2,3-TRZ-heme interactions by installing the fragment on the opposing end (D-ring) of the core steroid structure. 17EE was successfully derivatized to 17α-(2H-2,3,4-triazolyl)-estradiol (17-click) and isolated in 60% yield using a one step click reaction (Materials and Methods). Equilibrium binding of 17EE and 17-click to purified CYP3A4 in 100 mM KPi (pH = 7.4, + 20% glycerol) were measured by UV/vis absorbance at 25°C (Figure 4). 17EE binding induced a type I spectrum, typical of efficiently oxidized CYP substrates that are thought to displace the water molecule at the 6th axial position and favor the high spin enzyme species(39). The calculated 17EE binding isotherm displayed obvious sigmoidal behavior that is corroborated by pronounced curvature observed in an Eadie-Hofstee transformation of the data (not shown), suggesting the stoichiometry of binding is greater than unity (Figure 4). A simple Hill plot (not shown) was used for non-linear regression analysis and exhibited a Ks = 37.7 ± 1.7 μM and Hill coefficient of 1.75. The 17EE binding data were also fit to a two site sequential binding model (Figure 4) (Materials an Methods, equation 4) and the best fit yielded KD1 = 50 ± 25 μM for the first 17EE equivalent, which did not induce a significant heme spin state change, followed by binding of a second equivalent that perturbs the spin state with KD2 = 19 ± 8 μM. This was not entirely unexpected, as similar CYP3A4-steroid spin-silent binding interactions have already been documented in our lab for testosterone(42), and are suggested by crystallographic results with progesterone bound to CYP3A4 at a peripheral site(43).
Figure 4.

Optical titrations and binding isotherms for 17EE and 17-click binding to CYP3A4. Installment of 1,2,3-TRZ fragment on the 17EE framework to yield 17-click induces a type I (17EE, top left) to type II (17-click, top right) equilibrium binding conversion. Titrations were conducted in the presence of 2 μM CYP3A4 (100mM KPi + 20% glycerol, pH=7.4; 20 °C).,+ 17EE (left), + 17-click (right). Calculated binding isotherm for 17EE (bottom left) fitted best to a sequential two-site binding model where the binding of the first 17EE equivalent is ‘spin silent,’ while the second ‘spin-audible’ equivalent binds with positive cooperativity. 17-click binding isotherm (bottom right) displays hyperbolic binding behavior with similar affinity to parent 17EE.
In contrast, 17-click binding to CYP3A4 displays a dramatic reversal of the spin state towards low spin heme, as evidenced by the type II difference spectrum shown in Figure 4. This result suggests that despite the inferior azole-heme binding energetics of 1,2,3-TRZ compared with similar azole heterocycles IMZ and 1,2,4-TRZ, the 1,2,3-TRZ fragment is still capable of weighting the preferred 17EE binding poses to accommodate an additional type II binding interaction with heme iron. 17-click binding was best described using a hyperbolic binding model with KD = 45.8 ± 3.1 μM. Interestingly, further inspection of the type II optical signature at near protein saturation reveals that the interaction may be ‘strained’ or ‘incomplete’ (Table 3). This assessment is based on the sub-maximal Soret red-shift (2 nm) and decrease in CYP3A4 high spin fraction (ΔAbs390nm) observed in the absolute absorbance spectrum, and most evident by the diminished peak to trough intensity difference observed in the calculated difference spectrum relative to what is observed for the 1,2,3-TRZ heme complex. Others have suggested that UV/vis absorbance spectra for azole fragment-CYP complexes can be useful references for detecting sub-optimal heme ligation for type II species bearing similar fragments(23).
Table 3.
Comparison of normalized (1μM) UV/vis spectral data for 1,2,3-TRZ and 17-click binding CYP3A4.
Structural analysis of CYP3A4·17-click low spin complex by CW-EPR and HYSCORE spectroscopy
On the basis of DFT calculations and equilibrium binding studies conducted here, 1,2,3-TRZ fragment-heme interactions are anticipated to be weaker than imidazole-heme and 1,2,4-TRZ-heme interactions, but sufficient to modulate substrate orientation, at least in the case of 17-click. However, the incomplete spectral conversion with 17-click, compared to the larger spin state conversion with the fragment 1,2,3-TRZ, suggested the possibility that the heme ligation is not complete in the former case. Speculatively, the incomplete difference spectrum described above for 17-click could be due to conformational heterogeneity or axial ligand heterogeneity on the heme.
Therefore, to test this hypothesis, CW and pulsed EPR techniques were used to examine the type II CYP3A4 complexes of 1,2,3-TRZ and 17-click. For these experiments, the concentration of KPi was increased to 200 mM to achieve the high CYP3A4 concentration necessary for detectable EPR signals. Binding affinity of 17-click was demonstrated to be minimally affected by this change in ionic strength (KD100mM = 46 μM, vs. KD200mM = 78 μM) and the UV/vis absorbance spectra at saturation were identical to that observed previously (Data not shown). Figure 5 shows the CW EPR spectra for ligand-free CYP3A4, and CYP3A4 with 1,2,3-TRZ and 17-click. The EPR spectra for ligand-free CYP3A4 have g-values 2.421 (gz), 2.249 (gy) and 1.921 (gx). 1,2,3-TRZ shifted the EPR g-values of this complex, with two unique sets identified by fitting the CW-EPR spectrum (Materials and Methods): 2.505 (gz), 2.259 (gy) and 1.874 (gx); and 2.459 (gz), 2.261 (gy), and 1.890 (gx). Both species of 1,2,3-TRZ-bound CYP3A4 detected from the fit of the data have larger gz values than ligand free enzyme. It is speculated here that two unique sets of gvalues for the [1,2,3-TRZ • CYP3A4] complex might correspond to two possible heme-ligation states of 1,2,3-TRZ involving either N1(3) or N2, each producing distinct EPR spectra. Interestingly, 17-click produced rather small shifts in EPR g-values from those of the ligand-free protein: 2.415 (gz), 2.249(gy) and 1.922(gx), with gz being shifted to a smaller value than the ligand free enzyme. The results suggest that 1,2,3-TRZ and 17-click, both of which form low spin CYP3A4 complexes based on optical spectra, do so with distinctly different interactions with the heme-iron.
Figure 5.
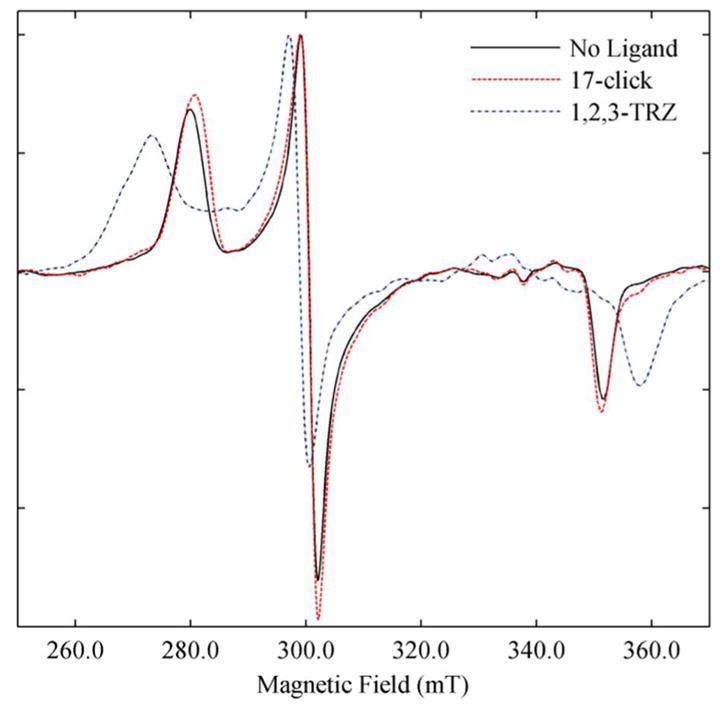
X-band CW EPR spectra of ligand free and ligand bound low spin CYP3A4. Spectra of ligand free (Black), 1,2,3-TRZ (Blue) saturated, and 17-click saturated (Red). For all spectra the protein concentration was 45 μM in 0.2 M KPi (pH = 7.4, + 20% glycerol). All experiments were conducted at 15 K and the EPR intensities were normalized.
Hyperfine sublevel correlation spectroscopy (HYSCORE) was used to probe further the environment of heme-Fe(III) with different ligands using the hyperfine interaction between the unpaired spin on heme iron and nearby nuclei. HYSCORE is a two-dimensional EPR method that allows for detection of the effects of neighboring nuclei on EPR electronic transitions. Specifically, we investigated the protons of the axial H2O coordinated to the heme that were assigned in previous studies of ligand-free CYP2C9 and CYP101A1 (P450cam)(44, 45). Figure 6 shows the HYSCORE spectra of CYP3A4 with no ligand at the magnetic field corresponding to gz for 17-click complex. The peaks/arcs extending between frequencies [(9.3, 16.0) and (10.7, 14.5)], and [(16.0, 9.5) and (14.5, 10.9)] MHz indicated by arrows have isotropic hyperfine splitting values of 4.31 MHz, estimated from the spectral simulation, that are similar to values reported for protons of the axial water in Fe(III) CYP2C9 and CYP101(44, 45). Detailed analysis of the spectral line shapes(33, 34) show that these signals arise from protons at a distance of 2.65 A from the heme Fe(III). The remaining signals arise from other protons near the heme. A second set of peaks/arcs extending between frequencies [(10.5, 14.0) and (12.2, 12.5)] and [(12.8, 11.8) and (14.0, 10.8)] MHz has isotropic hyperfine splitting value of −3.82 MHz, and lies at a distance of 3.05 A from the heme Fe(III). These signals are tentatively assigned to the thiolate β-protons (Fe(III)-S-CH2-R) of the cysteine residue axially bound to the heme Fe(III). As a positive type II binding control, the HYSCORE spectrum for CYP3A4·1,2,3-TRZ complex was obtained, and the proton frequency region of the HYSCORE spectrum at magnetic field strength corresponding to gz for this complex is also depicted in Figure 6. The HYSCORE spectrum clearly demonstrates displacement of the axial 6th water molecule by 1,2,3-TRZ fragment by the disappearance of the previously characterized proton peaks/arcs. Unexpectedly, the HYSCORE spectra of CYP3A4 with saturating concentration of 17-click retain the axial-H2O proton peaks/arcs, with only slight frequency shifts [(9.3, 16.0) and (10.5, 15.0)] and [(15.0, 10.3) and (16.0, 9.5)]. In addition, no new nitrogen interactions were detected by comparison with the ligand-free control spectra. The isotropic hyperfine coupling value obtained from spectral simulation of the shifted proton arcs is −4.02 MHz, which correspond to axially bound water protons at a distance of 2.75 A from heme Fe(III), a slight increase from the ligand free state.
Figure 6.
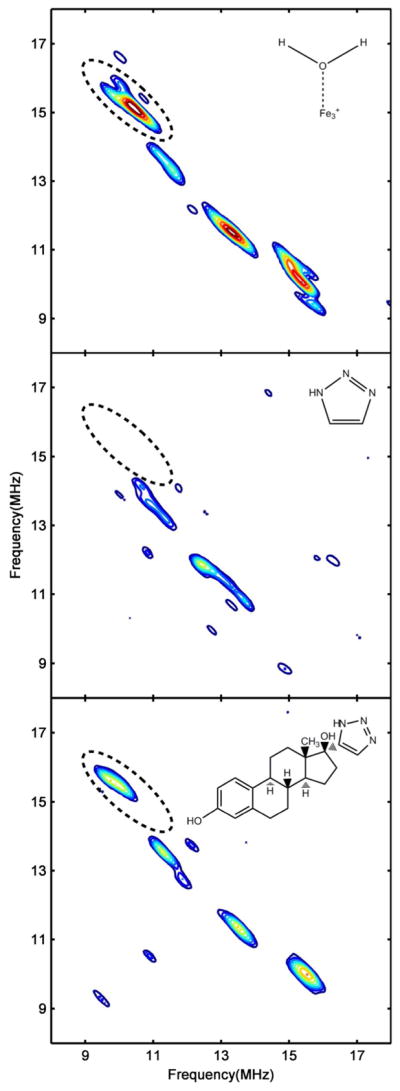
HYSCORE spectra of ligand free and ligand bound low spin CYP3A4 corresponding to gz. Shown is the proton frequency region of the HYSCORE spectra from ligand free CYP3A4 (Top), CYP3A4 + 50mM 1,2,3-TRZ (Middle), and CYP3A4 + 225 μM 17-click (Bottom). Proton signals corresponding to the axial H2O protons in ligand free CYP3A4 (Top) are defined by an ellipse, and are displaced by the type II binding 1,2,3-TRZ fragment (Middle). Despite type II binding observed optically, HYSCORE reveals that 17-click binding (Bottom) does not displace H2O from the heme, but instead alters its ligand field strength. Protein concentration was 45 μM for all samples. Magnetic field strength: ligand free = 288.0 mT; + 1,2,3-TRZ = 283.0 mT; + 17-click = 287.0 mT. All the spectra were normalized to the peaks of highest intensity (i.e. the N2 peak in the (-,+) quadrant).
These experimental results present a paradox. On one hand, 17-click induces a type II optical spectrum suggestive of direct interaction with the heme iron and displacement of the axial water. Yet, HYSCORE shows that the protons of the axial water (and the cysteine) are retained when 17-click binds, with small but readily detectable perturbation of both the protons and the EPR g-values. 17-click causes changes in electronic interactions of the heme while the axial water and cysteine remain. It is likely that the field strength of the axial water ligand is altered by hydrogen bonding interactions with the 1,2,3-TRZ moiety of 17-click, which is supported by the lengthening of the OH2···Fe(III) distance in the 17-click-bound HYSCORE spectra. Possible structural interpretations of this are suggested in the Discussion.
17-click functional inhibition of CYP3A4 and metabolic stability
17-click inhibited CYP3A4-mediated 6β-hydroxylation of testosterone in Supersomes with an IC50 = 17 ± 6 μM as determined by LC-MS/MS assay (Fig. S1). To assess metabolic stability, substrate depletion of 2.5 μM 17-click was also monitored, and demonstrated facile turnover of the type II ligand with only ~50% of the compound remaining after 25 min incubation in the presence of 100 nM CYP3A4 in Supersomes (Data not shown). A representative ion current chromatogram for m/z = 340 + 16 (O) = 356 from LC-MS/MS analysis of a larger scale incubation is shown in Figure 7 and clearly shows formation of at least four NADPH-dependent metabolites.
Figure 7.
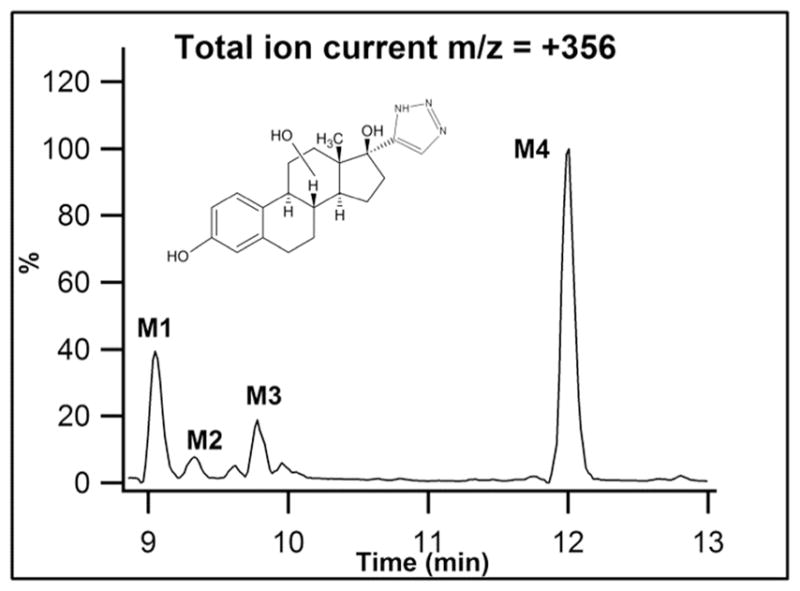
17-click Metabolites detected during LC-MS/MS analysis (m/z = 340 +16 (O) = 356) of incubations of 17-click in CYP3A4 Supersomes.
To assess the rate of CYP3A4-mediated 17-click turnover, KM and kcat for total 17-click metabolism were measured by LC-MS/MS using the methods of substrate depletion(18) and found to be 2.5 ± 0.4 μM, and 2.1 ± 0.3 min−1, respectively (Fig. S2). Our attempts to quantify kinetic parameters for 17EE using the substrate depletion approach were unsuccessful; however, it was observed that depletion rates were between 3–10 fold faster for 17EE relative to 17-click at low substrate concentrations, and that significant depletion was still evident for 17EE at high concentrations that render depletion of 17-click negligible (Fig. S3). Wang et al. have reported a KM = 2.5 ± 1.1 μM and kcat = 0.052 ± 0.007 min−1 for CYP3A4-mediated formation of 2-OH 17EE in Supersomes under conditions determined to be linear with respect to both assay time and enzyme concentration(46). In that study it was stated that 2-OH 17EE represents greater than 90% of total metabolism under the conditions employed. Our results suggest that induction of type II binding by the addition of a 1,2,3-TRZ fragment to 17EE has most likely slowed metabolism relative to the parent molecule by 3 – 10 fold, but that 17-click remains highly susceptible to oxidative turnover by CYP3A4.
Metabolic tailoring: 1,2,3-TRZ influence on oxidative regioselectivity in CYP3A4
Qualitatively, the number and relative abundance of CYP3A4-mediated 17-click metabolites are similar to that reported by both Lin et al.(47) and Geungerich(48) for 17EE. In order to compare the regioselectivity of 17EE oxidation with 17-click oxidation, authentic 2-OH and 4-OH 17-click metabolite standards (primary oxidation products for CYP3A4-mediated 17EE turnover(46, 47, 49)) were synthesized and isolated in 15% (2-OH) and 3% (4-OH) overall yield using procedures adapted from the literature (Materials and Methods)(14, 15). The retention times for the 2-OH and 4-OH 17-click standards did not match that for any of the metabolites during LC-qTOF MS anlaysis (Data not shown). Thus, metabolism at the preferred 2 and 4 positions of the 17EE steroid A-ring (Figure 8) is abolished by addition of 1,2,3-TRZ to the drug’s scaffold. Representative mass spectra obtained from MSE analysis (Materials and Methods) of m/z = 356.1974 for the 2-OH standard (similar to 4-OH 17-click spectrum, data not shown), and predominant oxidized metabolite, M4, observed in the Supersome incubation extract are shown in figure 9. Also shown for comparison in figure 9 is the MSE spectrum for the remaining 17-click parent from the incubation, corresponding to m/z = 340.2025. Table 4 lists the elemental composition determined from the observed mass of important fragments from each spectrum (vide infra). The spectra for M1, M2 and M3 were similar (Figure S4) to that shown for the most abundant metabolite, M4, with the exception of a prominent fragment ion unique to M4 at m/z = 213.1277, consistent with the molecular formula C15H17O (calculated m/z = 213.1279, −0.94 ppm).
Figure 8.
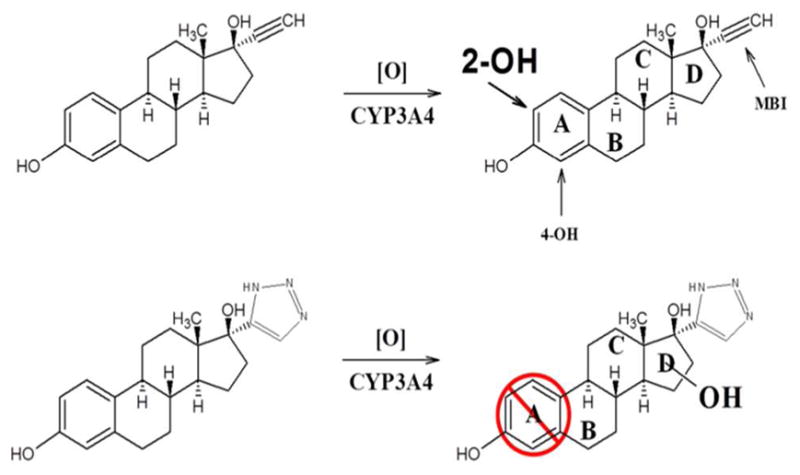
Schematic showing primary sites of oxidative metabolism for parent 17EE and the effect of 1,2,3-TRZ fragment addition on regioselectivity. When the triazole is added no metabolism occurs on the A-ring but metabolism is directed toward the triazole. Note: the Markush bonded hydroxyl group represents several possible sites of oxidation on or in the vicinity of the steroid D-ring as determined by mass spectrometry (See Results).
Figure 9.

MSE fragment ion spectra. 17-click (m/z = 340.2025 (C20H26N3O2)) (Top), 2-OH 17-click (Middle), and metabolite M4 (m/z = 356.1974 (C20H26N3O3)) (Bottom). M4 represents the most abundant metabolite species and closest in retention time to the synthesized 2-OH and 4-OH 17-click. High resolution mass spectra were acquired using a SYNAPT qTOF mass spectrometer in MSE mode with a collision energy ramp of 15–45V (Materials and Methods). Structural proposals for highlighted fragments were obtained using MassFragment in MassLynx MS analysis software. Note: the illustrated D-ring location of CYP3A4-mediated oxidation for M4 is indefinite, and merely represents one of few probable turnover positions near/on the steroid D-ring based on results from ion trapping (vide infra).
Table 4.
Elemental composition of key ion determined by qTOF MSE anlaysis of 17-click, 2-OH 17-click, and predominant metabolite M1.
| Formula | Relative Abundance (%) | Theoretical | Actual | σ (ppm) | |
|---|---|---|---|---|---|
| 17-click | C11H9O | 30 | 157.0653 | 157.0657 | 2.5 |
| C11H11O | 50 | 159.0810 | 159.0809 | −0.6 | |
| C20H24NO | 20 | 294.1858 | 294.1844 | −4.8 | |
| C20H24N3O | 100 | 322.1919 | 322.1909 | −3.1 | |
| C20H26N3O2 | 30 | 340.2025 | 340.2021 | −1.2 | |
| 2-OH 17-click | C11H9O2 | 30 | 173.0603 | 173.0602 | −0.6 |
| C11H11O2 | 75 | 175.0759 | 175.0759 | 0.0 | |
| C20H24NO2 | 25 | 310.1807 | 310.1801 | −1.9 | |
| C20H24N3O2 | 100 | 338.1869 | 338.1858 | −3.3 | |
| C20H26N3O3 | 5 | 356.1974 | 356.1963 | −3.1 | |
| M4 | C11H9O | 40 | 157.0653 | 157.0659 | 3.8 |
| C15H17O | 100 | 213.1279 | 213.1277 | −0.9 | |
| C20H22NO | 10 | 292.1701 | 292.1698 | −1.0 | |
| C20H22N3O | 15 | 320.1763 | 320.1761 | −0.6 | |
| C20H24N3O2 | 60 | 338.1869 | 338.1859 | −3.0 | |
| C20H26N3O3 | 10 | 356.1974 | 356.1971 | −0.8 |
The MS fragment data allow for some additional conclusions to be drawn regarding the structure of the 17-click metabolites. First, while all species investigated (including 17-click) were observed to dehydrate readily (ion at m/z = 356 (340) − 18 = 338 (322)), a second facile dehydration step was observed only for the metabolites (Figure S4) for M1, M2, and M3 spectra), and indicated by a prominent ion at m/z = 320 (20–100 rel. abundance), which is completely absent from the MSE spectrum of both 2-OH (Figure 9) and 4-OH (Data not shown) 17-click. Fragmentation of the metabolites favors two facile water losses and loss of N2 (m/z = 292), in stark contrast to fragmentation of the 2-OH/4-OH 17-click (and 17-click) that undergo a single dehydration and loss of N2 (m/z = 310 (M1–M4), m/z = 294 (17-click)), a species nearly absent from all metabolite fragment ion spectra. In addition, there are spectral shifts from m/z = 157.0657 (C11H9O, +2.5 ppm) and m/z = 159.0809 (C11H11O, −0.6 ppm) present in spectra of 17-click, to m/z = 173.0602 (C11H9O2, −0.6 ppm) and 175.0759 (C11H11O2, 0 ppm) observed in spectrum of the 2-OH (and 4-OH) 17-click. These observations suggest that hydroxyl groups are retained at estrogen A-ring positions during fragmentation under the experimental conditions used here. Thus, we conclude that CYP3A4-mediated oxidation of 17-click does not occur at the remaining A-ring position to form 1-OH 17-click, a species of which we had no standard for comparison. In further support of this conclusion is the fact that the spectra for the metabolites are very similar (excepting m/z = 213 in M4 spectra, vide infra) and if the 1-OH 17-click species were for some reason susceptible to facile A-ring dehydration during fragmentation (mass spectrum lacking prominent ions at m/z = 173,175, and 310) it would be anticipated to display a distinct fragment ion spectrum due to disruption of aromaticity. Plausible structures determined with MassFragment software (see Materials and Methods) for signature fragments that contain the steroid A-ring are shown in the spectra of Figure 9.
Additional structural insight into the identity of the primary metabolite, M4, was made possible via MSn analysis using an LTQ ion trap instrument. Based on two unique ion trapping sequences: m/z = 356→338→320→fragments, and m/z = 356→338→213→fragments, it is concluded that m/z = 213 is a kinetic product formed during fragmentation of m/z = 338, the formation of this species being entirely dependent on the loss of a second equivalent of water from M4 (Scheme 1). The overwhelming relative abundance of m/z = 213 in the fragment ion spectrum of M4, coupled with the complete absence of this (or equivalent) species in fragment spectra of 17-click, suggests that the required dehydration step to form m/z = 213 involves the oxygen incorporated into the molecule during CYP3A4-mediated turnover. Thus, the structural identity of this fragment should offer insight as to the location of oxidative turnover of 17-click. To this end, MassFragment software was used to generate a structural proposal (Figure 9) for fragment ion C15H17O (calculated m/z = 213.1279, −0.94 ppm), and a structure characterized by fragmentation of the estrogen D-ring of 17-click was determined to be the only plausible solution given the elemental composition of this fragment (see Discussion). In conclusion, addition of a 1,2,3-TRZ-heme interaction in the CYP3A4·17EE complex has eliminated the preferred sites of estrogen metabolism entirely (A-ring), and steered the metabolism predominantly towards the D-ring bearing the triazole moiety.
Scheme 1.
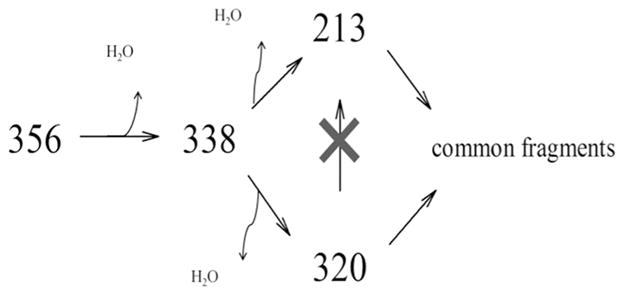
MS4 analysis of primary CYP3A4-mediated metabolite of 17-click (M4) reveals predominant M1daughter ion, m/z = 213, to be a kinetic product of the second dehydration step: m/z = 213 was entirely absent from MS4 spectra using the sequence 356→338→320→ fragment, while the ion remained in 80% rel. abundance (data not shown) using the sequence 356→338→213→ fragment.
Discussion
1,2,3-TRZ has received great attention in the drug-design arena(8, 9) since the discovery in 2001 of novel catalysts for the well-known 1,3-dipolar cyclo-addition reaction between organic azides and alkynes, or the ‘Click’ reaction. The synthetic advantages to the medicinal chemist, via the methods of click chemistry, have prompted widespread use of 1,2,3-TRZ as a central scaffold moiety in fragment-based drug design for a wide range of targets(8, 50). Interestingly, despite the prevalence in CYP-targeted drugs of the closely related azole congeners IMZ and 1,2,4-TRZ, the efficacy of 1,2,3-TRZ as a CYP pharmacophore has not been established. However, high-affinity interactions of 1,2,3-TRZ with transition metals are known; in fact, 1,2,3-TRZ forms complexes with a variety of transition metals including both ferrous and ferric iron species(11). Thus, the first goal of the present study was to employ a fragment-based systematic investigation of the relative heme-binding affinity, CYP-binding thermodynamics, and spectral behavior of 1,2,3-TRZ in comparison to established azole CYP-pharmacophores IMZ and 1,2,4-TRZ.
Our DFT results suggest that 1,2,3-TRZ interacts less favorably than the other azoles but it is competent for type II binding in as much as it can be a stronger ligand than water. The in silico energies for the complexes trend with aqueous basicity of the fragments (IMZ (pKa = 6.99) > 1,2,4-TRZ (pKa = 2.45) > 1,2,3-TRZ (pKa = 1.15)(37)); it was not entirely obvious that this trend should exist because very high basicity (nucleophilicy) for a distal heme ligand might disfavor heme coordination due to charge donation from the proximal thiolate(51). 1,2,3-TRZ-heme complexes were also calculated to have the longest bond length, as well as lowest bond order compared with the other fragments (IMZ and 1,2,4-TRZ). One interesting conclusion from the computational studies is that 1,2,3-TRZ is predicted to ligate heme-iron at both N1and N2, suggesting the possibility that two different routes for heme coordination could be active when binding CYP. Recently, an usually high affinity (0.018 μM, ligand efficiency = 0.88) 1,2,3-TRZ fragment species, 4-(3-methylphenyl)-1H-1,2,3-triazole, was discovered to coordinate two Co+2 ions simultaneously in human Methionine Amino Peptidase 2 via N1 and N2 during a fragmentbased drug screen(10). Thus, our DFT results and published work suggest that 1,2,3-TRZ could ligate the heme iron of CYPs in two different modes, with the possibility of substituent effects ‘steering’ ligation to either N1 or N2.
Experimental measures of CYP binding affinity and UV/vis absorbance spectral behavior for 1,2,3-TRZ provided further insight for this azole fragment. In buffered aqueous solution, 1,2,3-TRZ bound with modest affinity (up to 367-fold lower compared with 1,2,4-TRZ binding CYP3A4) relative to the other azoles when screened against CYP3A4 and CYP2C9. Spectrally, 1,2,3-TRZ displayed smaller red-shifts in both the Soret and alpha/beta band regions, as well as decreased effects on the alpha:beta ratio. The relative spectral behavior of 1,2,3-TRZ·CYP complexes appears to correlate with lower CYP binding affinity for 1,2,3-TRZ and is likely related to the inferior heme binding energetics observed in our calculations. Interestingly, this comparison highlights a perplexing aspect of the binding mechanism for small hydrophilic azole fragments to CYPs. Additional equilibrium binding complexity was observed in the isotherms of 1,2,4-TRZ binding both CYP2C9 and CYP3A4, corroborating a previous type II binding study by Locuson et al.(23). In that study the authors speculated that the multiple binding phases observed during equilibrium titration might be explained by considering the multiple nitrogen-ligation states that exist for 1,2,4-TRZ. However, similar binding complexity (and similar binding affinity) observed by the authors for the N(1)-methyl fragment derivative would suggest otherwise; the modification should sterically limit heme ligation to N(4) only. The authors also proposed that 1,2,4-TRZ·CYP complexes may exist as a mixture of directly-ligated and axial water-bridged complexes at equilibrium, which are anticipated to perturb the heme differentially as was shown in this study for a ligand bearing 1,2,3-TRZ. However, these hypotheses to explain complex equilibrium binding behavior would require a heterogeneous, slowly-equilibrating protein population and so multiple binding of 1,2,4-TRZ fragment is the more likely explanation. Also described in the study by Locuson et al. was binding isotherm complexity for IMZ binding CYP2C9 (WT) at pH = 7.4 that could be linearized via adjustment of the pH significantly above the fragment pKa (6.99,(30)). This behavior is not observed for IMZ binding to the hepta mutant CYP2C9 used here. This discrepancy may reflect modest differences in active site structure for the wild type vs. the mutant proteins. Also puzzling is the fact that the rank order CYP3A4 affinity measured here for IMZ and 1,2,4-TRZ was reversed relative to the DFT calculations, with the first equivalent (two-site sequential model) of 1,2,4-TRZ binding with greater affinity than IMZ. These results, together with the thermodynamic studies, highlight the important contributions of protein architecture, to fragment-CYP binding energetics.
The chemical similarity between IMZ and 1,2,3-TRZ allows for a meaningful comparison of enthalpic (ΔH°) and entropic (ΔS°) components of their binding affinity’s. Therefore, van ’t Hoff analysis was employed to obtain thermodynamic signatures for the CYP3A4 binding interaction. We observed a linear temperature dependence of equilibrium binding constants for these small hydrophilic azole fragments, suggesting that heat capacity changes (ΔC°p) for fragment-bound complexes are negligible, at least over the temperature range studied here. A negligible heat capacity change (constant ΔH° and ΔS°) upon formation of azole fragment·CYP3A4 complexes is consistent with the hydrophilic nature of IMZ and 1,2,3-TRZ, as well as their small size which is not expected to induce conformational changes when ligated to heme iron(43). Our thermodynamic results are informative on several levels. First, we found that the large discrepancy in ΔG° between IMZ and 1,2,3-TRZ (−5.1 vs. −2.3 kcal/mol at 25°C) binding CYP3A4 in 100 mM KPi is dictated by a much larger entropic penalty for binding in the case of 1,2,3-TRZ; surprisingly, the ΔH term is slightly more favorable for 1,2,3-TRZ binding than IMZ, but the entropic difference is a major contribution to their differential affinity’s. It is tempting to speculate that this phenomenon could be related to the well-documented 1,2,3-TRZ tautomeric complexity. 1,2,3-TRZ tautomerism is known to be highly dependent on solvent type, polarity, temperature, and 1,2,3-TRZ concentration(40, 41, 52, 53). In dilute aqueous environments, 1,2,3-TRZ exists at ~1:1 ratio of degenerate N1(3)-H tautomers, and the N2-H tautomer(41). 1,2,3-TRZ fragment binding to protein may disrupt this equilibrium mixture towards the selection of a single tautomeric form, presumably the symmetric N2-H tautomer that predominates in solvents of low dielectric. Additionally, if ligation to heme iron is favored at the more basic nitrogen positions (N1 and N3), then a tautomeric shift opposite to that induced by fragment binding can be expected. A large unfavorable ΔS° value of −16.5 cal/mol/K has been reported for the tautomerization towards the asymmetric N1(N3)-H tautomer in toluene(40). Next, the magnitudes of ΔH° and ΔS° measured for 1,2,3-TRZ and IMZ fragments binding CYP3A4 in KPi are comparable to those values reported in a previous solution thermodynamic study of small, IMZ-based ligands for CYP2B4(54). Notably, in that study, the enthalpic signatures (25°C) of phenylimidazole (PI) species 1-PI and 4-PI were either minimally effected (1-CPI, ΔΔH°binding = −0.07 kcal/mol) or dramatically attenuated (4-CPI ΔΔH°binding = + 2.12 kcal/mol) by chloro substitution, suggesting that addition of relatively lipophilic peripheral substituents contributes mostly toward increased, favorable, ΔS°binding, often at the expense of ΔH°binding, a parameter that, in the case of small, rigid, type II ligands such as 1,2,3-TRZ and IMZ, may be expected to be dominated by coordinate bond formation. An additional source of the larger entropic penalty for 1,2,3-TRZ may be the remaining localized water molecule observed by HYSCORE. These results have particular importance in the context of fragment based drug design strategies that incorporate click chemistry, for CYP inhibitors as well as other targets. The observation that the difference in binding free energy between IMZ and 1,2,3-TRZ is largely entropic provides an obvious strategy to incrementally improve the affinity of 1,2,3-TRZs; incorporation of hydrophobic groups would likely decrease the entropic cost of binding and increase their affinity very substantially as observed with CYP2B4 and phenylimidazoles.
To test whether 1,2,3-TRZ-heme binding interactions are sufficient to dictate overall ligand positioning of drug-like molecules, we sought an ideal molecular framework for 1,2,3-TRZ derivatization to use as a model. 17α-ethynylestradiol (17EE) was selected for derivatization because of its well described metabolic profile that allows for some assumptions concerning the molecular details of binding to be inferred: First, 17EE binds in multiple orientations, based on the reported oxidation profile that includes oxygenation on both the estrogen A-ring (2-OH predominant)(46, 48, 49) and the acetylenic function of the steroid D-ring(47), which imparts its behavior as a mechanism-based inactivator. However, the combination of the ligand’s relatively modest partition ratio of ~50, low off-target protein adduction, the lack of reported acetylene-derived carboxylic acid metabolites(47), and overwhelming production of 2-OH 17EE(46–48), all suggest that the estrogen prefers to bind with its A-ring nearest heme.
The UV/vis binding results for 17-click are striking. Based on historical precedent with nitrogen heterocycles, the type II spectrum induced suggests that the D-ring portion of the molecule now lies nearest the heme to yield an additional coordinate bonding interaction that is not possible in 17EE. In the context of a single heme binding site, this would suggest that the addition of the 1,2,3-TRZ causes the estradiol to adopt a different orientation with the TRZ moiety near the heme iron. However, our results are complicated by multiple binding for 17EE; yet binding of the first equivalent of 17EE induces a negligible heme spin shift suggesting our comparison is localized at the catalytically relevant site. Regardless of this complexity it is completely clear that throughout the binding isotherm the high spin state is favored. Regardless of the correct binding model, it is interesting that, despite the dramatic reversal of the UV/vis binding spectrum to type II for 17-click, no significant increase in ligand affinity was observed. Thus, the TRZ group does not significantly add binding affinity, but rather changes the binding mode of the steroid nucleus in a nearly isoenergetic complex. An additional observation of some importance is that the 17-click low spin complex yields a low spin difference spectrum with lower intensity than the 1,2,3-TRZ fragment. We considered the possibility that this could be due to ‘incomplete’ heme coordination, as suggested by others(23). The difference spectra for 17EE is less redshifted and less intense than the spectra obtained from the fragment (Table 3).
CW and pulsed EPR techniques proved to be invaluable for understanding the binding and turnover behavior of the [17-click • CYP3A4] complex. The CW EPR spectra show that 1,2,3-TRZ and 17-click induce entirely different perturbations in the EPR g-values, suggesting that 1,2,3-TRZ-heme interactions are not conserved when present on a drug scaffold. Most profound were the results of HYSCORE analysis which showed conclusively that the 17-click low spin complex perturbs, yet retains, its axial water ligand. The perturbations of the axial water ligand protons hyperfine splitting values reveal a significant increase in the Fe(III) – H distance by 0.1 A (2.65 Å → 2.75 Å), consistent with hydrogen bonding between the 1,2,3-TRZ fragment of 17-click and axial water. Thus, the [17-click • CYP3A4] complex is speculated to exist at equilibrium as a water-bridged type II ternary complex in which the heme field is perturbed indirectly towards the low spin state via alteration of the axial water ligand field strength. This implies that the type II optical spectrum observed is the result of the axial water ligand with increased basicity (stronger field ligand) in the presence of 17-click. A similar water-ligated type II complex was observed by Seward et al. between 1,2,4-TRZ antifungal drug fluconazole(55) and CYP121 of Mycobacterium tuberculosis. In that study, a mixture of water-bridged and directly ligated ligand-bound forms was observed in the crystal structure at 1.90 A resolution. Notably, our EPR and HYSCORE results reveal a homogeneous low-spin protein fraction, suggesting a preferred ligation state for 17-click. It is intriguing to speculate that this phenomenon might be more common than previously believed based on the type II spectrum obtained by UV/vis absorbance, which is not distinguishable from many type II spectra in the literature. Additionally, a review of the earlier P450cam literature reveals similar EPR behavior reported for binding of sterically hindered nitrogen heterocycles, at the time classified as ‘abnormal’ type II ligands(56). The CW EPR and HYSCORE results are thus entirely consistent with a lack of affinity increase upon derivatization of 17EE.
The metabolism of 17-click was studied to determine the functional consequences of this unusual heme ligation. In contrast to our expectation that 17-click would dramatically inhibit CYP activity, the estimated turnover number (kcat) of 2.5 min−1 for total metabolism demonstrates that 17-click is a good substrate from CYP3A4. In fact, comparison of depletion data obtained for CYP3A4-mediated 17EE oxidation suggests that the rate of 17-click turnover is only moderately slower for the low spin ligand. This result is similar to recent work by Jones et al., wherein nitrogen-ligated, type II spectral complexes are rapidly metabolized(5). Because the spectral results demonstrate that the 1,2,3-TRZ moiety of 17-click interacts with heme via H2O, we found it surprising that the number and relative abundance of the metabolites initially observed were so similar to that observed by others for 17EE.
A combination of high mass accuracy qTOF, and ion trapping (LTQ) mass spectrometries were used to determine structural features of the metabolites. First, comparison of the fragmentation behavior of 17-click and synthesized 2-OH and 4-OH 17-click standards with that of the observed 17-click metabolites allowed us to conclude that steroid A-ring metabolism had been completely abolished for the 1,2,3-TRZ derivative. Second, through ion-trapping it was determined that the predominant ion observed in the fragment ion spectrum of M4 originated via loss of the metabolically inserted oxygen atom, and thus probable structures for this fragment ion that were generated based on elemental composition of this fragment via high mass accuracy qTOF measurements were used to infer the regioselectivity of CYP3A4-mediated oxidation of 17-click. A structure resulting from oxidation in the vicinity of the steroid D-ring was determined to be the only plausible solution for the primary metabolite, M4. Therefore, the mass spectral data support the conclusion that 1,2,3-TRZ installation upon the 17EE scaffold alters ligand positioning.
In summary, the 1,2,3-TRZ fragment that is isosteric with imidazole and 1,2,4-TRZ fragments found in a wide range of drugs, including those intended to inhibit CYPs, is chemically competent to ligate to the heme of CYPs. However, when incorporated on the scaffold of 17EE, EPR spectra indicate that there is not direct nitrogen ligation. Rather, in the presence of the 1,2,3-TRZ-containing 17-click, the sixth axial water is retained and is likely hydrogen bonded to the triazole. This complex, surprisingly, undergoes rapid metabolism, with different regioselectivity than 17EE. Together the results indicate the potential for novel heme coordination by 1,2,3-TRZ when installed on a CYP substrate. These results, combined with recent findings published by others(5, 6), suggest that low spin, ‘type II’ complexes should not be assumed to be inhibitory. Most importantly, we present only the second case, and the first direct EPRbased evidence in the form of HYSCORE, of a triazole ligand that elicits a classic type II UV/vis signature indirectly via heme interactions mediated by the axial H2O ligand. Further studies are required to determine whether the bridged water-heme ligation structure is a general feature of ligands containing the 1,2,3-TRZ fragment.
Supplementary Material
Acknowledgments
Funding Sources: This work was supported by NIHGMS P0132165 and NIH TG07752.
The authors would like to thank Josh T. Pearson for his generous contribution of experimental materials and use of instrumentation, as well as helpful discussion.
ABBREVIATIONS
- KPi
Potassium Phosphate
- 1,2,3-TRZ
1H-1,2,3-triazole
- 1,2,4-TRZ
1H-1,2,4-triazole
- IMZ
imidazole
- HYSCORE
Hyperfine sublevel correlation spectroscopy
- EPR
electron paramagnetic spectroscopy
Footnotes
Supporting Information Available
Plots for IC50, and Km and Vmax determination by substrate depletion for 17-click; depletion time courses for 17-click and 17EE. Representative MSMS spectra for 17-click metabolites M1-M3. This material is available free of charge via the Internet at http://pubs.acs.org.
References
- 1.Cole PA, Robinson CH. Mechanism and inhibition of cytochrome P-450 aromatase. J Med Chem. 1990;33:2933–2942. doi: 10.1021/jm00173a001. [DOI] [PubMed] [Google Scholar]
- 2.Koltin Y, Hitchcock CA. The search for new triazole antifungal agents. Curr Opin Chem Biol. 1997;1:176–182. doi: 10.1016/s1367-5931(97)80007-5. [DOI] [PubMed] [Google Scholar]
- 3.Ouellet H, Johnston JB, Ortiz de Montellano PR. The Mycobacterium tuberculosis cytochrome P450 system. Arch Biochem Biophys. 2010;493:82–95. doi: 10.1016/j.abb.2009.07.011. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Olkkola KT, Ahonen J, Neuvonen PJ. The effects of the systemic antimycotics, itraconazole and fluconazole, on the pharmacokinetics and pharmacodynamics of intravenous and oral midazolam. Anesth Analg. 1996;82:511–516. doi: 10.1097/00000539-199603000-00015. [DOI] [PubMed] [Google Scholar]
- 5.Peng CC, Pearson JT, Rock DA, Joswig-Jones CA, Jones JP. The effects of type II binding on metabolic stability and binding affinity in cytochrome P450 CYP3A4. Arch Biochem Biophys. 2010;497:68–81. doi: 10.1016/j.abb.2010.03.011. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Pearson J, Dahal UP, Rock D, Peng C-C, Schenk JO, Joswig-Jones C, Jones JP. The kinetic mechanism for cytochrome P450 metabolism of Type II binding compounds: Evidence supporting direct reduction. Arch Biochem Biophys. 2011;511:69–79. doi: 10.1016/j.abb.2011.04.008. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Peng CC, Shi W, Lutz JD, Kunze KL, Liu JO, Nelson WL, Isoherranen N. Stereospecific metabolism of itraconazole by CYP3A4: Dioxolane ring scission of azole antifungals. Drug Metab Dispos. 2012;40:426–435. doi: 10.1124/dmd.111.042739. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Meldal M, Tornoe CW. Cu-Catalyzed azide alkyne cycloaddition. Chem Rev. 2008;108:2952–3015. doi: 10.1021/cr0783479. [DOI] [PubMed] [Google Scholar]
- 9.Kolb HC, Sharpless KB. The growing impact of click chemistry on drug discovery. Drug Discovery Today. 2003;8:1128–1137. doi: 10.1016/s1359-6446(03)02933-7. [DOI] [PubMed] [Google Scholar]
- 10.Congreve M, Chessari G, Tisi D, Woodhead AJ. Recent developments in fragment-based drug discovery. J Med Chem. 2008;51:3661–3680. doi: 10.1021/jm8000373. [DOI] [PubMed] [Google Scholar]
- 11.Moore DS, Robinson SD. Catenated Nitrogen Ligands Part II: Transition Metal Derivatives of Triazoles, Tetrazoles, Pentazoles, and Hexazine. In: Sykes AG, editor. Adv Inorg Chem. Academic Press; 1988. pp. 171–239. [Google Scholar]
- 12.Woods CM, Fernandez C, Kunze KL, Atkins WM. Allosteric activation of cytochrome P450 3A4 by alpha-naphthoflavone: Branch point regulation revealed by isotope dilution analysis. Biochemistry. 2012;50:10041–10051. doi: 10.1021/bi2013454. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Williams PA, Cosme J, Ward A, Angove HC, Matak Vinkovic D, Jhoti H. Crystal structure of human cytochrome P450 2C9 with bound warfarin. Nature. 2003;424:464–468. doi: 10.1038/nature01862. [DOI] [PubMed] [Google Scholar]
- 14.Tienan J, Shin K, Yoshinori Y. Copper-catalyzed synthesis of N-unsubstituted 1,2,3- triazoles from nonactivated terminal alkynes. Eur J Org Chem. 2004;2004:3789–3791. [Google Scholar]
- 15.Rugang X, Qiuyun C, Jin X, Huaming Z. A new efficient synthetic method for 2- and 4-hydroxy-17[alpha]-ethynyIestradiol. Steroids. 1990;55:488–490. doi: 10.1016/0039-128x(90)90085-p. [DOI] [PubMed] [Google Scholar]
- 16.Noble RW, Gibson QH. The Reaction of Ferrous Horseradish Peroxidase with Hydrogen Peroxide. J Biol Chem. 1970;245:2409–2413. [PubMed] [Google Scholar]
- 17.Obach RS, Reed-Hagen AE. Measurement of Michaelis constants for cytochrome P450-mediated biotransformation reactions using a substrate depletion approach. Drug Metab Dispos. 2002;30:831–837. doi: 10.1124/dmd.30.7.831. [DOI] [PubMed] [Google Scholar]
- 18.Nath A, Atkins WM. A theoretical validation of the ssubstrate depletion approach to determining kinetic parameters. Drug Metab Dispos. 2006;34:1433–1435. doi: 10.1124/dmd.106.010777. [DOI] [PubMed] [Google Scholar]
- 19.Zhao Y, Truhlar DG. Density Functionals with Broad Applicability in Chemistry. Acc Chem Res. 2008;41:157–167. doi: 10.1021/ar700111a. [DOI] [PubMed] [Google Scholar]
- 20.Swart M, Guell M, Luis JM, Sola M. Spin-state-corrected Gaussian-type orbital basis sets. J Phys Chem A. 2010;114:7191–7197. doi: 10.1021/jp102712z. [DOI] [PubMed] [Google Scholar]
- 21.Bode BM, Gordon MS. Macmolplt: a graphical user interface for GAMESS. J Mol Graphics Modell. 1998;16:133–138. doi: 10.1016/s1093-3263(99)00002-9. [DOI] [PubMed] [Google Scholar]
- 22.Das A, Zhao J, Schatz GC, Sligar SG, Van Duyne RP. Screening of type I and II drug binding to human cytochrome P450-3A4 in nanodiscs by localized surface plasmon resonance spectroscopy. Anal Chem. 2009;81:3754–3759. doi: 10.1021/ac802612z. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Locuson CW, Hutzler JM, Tracy TS. Visible spectra of type II cytochrome P450- drug complexes: Evidence that “incomplete” heme coordination is common. Drug Metab Dispos. 2007;35:614–622. doi: 10.1124/dmd.106.012609. [DOI] [PubMed] [Google Scholar]
- 24.Schenkman JB. Nature of the type I and II spectral changes in liver microsomes. Biochemistry. 1970;9:2081–2091. doi: 10.1021/bi00812a009. [DOI] [PubMed] [Google Scholar]
- 25.Roberts AG, Sjogren SEA, Fomina N, Vu KT, Almutairi A, Halpert JR. NMR-derived models of amidopyrine and its metabolites in complexes with rabbit cytochrome P450 2B4 reveal a structural mechanism of sequential N-dealkylation. Biochemistry. 2011;50:2123–2134. doi: 10.1021/bi101797v. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Roberts AG, Yang J, Halpert JR, Nelson SD, Thummel KT, Atkins WM. The structural basis for homotropic and heterotropic cooperativity of midazolam metabolism by human cytochrome P450 3A4. Biochemistry. 2011;50:10804–10818. doi: 10.1021/bi200924t. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Davydov DR, Deprez E, Hoa GHB, Knyushko TV, Kuznetsova GP, Koen YM, Archakov AI. High-pressure-induced transitions in microsomal cytochrome P450 2B4 in solution: Evidence for conformational inhomogeneity in the oligomers. Arch Biochem Biophys. 1995;320:330–344. doi: 10.1016/0003-9861(95)90017-9. [DOI] [PubMed] [Google Scholar]
- 28.Fukada H, Takahashi K. Enthalpy and heat capacity changes for the proton dissociation of various buffer components in 0.1 M potassium chloride. Proteins: Struct Funct, Bioinf. 1998;33:159–166. [PubMed] [Google Scholar]
- 29.Helgeson HC, Kirkham DH. Theoretical prediction of the thermodynamic behavior of aqueous electrolytes at high pressures and temperatures; II, Debye-Huckel parameters for activity coefficients and relative partial molal properties. Am J Sci. 1974;274:1199–1261. [Google Scholar]
- 30.Robert NG, Nand K, Rebecca ML. Thermodynamic quantities for the ionization reactions of buffers. J Phys Chem Ref Data. 2002;31:231–370. [Google Scholar]
- 31.Clarke ECW, Glew DN. Evaluation of thermodynamic functions from equilibrium constants. Trans Faraday Soc. 1966:62. [Google Scholar]
- 32.Bowman MK. Pulsed Electron Paramagnetic Resonance. In: Brustalon M, Giamello E, editors. Electron Paramagnetic Resonance - A Practitioner’s Toolkit. John Wiley & Sons; Hoboken: 2009. pp. 159–194. [Google Scholar]
- 33.Dikanov SA, Bowman MK. Cross-peak lineshape of two-dimensional ESEEM spectra in disordered S = 1/2, I = 1/2 spin system. J Magn Reson, Ser A. 1995;116:125–128. [Google Scholar]
- 34.Dikanov SA, Tyryshkin AM, Bowman MK. Intensity of cross-peaks in HYSCORE spectra of S = 1/2, I = 1/2 spin systems. J Magn Reson. 2000;144:228–242. doi: 10.1006/jmre.2000.2055. [DOI] [PubMed] [Google Scholar]
- 35.Stoll S, Schweiger A. EasySpin, a comprehensive software package for spectral simulation and analysis in EPR. J Magn Reson. 2006;178:42–55. doi: 10.1016/j.jmr.2005.08.013. [DOI] [PubMed] [Google Scholar]
- 36.Hariharan PC, Pople JA. The influence of polarization functions on molecular orbital hydrogenation energies. Theor Chem Acc: Theory, Comput, Model (Theoretica Chimica Acta) 1973;28:213–222. [Google Scholar]
- 37.Catalan J, Elguero J. Advances in Heterocyclic Chemistry. Vol. 41. Academic Press; 1987. Basicity and acidity of azoles; pp. 187–274. [Google Scholar]
- 38.Gillam EMJ, Baba T, Kim BR, Ohmori S, Guengerich FP. Expression of modified human cytochrome P450 3A4 in escherichia coli and purification and reconstitution of the enzyme. Arch Biochem Biophys. 1993;305:123–131. doi: 10.1006/abbi.1993.1401. [DOI] [PubMed] [Google Scholar]
- 39.Peng CC, Cape JL, Rushmore T, Crouch GJ, Jones JP. Cytochrome P450 2C9 type II binding studies on quinoline-4-carboxamide analogues. J Med Chem. 2008;51:8000–8011. doi: 10.1021/jm8011257. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40.Lunazzi L, Parisi F, Macciantelli D. Conformational studies by dynamic nuclear magnetic resonance spectroscopy. Part 27 Kinetics and mechanism of annular tautomerism in isomeric triazoles. J Chem Soc, Perkin Trans. 1984;2:1025–1028. [Google Scholar]
- 41.Minkin V, EJ, Katritzky A, Garnovskii A, Denisko O. Advances in Heterocyclic Chemistry. Vol. 76. Academic Press; 2000. The Tautomerism of Heterocycles: Five-Membered Rings with Two or More Heteroatoms; pp. 159–214. [Google Scholar]
- 42.Roberts AG, Campbell AP, Atkins WM. The thermodynamic landscape of testosterone binding to cytochrome P450 3A4: Ligand binding and spin state equilibria. Biochemistry. 2005;44:1353–1366. doi: 10.1021/bi0481390. [DOI] [PubMed] [Google Scholar]
- 43.Williams PA, Cosme J, Vinkovic DM, Ward A, Angove HC, Day PJ, Vonrhein C, Tickle IJ, Jhoti H. Crystal structures of human cytochrome P450 3A4 bound to metyrapone and progesterone. Science. 2004;305:683–686. doi: 10.1126/science.1099736. [DOI] [PubMed] [Google Scholar]
- 44.Goldfarb D, Bernardo M, Thomann H, Kroneck PMH, Ullrich V. Study of water binding to low-spin Fe(III) in cytochrome P450 by pulsed ENDOR and four-pulse ESEEM spectroscopies. J Am Chem Soc. 1996;118:2686–2693. [Google Scholar]
- 45.Roberts AG, Cheesman MJ, Primak A, Bowman MK, Atkins WM, Rettie AE. Intramolecular heme ligation of the cytochrome P450 2C9 R108H mutant demonstrates pronounced conformational flexibility of the B-C loop region: Implications for substrate binding. Biochemistry. 2010;49:8700–8708. doi: 10.1021/bi100911q. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 46.Wang B, Sanchez RI, Franklin RB, Evans DC, Huskey SEW. The Involvment of CYP3A4 and CYP2C9 in the metabolism of 17-ethinylestradiol. Drug Metab Dispos. 2004;32:1209–1212. doi: 10.1124/dmd.104.000182. [DOI] [PubMed] [Google Scholar]
- 47.Lin H-l, Kent UM, Hollenberg PF. Mechanism-based inactivation of cytochrome P450 3A4 by 17alpha-ethynylestradiol: Evidence for heme destruction and covalent binding to protein. J Pharmacol Exp Ther. 2002;301:160–167. doi: 10.1124/jpet.301.1.160. [DOI] [PubMed] [Google Scholar]
- 48.Guengerich FP. Oxidation of 17-alpha-ethynylestradiol by human liver cytochrome P- 450. Mol Pharmacol. 1988;33:500–508. [PubMed] [Google Scholar]
- 49.Peter Guengerich F. Metabolism of 17-alpha-ethynylestradiol in humans. Life Sci. 1990;47:1981–1988. doi: 10.1016/0024-3205(90)90431-p. [DOI] [PubMed] [Google Scholar]
- 50.Krasinski A, Radic Z, Manetsch R, Raushel J, Taylor P, Sharpless KB, Kolb HC. In situ selection of lead compounds by Click chemistry: Target-guided optimization of acetylcholinesterase inhibitors. J Am Chem Soc. 2005;127:6686–6692. doi: 10.1021/ja043031t. [DOI] [PubMed] [Google Scholar]
- 51.Sono M, Dawson JH. Formation of low spin complexes of ferric cytochrome P-450-CAM with anionic ligands. Spin state and ligand affinity comparison to myoglobin. J Biol Chem. 1982;257:5496–5502. [PubMed] [Google Scholar]
- 52.Tomas F, Abboud JLM, Laynez J, Notario R, Santos L, Nilsson SO, Catalan J, Claramunt RM, Elguero J. Tautomerism and aromaticity in 1,2,3-triazoles: the case of benzotriazole. J Am Chem Soc. 1989;111:7348–7353. [Google Scholar]
- 53.Wofford DS, Forkey DM, Russell JG. Nitrogen-15 NMR spectroscopy: prototropic tautomerism of azoles. J Org Chem. 1982;47:5132–5137. [Google Scholar]
- 54.Muralidhara BK, Negi S, Chin CC, Braun W, Halpert JR. Conformational flexibility of mammalian cytochrome P4502B4 in binding imidazole inhibitors with different ring chemistry and side chains - Solution thermodynamics and molecular modeling. J Biol Chem. 2006;281:8051–8061. doi: 10.1074/jbc.M509696200. [DOI] [PubMed] [Google Scholar]
- 55.Seward HE, Roujeinikova A, McLean KJ, Munro AW, Leys D. Crystal structure of the mycobacterium tuberculosis P450 CYP121-Fluconazole complex reveals new azole drug-P450 binding mode. J Biol Chem. 2006;281:39437–39443. doi: 10.1074/jbc.M607665200. [DOI] [PubMed] [Google Scholar]
- 56.Dawson JH, Andersson LA, Sono M. Spectroscopic investigations of ferric cytochrome P-450-CAM ligand complexes. Identification of the ligand trans to cysteinate in the native enzyme. J Biol Chem. 1982;257:3606–3617. [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.